

Abstract
Cardiac glycosides have been used for the treatment of heart failure because of their capabilities of inhibiting Na+/K+ ATPase (NKA), which raises [Na+]i and attenuates Ca2+ extrusion via the Na+/Ca2+ exchanger (NCX), causing [Ca2+]i elevation. The resulting [Ca2+]i accumulation further enhances Ca2+-induced Ca2+ release, generating the positive inotropic effect. However, cardiac glycosides have some toxic and side effects such as arrhythmogenesis, confining their extensive clinical applications. The mechanisms underlying the proarrhythmic effect of glycosides are not fully understood. Here we investigated the mechanisms by which glycosides could cause cardiac arrhythmias via impairing mitochondrial energetics using an integrative computational cardiomyocyte model. In the simulations, the effect of glycosides was mimicked by blocking NKA activity. Results showed that inhibiting NKA not only impaired mitochondrial Ca2+ retention (thus suppressed reactive oxygen species (ROS) scavenging) but also enhanced oxidative phosphorylation (thus increased ROS production) during the transition of increasing workload, causing oxidative stress. Moreover, concurrent blocking of mitochondrial Na+/Ca2+ exchanger, but not enhancing of Ca2+ uniporter, alleviated the adverse effects of NKA inhibition. Intriguingly, NKA inhibition elicited Ca2+ transient and action potential alternans under more stressed conditions such as severe ATP depletion, augmenting its proarrhythmic effect. This computational study provides new insights into the mechanisms underlying cardiac glycoside-induced arrhythmogenesis. The findings suggest that targeting both ion handling and mitochondria could be a very promising strategy to develop new glycoside-based therapies in the treatment of heart failure.
Introduction
Glycosides are capable of inhibiting sarcolemmal Na+/K+-ATPase (NKA), which blocks the extrusion of Na+ and results in cytosolic Na+ accumulation [1], [2]. Elevation of Na+ consequently suppresses Na+/Ca2+ exchanger (NCX), the primary Ca2+ efflux pathway in cardiac myocytes, leading to Ca2+ overload and increased sarcoplasmic reticulum (SR) Ca2+ uptake. The resultant greater Ca2+-induced Ca2+ release (CICR) allows for more powerful contractions by cross-bridge cycling in response to stimulations [3]. Because of their positive inotropic effects, cardiac glycosides, such as digoxin, have been widely used in the treatment of congestive heart failure [2], [4]. However, recently the use of glycoside treatment in HF patients has been largely supplanted by other drugs (e.g., angiotensin-converting enzyme (ACE) inhibitors, β-blockers and aldosterone antagonists) and cardiac resynchronization therapies [5], [6]. The diminished use of glycosides in the clinic was partially due to their well-known side effects such as cardiac arrhythmias, gastrointestinal symptoms, and central nervous system abnormalities [2], [5], [7], [8]. Whereas the proarrhythmic effect of glycosides largely confines their clinic applications, the detailed underlying molecular mechanisms are not completely understood.
A classical hypothesis on the proarrhythmic effect of glycosides is that when SR Ca2+ stores become too high, some Ca2+ might be released spontaneously through ryanodine receptors (RyRs), causing early or delayed afterdepolarizations or triggered activity [1], [8], [9]. However, emerging evidence suggests that SR Ca2+ overload is not the only root cause of glycoside-induced cardiac arrhythmias [5]. A series of work by Dr. O’Rourke’s group has suggested that glycosides (e.g. ouabain) may impair mitochondrial energy metabolism and raise oxidative stress in guinea pig cardiomyocytes, especially those at increased workload [1], [10], [11]. Particularly, their studies showed that ouabain-induced cytosolic Na+ accumulation caused mitochondrial Ca2+ deficiency, NADH imbalance, and increased reactive oxygen species (ROS) accumulation. Since many ion channels/exchangers underlying the action potential (AP) or involved in Ca2+ handling (e.g., the fast Na+ channels, RyRs, and SR Ca2+ ATPase) are redox and/or ATP sensitive, ouabain-induced mitochondrial dysfunction can disturb Ca2+ cycling and elicit erratic action potentials. Indeed, Liu et al. have demonstrated that ouabain caused mitochondrial oxidative stress and DADs in guinea pig ventricular myocytes [1]. They also showed that concurrent application of CGP-37157 (a mitochondrial Na+/Ca2+ exchanger, a.k.a. mNCE, inhibitor) with ouabain retained mitochondrial Ca2+ and NADH levels, suppressed ROS production and prevented ouabain-induced DADs [1], [10]. These findings highlight the important roles of mitochondrial Ca2+ and NADH homeostasis in glycoside-induced oxidative stress and cardiac arrhythmogenesis.
In cardiac cells, mitochondrial Ca2+ is regulated not only by mNCE but also by mitochondrial Ca2+ uniporters (MCU), the primary pathway of mitochondrial Ca2+ uptake. While the molecular identity of MCU has been revealed recently [12], [13], the mechanisms governing MCU Ca2+ uptake are still incompletely understood [14]–[16]. Consequently, the role of MCU in regulating Ca2+ cycling and excitation-contraction (E-C) coupling remains controversial. The major restriction of mitochondrial Ca2+ uptake is attributed to the low affinity of MCU to Ca2+. Specifically, the Ca2+ concentration for half-maximal mitochondrial Ca2+ uptake via MCU was reported as ∼10–20 μM in isolated mitochondria [17], [18], which substantially exceeds the cytosolic Ca2+ concentration during E-C coupling (0.5–1.5 μM). Therefore, it has been argued that mitochondria may have a minor role in regulating cellular Ca2+ cycling and E-C coupling [14], [19], [20]. On the contrary, other evidence suggests that mitochondria are functionally and physically tethered to SR by a mitochondrial fusion protein (namely mitofusin 2, a.k.a mfn2) [21]–[23]. The proximity between mitochondria and SR [24]–[28] can form a high Ca2+ microdomain, facilitating dynamic interorganellar coupling (e.g. rapid, beat-to-beat MCU Ca2+ uptake [10], [15], [29]) and the modulation of SR Ca2+ release by mitochondria [30], [31]. For instance, several studies have shown that the impairment of mitochondrial function, such as dissipation of mitochondrial membrane potential or inhibition of ATP synthesis, promotes proarrhythmic Ca2+ alternans [32]–[34]. How glycoside-induced mitochondrial dysfunction affects Ca2+ handling and how the altered Ca2+ cycling influences energy metabolism through feedback mechanisms are not well understood and difficult to examine experimentally due to the complex and nonlinear interactions formed by the participating subcellular components.
In this study, we incorporated a detailed antioxidant system into a recently published computational model of the cardiomyocyte to quantitatively investigate the mechanisms underlying cardiac glycoside-induced mitochondrial energetic dysfunction and glycoside- mediated abnormal Ca2+ cycling and automaticity. Our simulation work showed that NKA inhibition caused cytosolic Na+ and Ca2+ accumulations that adversely affected mitochondrial Ca2+ retention and energetics (such as reduced NADH and ATP production) similar to experimental observations. In addition, the impaired mitochondrial function could be significantly alleviated by inhibiting mNCE but barely ameliorated by enhancing MCU. Finally both Ca2+ and AP alternans could be elicited when severe ATP depletion occurred as the result of high frequency pacing and concurrent NKA inhibition, implicating a novel mechanism underlying the arrhythmogenic effect of cardiac glycosides.
Methods
Model Development
A multiscale guinea pig cardiomyocyte model was developed in order to systematically examine the effects of glycoside-induced high cytosolic Na+ ([Na+]i) on the interaction between mitochondrial energetics and cellular electrophysiology. The model was based on our recently published ECME-RIRR model [35] and consisted of major ion channels/transporters underlying the action potential, E-C coupling and Ca2+ handling, as well as mitochondrial energetics, ROS-induced ROS release (RIRR) and intramitochondrial Ca2+ regulation. To replicate mitochondrial Ca2+ and Na+ handling, a refined mitochondrial model [36] consisting of Na+, H+ and Pi + channels was incorporated. Moreover, a detailed antioxidant subsystem developed by Kembro et al. [37] was modified and added to account for the regulation of mitochondrial energetics (e.g. NADH) on ROS production and scavenging. Specifically, the expanded ROS metabolism pathway is comprised of: (i) glutathione peroxidase (GPX), Mn superoxide dismutase (MnSOD), glutaredoxin (GRX), and thioredoxin (Trx) subsystems in mitochondrial matrix; (ii) GPX, catalase (CAT), and Cu,Zn superoxide dismutase (Cu,ZnSOD) subsystems in the extra-mitochondrial space; and (iii) transhydrogenase (THD) on mitochondrial inner membrane. For simplicity, the pathway of isocitrate dehydrogenase 2 (IDH2) and diffusion of reduced glutathione (GSH) in the Kembro model [37] were not considered in the current work. The general model scheme is shown in Figure 1 and the complete model equations and parameters are listed in the Supplemental Information (File S1 and S2, respectively).
Figure 1. General scheme of guinea pig cardiomyocyte model.

The electrophysiological module describes major ion channels underlying the action potential, E-C coupling and Ca2+ handling. The mitochondrial module accounts for major components of mitochondrial energetics such as TCA cycle. The ROS-induced ROS release (RIRR) module describes ROS production, transport and scavenging. The mitochondrial energetics and RIRR are linked to Ca2+ handling and cellular electrophysiology through signaling ions (e.g., Ca2+ and Na+) and metabolic intermediates (e.g. ATP). In the figure, shunt represents the fraction of ROS production from the electron transport chain, and p1 and p2 represent the fraction of Ca2+ that MCU take up from the cytosol and the mitochondria-SR microdomain, respectively.
Modeling Mitochondrial Ca2+ Uptake through MCU
It is still debated how mitochondria take up Ca2+ in the cardiomyocyte. Some studies suggest that mitochondria take up Ca2+ solely from the cytoplasm [20], while others propose that MCU channels are tightly coupled to the SR and thus the main uptake is from the SR-mitochondria Ca2+ microdomain [10], [16]. We hypothesized that MCU distribute non-uniformly along the mitochondrial membrane, with expression abundant in areas facing the microdomain and scarce on membrane away from the SR. Consequently, MCU were modeled to draw Ca2+ from both the cytosol and the microdomain. The kinetics formulas and parameters of Ca2+ uptake remained the same as those in the ECME-RIRR model [35], except 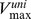 (the maximal rate of MCU) was split as
(the maximal rate of MCU) was split as 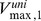 (representing uptake from the cytosol) and
(representing uptake from the cytosol) and 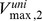 (representing uptake from the microdomain). For details see equations E168.1 and E168.2 in the Supplemental Information. The values of
(representing uptake from the microdomain). For details see equations E168.1 and E168.2 in the Supplemental Information. The values of 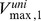 and
and 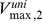 were adjusted accordingly to ensure that the total steady-state mitochondrial Ca2+ uptake flux equals the flux in the ECME-RIRR model:
were adjusted accordingly to ensure that the total steady-state mitochondrial Ca2+ uptake flux equals the flux in the ECME-RIRR model: 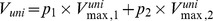 , where Vuni, Vuni,1 and Vuni,2 are the flux of the total Ca2+ uptake, the Ca2+ uptake from the cytosol, and the Ca2+ uptake from the microdomain, respectively. p1 and p2 represent the percentages of Ca2+ uptake flux from each pool, and their values are determined by the distribution of MCU proteins on the mitochondrial membrane.
, where Vuni, Vuni,1 and Vuni,2 are the flux of the total Ca2+ uptake, the Ca2+ uptake from the cytosol, and the Ca2+ uptake from the microdomain, respectively. p1 and p2 represent the percentages of Ca2+ uptake flux from each pool, and their values are determined by the distribution of MCU proteins on the mitochondrial membrane.
Model Simulation Protocol
The formulas of other processes, such as membrane potential, ion channels and metabolic reactions, and model parameters were the same as those in the ECME-RIRR model [35] (see the Supplemental Information). The code of the whole cell model was written in C++ (Visual Studio; Microsoft, Redmond, WA). The nonlinear ordinary differential equations were integrated numerically with CVODE as described previously [35], [38].
The cardiomyocyte was stimulated at 0.25 Hz until the steady state was reached. The steady state values were then used as initial conditions for all simulations. In the model, the effect of cardiac glycosides was mimicked by inhibiting NKA activity by 50%, which caused a 2.4-fold increase in cytosolic Na+ concentration in 3 mins. This amount of Na+ increase is comparable to that caused by 1 μM ouabain in the experimental study [1]. The cell was paced at 0.25 Hz for 1 min and then at an increasing rate (e.g., 2 Hz or 4 Hz) for 3 mins with or without NKA inhibition. We first simulated the dynamics of mitochondrial Ca2+ uptake, energy production and ROS metabolism during the transition of increasing workload. Various p1:p2 ratios (ranging from 1∶0 to 0∶1) were evaluated to match the experimental data [1]. The defined p1:p2 ratio was then used in the subsequent simulations. We then examined the effect of NKA inhibition-induced mitochondrial dysfunction on Ca2+ transients and AP under low (i.e. the fraction of electron transport chain O2 − production, shunt, = 1%) and relatively high (i.e. shunt = 2.5%) O2 − production conditions. We also explored whether blocking mNCE or enhancing MCU alleviates NKA inhibition-induced mitochondrial energetics impairment. Finally we examined whether NKA inhibition and associated mitochondrial dysfunction are capable of triggering Ca2+ alternans in a paced cardiomyocyte. The simulation results were post-processed and plotted using Origin 8.6 (OriginLab, Northampton, MA).
Results
Effects of MCU Ca2+ Pools and NKA Inhibition on Intracellular Ca2+ Dynamics during the Transition of Increasing Workload
We first examined the impacts of different subcellular mitochondrial Ca2+ uptake pools (i.e. cytosol vs. mitochondria-SR microdomain or p1:p2 ratio) on Ca2+ dynamics during the transition of increasing workload with or without NKA inhibition. As expected, increasing pacing frequency (from 0.25 Hz to 2 Hz) caused significant increase in both cytosolic and mitochondrial [Ca2+] (Fig. 2A and 2C, respectively). Interestingly, these elevations were differentially influenced by the p1:p2 ratio. Particularly, when MCU were modeled to take up Ca2+ completely from the cytoplasmic pool (i.e. p1 = 1 & p2 = 0), [Ca2+]i had the smallest elevation (e.g. systolic [Ca2+]i increased by 62.5%) in response to the increasing workload (Fig. 2A). It is clear that shifting the portion of Ca2+ uptake from the cytosol to the microdomain (or reducing p1:p2 ratio) enhanced [Ca2+]i accumulation. When p1 = 0 & p2 = 1 (i.e. completely from the microdomain), the increase of [Ca2+]i was the greatest (Fig. 2A). The effect of p1:p2 ratio on mitochondrial Ca2+ ([Ca2+]m) was similar to that of [Ca2+]i, except when [Ca2+]m was completely taken from the microdomain (i.e. p1 = 0 & p2 = 1), in which case [Ca2+]m increased first and then decreased slightly (Fig. 2C).
Figure 2. Model simulated effects of MCU Ca2+ uptake pool on [Ca2+]i and [Ca2+]m under control (A and C) and NKA inhibition (B and D) conditions during the transition of increasing pacing (from 0.25 Hz to 2 Hz).

Different colors represent different p1:p2 ratios: red (1∶0); green (3∶1); black (1∶1); blue (1∶3); and pink (0∶1). (E). Summarized effect of p1:p2 ratio on [Ca2+]m accumulation in response to increased energy demand. (F) Experimental data of the effect of NKA inhibition on [Ca2+]m accumulation (Reproduced from [1] with permission). The pacing frequency was increased to 1 Hz at 60 s.
NKA inhibition (by 50%) monotonically enhanced the accumulation of [Ca2+]i and amplified the influence of MCU Ca2+ uptake pool (Fig. 2B). The effect of blocking NKA on pacing-induced [Ca2+]m elevation was complex and dependent upon the p1:p2 ratio (Fig. 2D). As summarized in Figure 1E, NKA inhibition potentiated [Ca2+]m accumulation when mitochondria were modeled to sequestrate all Ca2+ from the cytosol (i.e. p1 = 1). NKA inhibition started to suppress the pacing-induced [Ca2+]m elevation with the pool shifting to the microdomain (i.e. reducing p1:p2 ratio). Specifically, when p1:p2 = 1∶3, NKA inhibition caused about 15% reduction in [Ca2+]m accumulation, which was comparable to the experimental data (18% reduction in systolic [Ca2+]m and 16% in diastolic [Ca2+]m) (Fig. 2F, reproduced from [1]). Therefore, we proposed that mitochondria take up Ca2+ ions mainly, if not exclusively, from mitochondrial-SR microdomain. The p1:p2 ratio of 1∶3 was used in all subsequent simulations. It is worth pointing out that in the experiments [1] the cardiomyocytes were treated with isoproterenol in addition to 1 Hz pacing. Thus, in our simulations, a higher (2 Hz) pacing frequency was used to produce comparable energy demand. In addition, the percentage of NKA activity inhibition caused by ouabain was not measured in the experiment, so the comparison was more qualitative than quantitative to some extent.
Effect of NKA Inhibition on Intracellular Ion Homeostasis and Mitochondrial Energetics
Next, the effects of NKA inhibition on ionic dynamics and mitochondrial energetics during the transition of increasing workload were simulated under the physiological ROS production conditions (e.g. ROS production fraction, shunt = 1%). Increasing pacing frequency from 0.25 Hz to 2 Hz caused a 1.6-fold elevation in [Na+]i and a 1.5-fold increase in mitochondrial Na+ concentration ([Na+]m), respectively (Fig. 3A and 3B). The NADH concentration decreased transiently and then returned to the basal level (Fig. 3E), which is consistent with the experimental data when workload is acutely increased [10], [39]. Increasing pacing rate alone caused a slight increase in ROS (Fig. 3F) and had minor effects on mitochondrial membrane potential (ΔΨm) (Fig. 3G), [ATP]i and [ATP]m (Fig. S1). Inhibiting NKA led to further increases of [Na+]i and [Na+]m (by 2.4-fold and 2.1-fold, respectively, Fig. 3A and 3B) and enhanced [Ca2+]i accumulation and [Ca2+]m attenuation (Fig. 3C and 3D). The elevation of [Na+]i was comparable to that caused by 1 μM ouabain in the experimental study [1]. The NADH level decreased by 9% and did not return to the basal level (Fig. 3E), along with a large (∼30- fold) increase of ROS (Fig. 3F) and small loss of ΔΨm (Fig. 3G) and ATP (Fig. S1). These simulations were comparable to experimental results (Inserts in Fig. 3; reproduced from [1] with permission).
Figure 3. Effects of blocking NKA on ion homeostasis and mitochondrial energetics under low O2 − production conditions with or without concurrent mNCE inhibition.

The cell was paced at 0.25: control; Red: 50% NKA inhibition; Dark green: 50% NKA inhibition+60% mNCE inhibition. (A): [Na+]i; (B): [Na+]m; (C): [Ca2+]i; (D): [Ca2+]m; (E): NADH; (F): ROS; and (G): mitochondrial membrane potential (ΔΨm). In this simulation, shunt = 1% and p1:p2 = 1∶3. Inserts: Experimental data of effects of ouabain (red) and CGP-37157 (dark green) on diastolic [Ca2+]m (Insert D), NADH (Insert E), and oxidative stress (Insert F) (Reproduced from [1]). In insert F, ouabain and CGP-37157 were applied at 1 min and pacing frequency was increased to 1 Hz at 5 min.
As heart failure often associates with increased ROS production, we simulated the effect of NKA inhibition on mitochondrial energetics under a relatively higher ROS production condition (e.g. shunt = 2.5%). As shown in Figure 4, the small increase of shunt per se did not significantly alter ion concentrations and mitochondrial energetics under control condition. However, simultaneously blocking NKA (by 50%) and increasing energy demand (from 0.25 Hz to 2 Hz) caused detriment on mitochondrial energetics under the higher ROS production condition. Specifically, NKA inhibition triggered sustained mitochondrial oscillations, including NADH (Fig. 4E), ROS production (Fig. 4F) and ΔΨm (Fig. 4G). The oscillations also included Ca2+ and Na+ ions in both cytosol and mitochondria (Fig. 4A–4D).
Figure 4. Effects of blocking NKA on ion homeostasis and mitochondrial energetics under higher O2 − production conditions with or without concurrent mNCE inhibition.

Black: control; Red: 50% NKA inhibition; Blue grey: 50% NKA inhibition+60% mNCE inhibition; Dark green: 50% NKA inhibition+90% mNCE inhibition. (A): [Na+]i; (B): [Na+]m; (C): [Ca2+]i; (D): [Ca2+]m; (E): NADH; (F): ROS; and (G): mitochondrial membrane potential (ΔΨm). In this simulation, shunt = 2.5% and p1:p2 = 1∶3.
The Effect of Modulating mNCE or MCU on NKA Inhibition-induced Mitochondrial Dysfunction
In cardiomyocyte, mitochondrial energetics largely relies on mitochondrial Ca2+ retention and NADH homeostasis mediated by that Ca2+ retention, especially during the transition of increasing energy demand [38], [40], [41]. Therefore, we examined whether modulating mitochondrial Ca2+ handling channels, namely mNCE and MCU, could influence the effects of NKA inhibition on mitochondrial energetics. Our results showed that the concurrent inhibition of mNCE (e.g. by 60%) significantly ameliorated NKA inhibition-induced mitochondrial dysfunction, without abolishing the inotropic effect of NKA inhibition (Fig. 3). Specifically, NKA inhibition-induced [Na+]i and [Na+]m elevations were slightly attenuated and [Ca2+]i accumulation was reduced by 13%. mNCE inhibition also overturned the blunted [Ca2+]m retention and restored NADH concentration, as wells as suppressed ROS production and preserved ΔΨm (Fig. 3, dark green curves) and ATP (Fig. S1). The beneficial effect of mNCE inhibition was consistent with previous experimental findings that application of CGP, an mNCE inhibitor, prevented ouabain-induced mitochondrial dysfunction and resultant oxidative stress (Fig. 3 Inserts) [1], [10]. While moderate inhibition of mNCE was beneficial, too much mNCE blocking (e.g. by 90%) completely suppressed NKA inhibition-induced [Ca2+]i elevation (and thus the inotropic effect) (data not shown). At the higher ROS production rate condition (e.g. shunt = 2.5%), blocking mNCE by 60% significantly delayed NKA inhibition-induced mitochondrial oscillations (Fig. 4, sky grey curves). Again, 90% mNCE inhibition counteracted the inotropic effect of glycoside, although it successfully prevented NKA inhibition-induced oxidative stress and mitochondrial oscillations (Fig. 4, dark green curves).
Another way to compensate NKA inhibition-induced mitochondrial Ca2+ decline is to increase mitochondrial Ca2+ uptake via MCU. Therefore, we examined whether enhancing MCU can alleviate NKA inhibition-induced mitochondrial energetics dysfunction similarly to mNCE inhibition. As expected, increasing MCU rate (e.g. by 100%) significantly increased [Ca2+]m (Fig. 5D). However, its effects on NKA inhibition-induced [Na+]i, [Na+]m, and [Ca2+]i alterations were not significant (Fig. 5A–5C). Enhancing MCU barely alleviated NKA inhibition-induced mitochondrial energetic impairments. For instance, NADH level slightly increased but did not completely recover (Fig. 5E), ROS still accumulated (Fig. 5F), and ΔΨm still dropped (Fig. 5G). Further enhancing MCU (e.g. by 4-fold) could increase mitochondrial Ca2+ accumulation to a level comparable to that caused by mNCE inhibition, but did not completely abolish NKA inhibition-induced mitochondrial energetics dysfunction (data not shown).
Figure 5. Effect of enhancing MCU on NKA inhibition-induced mitochondrial energetic dysfunction.

Black: control; Red: 50% NKA inhibition; Dark green: 50% NKA inhibition+100% MCU enhancement. (A): [Na+]i; (B): [Na+]m; (C): [Ca2+]i; (D): [Ca2+]m; (E): NADH; (F): ROS; and (G): mitochondrial membrane potential (ΔΨm). In this simulation, shunt = 1% and p1:p2 = 1∶3.
In addition to blocking mNCE and enhancing MCU, we also examined the effects of concurrently enhancing mNCE or inhibiting MCU with NKA inhibition. Increasing mNCE (100%) slightly enhanced NKA inhibition-induced [Ca2+]i elevation (Fig. S2A) and significantly exacerbated [Ca2+]m decline (Fig. S2B), NADH reduction (Fig. S2C) and ROS elevation (Fig. S2D). Concurrently blocking MCU (60%) with NKA inhibition had similar effects except it didn’t substantially promote ROS elevation (Fig. S2D). Further blocking MCU (100%) induced a more profound ROS elevation (data not shown).
Effect of NKA Inhibition on Ca2+ Cycling and AP under more Stressed Conditions
Finally, we investigated the effect of NKA inhibition on mitochondrial energetics and cellular electrophysiology under more stressed conditions, such as more severe NKA inhibition and/or higher energy demand. Increasing pacing rate from 0.25 Hz to 4 Hz resulted in mitochondrial depolarization and oscillations when the shunt was set to 2.5% (Fig. 6A), as well as cyclic ATP depletions (Fig. 6B). Similar results have been reported in our previous study [35]. Concurrent induction of NKA inhibition by 50% prompted mitochondrial depolarization (Fig. 6A red curve) and accelerated ATP consumption, resulting in further decline of ATP (Fig. 6B red curve). Figures 6C–6F present the dynamics of SERCA Ca2+ uptake (Jup), SR Ca2+ content, Ca2+ transients and action potential (AP) at the last second (i.e. 249 s) of (the 4 Hz) simulations. It is clear that the depletion of ATP significantly impaired SERCA Ca2+ uptake (Fig. 6C) and caused imbalance of SR Ca2+ cycling, leading to reducing and alternating SR Ca2+ content (Fig. 6D), as well as Ca2+ transient alternans with significantly increased amplitude of the larger Ca2+ transients within the large-small Ca2+ alternations (Fig. 6E). AP alternans with elevated resting potential and suppressed amplitude was also observed (Fig. 6F). More severe NKA inhibition (e.g. by 90%) accelerated ATP depletion, resulting in earlier occurrence of alternans (data not shown). Concurrent inhibition of mNCE (by 90%) retarded mitochondrial oscillations and mitigated cytosolic ATP depletion (Fig. 6A and 6B, green curve), which consequently augmented SR Ca2+ uptake (Fig. 6C), leading to a higher SR Ca2+ concentration (Fig. 6D). The stabilization and restoration of SR Ca2+ content abolished Ca2+ and AP alternans, producing a smaller Ca2+ transient and a larger AP amplitude (Fig. 6E and 6F).
Figure 6. Action potential (AP) and Ca2+ alternans induced by NKA inhibition.

The cell was paced at 0.25(by 50%). AP and Ca2+ alternans were observed at the end of pacing. Black: control; Red: 50% NKA inhibition; Dark green: 50% NKA inhibition+90% mNCE inhibition. (A): mitochondrial membrane potential (ΔΨm); (B): ATPi; (C): SERCA Ca2+ uptake; (D): [Ca2+]SR; (E): [Ca2+]i and (F): AP. In this simulation, shunt = 2.5% and p1:p2 = 1∶3.
To further explore the development of NKA inhibition-induced Ca2+ and AP alternans, the dynamics of ATP, [Ca2+]i, [Ca2+]SR and AP before, during and after the 5th and 9th mitochondrial depolarization (indicated by arrows in Fig. 6A) were analyzed. Within the first 4 oscillations, the morphology of APs did not change significantly, although their amplitudes dropped gradually (data not shown). At the 5th mitochondrial depolarization, [ATP]i decreased dramatically from 4.8 mM to 2.1 mM (Fig. 7A). Consequently, SERCA Ca2+ uptake was significantly impaired and the SR Ca2+ release-uptake cycle was disturbed, resulting in a large [Ca2+]SR depletion and [Ca2+]i accumulation (Fig. 7C and 7E). The elevated intracellular Ca2+ stimulated Na+/Ca2+ exchanger and caused APD prolongation (Fig. 7G). As a result, the AP could not fully repolarize and was still in the refractory period when the next electrical stimulation fired, resulting in EAD-like abnormal APs (Fig. 7G). Nonetheless, when ΔΨm recovered and [ATP]i rebounded to a relative higher level (Fig. 7A, dark green curve), the disturbed AP and Ca2+ transients could return to normal rhythms (Fig. 7E and 7G, dark green curves).
Figure 7. Comparisons of ATPi (A and B), [Ca2+]SR (C and D), [Ca2+]i (E and F), and action potential (AP) (G and H) before (Black), during (Red) and after (Dark green) the 5th (A,C,E,G) and 9th (B,D,F,H) mitochondrial depolarization (marked with arrows in Fig. 6A).

In this simulation, shunt = 2.5%, p1:p2 = 1∶3, and NKA inhibition was 50%.
As mitochondrial oscillations continued, ATP (Fig. 7B) and SR Ca2+ content further declined (Fig. 7D), the morphologies of Ca2+ transients and AP became more distorted. For instance, right before the 9th depolarization, [ATP]i and basal [Ca2+]SR were already dramatically low (1.6 mM and 0.36 mM, respectively) (Fig. 7B and 7D, black curves). The subsequent ΔΨm depolarization caused extra decreases of [ATP]i and SR Ca2+ (Fig. 7B and 7D, red curves) and a much larger Ca2+ transient that spanned several cycle lengths (Fig. 7F, red curve). The APs also displayed erratic morphologies, with a significantly prolonged AP followed by several ones with much smaller AP amplitudes (Fig. 7H, red curve). Interestingly, the Ca2+ transients and AP developed alternating behaviors (i.e., alternans) rather than recovering to normal rhythm (Fig. 7F and 7H, dark green curves) when ΔΨm repolarized and ATP restored (Fig. 7B).
Discussion
The present study provides, for the first time, a computational framework to quantitatively examine the mechanisms underlying mitochondrial dysfunction and cardiac arrhythmias induced by cardiac glycosides that are used in the treatment of heart failure. The main findings are: (i) NKA inhibition simultaneously raises [Ca2+]i and blunts [Ca2+]m retention, causing ATP depletion and ROS accumulation that disturb SR Ca2+ handling and elicit abnormal APs; (ii) NKA inhibition-induce adverse effects on mitochondrial energetics and Ca2+ cycling can be ameliorated by blocking mNCE, but not by enhancing MCU; (iii) under more severe conditions, NKA inhibition can cause proarrhythmic Ca2+ and AP alternans; and (iv) mitochondrial uniporter takes up Ca2+ mainly from the mitochondria-SR microdomain instead of the cytoplasm.
Glycosides and Mitochondrial Energetics Dysfunction: New Links Revealed by Model Simulations
In cardiomyocytes, mitochondria are the major sites of ROS production, and NADH plays a vital role in regulating both the production and scavenging of ROS. Previous studies showed that inhibiting mitochondrial Ca2+ uptake impaired NADH production and promoted ROS accumulation [1], [10]. Our simulations were able to reproduce these experimental observations. In addition, our data demonstrated for the first time that the blunted [Ca2+]m uptake (and resultant reduced NADH production) was not the sole reason causing oxidative stress during NKA inhibition, because ROS accumulation could occur even when NADH did not decline significantly (Fig. 8). It is known that increasing pacing frequency not only increases energy consumption but also causes ADP accumulation, which directly stimulates oxidative phosphorylation and the associated ROS production. Since NKA inhibition exacerbates [Ca2+]i overload and enhances ATP hydrolysis, the resulting further ADPm elevation can activate ATP synthesis and respiration, resulting in additional ROS production. Therefore, we propose that NKA inhibition such as by cardiac glycosides causes mitochondrial oxidative stress through two separate but concomitant pathways: (i) NKA inhibition blunting [Ca2+]m accumulation, which reduces NADH production and therefore ROS removal, and (ii) NKA inhibition increasing [Ca2+]i and ATP hydrolysis, which produces a large amount of ADP that stimulates mitochondrial respiration and therefore ROS production.
Figure 8. Summary of [Ca2+]i and mitochondrial energetics under 0.25 Hz, 2 Hz, 2 Hz + NKA inhibition, and 2 Hz + NKA inhibition + mNCE inhibition conditions.
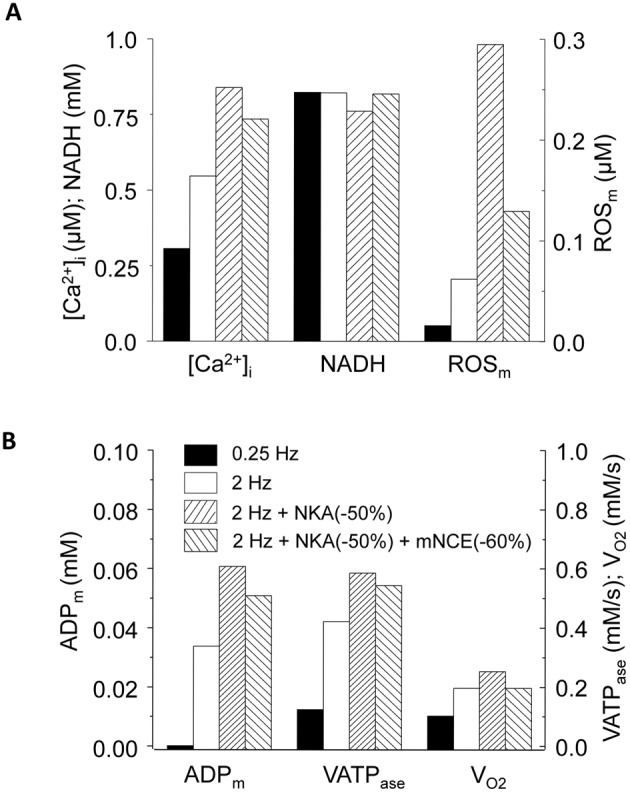
(A): [Ca2+]i, NADH and ROSm; and (B): ADPm, complex V activity (VATPase) and respiration rate (VO2). The data was recorded at the end point of simulations (i.e. 3 mins after increasing pacing). In this simulation shunt = 1%, p1:p2 = 1∶3, and NKA inhibition was 50%.
While the role of NKA inhibition in regulating mitochondrial ROS is well appreciated, the effect of ouabain on mitochondrial ATP synthesis remains elusive. A recent study showed that ouabain suppressed glucose-induced mitochondrial ATP production in rat pancreatic islets [42]. This inhibitory effect, caused by enhanced ROS production that impaired electron transport chain, could be observed only at high glucose concentration but not at the basal level of glucose. Similarly, another study reported that ouabain caused ATP reduction, a dramatic increase of intracellular Ca2+ (up to 40-fold) and enhanced ATP consumption in cultured chick ventricular cells [43]. The ATP reduction induced by ouabain was much less than that induced by cyanide and 2-deoxyglucosein, so the ATP loss observed in this experiment was likely caused by the increase of ATP consumption rather than the impairment of mitochondrial ATP synthesis. On the contrary, it has been shown that ouabain directly enhanced the rate of electron transport chain and the synthesis of ATP in cardiomyocytes via increasing the synthesis of ubiquinone [7]. These contradictory results might be due to differences in experimental conditions, animal species or tissue variations. Our results suggest that NKA inhibition differentially influence ATP production, depending on the severity of stress: (i) concurrent NKA inhibition and moderate increase of workload stimulates oxidative phosphorylation and ATP production, and (ii) under more severe conditions such as higher frequency pacing, NKA inhibition caused dramatic ROS accumulation, mitochondrial depolarization, and tremendous ATP depletion.
Glycosides-induced Ca2+ Alternans: A New Mitochondria-originated Arrhythmic Substrate
In previous animal and clinical studies, the arrhythmogenic effects of glycosides have been mainly attributed to NKA inhibition-induced Ca2+ overload and associated abnormal RyR Ca2+ release [2], [5], [7], [8], [44]. More recently, studies by Liu et al. suggested that mitochondrial dysfunction, particularly ouabain-induced ROS production, might also contribute to the arrhythmogenic effect of glycosides [1]. To better understand the mechanisms underlying glycoside-induced cardiac arrhythmias, we analyzed the dynamics of Ca2+ in the cytosol, mitochondria and SR, as well as the profiles of metabolic intermediates such as ATP and NADH. Our results showed that NKA inhibition caused a significant ROS accumulation during the transition of moderate workload increment. The mitochondria-derived ROS may lead to abnormal Ca2+ handling and erratic APs, as demonstrated in [1]. More importantly, the model revealed a novel phenomenon that has not reported before: under more stressed conditions such as higher frequency pacing, NKA inhibition causes Ca2+ and AP alternans by reducing ATP. It is worthy to point out that although in our simulations Ca2+ alternans could be induced only at higher pacing rate (i.e. 4 Hz), the mechanism underlying the genesis of this type of alternans is different from that of classical alternans induced by increasing pacing rate. Actually, in our hands pacing at 4 Hz per se did not induce Ca2+ alternans. Further analysis showed that the genesis of Ca2+ alternans was attributed to NKA inhibition-induced mitochondrial dysfunction. Particularly, severe ATP depletion impaired SERCA activity, reduced the SR Ca2+ content, and thereby generated SR release and uptake mismatch, causing intracellular Ca2+ alternans. A similar mitochondrial dysfunction-induced Ca2+ alternans has been reported by several experimental studies [45], [46]. The role of mitochondrial dysfunction in the genesis of Ca2+ alternans was further supported by the mNCE inhibition simulation. As shown in Figure 5, blocking mNCE ameliorated NKA inhibition-induced mitochondrial dysfunction, mitigating the decline of SR Ca2+ and suppressing the genesis of alternans. Thus, our in silico study revealed a new plausible mechanism responsible for glycoside-induced cardiac arrhythmias, which is different from the traditional paradigm of SR Ca2+ overload and associated abnormal RyR Ca2+ release. Our model also suggested that the increase of workload could potentiate the mismatch between energy supply and demand and increase the risk of NKA inhibition-mediated arrhythmias. Thus, digitalis toxic effects could be more problematic at increased heart rate.
Can Modulating Mitochondrial Ca2+ Handling Alleviate Glycoside-induced Arrhythmogenesis?
Studies have shown that CGP-17157, a mNCE inhibitor, mitigated ouabain-induced adverse effects on mitochondrial function and lowered the occurrence of cardiac arrhythmias in guinea pigs [1], [10]. The beneficial effect of CGP was thought due to its capability of retaining [Ca2+]m accumulation that preserves mitochondrial NADH homeostasis and reduces ROS production. Our simulations are quantitatively comparable to these experimental results. Moreover, we showed that the mNCE inhibition attenuated cytosolic Ca2+ accumulation, which contributed to the anti-arrhythmic effect of CGP-17157. In another word, the therapeutic effect of mNCE inhibitors was attributed to not only preserving [Ca2+]m retention and mitochondrial function, but also attenuating glycoside-induced intracellular Ca2+ overload. One should be aware though that it is the [Ca2+]i elevation that is primarily responsible for the (beneficial) inotropic effect of glycosides treatment. Therefore, the dosage of the mNCE inhibitor is likely to be critical to counteracting the adverse effects of glycosides. Actually, we showed that too much mNCE inhibition, such as by 90%, completely abolished NKA inhibition-induced Ca2+ accumulation, although it significantly improved mitochondrial function and suppressed oxidative stress.
While inhibiting mNCE efficiently relieved NKA inhibition-induced adverse influence on mitochondrial energetics, the effect of increasing mitochondrial Ca2+ uniporters (MCU) activity was not evident, which might be caused by the substrate compartmentation of MCU. Our model analysis showed that MCU need to take up most of Ca2+ (>70%) from the microdomain in order to reproduce NKA inhibition-induced [Ca2+]m attenuation observed in the experimental studies [1], [10]. These results indicate that the majority of Ca2+ taken up by MCU is from the microdomain instead of cytoplasm. Thus, enhancing MCU activity increased [Ca2+]m uptake and retention, but it did not substantially reduce NKA inhibition-induced [Ca2+]i overload and ROS production. These in silico results also imply that MCU are not uniformly distributed across the mitochondrial membrane, but rather are concentrated at mitochondria-SR contacting sites. The non-homogenous localized expression of ion channels on the sarcoplasmic or intracellular membranes has been reported previously. For instance, studies showed that L-type Ca2+ channels are concentrated in the neighborhood of the junctional SR at T-tubules [47], and this unique microstructure allows a correlation between ionic channel function and distribution. Similarly, abundant expression of MCU at the mitochondria-SR contacting sites can facilitate fast mitochondrial Ca2+ uptake in response to increased energy demand. With the molecular identity of MCU emerging [13], experimentally verifying the non-homogenous distribution of MCU in cardiac mitochondria is no longer far-fetched.
Study Limitations
The model developed in this study incorporates subcellular Ca2+ compartmentation for MCU, therefore providing advantages over existing cardiomyocyte models [48]–[51]. However, it also has some limitations that need be addressed. (i) The current model lacks an isoproterenol signaling pathway, which impedes the direct comparison between model simulations and experimental data of Liu et al. [1]. However, this limitation would not alter our conclusion on the NKA inhibition-induced mitochondrial dysfunction. (ii) Previous studies have suggested that Ca2+ wave propagation plays a critical role in intracellular Ca2+ regulation and genesis of Ca2+ alternans [45], [46]. The “mitochondria-SR Ca2+ microdomain” notion proposed in this study (and by others elsewhere) may be involved in regulating Ca2+ wave propagation. Incorporating spatial resolution of local Ca2+ distribution in the microdomain would allow examination of its role in regulating cellular electrophysiology and mitochondrial energetics. (iii) The current model did not incorporate the direct link between mitochondrial ROS and redox sensitive SR Ca2+ proteins. It has been shown that oxidative stress can modulate SR Ca2+ handling proteins and cause abnormal electrical activities in cardiomyocytes [52]–[54]. The NKA inhibition-induced ROS production would exacerbate glycoside-induced abnormal cellular electrophysiology [1]. Adding mdROS modulation of redox sensitive ion channels/transporters is ongoing in our laboratory. (iv) In the current model, the equations of complexes of the electron transport chain are lumped, which makes it hard to investigate the specific role of the complexes of electron transport chain (ETC) in mitochondrial dysfunction. A more detailed ETC model [55] can be incorporated into the ECME-RIRR model to examine the effect of ETC complexes on mitochondrial function. (v) The present model did not consider the effect of energy depletion on ATP-sensitive potassium channels (KATP), as our main focus is on mitochondrial dysfunction and Ca2+ handling. It is expected that incorporating KATP would exacerbate the deleterious effect of NKA inhibition-induced mitochondrial dysfunction on cardiac action potentials. As shown before, depletion of ATP activates KATP currents, resulting in AP shortening or even cellular inexcitability [35], [56].
In summary, the present study provides a novel computational tool to quantitatively investigate the proarrhythmic effects of glycosides (and resultant NKA inhibition) in the cardiomyocyte. The simulations reveal that NKA inhibition can cause mitochondrial oxidative stress through two separate and concomitant pathways, which involve an intricate interplay between cardiac electrophysiology and mitochondrial energetics. Moreover, the model suggests that under more severe conditions, NKA inhibition can induce Ca2+ alternans, contributing to its proarrhythmic effect. Finally, the study verifies the role of mitochondria-SR Ca2+ microdomain in regulating mitochondrial Ca2+ uptake and energetics. Taken together, this work underscores the importance of targeting both intracellular ion handling and mitochondrial energetics in developing new glycoside-based heart failure therapies.
Supporting Information
Effects of blocking NKA on cytosolic and mitochondrial ATP under low O2− production conditions with or without concurrent mNCE inhibition. The cell was paced at 0.25 Hz for 1 min then at 2 Hz for 3 mins. Black: control; Red: 50% NKA inhibition; Dark green: 50% NKA inhibition+60% mNCE inhibition. (A): [ATP]i; (B): [ATP]m. shunt = 1% and p1:p2 = 1∶3.
(TIF)
Effect of blocking MCU or enhancing mNCE on NKA inhibition-induced Ca2+ and mitochondrial energetics changes. Black: control; Red: 50% NKA inhibition; Dark green: 50% NKA inhibition+60% MCU inhibition; Blue: 50% NKA inhibition+100% mNCE enhancement. (A): [Ca2+]i; (B): [Ca2+]m; (C): NADH; (D): ROS. shunt = 1% and p1:p2 = 1∶3.
(TIF)
Supplemental Information model equations.
(DOCX)
Supplemental Information model parameters.
(DOCX)
Acknowledgments
The authors thank Dr. X. Margaret Liu for reading and insightful criticisms.
Funding Statement
This work is supported by NIH/NHLBI R00HL095648 and University of Alabama at Birmingham Comprehensive Cardiovascular Center. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Liu T, Brown DA, O’Rourke B (2010) Role of mitochondrial dysfunction in cardiac glycoside toxicity. J Mol Cell Cardiol 49: 728–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rahimtoola SH (2004) Digitalis therapy for patients in clinical heart failure. Circulation 109: 2942–2946. [DOI] [PubMed] [Google Scholar]
- 3. Bers DM, Despa S (2009) Na+ transport in cardiac myocytes; Implications for excitation-contraction coupling. IUBMB Life 61: 215–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hashim T, Elbaz S, Patel K, Morgan CJ, Fonarow GC, et al.. (2013) Digoxin and 30-day All-cause Hospital Admission in Older Patients with Chronic Diastolic Heart Failure. The American journal of medicine. [DOI] [PMC free article] [PubMed]
- 5. Gonano LA, Sepulveda M, Rico Y, Kaetzel M, Valverde CA, et al. (2011) Calcium-calmodulin kinase II mediates digitalis-induced arrhythmias. Circ Arrhythm Electrophysiol 4: 947–957. [DOI] [PubMed] [Google Scholar]
- 6. Rathore SS, Curtis JP, Wang Y, Bristow MR, Krumholz HM (2003) Association of serum digoxin concentration and outcomes in patients with heart failure. JAMA 289: 871–878. [DOI] [PubMed] [Google Scholar]
- 7. Campia I, Sala V, Kopecka J, Leo C, Mitro N, et al. (2012) Digoxin and ouabain induce the efflux of cholesterol via liver X receptor signalling and the synthesis of ATP in cardiomyocytes. Biochem J 447: 301–311. [DOI] [PubMed] [Google Scholar]
- 8. Ferrier GR (1977) Digitalis arrhythmias: role of oscillatory afterpotentials. Prog Cardiovasc Dis 19: 459–474. [DOI] [PubMed] [Google Scholar]
- 9.Roberts BN, Christini DJ (2011) NHE Inhibition Does Not Improve Na+ or Ca2+ Overload During Reperfusion: Using Modeling to Illuminate the Mechanisms Underlying a Therapeutic Failure. Plos Computational Biology 7. [DOI] [PMC free article] [PubMed]
- 10. Maack C, Cortassa S, Aon MA, Ganesan AN, Liu T, et al. (2006) Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ Res 99: 172–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu T, O’Rourke B (2008) Enhancing mitochondrial Ca2+ uptake in myocytes from failing hearts restores energy supply and demand matching. Circ Res 103: 279–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R (2011) A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476: 336–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, et al. (2011) Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476: 341–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dedkova EN, Blatter LA (2008) Mitochondrial Ca2+ and the heart. Cell Calcium 44: 77–91. [DOI] [PubMed] [Google Scholar]
- 15. Liu T, O’Rourke B (2009) Regulation of mitochondrial Ca2+ and its effects on energetics and redox balance in normal and failing heart. J Bioenerg Biomembr 41: 127–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. O’Rourke B, Blatter LA (2009) Mitochondrial Ca2+ uptake: tortoise or hare? J Mol Cell Cardiol 46: 767–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Denton RM, McCormack JG (1985) Ca2+ transport by mammalian mitochondria and its role in hormone action. Am J Physiol 249: E543–554. [DOI] [PubMed] [Google Scholar]
- 18. Huser J, Blatter LA, Sheu SS (2000) Mitochondrial calcium in heart cells: beat-to-beat oscillations or slow integration of cytosolic transients? J Bioenerg Biomembr 32: 27–33. [DOI] [PubMed] [Google Scholar]
- 19. Bassani JW, Bassani RA, Bers DM (1994) Relaxation in rabbit and rat cardiac cells: species-dependent differences in cellular mechanisms. J Physiol 476: 279–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bassani RA, Bassani JW, Bers DM (1992) Mitochondrial and sarcolemmal Ca2+ transport reduce [Ca2+]i during caffeine contractures in rabbit cardiac myocytes. J Physiol 453: 591–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen Y, Csordas G, Jowdy C, Schneider TG, Csordas N, et al. (2012) Mitofusin 2-containing mitochondrial-reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca2+ crosstalk. Circ Res 111: 863–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Csordas G, Hajnoczky G (2009) SR/ER-mitochondrial local communication: calcium and ROS. Biochim Biophys Acta 1787: 1352–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Csordás G, Hajnóczky G (2009) SR/ER–mitochondrial local communication: Calcium and ROS. Biochimica et Biophysica Acta (BBA) - Bioenergetics 1787: 1352–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sharma VK, Ramesh V, Franzini-Armstrong C, Sheu SS (2000) Transport of Ca2+ from sarcoplasmic reticulum to mitochondria in rat ventricular myocytes. J Bioenerg Biomembr 32: 97–104. [DOI] [PubMed] [Google Scholar]
- 25. Berridge MJ (2006) Calcium microdomains: organization and function. Cell Calcium 40: 405–412. [DOI] [PubMed] [Google Scholar]
- 26. Franzini-Armstrong C (2007) ER-mitochondria communication. How privileged? Physiology (Bethesda) 22: 261–268. [DOI] [PubMed] [Google Scholar]
- 27. Rizzuto R, Pozzan T (2006) Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol Rev 86: 369–408. [DOI] [PubMed] [Google Scholar]
- 28. Ramesh V, Sharma VK, Sheu SS, Franzini-Armstrong C (1998) Structural proximity of mitochondria to calcium release units in rat ventricular myocardium may suggest a role in Ca2+ sequestration. Ann NY Acad Sci 853: 341–344. [DOI] [PubMed] [Google Scholar]
- 29. Rizzuto R, Duchen MR, Pozzan T (2004) Flirting in little space: the ER/mitochondria Ca2+ liaison. Sci STKE 2004: re1. [DOI] [PubMed] [Google Scholar]
- 30. Yan Y, Liu J, Wei C, Li K, Xie W, et al. (2008) Bidirectional regulation of Ca2+ sparks by mitochondria-derived reactive oxygen species in cardiac myocytes. Cardiovasc Res 77: 432–441. [DOI] [PubMed] [Google Scholar]
- 31. Zhou L, Aon MA, Liu T, O’Rourke B (2011) Dynamic modulation of Ca(2+) sparks by mitochondrial oscillations in isolated guinea pig cardiomyocytes under oxidative stress. Journal of molecular and cellular cardiology 51: 632–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Florea SM, Blatter LA (2010) The role of mitochondria for the regulation of cardiac alternans. Front Physiol 1: 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Florea SM, Blatter LA (2012) Regulation of cardiac alternans by beta-adrenergic signaling pathways. Am J Physiol Heart Circ Physiol 303: H1047–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Smith RM, Visweswaran R, Talkachova I, Wothe JK, Tolkacheva EG (2013) Uncoupling the mitochondria facilitates alternans formation in the isolated rabbit heart. Am J Physiol Heart Circ Physiol 305: H9–H18. [DOI] [PubMed] [Google Scholar]
- 35. Zhou L, Cortassa S, Wei AC, Aon MA, Winslow RL, et al. (2009) Modeling cardiac action potential shortening driven by oxidative stress-induced mitochondrial oscillations in guinea pig cardiomyocytes. Biophys J 97: 1843–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wei AC, Aon MA, O’Rourke B, Winslow RL, Cortassa S (2011) Mitochondrial energetics, pH regulation, and ion dynamics: a computational-experimental approach. Biophys J 100: 2894–2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kembro JM, Aon MA, Winslow RL, O’Rourke B, Cortassa S (2013) Integrating mitochondrial energetics, redox and ROS metabolic networks: a two-compartment model. Biophys J 104: 332–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cortassa S, Aon MA, O’Rourke B, Jacques R, Tseng HJ, et al. (2006) A computational model integrating electrophysiology, contraction, and mitochondrial bioenergetics in the ventricular myocyte. Biophys J 91: 1564–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brandes R, Bers DM (1999) Analysis of the mechanisms of mitochondrial NADH regulation in cardiac trabeculae. Biophys J 77: 1666–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Brandes R, Bers DM (1997) Intracellular Ca2+ increases the mitochondrial NADH concentration during elevated work in intact cardiac muscle. Circ Res 80: 82–87. [DOI] [PubMed] [Google Scholar]
- 41. Brandes R, Bers DM (2002) Simultaneous measurements of mitochondrial NADH and Ca(2+) during increased work in intact rat heart trabeculae. Biophys J 83: 587–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kajikawa M, Tsuura Y, Mukai E, Takeda T, Hamamoto Y, et al. (2002) Ouabain Suppresses Glucose-Induced Mitochondrial ATP Production and Insulin Release by Generating Reactive Oxygen Species in Pancreatic Islets. Diabetes 51: 2522–2529. [DOI] [PubMed] [Google Scholar]
- 43. Ishida H, Kohmoto O, Bridge JH, Barry WH (1988) Alterations in cation homeostasis in cultured chick ventricular cells during and after recovery from adenosine triphosphate depletion. J Clin Invest 81: 1173–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fedida D, Noble D, Rankin AC, Spindler AJ (1987) The arrhythmogenic transient inward current iTI and related contraction in isolated guinea-pig ventricular myocytes. J Physiol 392: 523–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Li Q, O’Neill SC, Tao T, Li Y, Eisner D, et al. (2012) Mechanisms by which cytoplasmic calcium wave propagation and alternans are generated in cardiac atrial myocytes lacking T-tubules-insights from a simulation study. Biophys J 102: 1471–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mackenzie L, Roderick HL, Berridge MJ, Conway SJ, Bootman MD (2004) The spatial pattern of atrial cardiomyocyte calcium signalling modulates contraction. J Cell Sci 117: 6327–6337. [DOI] [PubMed] [Google Scholar]
- 47.Bers DM (2001) Excitation–Contraction Coupling and Cardiac Contractile Force edn 2 Kluwer Academic, Dordrecht, Netherlands.
- 48. Magnus G, Keizer J (1998) Model of β-cell mitochondrial calcium handling and electrical activity. I.Cytoplasmic variables. Am J Physiol Cell Physiol 274: C1158–1173. [DOI] [PubMed] [Google Scholar]
- 49. Matsuoka S, Sarai N, Jo H, Noma A (2004) Simulation of ATP metabolism in cardiac excitation-contraction coupling. Prog Biophys Mol Biol 85: 279–299. [DOI] [PubMed] [Google Scholar]
- 50. Greenstein JL, Hinch R, Winslow RL (2006) Mechanisms of excitation-contraction coupling in an integrative model of the cardiac ventricular myocyte. Biophys J 90: 77–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gauthier LD, Greenstein JL, Winslow RL (2012) Toward an integrative computational model of the Guinea pig cardiac myocyte. Front Physiol 3: 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Xie LH, Chen F, Karagueuzian HS, Weiss JN (2009) Oxidative-stress-induced afterdepolarizations and calmodulin kinase II signaling. Circ Res 104: 79–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Barrington PL, Meier CF Jr, Weglicki WB (1988) Abnormal electrical activity induced by free radical generating systems in isolated cardiocytes. J Mol Cell Cardiol 20: 1163–1178. [DOI] [PubMed] [Google Scholar]
- 54. Horackova M, Ponka P, Byczko Z (2000) The antioxidant effects of a novel iron chelator salicylaldehyde isonicotinoyl hydrazone in the prevention of H(2)O(2) injury in adult cardiomyocytes. Cardiovasc Res 47: 529–536. [DOI] [PubMed] [Google Scholar]
- 55. Gauthier LD, Greenstein JL, Cortassa S, O’Rourke B, Winslow RL (2013) A computational model of reactive oxygen species and redox balance in cardiac mitochondria. Biophys J 105: 1045–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Akar FG, Aon MA, Tomaselli GF, O’Rourke B (2005) The mitochondrial origin of postischemic arrhythmias. J Clin Invest 115: 3527–3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Effects of blocking NKA on cytosolic and mitochondrial ATP under low O2− production conditions with or without concurrent mNCE inhibition. The cell was paced at 0.25 Hz for 1 min then at 2 Hz for 3 mins. Black: control; Red: 50% NKA inhibition; Dark green: 50% NKA inhibition+60% mNCE inhibition. (A): [ATP]i; (B): [ATP]m. shunt = 1% and p1:p2 = 1∶3.
(TIF)
Effect of blocking MCU or enhancing mNCE on NKA inhibition-induced Ca2+ and mitochondrial energetics changes. Black: control; Red: 50% NKA inhibition; Dark green: 50% NKA inhibition+60% MCU inhibition; Blue: 50% NKA inhibition+100% mNCE enhancement. (A): [Ca2+]i; (B): [Ca2+]m; (C): NADH; (D): ROS. shunt = 1% and p1:p2 = 1∶3.
(TIF)
Supplemental Information model equations.
(DOCX)
Supplemental Information model parameters.
(DOCX)