

Abstract
In the present paper, trapped ion mobility spectrometry (TIMS) and theoretical calculations have been used to study carotenoid geometrical motifs generated by photoisomerization from the all-trans geometry. Multiple geometric isomers of the carotenoids lutein and zeaxanthin were separated using TIMS (R > 110) for [M]+, [M + H]+, and [M – 18]+ molecular species. Comparison of observed cross sections with those obtained from molecular dynamics calculations showed that the number of cis double bonds and s-cis single bonds in the polyene chain determine the topology space of the carotenoid. The intensities of IMS signals are correlated with the relative stability of these geometric isomers.1,2 The most stable isomer is the all-trans geometry regardless of the ionization state ([M – 18]+, [M]+, and [M + H]+), and structural stability decreases with the increasing number of cis and/or s-cis bonds in the polyene chain.
Carotenoid pigments are found throughout nature where they serve a variety of roles.3 Synthesized de novo only in plants, algae, bacteria, and fungi, they are essential to photosynthesis, function as antioxidants, and are involved in the coloration in many animals as well as plants.4 For animals including humans, conversion of pro-vitamin-A carotenoids to retinal is essential to health, and the hydroxyl-carotenoids (lutein, zeaxanthin, and meso-zeaxanthin) function to protect the central retina from photo-oxidative damage (see more details in reviews5−7). The biological function of carotenoids is intrinsically influenced by their end-group type, functionalization, and geometric topology as controlled by steroisomerism of polyene double bonds and conformational flexibility.6,8,9 In nature, carotenoids are found in a variety of environments ranging from hydrophobic and low dielectric lipid membranes and structures to sites at the interface with the polar aqueous medium of the cell.10−12 In aqueous environments, carotenoids are associated with specific binding proteins13 or general purpose lipid transport proteins.14 The structural features that dominate the functionality of carotenoids also influence their conformational dynamics.6 Although the conformational flexibility of carotenoids with respect to the ionone end groups has been well described,9 the conformational role associated with s-cis orientations of “single” bonds of the polyene chain have not been widely considered when describing the range in topologies of carotenoids and their functionality. In a given physiological environment, carotenoid topological space is determined by a number of factors (e.g., electrostatic interactions with the solvent, principally H-bonding with oxygen functionalities in xanthophylls, entropic effects, and isomerization of the polyene chain). There is considerable interest in understanding how structural diversity between carotenoids as well as the dynamic structural interconversion of geometric isomers15−17 and conformers dictate the biological selectivity leading to specific uptake, transport, and binding of these molecules.6,18,19
During the past decade, the use of ion mobility spectrometry (IMS) combined with molecular dynamic simulations has proven to be a versatile technique for the analysis of intermediate and equilibrium structures of biomolecules enabling the correlation of ion-neutral, collision cross sections (CCS) with candidate structures.20,21 In particular, it has been shown that IMS permits the inspection of the topological dynamics with the ability to follow structural changes as a function of the solvent conditions, as well as bath gas type and temperature over millisecond time scales.22,23 Using this approach, it has been shown that isomerization in the peptide backbone can define the topological space accessible to a biomolecule.21 Pierson et al. have recently demonstrated the influence of cis-trans (E/Z) isomers on peptide conformation for proline-containing peptides using ion mobility spectrometry.23 Analogous to this effect in peptides, here we expect that cis-trans geometrical isomerization of the carotenoid polyene chain will significantly influence the topology of these biological structures. In addition, the polyene chain of the carotenoids can exist in a large number of conformers originating from the s-cis/s-trans geometry associated with the single bonds of the conjugated system.24 The recent advent of trapped ion mobility spectrometry (TIMS)25 enables the study of small mobility ranges with high mobility resolution and is expected to permit a detailed characterization of the carotenoid geometrical isomers and conformers.
In this study, we present evidence that carotenoid topology differences of the cis-trans geometrical isomers and s-cis conformers are directly observable in two carotenoid systems: lutein and zeaxanthin. Despite the significant biological activity of carotenoids, the role played by their structural topologies associated with the cis-trans isomerization and s-cis conformer interconversion with the polyene chain remains a topic of investigation.18 Our experimental observations and theoretical analysis demonstrate that these cis-trans associated motifs of the polyene chain are the main factor governing the carotenoid structural topology, and their abundance parallels the relative stability of carotenoid geometric isomers.2,16
Experimental Section
Carotenoid Samples
Carotenoid samples were extracted from marigold flowers,26 a common commercial source of lutein and zeaxanthin. Lutein- and zeaxanthin-containing extracts were purified by preparative reversed-phase HPLC (Phenomenox ODS Ultracarb 3 μm, 250 mm × 4.6 mm column) with acetonitrile/methanol/TEA (85%/15%/0.1% v/v) as a mobile phase. Final purity of the individual all-trans carotenoids was confirmed by UV–vis detection (300–500 nm)7 and by mass spectrometry.
Structural Separation by TIMS-MS
Details regarding TIMS operation and differences from traditional IMS can be found elsewhere.25 TIMS mobility separation utilizes an electric field to hold ions stationary against a moving gas, so that the drift force is compensated by the electric field. This concept follows the idea of a parallel flow ion mobility analyzer,27 with the main difference being that ions are also confined radially using a quadrupolar field to guarantee higher ion transmission and sensitivity. The separation in a TIMS device can be described by the center of the mass frame using the same principles as in a conventional IMS drift tube.28,29 Because mobility separation is related to the number of ion-neutral collisions (or drift time in traditional drift tube cells), the mobility separation in a TIMS device depends on the bath gas drift velocity, ion confinement, and ion elution parameters. The mobility, K, of an ion in a TIMS cell is described by:
| 1 |
where vg, E, Velution, and Vbase are the velocity of the gas, applied electric field, elution voltage, and base voltage, respectively. The constant A, which accounts for the velocity of gas, can be determined using calibration standards of known mobilities. In TIMS operation, multiple geometric isomers/conformers are trapped simultaneously at different E values resulting from a voltage gradient applied across the IMS cell. After thermalization, geometrical isomers/conformers are eluted by decreasing the electric field in stepwise decrements (referred to as the “ramp”). Each isomer/conformer eluting from the TIMS cell can be described by a characteristic voltage gradient (i.e., Velution – Vbase). Eluted ions are then mass analyzed and detected by a maXis impact Q-ToF mass spectrometer (Bruker, Billerica, MA). The elution voltage, Velution, can be calculated from the elution time:
| 2a |
and
| 2b |
where V0 is the initial potential at the entrance to the TIMS analyzer, r is the rate at which the potential is ramped, Telute is the time at which the ion elutes, Tramp is the total ramp time, Ttotal is the total time for a single TIMS experiment, Ttrap is the time before the mobility analysis (i.e., to inject ions into the TIMS trap), and TOF is the time between elution of the ion and detection of the ion at the TOF detector.
The TIMS funnel was controlled using in-house software, written in National Instruments LabVIEW, and synchronized with the maXis Impact Q-ToF acquisition program (more details in ref (25)). Separation was performed using nitrogen as a bath gas at ≈300 K, and the gas flow velocity was controlled by the pressure difference between the front (P1) and back (P2) of the TIMS analyzer. P1 and P2 values were set to 2.6 and 1.0 mbar for all experiments. The same RF (880 kHz and 200–350 Vpp) was applied to all electrodes including the entrance funnel, the mobility separating section, and the exit funnel. An atmospheric pressure photoionization source (APPI, Apolo II Bruker Daltonics, Inc., MA) using a Kr lamp with main emission bands at 10.0 and 10.6 eV was used for all the analyses.
A Tuning Mix mass spectrometry standard (Tunemix, G2421A, Agilent Technologies, Santa Clara, CA) was used as a mobility calibration standard. Details on the Tunemix structures (e.g., m/z = 322 K0 = 1.376 cm2 V–1 s–1, m/z = 622 K0 = 1.013 cm2 V–1 s–1, and m/z = 922 K0 = 0.835 cm2 V–1 s–1) can be found in ref (30). Carotenoid samples (lutein and zeaxanthin) were reconstituted in a 70:30 acetonitrile/methanol solution to a final concentration of 1–10 nM. Toluene (8.8 eV IP), acetone (9.7 eV IP), or anisole (8.2 eV IP) were used as APPI additives at a concentration of 10% (v/v) to enhance ionization31 and to study the effect of solvent conditions on ionization patterns and relative proportions of geometrical isomers/conformers formed during the photoionization process. For simplicity, lutein samples were only analyzed with added anisole APPI. Mobility values (K) were correlated with CCS (Ω) using the equation:
| 3 |
where z is the charge of the ion, kB is the Boltzmann constant, N* is the number density, and mI and mb refer to the masses of the ion and bath gas, respectively.28
Instrumental parameters were optimized to achieve the highest IMS resolution for the carotenoid molecular ions. A peak width (i.e., fwhm of the mobility peak) that corresponds to a single isomer was measured using the sphere-like tune mix mobility standard series. In addition, to ensure that the IMS peak width was not influenced by the number of ions in the TIMS cell (i.e., columbic effects compromising ion trapping), a dilution series (1:10–1:103) of the tune mix was used to determine the mobility peak width as a function of the concentration in all APPI solvent conditions. No significant variation in the IMS peak width was observed beyond a 1:100 fold dilution. The 622 m/z (K0 = 1.013 cm2 V–1 s–1, CCS = 202 Å2) component yielded a 1.74 Å2 peak width and was used as a reference for the peaks observed for the carotenoid isomers. Under these experimental conditions, a mobility resolution of over 110 was obtained in the TIMS analyzer (more details in the Supporting Information).
Theoretical Calculations
A pool of candidate structures with varying numbers of cis bond geometries of the polyene chain (including s-cis and s-trans conformations) for the lutein and zeaxanthin molecular and fragment ions were generated and optimized at the B3LYP/G-311G level.24 Energies of formation were calculated for all the obtained structures after a series of simulated annealing and geometry optimization cycles using AMBER03 force field in YASARA32 (analogous to method A described in ref (21)). Candidate structures were then submitted to MOBCAL33 to determine theoretical K0 and CCS values (assuming helium as a bath gas) using the projection approximation (PA), the elastic hard sphere scattering (EHSS) and the trajectory (TM) methods (values are provided in the Supporting Information). It is known that for small molecules, deviations between these three methods due to the structural and elemental composition can be observed.33 In order to evaluate theoretical CCS in nitrogen, an empirical formula was obtained by correlating theoretical (in helium using the EHSS) and experimental (in nitrogen using TIMS) CCS of polyaromatic hydrocarbons (PAH) standards (12 compounds): CCS(N2) = 1.1606 × CCS(He) + 41.547 [Å2], (R2 = 0.9912). The use of PAH standards is justified by the composition and elemental similarity to carotenoids and by their small structural flexibility.
Results and Discussion
The TIMS-MS analysis demonstrates that ion abundance and ionization state vary with the APPI solvent condition. In particular, consistent with the report of Rivera et al.31 the observation of a carotenoid molecular ion, protonated ion, or fragment ion vary with the solvent conditions during the APPI process and is specific to each carotenoid (Figure 1). For zeaxanthin, [M]+ molecular ions were observed when 10% anisole and toluene were used as additives to the 70:30 acetonitrile/methanol solution, although [M + H]+ molecular ions were observed when 10% acetone was utilized. For lutein, a [M – 18]+ fragment ion predominates with a 70:30 acetonitrile/methanol solution containing 10% anisole. The predominance of the [M – 18]+ ion over that of [M]+ originates from the facile loss of water due to the allylic 3′-hydroxyl leading to the stabilized [M – 18]+ ion, a process unavailable to the secondary 3- and 3′-hydroxyls of zeaxanthin.12,34
Figure 1.

Typical MS profiles for zeaxanthin and lutein for APPI solvents conditions (acetone, anisole, and toluene in 70:30 acetonitrile/methanol). Notice the difference in molecular ionization as a function of the APPI solvent and the carotenoid structure.
Multiple cis geometric isomers and presumably also s-cis conformers of zeaxanthin and lutein are observed in the IMS analysis (Figure 2). Each peak assigned in Figure 2 (denoted by a Gaussian profile) has a broader IMS peak width than expected when considering a single analyte (e.g., from m/z = 622 of the tune mix series). The broadening of the peaks may be explained by the following: (i) the existence of multiple isomers with nearly identical CCSs that cannot be resolved or (ii) isomers that interconvert under the experimental conditions (e.g., effective temperature, experiment time scale, etc.). However, no variations in the IMS profiles were observed over different trapping times (e.g., 100–500 ms), which suggests that differences in the observed IMS profiles are attributable to a distribution of geometric isomers/conformers that exist as solvated carotenoid ions formed during the photoionization process (e.g., Figure 2a,c vs Figure 2b). That is, ion-neutral collisions during the TIMS separation with the residual gas does not seem to contribute to the IMS distribution, but solvent conditions during APPI do.
Figure 2.
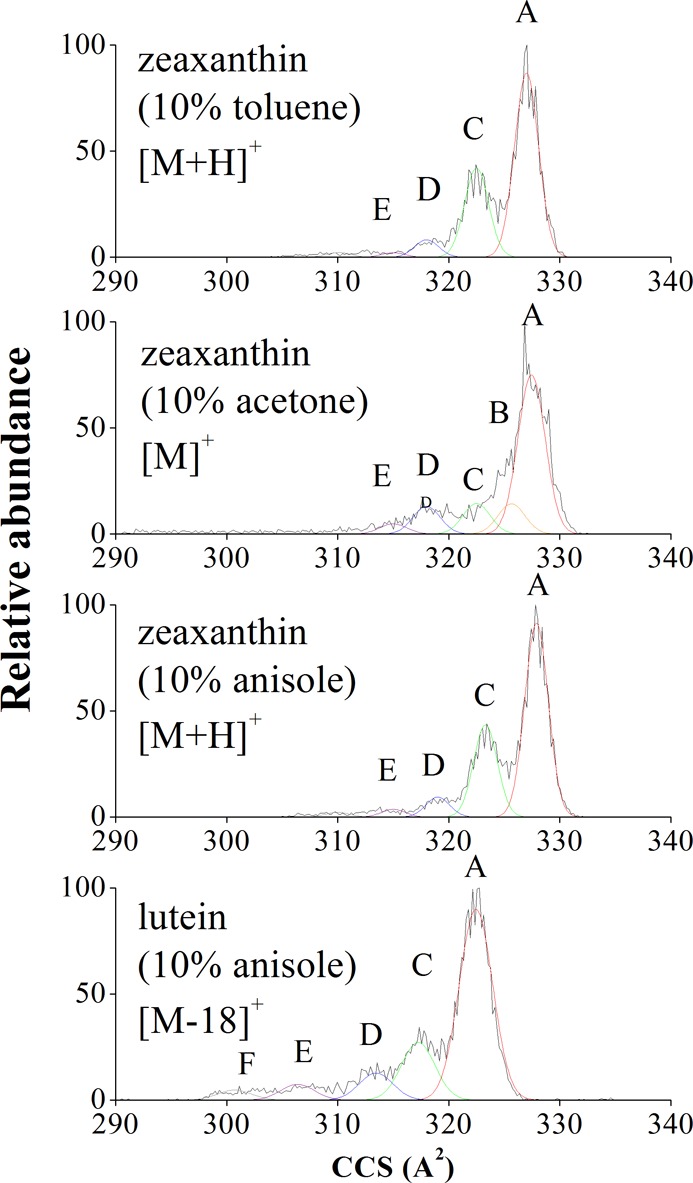
Typical IMS spectra of zeaxanthin and lutein with acetonitrile/methanol 70:30 APPI solvent conditions and various additives (from top to bottom): [M + H]+ zeaxanthin with acetone, [M]+ zeaxanthin with anisole, [M]+ zeaxanthin with toluene, and [M – 18]+ lutein with anisole. IMS peaks A–F have been designated in the spectra and are depicted using Gaussian profiles.
Candidate structures were proposed for the most abundant IMS peaks (see Table 1 and Figure 3). A general trend is that the most abundant IMS peak, A, corresponds to the all-trans geometric isomer for both carotenoids (the single isomer present in the starting solutions). Moreover, the predominance of the all-trans geometric isomer is also anticipated due to its higher stability relative to other isomers for both carotenoid ions (lutein and zeaxanthin).2,35 Other geometric isomers/conformers with increasing numbers of cis double bonds (and/or s-cis bonds) are assigned to the IMS peaks B–D and were formed during the APPI process. IMS peak B is assigned to the 13-cis and 15-cis geometric isomers which were calculated to have the same CCS values; however, the 15-cis isomer is energetically more stable, its low kinetic barrier conversion back to the all-trans isomer is facile, and its abundance in thermally equilibrated samples is small.17,18,36 Similarly, the IMS peaks C and D have been assigned as 9-cis and 9,13′-di-cis (or 13,9′-di-cis) candidate structures, respectively. Overlapping conformers having s-cis and di-s-cis geometries within the polyene chain are also assigned to these IMS bands and can contribute to the broadening. Isomer bands E and F are the result of the presence of multiple s-cis bonds and cis double bonds in the polyene chain (for additional information on potential candidate structures see the Supporting Information). The lower stability of the geometric isomers E and F translates to the low abundance observed in Figure 2. A similar trend was observed for [M – 18]+ lutein isomers. The IMS peak A′ can be correlated with the all-trans candidate structure, the IMS peaks C′ and D′ can be correlated with the 9-cis and 9,13′-di-cis (or 13, 9′-cis) candidate structures, respectively. Isomer bands E′ and F′ are the result of the presence of multiple s-cis and cis double bonds in the polyene chain (for additional information on potential candidate structures, see the Supporting Information). Although several potential candidate structures can be assigned to the isomer bands depicted in Figure 2, assignment has been made on the basis of structural stability.
Table 1. Candidate Structures Proposed for Zeaxanthin [M]+ and Lutein [M – 18]+ Carotenoidsa.
| theoretical
in N2 |
|||||
|---|---|---|---|---|---|
| carotenoid isomer | K0 (cm2/(V s)) | CCS (Å2) | ΔE (kJ mol–1) | experimental IMS peak (Å2) | |
| zeaxanthin [M]+ and [M+H]+ | all-trans | 0.6608 | 327 | 327 - A | |
| 15-cis | 0.6656 | 325 | 19 | 325 - B | |
| 13-cis | 0.6655 | 325 | 28 | 325 - B | |
| 9-cis | 0.668 | 324 | 15 | 323 - C | |
| 9,13′-di-cis | 0.6753 | 320 | 22 | 318 - D | |
| lutein [M-18]+ | all-trans | 0.6709 | 323 | 322 - A | |
| 9-cis | 0.6832 | 317 | 5 | 317 - C | |
| 9,13′-di-cis | 0.6883 | 315 | 3 | 315 - D | |
More details in the supplemental material. Energies were calculated relative to the most abundant isomer. Experimental CCS error is <5 Å2.
Figure 3.

Candidate structures of zeaxanthin [M]+ with varying number of cis-trans isomerization in the polyene chain.
Overall, the stability of the cis-trans geometrical isomers/s-cis conformational structures parallels the relative abundances of the IMS spectra. Slight variations are to be expected, because the geometrical isomers were generated by the photoionization process from the all-trans form of the lutein and zeaxanthin isomers (e.g., incomplete photoisomerization). Solvent composition may be expected to influence the abundances of ions generated during photoionization; nevertheless, isomers of higher stability will be energetically more favorable. For example, IMS peak B in the analysis of zeaxanthin varied in abundance under different solvent conditions. A lower relative abundance of the 13-cis and 15-cis isomers (isomer band B) was observed when using solvent containing 10% (v/v) anisole or toluene as compared to a solvent system lacking this aromatic component. This example shows the influence of the initial APPI ionization state on the distribution of zeaxanthin geometrical isomers/conformers and consequently on the IMS profile. That is, experimental data suggest that the distribution of carotenoid geometrical isomers/conformers observed in these TIMS experiments depend on the presence of the aromatic hydrocarbon additive. Relative geometrical isomer/conformer abundances may be modulated by the probability of ionization of a given isomer during the APPI process31 (e.g., [M]+ or [M + H]+), and this could define the relative isomer/conformer abundances; although the process of ionization was not addressed directly in the present study, the differences of the theoretical CCS values of [M + H]+ and [M]+ are much smaller than the differences in CCS attributable to the number of cis bonds in the polyene chain of the proposed candidate structures. Similar IMS trends between [M + H]+ zeaxanthin and [M – 18]+ lutein suggest that the loss of hydroxyl group does not influence the distribution of observed species that are attributable entirely to the carotenoid backbone geometry. A comparison with conclusions obtained from NMR solution studies of zeaxanthin and lutein geometrical isomers is in good agreement with the most abundant candidate structures proposed in this study.37
Carotenoids are often bound within proteins where they serve a variety of functions.13,38 The versatility that results from the ability of the polyene chain to exist in a variety of topologically distinct forms separated by low energetic barriers provides these molecules the ability to adjust topologically to protein constraints and also presents a subtle means to influence the electronic energy levels of the carotenoid.10,38,39 Within the crystal structure of apocarotenoid-15,15-oxygenase, the substrate carotenoid is found to be bound in a cis-isomeric geometry. Upon binding, three adjacent double bonds undergo isomerization from a trans-trans-trans to a cis-trans-cis geometry.38 The cleavage enzyme responsible for the formation of 9-cis -violoxanthin and 9′-cis-neoxanthin is another example of a protein in which the carotenoid binds to a protein.40 Numerous other examples of carotenoid binding proteins include light harvesting protein (to which four carotenoids 2 luteins, zeaxanthin, and 9-cis-neoxanthin are bound),41 α-crustacyanin from lobster (which binds astaxanthin),42 human steroidal acute regulatory domain protein (STAR D 3, which binds lutein),43 and the human glutathione transferase Pi I (which binds zeaxanthin).44 In addition, the inspection of X-ray structures of hydroxyl carotenoids within three proteins (IM98,45 3PL9,46 and 1RWT47) shows that the hydrogen bonds link the terminal ring of the carotenoid molecule to the protein binding site. That is, the topological space of the carotenoids in biological processes can adopt multiple forms, and evidence suggests that they play a dominant role in the biological function of carotenoid binding proteins.
Conclusions
In the current study, TIMS-MS combined with theoretical calculations demonstrates that carotenoid geometric isomers/conformers of lutein and zeaxanthin are separately observable. For the first time, ion-neutral CCSs of zeaxanthin and lutein geometric isomers/conformers are reported and compared with theoretical calculations of candidate structures. The abundance of the individual carotenoid isomers/conformers were observed to be consistent with their relative stability: the larger the intensity in the IMS profile, the more stable the structure. As anticipated, larger numbers of cis and/or s-cis bonds generally correspond to smaller CCS values. This study highlights the potential of TIMS-MS combined with theoretical calculations to describe the topological space for low molecular weight biological molecules.
Acknowledgments
This work was supported by the National Institute of Health (Grant No. R00GM106414) and a FFL Bruker Daltonics, Inc. fellowship. The authors wish to acknowledge Dr. Desmond Kaplan from Bruker Daltonics, Inc. for the development of TIMS acquisition software. The authors would like to thank Carlos Torres and Industrial Organica S.A. de C.V. for the generous gift of carotenoid samples used in this study. J.L. and V.M. would like to thank Four Leaf Japan for partial support.
Supporting Information Available
Additional information as noted in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Hernandez-Marin E.; Galano A.; Martinez A. J. Phys. Chem. B 2013, 117, 4050–4061. [DOI] [PubMed] [Google Scholar]; Weedon B. C. L. In Carotenoids; Isler O., Ed.; Birkauser Verlag: Basel, 1971; pp 267–323. [Google Scholar]
- Zechmeister L.Cis-Trans Isomeric Carotenoids, Vitamin A, and Arylpolyenes; Verlag: Vienna, 1962. [Google Scholar]
- Britton G.; Liaaen-Jensen S.; Pfander H.. Carotenoids, Vol. 3: Biosynthesis and Metabolism Birkhauser-Verlag: Basel, 2004; p 670. [Google Scholar]
- Isler O.Carotenoids; Birkhauser Verlag: Basel, 1971. [Google Scholar]
- Rao A. V.; Rao L. G. Pharmacol. Res. 2007, 55, 207–16. [DOI] [PubMed] [Google Scholar]; Landrum J. T.; Nolan J.. CRC Press: Boca Raton, 2013; p 272. [Google Scholar]; Landrum J. T.CRC Press: Boca Raton, 2010; p 544. [Google Scholar]
- Landrum J. T.; Chatfield D. C.; Mebel A. M.; Alvarez-Calderon F.; Fernandez M. V. Arch. Biochem. Biophys. 2010, 493, 169–174. [DOI] [PubMed] [Google Scholar]
- Landrum J. T.; Bone R. A. Arch. Biochem. Biophys. 2001, 385, 28–40. [DOI] [PubMed] [Google Scholar]
- Bone R. A.; Landrum J. T.; Hime G. W.; Cains A.; Zamor J. Invest. Ophthalmol. Vis. Sci. 1993, 34, 2033–2040. [PubMed] [Google Scholar]
- Cerezo J.; Zuniga J.; Bastida A.; Requena A.; Ceron-Carrasco J. P. J. Phys. Chem. 2013, 15, 6527–6538. [DOI] [PubMed] [Google Scholar]
- Amunts A.; Toporik H.; Borovikova A.; Nelson N. J. Biol. Chem. 2010, 285, 3478–3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiedt K. In Biosynthesis and Metabolism, Britton G.; Liaaen-Jensen S.; Pfander H., Eds.; Birkhauser Verlag: Basel, 1998; pp 285–358. [Google Scholar]; Weedon B. C. L. In Carotenoids; Isler O., Ed.; Birkhauser-Verlag: Basel, 1971; pp 29–59. [Google Scholar]; Davis B. H. Pure Appl. Chem. 1991, 63, 131–140. [Google Scholar]; Bone R. A.; Landrum J. T.; Tarsis S. L. Vision Res. 1985, 25, 1531–1535. [DOI] [PubMed] [Google Scholar]; Kirschfeld K. Proc. R. Soc. London, Ser. B 1982, 216, 71–85. [DOI] [PubMed] [Google Scholar]
- Bone R. A.; Landrum J. T.; Hime G. W.; Cains A.; Zamor J. Invest. Ophthalmol. Vis. Sci. 1993, 34, 2033–2040. [PubMed] [Google Scholar]
- Pilbrow J.; Garama D.; Carne A. Acta Biochim. Pol. 2012, 59, 163–165. [PubMed] [Google Scholar]
- During A.; Doraiswamy S.; Harrison E. H. J. Lipid Res. 2008, 49, 1715–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]; Kiefer C.; Sumser E.; Wernet M. F.; Von Lintig J. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 10581–10586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emenhiser C.; Sander L. C.; Schwartz S. J. J. Chromatogr. 1995, 707, 205–216. [Google Scholar]
- Emenhiser C.; Englert G.; Sander L. C.; Ludwig B.; Schwartz S. J. J. Chromatogr. A 1996, 719, 333–343. [DOI] [PubMed] [Google Scholar]
- Stahl W.; Schwarz W.; Sundquist A. R.; Sies H. Arch. Biochem. Biophys. 1992, 294, 173–177. [DOI] [PubMed] [Google Scholar]
- von Doering W.; Sotiriou-Leventis C.; Roth W. R. J. Am. Chem. Soc. 1995, 117, 2747–2757. [Google Scholar]
- Bhosale P.; Li B.; Sharifzadeh M.; Gellermann W.; Frederick J. M.; Tsuchida K.; Bernstein P. S. Biochemistry 2009, 48, 4798–4807. [DOI] [PubMed] [Google Scholar]
- Fernandez-Lima F. A.; Blase R. C.; Russell D. H. Int. J. Mass Spectrom. 2010, 298, 111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]; Valentine S. J.; Counterman A. E.; Clemmer D. E. J. Am. Soc. Mass Spectrom. 1999, 10, 1188–1211. [DOI] [PubMed] [Google Scholar]; Tao L.; McLean J. R.; McLean J. A.; Russell D. H. J. Am. Soc. Mass Spectrom. 2007, 18, 1232–1238. [DOI] [PubMed] [Google Scholar]; Bush M. F.; Hall Z.; Giles K.; Hoyes J.; Robinson C. V.; Ruotolo B. T. Anal. Chem. 2010, 82, 9557–9565. [DOI] [PubMed] [Google Scholar]; Jarrold M. F. Annu. Rev. Phys. Chem. 2000, 51, 179–207. [DOI] [PubMed] [Google Scholar]; Murray M. M.; Krone M. G.; Bernstein S. L.; Baumketner A.; Condron M. M.; Lazo N. D.; Teplow D. B.; Wyttenbach T.; Shea J.-E.; Bowers M. T. J. Phys. Chem. B 2009, 113, 6041–6046. [DOI] [PMC free article] [PubMed] [Google Scholar]; Baumketner A.; Bernstein S. L.; Wyttenbach T.; Bitan G.; Teplow D. B.; Bowers M. T.; Shea J.-E. Protein Sci. 2006, 15, 420–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Lima F. A.; Wei H.; Gao Y. Q.; Russell D. H. J. Phys. Chem. A 2009, 113, 8221–8234. [DOI] [PubMed] [Google Scholar]
- Kanu A. B.; Dwivedi P.; Tam M.; Matz L.; Hill H. H. J. Mass Spectrom. 2008, 43, 1–22. [DOI] [PubMed] [Google Scholar]; Kanu A. B.; Hill H. H. Jr Talanta 2007, 73, 692–699. [DOI] [PubMed] [Google Scholar]; Eiceman G. A.; Krylov E. V.; Krylova N. S.; Nazarov E. G.; Miller R. A. Anal. Chem. 2004, 76, 4937–4944. [DOI] [PubMed] [Google Scholar]; Pierson N. A.; Chen L.; Valentine S. J.; Russell D. H.; Clemmer D. E. J. Am. Chem. Soc. 2011, 133, 13810–13813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierson N. A.; Chen L.; Russell D. H.; Clemmer D. E. J. Am. Chem. Soc. 2013, 135, 3186–3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez V.Measurement of Carotenoid Levels in Human Serum and a Catalog of the Lutein Conformation Populations from Semi-empirical Calculations. Master’s Thesis. Florida International University, Miami, October, 2011. [Google Scholar]
- Fernandez-Lima F. A.; Kaplan D. A.; Park M. A. Rev. Sci. Instrum. 2011, 82, 126106. [DOI] [PMC free article] [PubMed] [Google Scholar]; Fernandez-Lima F.; Kaplan D.; Suetering J.; Park M. Int. J. Ion Mobility Spectrom. 2011, 14, 93–98. [Google Scholar]
- Barzana E.; Rubio D.; Santamaria R. I.; Garcia-Correa O.; Garcia F.; Ridaura Sanz V. E.; Lopez-Munguia A. J. Agric. Food Chem. 2002, 50, 4491–4496. [DOI] [PubMed] [Google Scholar]
- Zeleny J. Philos. Mag. 1898, 46, 120–154. [Google Scholar]
- McDaniel E. W.; Mason E. A.. Mobility and Diffusion of Ions in Gases; John Wiley and Sons, Inc., New York, NY, 1973; p 381. [Google Scholar]
- Ellis H. W.; Thackston M. G.; McDaniel E. W.; Mason E. A. At. Data Nucl. Data Tables 1984, 31, 113–151. [Google Scholar]
- Flanagan L. A.Mass spectrometry calibration using homogeneously substituted fluorinated triazatriphosphorines. U.S. Patent 5,872,357. Hewlett-Packard Company: Palo Alto, CA, February 16, 1999; p 19. [Google Scholar]
- Rivera S.; Vilaró F.; Canela R. Anal. Bioanal. Chem. 2011, 400, 1339–1346. [DOI] [PubMed] [Google Scholar]
- YASARA Biosciences: Molecular Graphics, Modeling, and Simulation Program (2013).
- Mesleh M. F.; Hunter J. M.; Shvartsburg A. A.; Schatz G. C.; Jarrold M. F. J. Phys. Chem. 1996, 100, 16082–16086. [Google Scholar]; Shvartsburg A. A.; Jarrold M. F. Chem. Phys. Lett. 1996, 261, 86–91. [Google Scholar]
- Dachtler M.; Glaser T.; Kohler K.; Albert K. Anal. Chem. 2001, 73, 667–674. [DOI] [PubMed] [Google Scholar]
- Weedon B. C. L. Rev. Pure Appl. Chem. 1970, 20, 51. [Google Scholar]
- Stahl W.; Sundquist A. R.; Hanusch M.; Schwarz W.; Sies H. Clin. Chem. 1993, 39, 810–814. [PubMed] [Google Scholar]
- Aman R.; Biehl J.; Carle R.; Conrad J.; Beifuss U.; Schieber A. Food Chem. 2005, 92, 753–763. [Google Scholar]
- Kloer D. P.; Ruch S.; Al-Babili S.; Beyer P.; Schulz G. E. Science 2005, 308, 267–269. [DOI] [PubMed] [Google Scholar]
- Kerfeld C. A.; Alexandre M.; Kirilovsky D. In Carotenoids: Physical, Chemical, and Biological Functions and Properties; Landrum J. T., Ed.; CRC Press: Boca Raton, 2010; pp 3–17. [Google Scholar]; Cogdell R. J.; Isaacs N. W.; Freer A. A.; Arrelano J.; Howard T. D.; Paziz M. Z.; Hawthornwaite-Lawless A. M.; Prince S. Prog. Biophys. Mol. Biol. 1997, 68, 1–27. [DOI] [PubMed] [Google Scholar]
- Schwartz S. H.; Tan B. C.; Gage D. A.; Zeevaart J. A.; McCarty D. R. Science 1997, 276, 1872–4. [DOI] [PubMed] [Google Scholar]
- Domonkos I.; Kis M.; Gombos Z.; Ughy B. Prog. Lipid Res. 2013, 52, 539–561. [DOI] [PubMed] [Google Scholar]
- Weesie R. J.; Jansen F. J.; Merlin J. C.; Lugtenburg J.; Britton G.; de Groot H. J. Biochemistry 1997, 36, 7288–7296. [DOI] [PubMed] [Google Scholar]
- Li B.; Vachali P.; Frederick J. M.; Bernstein P. S. Biochemistry 2011, 50, 2541–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhosale P.; Larson A. J.; Frederick J. M.; Southwick K.; Thulin C. D.; Bernstein P. S. J. Biol. Chem. 2004, 279, 49447–49454. [DOI] [PubMed] [Google Scholar]
- Kerfeld C. A.; Sawaya M. R.; Brahmandam V.; Cascio D.; Ki Ho K.; Trevithick-Sutton C. C.; Krogmann D. W.; Yeates T. O. Structure 2003, 11, 55–65. [DOI] [PubMed] [Google Scholar]
- Pan X.; Li M.; Wan T.; Wang L.; Jia C.; Hou Z.; Zhao X.; Zhang J.; Chang W. Nat. Struct. Mol. Biol. 2011, 18, 309–315. [DOI] [PubMed] [Google Scholar]
- Liu Z.; Yan H.; Wang K.; Kuang T.; Zhang J.; Gui L.; An X.; Chang W. Nature 2004, 428, 287–292. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.