

Abstract
Silk-elastin-like protein polymers (SELPs) combine the mechanical and biological properties of silk and elastin. These properties have led to the development of various SELP-based materials for drug delivery. However, SELPs have rarely been developed into nanoparticles, partially due to the complicated fabrication procedures, nor assessed for potential as an anticancer drug delivery system. We have recently constructed a series of SELPs (SE8Y, S2E8Y, and S4E8Y) with various ratios of silk to elastin blocks and described their capacity to form micellar-like nanoparticles upon thermal triggering. In this study, we demonstrate that doxorubicin, a hydrophobic antitumor drug, can efficiently trigger the self-assembly of SE8Y (SELPs with silk to elastin ratio of 1:8) into uniform micellar-like nanoparticles. The drug can be loaded in the SE8Y nanoparticles with an efficiency around 6.5% (65 ng doxorubicin/μg SE8Y), S2E8Y with 6%, and S4E8Y with 4%, respectively. In vitro studies with HeLa cell lines demonstrate that the protein polymers are not cytotoxic (IC50 > 200 μg/mL), while the doxorubicin-loaded SE8Y nanoparticles showed a 1.8-fold higher cytotoxicity than the free drug. Confocal laser scanning microscopy (CLSM) and flow cytometry indicate significant uptake of the SE8Y nanoparticles by the cells and suggest internalization of the nanoparticles through endocytosis. This study provides an all-aqueous, facile method to prepare nanoscale, drug-loaded SELPs packages with potential for tumor cell treatments.
1. Introduction
During the past three decades, various drug delivery vehicles (hydrogels, nanofibers, and nanoparticulate carriers), have been developed to improve therapeutic outcomes. These delivery vehicles decreased dosing, reduced toxicity and side effects, while improving the bioavailability of drugs.1−4 The material used for drug delivery generally should possess controlled structure, morphology, and function, as well as excellent mechanical properties.5 Therefore, biodegradable and biocompatible materials such as synthetic (aliphatic polyesters, polyglycolic acid, polylactic acid) and genetically engineered (polypeptides and proteins) polymers are preferred for encapsulation or binding of drugs.6,7
Synthetic polymers developed in recent years show good potential to control the release of the encapsulated drugs over time, but they usually need to be fabricated with organic solvents or harsh conditions, thus resulting in negative effects on the biocompatibility due to residual toxic solvents or degradation products.8 Most recently, genetically engineered protein polymers, consisting of repeating amino acid sequences from natural structural proteins or of de novo design, have been developed as diverse materials for controlled drug delivery. Compared with chemical synthesis, genetic engineering has enabled the protein materials with uniform composition, precisely controlled molecular weight and tunable structures for use in drug delivery.9,10 Repeating blocks from many natural structural proteins like collagen, silk, and elastin, with unique mechanical and biological properties, have been widely chosen as design units. For example, various elastin-like peptides have been constructed as block copolymers to form micellar-like nanoparticles for drug delivery,11−13 and lysine-modified chimeric spider silk proteins have been designed to generate nanoparticles for gene delivery.14 In addition, a series of silk-elastin-like protein polymers (SELPs) displaying unique mechanical properties have been produced, which combines repeating sequences derived from silk and elastin. Compared with elastin-based materials, the silk blocks in SELPs are able to crystallize into β-sheets via hydrogen bonding enabling robust materials formation. In addition, through tuning the ratio of silk and elastin blocks, material structures, strength and biodegradability can be controlled.15−18 In summary, SELPs form materials with unique mechanical properties, avoid a need for chemical cross-linking, can be processed in aqueous conditions, and have been explored for various biomedical applications.16
Although great progress has been made with SELPs for drug delivery, they have been mainly used as sol–gel systems for direct injection into solid tumors, which greatly limits their broader application.16 Fabricating SELPs into nanoparticulate carriers would be preferred because these forms can systemically administer drugs to target sites. Recently, Anumolu et al. generated highly uniform SELPs nanoparticles using an electrospray droplet evaporation technique and showed these nanoparticles encapsulated model therapeutic agents. However, the applied preparation technique is relatively sophisticated with limits to scalability.19,20 We have earlier reported the design of three genetically engineered silk elastin-like protein polymers (SE8Y, S2E8Y, S4E8Y) with silk (GAGAGS) to elastin block (GXGVP) ratios at 1:8, 1:4, and 1:2, respectively. These polymers spontaneously formed, or by thermal triggering, self-assemble into micellar-like nanoparticles either reversibly or irreversibly.21
The aim of this study was to investigate the applicability of genetically engineered SELP (SE8Y, S2E8Y, S4E8Y) nanoparticles as drug carriers. First, the hydrophobic molecule 1-anilinonaphthalene-8-sulfonic acid (1,8-ANS) was studied to determine the critical micelle concentration (CMC) and loading capacities of these protein polymers. Next, doxorubicin was loaded into the SELPs nanoparticles to characterize size and properties. Further, the cytotoxicity and cellular uptake of doxorubicin-encapsulated SELP nanoparticles against Hela cells was investigated.
2. Materials and Methods
2.1. Materials
All chemical reagents were purchased from Sigma-Aldrich (St. Louis, MO) or Fisher Scientific, Inc. (Pittsburgh, PA). Dulbecco’s phosphate-buffered saline (PBS), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), Dulbecco’s modified Eagle’s medium (DMEM), and fetal bovine serum (FBS) were purchased from Invitrogen (Carlsbad, CA). Doxorubicin hydrochloride was obtained from LC laboratories (Woburn, MA). To convert the drug into the hydrophobic form, doxorubicin hydrochloride was neutralized by phosphate buffer solution (0.1 M, pH 8.5) followed by centrifugation. The solid pellet was washed with water and lyophilized for drug loading experiment. Nickel-chelated sepharose resin was purchased from Qiagen (Valencia, CA). Methods used for the synthesis of recombinant silk-elastin-like protein polymers (SE8Y, S2E8Y and S4E8Y) with silk to elastin ratios at 1:8, 1:4, and 1:2, and molecular weights of 55.7, 53.0, and 47.8 kDa, respectively, have been described previously.21 The amino acid sequences of SE8Y is [(GAGAGS)(GVGVP)4(GYGVP)(GVGVP)3]14, S2E8Y is [(GAGAGS)2(GVGVP)4(GYGVP)(GVGVP)3]12 and S4E8Y is [(GAGAGS)4(GVGVP)4(GYGVP)(GVGVP)3]9. The purified proteins were dialyzed against deionized water for 5 days using Slide-A-Lyzer Dialysis cassettes (MWCO 3.5 kDa, Thermo Scientific) and then concentrated using Amicon Ultra-15 centrifugal filter units with Ultracel-30 membranes (Millipore, Billerica, MA). Protein concentrations were measured using a Pierce BCA Protein Assay kit (Product # 23225; Thermo Scientific, Rockford, IL) and the purity of the proteins was monitored via sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE).
2.2. Fluorescence Spectroscopy of Protein Polymers Mixed with 1,8-ANS and Critical Micelle Concentration (CMC) Measurement
Fluorescence measurements were performed using a Hitachi F-4500 fluorescence spectrophotometer equipped with a water-circulator cooled cell jacket. The 1,8-ANS was used as a hydrophobic fluorescent probe. Stock solutions of SELPs (10 mg/mL) and 1,8-ANS (8 mM) prepared with PBS were mixed together at various concentrations for the SELPs and 80 μM for 1,8-ANS. The mixture of SELPs and fluorescent probe was incubated at 25 °C for 10 min and emission spectra were recorded three times at an excitation wavelength of 370 nm.
Based on the 1,8-ANS excitation spectra, the fluorescence intensity was plotted against the logarithm of the protein polymers concentrations. The CMC was determined based on the crossover point.22
2.3. Preparation of Drug Loading Micellar-Like Nanoparticles
A 1 mg aliquot of doxorubicin was dissolved in 1 mL of silk-elastin-like protein polymer solution (1 mg/mL) and incubated at room temperature (25 °C) for 8 h under dynamic conditions (20 rpm). The drug-containing solution was then centrifuged at 6000 × g for 10 min to remove undissolved drug. The supernatant was positioned into a Slide-A-Lyzer Dialysis Cassette (MWCO 3.5 kDa, Thermo Scientific) and then subjected to dialysis against 1 L of distill water for 24 h to remove unloaded drugs. The water was refreshed every 4 h. The loading amount of doxorubicin was measured by UV absorbance at 480 nm, using a standard calibration curve experimentally obtained. The drug loading efficiency (DLE) was defined as follows: DLE = (mass of drug loaded in micelles/mass of drug-loaded micelles) × 100%.
2.4. Characterization of Drug-Loaded SELP Nanoparticles
Dynamic light scattering (DLS) was performed on a DynaPro Titan instrument (Wyatt Technology, Santa Barbara, CA) equipped with a temperature controller. Drug loading protein solutions (0.2 mg/mL) were filtered through a 0.45 μm filter prior to DLS measurements that were carried out at 25 and 37 °C, respectively. The samples were stabilized at the designated temperature for 10 min before measurement. To obtain the hydrodynamic radii (Rh), the intensity autocorrelation functions were analyzed by the Dynamics software (Wyatt Technology, Santa Barbara, CA).
The phase transition was determined by monitoring the absorbance of drug loading solutions (1 mg/mL) in PBS at 300 nm as a function of temperature on an Aviv 14DS UV–vis spectrophotometer (Aviv Biomedical, Lakewood, NJ). Absorbance was recorded after equilibrating a solution at the designated temperature for 30 s. Drug doxorubicin should not interfere with the measurement of the phase transition because the absorbance of doxorubicin at 300 nm would not change with the increase of temperature.
Cryogenic scanning electron microscope (cryo-SEM) was used to confirm the structure of the drug-loaded protein nanoparticles incubated at 37 °C. A drop (10 μL) of sample suspension was placed inside a custom-made copper holder and plunge-frozen in slushy nitrogen. After freezing, the sample was transferred under liquid nitrogen in a Leica EM VCT100 cryo transfer system (Leica Microsystems Inc., Buffalo Grove, IL) to a precooled Baltec MED020 high vacuum freeze-fracture coating system (Baltec, Baltzers, Liechtenstein, Germany) at −140 °C. The sample was then fractured and partially freeze-dried at −110 °C for 2 min, followed by coating with a thin layer (10 nm thick) of Pt/Pd prior to imaging in a precooled (−120 °C) cryo-SEM (Zeiss NVision 40, Carl Zeiss SMT Inc., Peabody, MA, U.S.A.).
2.5. Cytotoxicity Assay
HeLa cells were purchased from ATCC (Manassas, VA) and maintained in high glucose DMEM supplemented with 10% FBS and 1% penicillin/streptomycin at 37 °C in the presence of 5% CO2. Cells were seeded in a 96-well plate at a density of 10000 cells per well 24 h before the delivery experiments. At the day of delivery, varying doxorubicin encapsulated SELPs nanoparticles and free doxorubincin were added to cells directly. The cell viability was measured by MTT assay following a further 48 h of incubation. At the end of incubation, the cell culture medium was aspirated, and the cells were washed with PBS one time followed by incubation with 125 μL MTT solution (0.5 mg/mL in DMEM) for 4 h at 37 °C. The resulting formazan dyes were dissolved in 125 μL DMSO, and the absorbance of solutions was monitored at 595 nm on a SpectraMax M2 multimode microplate reader (Molecular Devices, Inc., Sunnyvale, CA).
2.6. Cellular Uptake of Doxorubicin Encapsulated SELP Nanoparticles
For the confocal laser scanning microscopy (CLSM) observations, HeLa cells were seeded in culture slides (BD Falcon) at a density of 50000 cells per vessel and incubated for 24 h. The cells were washed with PBS and incubated with doxorubicin encapsulated SE8Y (5 μM) or free doxorubicin (5 μM) in 0.5 mL of DMEM medium for 40 min or 4 h. At the end of incubation, DMEM medium was aspirated, and the cells were washed with PBS two times before fixed with 3.8% formalin solution, followed by cell nuclei staining with DAPI. The CLSM images were obtained on Axiovert 200 M inverted microscopes (Zeiss). For the FACS analysis of nanoparticle uptake, HeLa cells (10000 cells per well) were seeded in a 12-well plate a day before experiment and then incubated with doxorubicin encapsulated silk-elastin nanoparticles SE8Y for 4 h (5 μM) at 4 and 37 °C, respectively. At the end of incubation, cells were harvested, washed with PBS, and resuspended in PBS for FACS analysis on FACScalibur (BD Sciences).
3. Results and Discussion
3.1. Loading of Hydrophobic Fluorescent Molecules
In our previous study we constructed a series of silk elastin-like protein polymers (SELPs) with different ratios of silk (GAGAGS) to elastin (GXGVP) blocks and found these protein polymers could spontaneously form micellar-like nanoparticles.21 The assembly capability of these protein polymers depended on the ratio of silk to elastin blocks. In the current study using these proteins, we assessed the capacity of the nanoparticles to uptake hydrophobic molecules to investigate whether they could be employed for drug delivery. The three recombinant SELPs (SE8Y, S2E8Y, S4E8Y) with silk to elastin block ratios at 1:8, 1:4, and 1:2, were expressed and purified, and the purity of the proteins was confirmed by SDS-PAGE (Figure 1). Next, a hydrophobic fluorescent molecule, 1,8-ANS was introduced into the three protein solutions, respectively. Notably, 1,8-ANS fluoresces in a hydrophobic environment and has often been used to study the drug encapsulation capacity of amphiphilic block copolymers.12,22 The fluorescent spectra of 1,8-ANS with 0.5 mg/mL SE8Y, S2E8Y, and S4E8Y protein solutions at 25 °C is shown in Figure 1. The wavelengths of the emission peaks for SE8Y and S2E8Y were 475 ± 3 nm, and for S4E8Y was 509 ± 3 nm. The blue shift of the emission peak indicated the microscopic environments around 1,8-ANS were more hydrophobic.23 The fluorescence intensity of SE8Y at 475 ± 3 nm was around 2-fold higher than that of S2E8Y and 8-fold higher than S4E8Y, which indicated that the loading capacity of the hydrophobic molecule of SE8Y was significantly higher than that of S2E8Y and S4E8Y. This result may be related to the physical properties of the original proteins. Before adding 1,8-ANS, S4E8Y already formed a number of micellar-like nanoparticles with a dense cross-linked silk core,21 which might hinder the diffusion of the 1,8-ANS into the particles. In contrast, SE8Y solution was dominated by the free chains of the protein. The addition of 1,8-ANS might trigger the formation of micellar-like nanoparticles due to the change of hydrophobicity, leading to higher amounts of 1,8-ANS encapsulated into the particles. To verify this, the fluorescence spectra of urea (denaturant) added to SE8Y/ANS was also examined. As expected, the assembled structures were disrupted by urea as the fluorescence intensity dramatically decreased. To exclude the possibility of fluorescence of SELPs themselves, the protein solution of SE8Y was also included as a control (Figure 1).
Figure 1.

Fluorescence spectra of 1,8-ANS (80 μM) with 0.5 mg/mL SE8Y, S2E8Y, and S4E8Y in phosphate buffer saline at 25 °C. Fluorescence spectra of SE8Y and SE8Y in 8 M urea solution were included for controls. The inset shows the SDS-PAGE gel analysis of purified SE8Y, S2E8Y, and S4E8Y proteins.
Furthermore, the fluorescence intensity increased with the protein polymer concentration, which could be explained by an increased number of nanoparticles in the solution leading to a larger reservoir for the hydrophobic fluorophore. From the plot of fluorescence intensity versus protein polymer concentration, an abrupt increase of fluorescence value at 475 nm can be detected upon increasing protein polymer concentrations, indicating the formation of micelles and the transfer of 1,8-ANS into the micellar-like nanoparticles (Figure 2a–c). As illustrated in Figure 2d, the critical micelle concentration (CMC) for the SE8Y and S2E8Y was 0.125 (2.24 μM) and 0.25 mg/mL (4.72 μM), respectively (Figure 2d), which suggests stable micellar-like structures formed.23 S4E8Y did not show an abrupt increase of fluorescence upon increasing protein concentrations, indicating few particles formed. In summary, SE8Y showed the highest loading capacity for 1,8-ANS and the lowest CMC value.
Figure 2.

Fluorescence spectra of 1,8-ANS (80 μM) with varying concentration of SE8Y (a), S2E8Y (b), and S4E8Y (c) in PBS at 25 °C. (d) Fluorescence intensity of 1,8-ANS as a function of the logarithmic concentration of the SE8Y (closed dot) and S2E8Y (open dot). Arrows indicate the critical micellar concentrations (CMC).
3.2. Preparation of Drug Loading Protein Nanoparticles
Next, we examined whether these SELPs can be served as a potential drug delivery system. Hydrophobic drug doxorubicin was added to SELP solution followed by incubation at 25 °C. The drug loading efficiency value of SE8Y was 6.5%, whereas S2E8Y was 6% and S4E8Y was 4%. The difference in encapsulation efficiency of doxorubicin between SE8Y and S4E8Y was less than 2-fold, whereas the difference in the encapsulation of 1,8-ANS was about 8-fold. This could be explained by the different encapsulation mechanism and measurement of loading capacity for doxorubicin and 1,8-ANS. The average hydrodynamic radii (Rh) of Dox-loaded nanoparticles were 50 ± 10 (SE8Y), 72 ± 11 (S2E8Y), and 142 ± 10 nm (S4E8Y), respectively, at 25 °C (Figure 3a). Before adding Dox, the average Rh of SE8Y was only 5.2 ± 1.8 nm, which is suggestive of free chains. This result demonstrated that Dox triggered the self-assembly of SE8Y into nanoparticles, the same as 1,8-ANS. Most importantly, these triggers might be expanded to other hydrophobic molecules. Based on our previous studies, SELPs tended to assemble into micellar-like particles with silk blocks buried in the core, thus, the hydrophobic compounds likely bind to the silk blocks (Figure 3b). The average Rh of S2E8Y and S4E8Y were 29 ± 9.8 and 89 ± 7.3 nm, respectively, which was much smaller than that of Dox-loaded nanoparticles. This might be due to the hydrophobic association of small particles into larger ones. Notably, when we added hydrophilic doxorubicin hydrochloride into SELP protein solutions, much less doxorubicin was encapsulated into the protein nanoparticles (DLE < 0.1%), which indicated that hydrophobic interactions played a dominant role in the encapsulation process instead of other interactions including hydrogen bonding.
Figure 3.

Characterization of doxorubicin-encapsulated nanoparticles. (a) Sizes of SE8Y/Dox, S2E8Y/Dox, and S4E8Y/Dox complexes at 25 °C (gray bar) and 37 °C (black bar) with respective protein solutions as controls (white bar). (b) Turbidity profiles of the SE8Y/Dox (S1/Dox), S2E8Y/Dox (S2/Dox), and S4E8Y/Dox (S4/Dox) complexes at 1 mg/mL as a function of temperature. (c) Doxorubicin-triggered self-assembly of SE8Y into micellar-like nanoparticles. (d) Representative cryogenic scanning electron microscope (cryo-SEM) of Dox-loaded SE8Y nanoparticles.
When Dox-loaded nanoparticles of SELPs were incubated at 37 °C (physical temperature), the average Rh of SE8Y particles increased to 139 ± 1.8 nm. In contrast, the sizes of nanoparticles of S2E8Y and S4E8Y did not change significantly. This might be explained by the lower phase transition temperature (∼35 °C) of drug-loaded SE8Y (Figure 3c). Notably, the phase transition temperature (Tt) of the SELPs was significantly decreased following encapsulation of doxorubicin. Tt value usually changes with varying hydrophobicity of the guest residue (X) of the elastin blocks (GXGVP). Hydrophobic guest residues typically depress the Tt value, and hydrophilic residues elevate the Tt value.23−25 Similarly, the encapsulation of hydrophobic molecules doxorubicin might increase the hydrophobicity of protein solutions, thus, down-regulating the Tt. Interestingly, SE8Y and S2E8Y were found to exhibit a two-step thermal transition in our previous study. The first transition was believed to be caused by the self-assembly of the free chains of SELPs into small particles.21 Following encapsulation of doxorubicin, the free chains of SELPs have been induced to assemble into particles (Figure 3a). Thus, the first transition of doxorubicin-loaded SE8Y and S2E8Y was not observed. To directly visualize the morphology and size of the Dox-loaded SE8Y nanoparticles incubated at 37 °C, we imaged the samples by cryogenic scanning electron microscope (cryo-SEM). SEM imaging confirmed the morphology of Dox-loaded nanoparticles that displayed nanoparticle spheres with average diameter ranging from 250 to 300 nm, which was consistent with that determined by DLS at 37 °C (Figure 3d).
The stability of drug-loaded SE8Y nanoparticles under physiological conditions was also examined. After 48 h incubation in PBS supplemented with 10% FBS, the average Rh of drug-loaded SE8Y nanoparticles increased from 142 to 181 nm (Figure 4) with an apparent increase of polydispersity, which might be due to the interactions of particles with serum proteins. However, no obvious disruption of particles was observed during the incubation.26 The stability of these nanoparticles might be due to the physical cross-linking of silk units in SELPs, which avoids the need to introduce other cross-linking agents.
Figure 4.

DLS size distribution profiles for doxorubicin encapsulated SE8Y (S1/Dox) nanoparticles in phosphate buffer saline with 10% FBS at 37 °C over time.
3.3. In Vitro Cytotoxicity
As a good drug delivery vehicle, low cytotoxicity of the vehicle itself is important for biomedical applications.27,28 Cytotoxicity of SELPs in vitro was determined against HeLa cells via MTT assay. As shown in Figure 5a, cell viability was still above 90% when the concentration of protein polymers was increased to 200 μg/mL, indicating the low cytotoxicity and high potential of SELPs as chemotherapy drug carrier. When the cells were treated by doxorubicin encapsulated SELPs nanoparticles, cell viability was significantly decreased in comparison with DMEM treated controls. The IC50 (the concentration required for 50% inhibition of cellular growth) values were determined as 0.95 μM for free Dox, 0.55 μM for doxorubicin encapsulated SE8Y (Dox-S1), 0.61 μM for doxorubicin encapsulated S2E8Y (Dox-S2), and 1.35 μM for doxorubicin encapsulated S4E8Y (Dox-S4), respectively (Figure 5b,c). Free doxorubicin is considered to directly diffuse through the membranes into the cell and nucleus. In contrast, the slightly lower level of toxicity for S4/dox compared to free doxorubicin might be due to the additional steps required for S4/dox to be taken up by the cells, along with the associated complex process for drug release into the cytosol and nucleus. Therefore, 48 h might be not enough for the complete release of doxorubicin from the S4/dox inside a cell. Importantly, Dox-S1 and Dox-S2 showed a higher cytotoxicity than the free drug. This might be explained by the controlled release of drugs leading to a higher concentration of doxorubicin entering the nuclei. However, the detailed drug release mechanism was not clear. Notably, in the present study, in vitro release experiments cannot adequately mimic in vivo systems. We tried various PBS buffers with pHs from 4.0 to 8.0 and with or without FBS for the in vitro release experiments, but we did not find any significant release of drug over 4 days. Only when we added proteases like elastase did we see small amounts of drug release. Therefore, enzymatic degradation of SELPs contributes to the release of the doxorubicin. This inference can also be supported by the report on the degradation of the elastin block-based nanoparticles by proteases (elastase and collagenase), and comparable proteolysis occurs after cellular uptake of nanoparticles by murine hepatocytes.29
Figure 5.

(a) Cytotoxicity of the SE8Y (S1), S2E8Y (S2), and S4E8Y (S4) protein polymers against Hela cells. (b) In vitro cytotoxicity of SE8Y/Dox (S1/Dox), S2E8Y/Dox (S2/Dox), and S4E8Y/Dox (S4/Dox) complexes and free doxorubicin against HeLa cells. (c) IC50 values of S1/Dox, S2/Dox, and S4/Dox complexes and free doxorubicin against HeLa cells.
3.4. Cellular Uptake and Intracellular Trafficking of Dox-Loaded SE8Y Nanoparticles (S1/Dox)
Due to the higher drug loading capacity of SE8Y and the cytotoxicity of its Dox-loaded nanoparticles, the intracellular trafficking and fate of S1/Dox were monitored by confocal laser scanning microscopy. CLSM images (Figure 6) of HeLa cells treated with S1/Dox and free Dox indicated the different uptake pathways of nanoparticles and free Dox. At 40 min of incubation, free doxorubicin accumulated in cell nuclei, which is understandable because free Dox enters cells quickly via a membrane diffusion pathway. S1/Dox treated cells, however, mostly accumulated the nanoparticles in cytoplasm, which indicates that nanoparticles might enter cells via an endocytosis pathway. It is worth to note that the cellular uptake of drug encapsulated nanoparticles via endocytosis can be a rapid process, even within 15 min.30 A longer incubation of S1/Dox with HeLa cells facilitated the diffusion of Dox into nuclei, which may arise from the gradual intracellular release of DOX from the S1/Dox nanoparticles. We believe that the controlled release of DOX from S1/Dox could contribute to the higher delivery efficiency of SE8Y nanoparticles.31,32 The internalization and endocytosis of S1/DOX nanoparticles were further confirmed and studied by a FACS quantification of nanoparticle uptake. Endocytosis is known as an energy-dependent process, the incubation of nanoparticles with cells at a lower temperature can inhibit the efficient cellular uptake of nanoparticles.33 As shown in Figure 7, a decreased DOX fluorescence intensity was observed for cells incubated with S1-Dox at 4 °C compared to that at 37 °C, suggesting the lower cellular uptake of S1-Dox nanoparticles at 4 °C. This method has been widely used to characterize the uptake/endocytosis of other types of doxorubicin encapsulated nanoparticles.34
Figure 6.

Delivery of doxorubicin into HeLa cells via SE8Y nanoparticles with different times: (a) 40 min and (b) 4 h by CLSM. Nucleus was stained with DAPI (blue signal). The scale bar is 10 μm.
Figure 7.
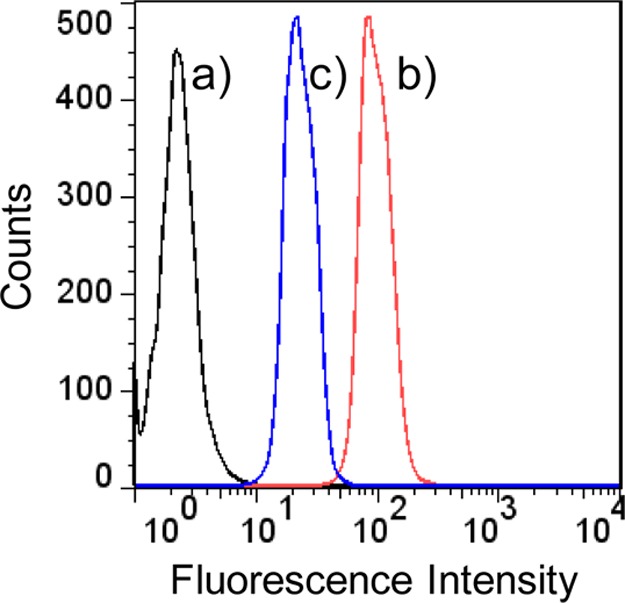
Flow cytometry analysis of HeLa cells treated with PBS (a) and S1-Dox nanoparticles at 37 °C (b) and 4 °C (c). The weaker doxorubicin fluorescence observed at 4 °C compared to that at 37°, indicating a decreased cellular uptake of S1-Dox nanoparticles at a lower temperature and suggesting the uptake of S1-Dox is an energy-dependent process.
4. Conclusions
Engineered silk-elastin-like protein polymers, triggered by hydrophobic molecules, assembled into nanoparticles with potential for use for the delivery of hydrophobic drugs. A major advantage of SELPs system is that nanoparticles, fully biocompatible, can be fabricated and loaded with an all-aqueous process under mild conditions, which is important for the encapsulation of unstable drugs. In addition, genetically encoded synthesis provides a simple and accurate method to control particle size, capacity of drug loading and incorporate other biologically active domains. We anticipate that this hydrophobic drug triggered SELPs nanoparticle system possesses good potential for the future direction related to cancer treatments.
Acknowledgments
This work was supported by the NIH P41 Tissue Engineering Resource Center (P41 EB002520) and the Shanghai Pujiang Program (13PJ1404800). Further support by the Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning is appreciated. This work was performed in part at the Center for Nanoscale Systems (CNS), a member of the National Nanotechnology Infrastructure Network (NNIN). CNS is part of the Faculty of Arts and Sciences at Harvard University.
Author Contributions
† These authors contributed equally to this work (X.-X.X. and M.W.).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Uhrich K. E.; Cannizzaro S. M.; Langer R. S.; Shakesheff K. M. Chem. Rev. 1999, 99, 3181–3198. [DOI] [PubMed] [Google Scholar]
- Frandsen J. L.; Ghandehari H. Chem. Soc. Rev. 2012, 41, 2696–2706. [DOI] [PubMed] [Google Scholar]
- Allen T. M.; Cullis P. R. Science 2004, 303, 1818–1822. [DOI] [PubMed] [Google Scholar]
- Saha R. N.; Vasanthakumar S.; Bende G.; Snehalatha M. Mol. Membr. Biol. 2010, 27, 215–231. [DOI] [PubMed] [Google Scholar]
- Altman G. H.; Diaz F.; Jakuba C.; Calabro T.; Horan R. L.; Chen J.; Lu H.; Richmond J.; Kaplan D. L. Biomaterials 2003, 24, 401–416. [DOI] [PubMed] [Google Scholar]
- Langer R.; Peppas N. A. AIChE J. 2003, 49, 2990–3006. [Google Scholar]
- Liechty W. B.; Kryscio D. R.; Slaughter B. V.; Peppas N. A. Annu. Rev. Chem. Biomol. Eng. 2010, 1, 149–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morlock M.; Koll H.; Winter G.; Kissel T. Eur. J. Pharm. Biopharm. 1997, 43, 29–36. [DOI] [PubMed] [Google Scholar]
- Maskarinec S. A.; Tirrell D. A. Curr. Opin. Biotechnol. 2005, 16, 422–426. [DOI] [PubMed] [Google Scholar]
- Langer R.; Tirrell D. A. Nature 2004, 428, 487–492. [DOI] [PubMed] [Google Scholar]
- Kim W.; Chaikof E. L. Adv. Drug Delivery Rev. 2010, 62, 1468–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita Y.; Mie M.; Kobatake E. Biomaterials 2009, 30, 3450–3457. [DOI] [PubMed] [Google Scholar]
- Chilkoti A.; Dreher M. R.; Meyer D. E. Adv. Drug Delivery Rev. 2002, 54, 1093–1111. [DOI] [PubMed] [Google Scholar]
- Numata K.; Hamasaki J.; Subramanian B.; Kaplan D. L. J. Controlled Release 2010, 146, 136–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megeed Z.; Cappello J.; Ghandehari H. Adv. Drug Delivery Rev. 2002, 54, 1075–1091. [DOI] [PubMed] [Google Scholar]
- Gustafson J. A.; Ghandehari H. Adv. Drug Delivery Rev. 2010, 62, 1509–1523. [DOI] [PubMed] [Google Scholar]
- Hu X.; Wang X.; Rnjak J.; Weiss A. S.; Kaplan D. L. Biomaterials 2010, 31, 8121–8131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X.; Lu Q.; Sun L.; Cebe P.; Wang X.; Zhang X.; Kaplan D. L. Biomacromolecules 2010, 11, 3178–3188. [DOI] [PubMed] [Google Scholar]
- Anumolu R.; Gustafson J. A.; Magda J. J.; Cappello J.; Ghandehari H.; Pease L. F. ACS Nano 2011, 5, 5374–5382. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Jaworek A. Powder Technol. 2007, 176, 18–35. [Google Scholar]
- Xia X. X.; Xu Q.; Hu X.; Qin G.; Kaplan D. L. Biomacromolecules 2011, 12, 3844–3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W.; Thévenot J.; Ibarboure E.; Lecommandoux S.; Chaikof E. L. Angew. Chem., Int. Ed. 2010, 49, 4257–4260. [DOI] [PubMed] [Google Scholar]
- Urry D. W.; Urry K. D.; Szaflarski W.; Nowicki M. Adv. Drug Delivery Rev. 2010, 62, 1404–1455. [DOI] [PubMed] [Google Scholar]
- Urry D. W.; Hugel T.; Seitz M.; Gaub H. E.; Sheiba L.; Dea J.; Xu J.; Parker T. Philos. Trans. R. Soc., B 2002, 357, 169–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreher M. R.; Simnick A. J.; Fischer K.; Smith R. J.; Patel A.; Schmidt M.; Chilkoti A. J. Am. Chem. Soc. 2008, 130, 687–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaniel J. R.; Bhattacharyya J.; Vargo K. B.; Hassouneh W.; Hammer D. A.; Chilkoti A. Angew. Chem., Int. Ed. 2013, 52, 1683–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKay J. A.; Chen M.; McDaniel J. R.; Liu W.; Simnick A. J.; Chilkoti A. Nat. Mater. 2009, 8, 993–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seib F. P.; Jones G. T.; Rnjak-Kovacina J.; Lin Y.; Kaplan D. L. Adv. Healthcare Mater. 2013, 2, 1606–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah M.; Hsueh P. Y.; Sun G.; Chang H. Y.; Janib S. M.; MacKay J. A. Protein Sci. 2012, 21, 743–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi C. H.; Hao L.; Narayan S. P.; Auyeung E.; Mirkin C. A. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 7625–7630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren D.; Kratz F.; Wang S. W. Small 2011, 7, 1051–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- dos Santos T.; Varela J.; Lynch I.; Salvati A.; Dawson K. A. Small 2011, 7, 3341–3349. [DOI] [PubMed] [Google Scholar]
- Canton I.; Battaglia G. Chem. Soc. Rev. 2012, 41, 2718–2739. [DOI] [PubMed] [Google Scholar]
- Arora H. C.; Jensen M. P.; Yuan Y.; Wu A.; Vogt S.; Paunesku T.; Woloschak G. E. Cancer Res. 2012, 72, 769–778. [DOI] [PMC free article] [PubMed] [Google Scholar]