

Abstract
Here we investigated whether changes in neurogenesis and BDNF expression are possible mechanisms involved in the depression-like symptom during the withdrawal/abstinence period after chronic binge-pattern alcohol consumption given the limited number of studies addressing the link between these factors in the adolescent brain. Forty-seven male Sprague-Dawley rats were used in the study and the experimental protocol started when rats were 25-days old. Rats were assigned to either: (a) ethanol or (b) control group. Animals in each group were further randomized to receive either: brain-derived neurotrophic factor (BDNF) receptor agonist or vehicle. Rats were trained to self-administer ethanol and the binge protocol consisted of daily 30-min experimental sessions 4 hours into the dark period for 12 days. Two days after the last drinking session, rats were tested in the sucrose preference test to evaluate anhedonia and the open field test after habituation to evaluate behavioral despair. Our data showed that: (1) self-administration of alcohol in a binge-like pattern causes inebriation as defined by the National Institute on Alcohol Abuse and Alcoholism (NIAAA) and this pattern of alcohol exposure is associated with the development of depression-like symptom; (2) no significant difference in blood alcohol levels between the 2 ethanol groups; and (3) chronic binge drinking resulted in the development of depressive phenotype, decrease survival and neuronal differentiation of neural progenitor cells in the hippocampus, and decrease BDNF effect during the withdrawal period. But the most important finding in our study is that augmenting BDNF actions through the use of tyrosine kinase receptor B (TrkB, a BDNF receptor) agonist restored neurogenesis and abolished the alcohol-induced anhedonia and despair behaviors seen during the withdrawal/abstinence period. Our results suggest that BDNF might be a molecule that can be targeted for interventions in alcoholism–depression co-incidence.
Keywords: sucrose preference test, open field test, hippocampus, TrkB, immunohistochemistry, doublecortin
Binge drinking in young people is on the rise in the United States (Naimi et al., 2003), Europe (Vicki et al., 2010), and in most developing countries throughout the world (Courtney and Polich, 2009). Reports indicate that alcohol consumption during adolescence is approximately 12% in eighth graders, 22% in tenth graders and 28% in twelfth-graders (Johnston et al., 2004, Masten et al., 2008). Moreover, it has been reported that more than half of 12 to 17 year-olds consume alcohol in a binge-like fashion (Koob and Le Moal, 1997, Crews and Nixon, 2009) making it a common pattern of drinking among teenagers (Johnston et al., 2004). Although some adolescents may consider alcohol use as a “rite of passage,” chronic alcohol consumption especially in a binge-like pattern is not a benign condition that usually resolves with age. In fact, there is increasing evidence that initiation of alcohol consumption during adolescence increases the probability of developing alcohol abuse and dependence in adulthood (Grant and Dawson, 1997).
Heavy drinking during adolescence is a concern because their brain responds differently to the effects of alcohol than the adult brain, probably because it is still in a developmental state (Spear, 2000, Bava et al., 2010). Indeed, clinical studies in binge drinking and alcohol-dependent adolescents’ show hippocampal volume reductions seen in magnetic resonance imaging as well as functional impairments in learning and memory processes (De Bellis et al., 2000, Nagel et al., 2005, Medina et al., 2007, Schulteis et al., 2008, Schweinsburg et al., 2010). Several reports consistently show that the adolescent limbic system is susceptibility to alcohol’s memory-impairing and mood-altering properties (Crews et al., 2000, White and Swartzwelder, 2005). The drive for excessive alcohol consumption to neurotoxic levels seen in adolescents during binge drinking may be because they are resistant to the motor-impairing and sedative effects of alcohol but sensitive to its rewarding and reinforcing properties (Doremus et al., 2010), when compared to adults. Clinical reports show that young people between 16 to 19 years of age who engage in alcohol use in a binge-like pattern are six times more likely to experience depression compared to their non-drinking counterparts (Deykin et al., 1987, Boden and Fergusson, 2011). Taken together, these reports suggest that structural changes in the limbic area of the brain may underlie the cognitive and affective impairments seen as a consequence of chronic alcohol use.
A key limbic structure particularly susceptible to alcohol-related damage is the hippocampal formation (De Bellis et al., 2000, Nagel et al., 2005, Boden and Fergusson, 2011). Many believe that the structure and function of the hippocampus rely upon the neural progenitor cells (NPCs) present in the subgranular zone of the dentate gyrus and its role in constitutive neurogenesis (Kempermann et al., 2004, Imayoshi et al., 2008). Accumulating evidence indicates that hippocampal neurogenesis play a role in learning and memory as well as depression [reviewed in (Gould et al., 1999)] suggesting the possibility that alcohol effects on neurogenesis may be a contributing factor in the cognitive and affective impairments seen in alcohol use.
Structural changes that occur in the brain during alcohol consumption and withdrawal are accompanied by several molecular mechanisms. Brain-derived neurotrophic factor (BDNF) is a pivotal molecule identified as a major regulator of structural changes in the brain in response to alcohol use. For example, BDNF is implicated in neurogenesis (Lee and Son, 2009, Shen and Cowan, 2010), depression (Duman and Monteggia, 2006, Neto et al., 2011), and the synaptic changes seen in the adult brain associated with drug addiction (Pandey et al., 2006). In the this study we examined whether changes in neurogenesis and BDNF expression are possible mechanisms involved in the depression-like symptom during the withdrawal/abstinence period induced by chronic binge-pattern alcohol consumption given the limited number of studies addressing the link between these factors in the adolescent brain.
Experimental Procedures
Animal Model
A total of forty-seven male Sprague-Dawley rats were used in the study, housed in pairs upon arrival from the breeder, and acclimated to their new environment for one week before starting the study to provide them the advantage of dealing with the potential stress related to the experimental manipulations. The experimental protocol (Fig. 1) started when rats were 25 days old to closely parallel the human adolescent population. Rats were randomly assigned to either: (a) ethanol + tyrosine kinase B (TrkB) agonist (n=12), (b) ethanol group + vehicle (n=12), or (c) control + TrkB agonist (n=12), or (d) control + vehicle (n = 11). Rats in the ethanol + TrkB agonist (ETOH + TrkB) group were trained to self-administer sweetened ethanol and given the TrkB receptor agonist intraperitoneally (i.p.). Rats in the ethanol + vehicle (ETOH + vehicle) group were trained to self-administer sweetened ethanol and given equal volume of vehicle solution i.p. Animals in the control (CON) groups received water but the TrkB group received the receptor agonist i.p. while the vehicle group received equal volume of vehicle injections. Animals had free access to food and water and handled daily throughout the study. The animal housing room was maintained at 24ºC ± 1.5°C, room humidity controlled and a 14:10 hour light:dark cycle was maintained. All animals were housed in the same room so that temperature, humidity, and lighting conditions were similar for all groups. All experimental protocols were approved by the Institutional Animal Care and Use Committee and in accordance with the National Institutes of Health guidelines.
Figure 1. Study Timeline.
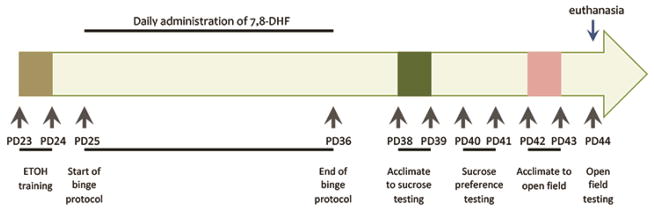
PD – postnatal day, ETOH – alcohol.
Ethanol Oral Self-administration
Rats were trained to self-administer ethanol versus water in a two-bottle choice home cage situation. Training consisted of 2 daily sessions starting 4 hours into the dark cycle where food was removed and two-bottles were placed on the cage lid: one containing water and the other sweetened ethanol. During training, rats remained in their home cage and allowed to drink from the 2-bottle system throughout the entire dark cycle. Sweetened ethanol (10% w/v) was prepared with 95% ethyl alcohol and tap water + 3% glucose and 0.125% saccharin (Sigma Aldrich, St. Louis, MO). All rats acquired the behavior after the training sessions and the experimental period started the following day. During the experimental period, food was removed and bottles with ethanol and water were attached to the cage lid with stainless steel springs to reduce spilling staring 4 hours into the dark cycle and rats were allowed to drink for 30 minutes. The position of the bottles was alternated daily to avoid possible side preference. All bottles were weighed immediately before and after the 30-min drinking sessions. Differences in bottle weights were converted to volume intakes by accounting for the ethanol solution density (weight in grams/0.9868). The binge drinking protocol was conducted for 12 consecutive days. Level of intoxication was assessed using the behavioral intoxication scale (Knapp and Crews, 1999): 0 – normal; 1 – hypoactive; 2 – presence of ataxia; 3 – ataxia accompanied by delayed righting reflex; 4 – loss of righting reflex; 5 – loss of eye blink.
Drug Administration
The selective BDNF TrkB receptor agonist 7,8-dihydroflavone (7,8-DHF) was given i.p. at 5 mg/Kg. The drug was purchased from TCI America (Portland, OR) and dissolved in dimethyl sulfoxide (DMSO) and diluted with a final DMSO concentration of 0.02%. Vehicle solution consisted of the same DMSO concentration and equal volume of injection was given. Vehicle or 7,8-DHF was given daily 30 minutes prior to the 12-day drinking sessions. The TrkB receptor agonist, 7,8-DHF has been shown to cross the blood-brain barrier and induces dimerization and autophosphorylation of the tyrosine receptor leading to activation of its downstream signaling (Jang et al., 2010). Moreover, 7,8-DHF presents better pharmacokinetic properties than BDNF and higher TrkB binding affinity, thus it mimics BDNF ligand function.
Bromodeoxyuridine (BrdU) Injections
The thymidine analog BrdU (Chemicon, Temecula, CA) was used to label proliferating cells. BrdU is incorporated into the genetic materials on mitotic division within 2 hours after injection, after which it can be detected immunohistochemically in the daughter cells (Kuhn et al., 1996). BrdU was dissolved in 0.9% sterile NaCl and filtered at 22 μm. The resulting solution was injected at 75 mg/kg intraperitoneally. BrdU injections were given for 3 consecutive days starting on postnatal day 25 (start of experimental protocol).
Testing for ‘depressive phenotype’
To assess depression-like symptoms, the sucrose preference and open field tests were conducted 2 days after the last drinking session. For the sucrose preference test, rats were acclimated to a two-bottle choice of drinking water and 1% sucrose solution for 2 days followed by 2 days of testing. On the day of testing, two pre-weighted bottles of: 5% sucrose solution and tap water were presented. To prevent possible effects of side preference in drinking behavior, the position of the bottles was switched after 24 hours of testing. No food or water deprivation was applied before or during the test. Liquid consumption from each bottle corrected by body weight was used to calculate sucrose solution intake, water intake and total consumption by the end of the 48-hour period. Sucrose preference was calculated using the following equation: sucrose preference (%) = sucrose intake/(sucrose intake + water intake) × 100.
The open field test was conducted the day after sucrose preference test between 10 a.m. to 12 p.m. The test was conducted using a black Plexiglas square box (100 × 100 × 50 cm). Before testing, rats were habituated to the testing apparatus by placing them individually in the box for 20 minutes × 2 days then tested the day after. The box was cleaned with 70% ethanol after each rat. The box was divided into two areas: peripheral area and center area (60 × 60 cm). During testing, rats were placed in the peripheral area then allowed to freely explore for 5 minutes. Activity in the open field was recorded with a camera connected to a computerized videotracking system (EthoVision® XT, v8.5; Noldus Technology, Leesburg, VA), which provided measures of immobility time, horizontal movement (distance covered during locomotion), and number of entries in the center area.
Blood Alcohol Concentration
Blood samples were collected from the saphenous vein 60 minutes following the 30-min binge session. Samples (~ 0.5 ml) were collected every other day then centrifuged for 5 minutes at 1,800 × g to separate the plasma and then stored at −20°C until time of assay. Serum BAC levels were determined using the Blood Ethanol Analyzer Model GL5 (Analox Instruments, Lunenburg, MA) calibrated with a 300 mg/dL external standard. Measurements were performed in triplicate, averaged and used as one individual data point.
Tissue Preparation
At the end of the experimental protocol, all rats were euthanized. Euthanasia involved use of CO2 inhalation followed by decapitation once rats became unresponsive. At the time of sacrifice, hippocampal tissues used for Western blot and ELISA were dissected and placed in liquid nitrogen until processed. Brains used for immunohistochemistry were placed in a fixative solution (4% paraformaldehyde, pH 7.4) overnight then cryoprotected with 30% sucrose in 0.1 M phosphate-buffered saline solution containing 25% ethylene glycol and stored at −20 °C until they were sectioned. Coronal sections were obtained throughout the entire hippocampal formation using a cryostat at 20μm thickness.
Immunohistochemistry
The free-floating section method was used for to examine neurogenesis in the dentate gyrus (Briones et al., 2005, Briones and Woods, 2011). For detection of BrdU-labeled nuclei, DNA was denatured to expose the antigen before incubation in anti-BrdU primary antibody. Briefly, free-floating sections were pretreated in 50% formamide/50% 2× saline-sodium citrate buffer (SSC) at 65 °C for 2 h, rinsed in 2× SSC, and then incubated in 2 N HCl at 37 °C for 30 min. Tissues were rinsed in borate buffer (pH 8.5) for 15 minutes and placed in 0.6% H2O2 in Tris-buffered saline (TBS) for 30 minutes to block endogenous peroxidase, followed by several rinses in TBS (pH 7.5). Tissues were placed in TBS/0.1% Triton X-100/3% donkey serum (TBS-TS) for 1 hour followed by incubation with anti-BrdU primary antibodies at a concentration of 1:400 (monoclonal mouse; Boehringer Mannheim; Indianapolis, IN) in TBS-TS overnight at 4 °C. The following day, the primary antibody was detected using biotinylated immunoglobulin G (IgG) donkey anti-mouse secondary antibodies (Vector Laboratories; Burlingame, CA) at a concentration of 1:200 for 2 hours. Tissues were rinsed in TBS and incubated in avidin-biotin complex (ABC kit; Vector Laboratories) for 1 hour at room temperature. Immunoreactions were visualized by treatment of section with hydrogen peroxide and 3,3 - diaminobenzidine tetrahydrochloride in Tris buffer (pH 7.3). After thorough rinsing, the tissue sections were mounted on gelatin-coated slides and dried, and coverslips were applied. To minimize intergroup and interbrain staining variability and to ensure reproducibility of results, tissues from all experimental groups were run simultaneously and under identical conditions.
To determine survival and differentiation of newly formed cells, the total number of BrdU-positive cells in the granule cell layer and its corresponding sample volume were determined in 8 coronal sections, 240 μm apart, using the optical disector method (StereoInvestigator, MicroBrightfield, Colchester, VT). Briefly, each section was examined at a magnification of 40x, and an unbiased counting frame was positioned randomly across the dentate gyrus area. The number of labeled cells was related to the number of sections counted multiplied by the reference volume to provide an unbiased estimation of the total number of BrdU-positive cells as previously described (Briones et al., 2005, Briones and Woods, 2011). To determine phenotype of surviving newly formed cells, the antibody anti-doublecortin (1:400; Santa Cruz Biotechnology), marker for immature neurons, was used. Doublecortin immunoreactivity was quantified by optical densitometry with an image analysis software (Bioquant Image Analysis, R&M Biometric, Nashville, TN). Densitometry measurements were divided by the area of the tissue section and expressed as pixels/mm2.
Enzyme-linked Immunosorbent Assay (ELISA)
To detect BDNF level, ELISA was carried out using the BDNF Emax ImmunoAssay System kit (Promega, Madison, WI) according to manufacturers specifications. Frozen hippocampal tissues (0.2 g) was homogenized with a glass homogenizer in extraction buffer (pH 7.5) containing Tris-acetate 20 mM, NaCl 150 mM, 1 mM EDTA, 1 mM Na pyrophosphate, 100 mM PMSF, leupeptin 1 mg/ml and centrifuged at 12,000 × g for 20 minutes at 4°C. The supernatant was collected and total protein was determined by bicinchoninic acid (BCA) protein assay reagent kit (Pierce, Milwaukee, WI). Briefly, 96-well plate precoated with monoclonal antibody specific for BDNF were incubated at 4°C overnight on an orbital plate shaker (≈250 rpm) then washed 2× with wash buffer (pH 7.6) containing Tris HCl 20 mM, NaCl 150 mM, and 0.05% Tween. The blocking buffer (200 μl) provided by the manufacturer was then added to the well together with the sample (100 μl) then incubated at room temperature for 1 hour followed by several washes. After washing, the immobilized antigen was incubated with monoclonal anti-BDNF overnight at 4°C. The plates were washed again and then incubated with anti-mouse IgG horseradish peroxidase conjugate for 2 hours at room temperature. Tetramethyl benzidine/peroxidase substrate solution was added to the wells after washing to produce the color reaction then incubated for 15 minutes. Color reaction was stopped with 1N HCl (100 μl) and reaction was read in a microplate reader (Bio-Tek, Winooski, VT) at a wavelength of 450 nm (650-nm reference wavelength). BDNF concentration was determined from the regression line for the BDNF standard corrected for the total amount of protein in the sample and assay sensitivity ranged from 7.8 to 500 pg/ml. Assay was performed in triplicates and measurements were averaged and used as one individual data point for statistical analysis.
Western Blot
To detect levels of TrkB and phosphorylated TrkB (p-TrkB), 0.5 g of frozen hippocampal tissues were homogenized and centrifuged at 25,000 × g for 20 minutes as previously described (Briones et al., 2006, Briones et al., 2009). Aliquots from the supernatant were removed for protein determination. Protein concentration in samples was determined using the BCA-Protein assay (Pierce, Rockford, IL). The primary antibodies used are: 1) anti-TrkB (1:750, BD Biosciences, San Jose, CA) and 2) anti-phospho-TrkB (1:1000, Santa Cruz Biotechnology, CA). For Western blot analyses, equal amounts of protein (40 μg) from each rat were loaded and separated by SDS-PAGE gel electrophoresis in 8% – 16% acrylamide gradient gels. The protein bands were transferred electrophoretically to nitrocellulose membranes (Amersham, Piscataway, NJ) stained with 0.5% Ponceau Red to visualize total proteins, then destained. Non-specific binding sites were blocked then nitrocellulose membranes were incubated overnight at 4°C with gentle agitation in the primary antibody. The secondary antibodies used are horseradish peroxidase-conjugated immunoglobulin (Sigma, St. Louis, MO) and the Super Signal chemiluminescense substrate kit (Pierce, Rockford, IL) was used to visualize immunoreactive bands. After visualization, the membranes were then stained with Amido-Black to qualitatively verify protein loading. Samples were analyzed in quadruplicates and measurements were averaged and used as one individual data point for statistical analysis. Quantification of differences in protein bands between samples was done using densitometric analysis (Scion Image Beta 4.0.2; Frederick MD). The internal control β-actin was used to standardize experimental values in densitometric analysis.
Statistical Analysis
The SAS general linear model (SAS Institute, North Carolina) procedures for analysis of variance (ANOVA) were used to examine effects of experimental conditions (ethanol vs. saline groups; TrkB agonist vs. vehicle) on neurogenesis, BDNF effects, sucrose preference test, and open-field test. Repeated measures ANOVA were used to examine effects of chronic binge-pattern alcohol exposure on BAC. All error bars represent ± standard error of the mean (SEM) of the sample size used in the study.
Results
Binge alcohol consumption causes intoxication
Blood alcohol concentration (BAC) was determined from the samples analyzed every other day and repeated measures analysis showed no significant difference in levels over the course of the ethanol exposure paradigm (Fig.2A). Also, no significant difference was seen in alcohol consumption between the ETOH + vehicle and ETOH + TrkB groups (Fig. 2B). On average, rats in the ethanol groups consumed greater than 0.75 g/Kg alcohol during the 30-minute drinking session and produced BAC ≥ 0.08 g%, which reflects the defined BAC threshold for binge drinking by the National Institute of Alcohol Abuse and Alcoholism (NIAAA, 2004). The BAC and ETOH intake values observed were highly correlated (Fig. 2C) and consistent throughout the 12 days period suggesting that the alcohol-treated groups were intoxicated to the same degree. Furthermore, the intoxication behavior score between the ethanol groups was not significantly different (Table 1) across the 12-day drinking period.
Figure 2. BAC Level.

Alcohol was administered in a binge-like pattern for 12 days and BAC was determined every other day. Both BAC (A) and alcohol intake (B) was not significantly different between the ETOH groups; and the increased BAC seen correspond with alcohol consumption as seen in the scatterplot illustrating a single day of drinking session (C). BAC – blood alcohol concentration.
Table 1. Intoxication score and liquid intake.
Intoxication scores taken during the binge drinking sessions were averaged. As well, liquid intake during the 2-day sucrose preference test was averaged. Both alcohol groups showed nearly identical intoxication scores. Similarly, liquid intake during the sucrose preference test showed no significant group differences.
| Group | Average Intoxication Score | Total Liquid Intake (%) |
|---|---|---|
| ETOH + vehicle | 2.2 ± 0.23 | 81.96 ± 9.2 |
| ETOH + TrkB | 2.0 ± 0.30 | 77.43 ± 8.01 |
| CON + vehicle | 0 | 79.23 ± 8.31 |
| CON + TrkB | 0 | 80.11 ± 7.32 |
Binge ETOH consumption causes depression-like symptoms
To evaluate if binge alcohol exposure can cause depression-like symptom and whether increasing BDNF signaling can attenuate the effects of alcohol in this affective behavior, we performed the sucrose preference and open field tests (Fig. 3). The sucrose preference test was given 48 hours after the last alcohol intake to evaluate anhedonia, a major feature of depressive disorder. We found a significant main effect of alcohol in that rats in the ETOH + vehicle group drank significantly less sucrose solution in comparison to the control groups and ETOH + TrkB group (F(3,43) = 6.81, p=0.031). Rats in the ETOH + vehicle group consumed approximately 28% and 26% less of the sucrose solution when compared to the control groups and the ETOH + TrkB groups respectively. Posthoc comparisons showed no significant difference in sucrose consumption between the ETOH + TrkB and control groups. Meanwhile, no significant difference was seen in the total liquid consumption between the groups (Table 1).
Figure 3. Test for Depressive-like Symptoms.
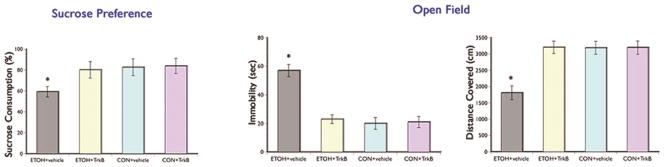
Significant decreased in hedonic response (sucrose preference test) and increased in behavioral despair (open field test) in the ETOH + vehicle group compared to the ETOH + TrkB and control groups. Augmenting BDNF signaling by giving the TrkB agonist inhibited the alcohol-induced depression phenotype. *p < 0.05
The open field is a widely used test for depressive-like symptoms (Che et al., 2013) because it provides information on ‘despair’ behavior (immobility), active behaviors (horizontal movement), and ‘anxiety’ behaviors (number of entries in the center area). Our results showed that the ETOH + vehicle group had significantly longer immobility time (F(3,43) = 6.01, p=0.030) and decreased horizontal movements (F(3,43) = 7.02, p=0.029) in the open field compared to the ETOH + TrkB and control groups. Post hoc comparisons showed no significant differences in behaviors in the open field between the ETOH + TrkB and control groups. No significant group differences were seen in the number of entries in the center area. Together, these results suggest that the depressive phenotype (i.e. anhedonia and despair) is manifested during the withdrawal/abstinence period from binge ethanol exposure but augmenting BDNF actions during ethanol consumption can abolish this negative effects of alcohol on affective behavior.
Binge alcohol consumption decreases survival and neuronal differentiation of NPCs
Since BrdU was injected at the onset of the binge drinking protocol, we were able to assess the number of dividing cells that survived until the brains were removed by quantifying the total number of BrdU-labeled cells in the dentate gyrus of the hippocampal region. We found that BrdU-positive cells appear as punctate staining and most labeled cells were in clusters present in both blades of the dentate gyrus. A significant main effect of alcohol (F(3,43) = 8.03, p=0.024) was seen in that the number of BrdU-positive cells per hippocampal volume in the ETOH + vehicle group decreased by approximately 26% and 31% in comparison to the ETOH + TrkB and control group that received vehicle, respectively (Fig. 4A). A significant main effect of 7,8-DHF (F(3,43) = 9.35, p=0.021) was also seen in that overall, the control group that received the TrkB agonist showed the highest number of BrdU-labeled cells. These results suggest that ETOH and BDNF have detrimental and enhancing effects, respectively, on cell proliferation.
Figure 4.
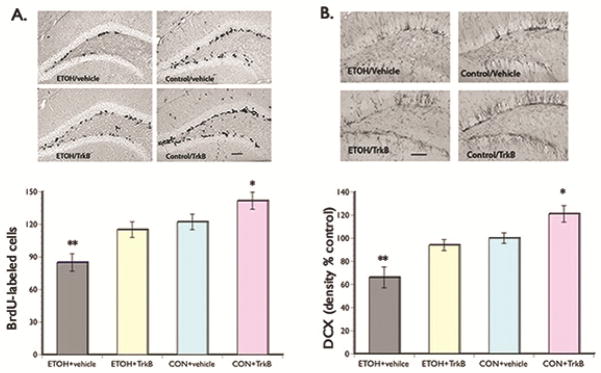
(A) BrdU Labeling. Upper panel: representative photomicrographs of BrdU staining scattered along both blades of the dentate gyrus. Lower panel: Bar graph shows significantly decreased survival of proliferating NPCs in the ETOH + vehicle group compared to the ETOH + TrkB and control rats. Overall, control rats that received the TrkB agonist showed the highest number of BrdU-labeled cells. These results suggest the role of BDNF in neuronal plasticity and its neuroprotective effects against the detrimental effects of alcohol. (B) DCX Labeling. Upper panel: representative photomicrographs of DCX staining in the dentate gyrus. Lower panel: Bar graph shows significantly decreased DCX immunoreactivity in the ETOH + vehicle group compared to the ETOH + TrkB and control rats. Control rats given the TrkB agonist showed the highest density of DCX staining. These results suggest the enhancing effects of BDNF signaling on the surviving newly formed cells to differentiate into neuronal phenotype. *p < 0.05; **p <0.01 (posthoc comparisons). Scale bar = 200 μm (left) and 100 μm (right).
We also examined the phenotype of the BrdU-labeled cells by using the marker doublecortin (DCX) for quantification of immature neurons. Doublecortin can provide information on the number of NPCs that proliferated, survived, differentiated into neurons, and begun to develop neurites (Couillard-Despres et al., 2005). We found significant main effect of alcohol (F(3,43) = 10.06, p=0.020) in that the density of DCX immunoreactivity was lower in the ETOH + vehicle group relative to the ETOH + TrkB and control groups (Fig. 4B). Meanwhile, no significant difference was seen in DCX immunoreactivity between the ethanol + TrkB and control group that received vehicle. A main effect of 7,8-DHF (F(3,43) = 10.24, p=0.018) was also seen in that the density of DCX immunoreactivity was significant increased overall in the control group that received the TrkB agonist. Collectively, these results suggest that neurotoxic substances such as alcohol can inhibit survival and differentiation of proliferating NPCs; however, enhancing BDNF actions increased neurogenesis and prevented the toxic effects of alcohol.
Binge alcohol affects BDNF function
To gain insight into a possible mechanism that mediate alcohol effects on neurogenesis and depression-like symptoms we examined BDNF function in the hippocampus. Our results showed decrease BDNF levels in the ETOH + vehicle group when compared to the control groups and ETOH + TrkB group (F(3,43) = 7.49, p=0.033). BDNF level in the ETOH + vehicle group is 30% and 21% lower when compared to the ETOH + TrkB and control group that received vehicle, respectively (Fig 5A). Posthoc comparisons showed no significant difference in BDNF expression between the ETOH + TrkB and control group that received vehicle. Comparison of the control groups showed significantly increase BDNF level in animals that received the TrkB agonist.
Figure 5. BDNF Action.

(A) ELISA results show significant decreased in BDNF protein in the ETOH + vehicle group in relation to the ETOH + TrkB and control groups. Control rats that received the TrkB agonist showed the highest levels of BDNF. Representative Western blot of Trk B expression using β-actin as an internal control (upper panel). No significant group differences were seen in total TrkB levels (B) but significant decreased in TrkB expression is evident in the ETOH + vehicle group compared to the ETOH + TrkB and control groups (C). The highest level of TrkB was seen in the control group that received 7,8-DHF. These results suggest that giving the TrkB agonist abolished the effects of alcohol on BDNF action. Representative Western blots are shown in the lower panel, right. *p <0.05 and **p <0.01 (posthoc comparisons).
As well, evaluation of the activation of the BDNF receptor TrkB, showed no significant groups differences in total TrkB level (Fig. 5B). However, analysis of levels of phosphorylated TrkB showed that expression in the ETOH + vehicle group is 30% and 32% lower compared to the ETOH + TrkB and control group that received vehicle, respectively (Fig. 5C). Posthoc comparisons revealed that levels of phosphorylated TrkB in the ETOH + TrkB and the control group that received vehicle are not significantly different. However, comparison of the control groups showed significantly increase phosphorylated TrkB level in animals that received the agonist. These results suggest that chronic ethanol exposure during adolescence caused a reduction in the expression of BDNF and its ability to activate the TrkB receptor. Giving TrkB agonist not only reversed alcohol effects on receptor activation but also increased BDNF expression.
Discussion
The primary objective of the study is to empirically test the possible mechanisms that underlie the depression-like symptom during the withdrawal/abstinence period after chronic binge alcohol drinking in adolescence. The impetus for the study are the epidemiological reports on the increased incidence of alcohol consumption in a binge-like pattern among adolescents (Naimi et al., 2003, Johnston et al., 2004, Masten et al., 2008, Courtney and Polich, 2009, Vicki et al., 2010), and the rise in suicide rates in this age group prompted by depression (Deykin et al., 1987, Boden and Fergusson, 2011) suggesting a link between these two factors. The key findings that emerged in this work are that: 1) binge-like drinking causes inebriation as defined by the National Institute on Alcohol Abuse and Alcoholism (NIAAA) and this pattern of alcohol exposure is associated with the development of depression-like symptom, and 2) the possible mechanisms involved in the development of depression-like symptom during the withdrawal/abstinence period after chronic binge alcohol exposure are decreased survival and differentiation of proliferating cells possible mediated by reductions in hippocampal BDNF function.
In our study, rats exposed to alcohol in a binge-pattern reached BAC ≈ 0.08 grams % during acute consumption, which is considered to be an intoxicated state (NIAAA, 2004). Alcohol impairs the functioning of a variety of behavioral domains including memory and judgment [reviewed in (Nelson et al., 1999)], and motor performance (Fogarty and Vogel-Sprott, 2002). Additionally, alcohol can also cause affective impairment such as anxiety (Pandey et al., 2006, Pandey et al., 2008) and depression (Caldwell et al., 2008, Hauser et al., 2011). In the present study we examined the effects of chronic alcohol exposure on depression-like symptom because of the dearth of information on this topic in the adolescent brain even though increasing evidence shows that incidence of depression is higher in adolescents compared to adults (Rao, 2006). Our data show decreased hedonic and despair behaviors in rats exposed to alcohol alone (ETOH + vehicle) during the withdrawal period where they demonstrate a significantly diminished preference for sweet substances in the sucrose preference test and increased immobility time, as well as decreased locomotor activity in the open field test in comparison to the ETOH + TrkB and control groups. The anhedonia and despair seen in the ETOH + vehicle rats in this study is consistent with those reported by others in adult rats (Hauser et al., 2011) and mice (Stevenson et al., 2009) given alcohol daily for several days and also in adult mice prenatally exposed to alcohol that demonstrate increased learned helplessness and immobility in the forced swimming test (Caldwell et al., 2008). Our results are also in line with epidemiological data that report chronic alcohol misuse and alcohol dependence in adults are associated with rates of major depressive disorder ranging from 30% to 50% (Regier et al., 1990, Sullivan et al., 2005). Clinical studies show that the development of depressive and addictive disorders among adolescents are considered to be even higher than that observed in adults (Regier et al., 1990, Rohde et al., 1991) with the highest incidence seen between 15 and 19 years of age (Sullivan et al., 2005). The development of depression during the withdrawal/abstinence period following chronic alcohol use may be attributed to the cyclic pattern of euphoria after alcohol consumption followed by the dysphoric mood that accompanies the withdrawal period. Thus, chronic binge drinking may increase the risk for the development of long-term affective impairment since it produces repeated euphoria and dysphoria.
Determining the neural mechanisms that may be involved in mediating depression in alcoholism is a crucial step in understanding these comorbid disorders. One of the mechanisms we examined in this study is neurogenesis and we demonstrate that rats exposed to alcohol alone (ETOH + vehicle) in a binge-like pattern for 12 days show significantly decreased number of BrdU-labeled cells and DCX immunoreactivity in the dentate gyrus when compared to the ETOH + TrkB and control groups during the withdrawal period. These data suggest that alcohol can inhibit the survival and neuronal differentiation of proliferating NPCs. Our results parallel the findings of others that report inhibition of NPC proliferation in the dentate gyrus following chronic alcohol exposure in adolescent rats (He et al., 2005, McClain et al., 2011) and mice (Stevenson et al., 2009), as well as adult rats when exposed to alcohol prenatally (Hamilton et al., 2011). It is possible that imaging reports of smaller hippocampal volume in adolescents who began drinking at an early age (De Bellis et al., 2000, Nagel et al., 2005) and on adults diagnosed with depression (Sheline et al., 2003, Sullivan et al., 2005) may be explained by decrease cell proliferation demonstrated in these studies. However, our results are in contrast with a previous study that reported increase cell proliferation during the withdrawal period (Nixon and Crews, 2004). The conflicting findings may be due to the mode of alcohol administration and the use a drug to reduce withdrawal severity. Namely, our study used the alcohol self-administration paradigm whereas the other study used the gastric gavage administration method and withheld food during the binge paradigm possibly leading to increase withdrawal severity. Indeed, the other study administered diazepam to control the severity of alcohol withdrawal; thus, the anti-depressive properties of diazepam in combination with greater withdrawal severity most likely led to withdrawal-induced increase cell proliferation as reported in the other study.
How alcohol affects neurogenesis is still poorly understood but it is possible that it: 1) alters the cell cycle (Morris et al., 2010) interfering with proliferation, and 2) decreases BDNF action that promotes cell survival and differentiation as evidenced in our data on the ETOH + TrkB group. The importance of BDNF in neurogenesis is evident from our results that show administration of the TrkB agonist in the saline control group resulted in increase cell proliferation and neuronal differentiation in the dentate gyrus. These findings are consistent with previous reports that demonstrate the role of BDNF/TrkB in brain plasticity. That is, BDNF/TrkB promotes the survival and differentiation of NPCs, as well as increase the branching of axons and dendrites and stabilizing synaptic contacts of existing neurites [reviewed in (Neto et al., 2011)]. What is not clear from our results however, but clearly plausible is whether the protection conferred by BDNF on the survival of proliferating NPCs may have been augmented by the reported compensatory burst in cell proliferation during the alcohol withdrawal period (Nixon and Crews, 2004). This issue can be addressed in future studies that make clear the time course of changes in various phases of neurogenesis during alcohol withdrawal following binge drinking.
Our study also show significantly decreased BDNF protein levels as well as reductions in TrkB expression in the hippocampus of rats given alcohol alone (ETOH + vehicle) in comparison to the ETOH + TrkB and control groups and this reduction is most likely due to decreased BDNF signaling. Our results are in line with other experimental studies that show significantly lower hippocampal BDNF levels in adult rats that demonstrate anhedonia following alcohol exposure, and adult mice that demonstrate learned helplessness when exposed to alcohol prenatally (Caldwell et al., 2008, Hauser et al., 2011). BDNF may be an important molecule in the hippocampus to mediate some of the functional consequences of ethanol exposure such as depression. Indeed, clinical studies show significant reductions in peripheral BDNF level in patients with alcohol dependence diagnosed with major depression [reviewed in (Duman and Monteggia, 2006)]. Collectively these data suggest a role for hippocampal BDNF effects in the depression-like symptom induced by alcohol.
Our results on BDNF expression however, is in contrast to those who report increased BDNF expression in the hippocampus of adult rats following 12 hours of alcohol withdrawal (Tapia-Arancibia et al., 2001). Some possible explanations for the difference in findings may be the alcohol paradigm used, age of the brain examined, and timing of measurement during the withdrawal period. Namely, our study used the chronic binge-pattern alcohol paradigm and we examined the adolescent brain 7 days after the last alcohol consumption. In contrast, the other study used the alcohol vapor paradigm and examined the adult brain 12 hours after the last alcohol exposure. Thus, it is possible that consuming large quantities of alcohol at a time such as those seen in the vapor paradigm, is more damaging to the developing adolescent brain. Similarly, our results are in conflict with reports of increased serum BDNF in adult alcoholics a week after their last alcohol consumption (Huang et al., 2008, Costa et al., 2011). An explanation for the difference in results between our study and the clinical reports of decreased serum BDNF in alcoholic patients weeks after withdrawal are the measurement used. That is, the clinical reports measured serum BDNF level while our study examined expression in hippocampal tissues. Peripheral levels of BDNF reflected in serum will likely differ from central expression.
Our results also differ from a recent study that reports no difference in BDNF expression in the adolescent brain between the ethanol and control group following a 4-day binge (McClain et al., 2013). A possible reason for these conflicting findings is that we measured BDNF expression after completion of behavioral testing while the recent study measured BDNF expression at different time points after alcohol administration. It possible that our results reflect the confounding effects of cognitive testing on BDNF expression but it is highly unlikely since all experimental groups underwent behavioral testing. However, a more likely explanation is that there may be differences on how BDNF expression was assessed. That is, we analyzed the sample from each rat and reported the data obtained as group means while it is not clear from the recent study whether the samples were pooled to evaluate BDNF expression where individual variability cannot be detected.
A critical finding in the present study is how BDNF action directly mediates alcohol-induced depressive-like symptoms by regulating neurogenesis. Our data showing that depressive-like symptoms during the alcohol withdrawal period is associated with diminished survival and neuronal differentiation of proliferating NPCs but enhancing BDNF signaling by giving the TrkB agonist during alcohol consumption can restore neurogenesis and abolish the anhedonia and despair behaviors points to this issue. It is likely that the behavioral testing in the present study may have influence TrkB expression but not its phosphorylation because even though no significant differences were seen in total TrkB level, data on phosphorylated TrkB level show differences associated with ETOH and 7,8-DHF. The next question that arises is what intracellular pathways may be involved in BDNF signaling activated through phosphorylation of TrkB receptors that can regulate alcohol effects on neurogenesis and affective behavior. Another related question that warrants further study are other molecular mechanisms that share common signaling pathways with BDNF that may regulate neurogenesis and depressive-like symptoms following chronic binge-pattern alcohol exposure.
Conclusion
The present investigation provides possible mechanisms that mediate the depression-like symptom seen during the withdrawal/abstinence period following chronic binge alcohol exposure. Our study implicates decreased survival and neuronal differentiation of proliferating NPCs mediated by reduced BDNF signaling in the hippocampus as factors that may be responsible for the predisposition to alcohol-related anhedonic and despair behaviors during the withdrawal period. The decrease survival of proliferating NPCs associated with alcohol exposure as demonstrated in our data may also explain the reduction in hippocampal volume seen in alcoholics and those diagnosed with depression. BDNF might be an important regulator involved in the neuroadaptation commonly reported during alcohol withdrawal because of its role in neural plasticity. Finally, since the common link between decreased neurogenesis and development of alcohol-induced depression-like symptom is BDNF as we demonstrate in this study, it might be a molecule that can be targeted for interventions in alcoholism–depression coincidence.
Highlights.
The binge-like pattern of alcohol exposure is associated with the development of depression-like symptom
Chronic binge drinking resulted in decrease hippocampal neurogenesis and BDNF expression during the withdrawal period
Augmenting BDNF restored neurogenesis and abolished the depression-like behaviors caused by chronic binge alcohol
Acknowledgments
This work was supported in part by the National Institutes of Health RO1 NR007666 (TLB) and Wayne State University Faville Endowment Funds (TLB). We are grateful for the assistance of Maria Palu in imaging and BrdU administration.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors maybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bava S, Thayer R, Jacobus J, Ward M, Jernigan TL, Tapert SF. Longitudinal characterization of white matter maturation during adolescence. Brain Res. 2010;1327:38–46. doi: 10.1016/j.brainres.2010.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boden JM, Fergusson DM. Alcohol and depression. Addiction. 2011;106:906–914. doi: 10.1111/j.1360-0443.2010.03351.x. [DOI] [PubMed] [Google Scholar]
- Briones TL, Rogozinska M, Woods J. Environmental experience modulates ischemia-induced amyloidogenesis and enhances functional recovery. J Neurotrauma. 2009;24:613–625. doi: 10.1089/neu.2008.0707. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Briones TL, Suh E, Hattar H, Wadowska M. Dentate gyrus neurogenesis after cerebral ischemia and behavioral training. Biol Res Nurs. 2005;6:167–179. doi: 10.1177/1099800404271328. [DOI] [PubMed] [Google Scholar]
- Briones TL, Woods J. Chemotherapy-induced cognitive impairment is associated with decreases in cell proliferation and histone modifications. BMC Neurosci. 2011;9:124. doi: 10.1186/1471-2202-12-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briones TL, Woods J, Wadowska M, Rogozinska M. Amelioration of cognitive impairment and changes in microtubule-associated protein 2 after transient global cerebral ischemia are influenced by complex environment experience. Behav Brain Res. 2006;168:261–271. doi: 10.1016/j.bbr.2005.11.015. [DOI] [PubMed] [Google Scholar]
- Caldwell K, Sheema S, Paz RD, Samudio-Ruiz SL, Laughlin MH, Spence NE, Roehlk MJ, Alcon SN, Allan AM. Fetal alcohol spectrum disorder-associated depression: evidence for reductions in the levels of brain-derived neurotrophic factor in a mouse model. Pharmacol Biochem Behav. 2008;90:614–624. doi: 10.1016/j.pbb.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Che Y, Cui Y-H, Tan H, Andreazza AC, Young LT, Wang J-F. Abstinence from repeated amphethamine treatment induces depressive-like behaviors and oxidative damage in rat brain. Psychopharmacology. 2013;227:605–614. doi: 10.1007/s00213-013-2993-0. [DOI] [PubMed] [Google Scholar]
- Costa M-A, Girard M, Dalmay F, Malauzat D. Brain-derived neurotrophic factor serum levels in alcohol-dependent subjects 6 months after alcohol withdrawal. Alcohol Clin Exp Res. 2011;35 doi: 10.1111/j.1530-0277.2011.01548.x. [DOI] [PubMed] [Google Scholar]
- Couillard-Despres S, Winner B, Schaubeck S, Aigner R, Vroemen M. Doublecortin expression levels in adult brain reflect neurogenesis. Eur J Neurosci. 2005;21:1–14. doi: 10.1111/j.1460-9568.2004.03813.x. [DOI] [PubMed] [Google Scholar]
- Courtney KE, Polich J. Binge drinking in young adults: data, definitions, and determinants. Psychological Bulletin. 2009;135:142–156. doi: 10.1037/a0014414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT, Braun CJ, Hoplight B, Switzer RC, Knapp DJ. Binge ethanol consumption causes differential brain damage in young adolescent rats compared with adult rats. Alcohol Clin Exp Res. 2000;24:1712–1723. [PubMed] [Google Scholar]
- Crews FT, Nixon K. Mechanisms of neurodegeneration and regeneration in alcoholism. Alcohol Alcohol. 2009;44:115–127. doi: 10.1093/alcalc/agn079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bellis MD, Clark DB, Beers SR, Soloff PH, Boring AM, Hall J, Kersh A, Keshavan MS. Hippocampal volume in adolescent-onset alcohol use disorders. Am J Psychiatry. 2000;157:737–744. doi: 10.1176/appi.ajp.157.5.737. [DOI] [PubMed] [Google Scholar]
- Deykin EY, Levy JC, Wells V. Adolescent depression, alcohol and drug use. Am J Public Health. 1987;77:178–182. doi: 10.2105/ajph.77.2.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doremus -FTL, Varlinskaya E, Spear LP. Motivational systems in adolescence: possible implications for age differences in substance abuse and risk-taking behaviors. Brain Cogn. 2010;72:114–123. doi: 10.1016/j.bandc.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biol Psychiatry. 2006;59:1116–1127. doi: 10.1016/j.biopsych.2006.02.013. [DOI] [PubMed] [Google Scholar]
- Fogarty JN, Vogel-Sprott M. Cognitive processes and motor skills differ in sensitivity to alcohol reinforcement. J Stud Alcohol. 2002;63:404–411. doi: 10.15288/jsa.2002.63.404. [DOI] [PubMed] [Google Scholar]
- Gould E, Tanapat P, Hastings NB, Shors TJ. Neurogenesis in adulthood: a possible role in learning. Trends in Cognitive Sciences. 1999;3:186–192. doi: 10.1016/s1364-6613(99)01310-8. [DOI] [PubMed] [Google Scholar]
- Grant BF, Dawson DA. Age at onset of alcohol use and its association with DSM-IV alcohol abuse and dependence: results from the national longitudinal alcohol epidemiologic survey. J Subst Abuse. 1997;9:103–110. doi: 10.1016/s0899-3289(97)90009-2. [DOI] [PubMed] [Google Scholar]
- Hamilton GF, Murawski NJ, St Cyr SA, Jablonski SA, Schiffino FL, Stanton ME, Klintsova AY. Neonatal alcohol exposure disrupts hippocampal neurogenesis and contextual fear conditioning in adult rats. Brain Res. 2011;1412:88–101. doi: 10.1016/j.brainres.2011.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser SR, Getachew B, Taylor RE, Tizabi Y. Alcohol induced depressive-like behavior is associated with a reduction in hippocampal BDNF. Pharmacol Biochem Behav. 2011;100:253–258. doi: 10.1016/j.pbb.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Nixon K, Shetty AK, Crews FT. Chronic alcohol exposure reduces hippocampal neurogenesis and dendritic growth on newborn neurons. Eur J Neurosci. 2005;21:2711–2720. doi: 10.1111/j.1460-9568.2005.04120.x. [DOI] [PubMed] [Google Scholar]
- Huang M-C, Chen C-H, Chen C, Liu S-C, Ho C-J, Shen WW, Leu S-J. Alterations of serum brain-derived neurotrophic factor levels in early alcohol withdrawal. Alcohol Alcohol. 2008;43:241–245. doi: 10.1093/alcalc/agm172. [DOI] [PubMed] [Google Scholar]
- Imayoshi I, Sakamoto M, Ohtsuka T, Takao K, Miyakawa T, Yamaguchi M, Mori K, Ikeda T, Itohara S, Kageyama R. Roles of continuous neurogenesis in the structural and functional integrity of the adult forebrain. Nat Neurosci. 2008;11:1153–1161. doi: 10.1038/nn.2185. [DOI] [PubMed] [Google Scholar]
- Jang SW, Liu X, Yepes M, Shepherd KR, Miller GW, Liu Y, Wilson WD, Xiao G, Blanchi B, Sun YE, Ye K. A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc Natl Acad Sci USA. 2010;107:2687–2692. doi: 10.1073/pnas.0913572107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston LD, O’Malley PM, Bachman JG, Schulenberg JE. Monitoring the Future, National Survey Results on Drug Use, 1975–2004. 2004 NIH Pub No. 05-5727 1. [Google Scholar]
- Kempermann G, Wiskott L, Gage FH. Functional significance of adult neurogenesis. Curr Opin Neurobiol. 2004;14:186–191. doi: 10.1016/j.conb.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Knapp DJ, Crews FT. Induction of cyclooxygenase-2 in brain during acute and chronic ethanol treatment and ethanol withdrawal. Alcohol Clin Exp Res. 1999;23:633–643. [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Drug abuse: hedonic homeostatic dysregulation. Science. 1997;278:52–58. doi: 10.1126/science.278.5335.52. [DOI] [PubMed] [Google Scholar]
- Kuhn H, Dickinson-Anson H, Gage FH. Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. J Neurosci. 1996;17:5820–5829. doi: 10.1523/JNEUROSCI.16-06-02027.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E, Son H. Adult hippocampal neurogenesis and related neurotrophic factors. BMB Rep. 2009;42:239–244. doi: 10.5483/bmbrep.2009.42.5.239. [DOI] [PubMed] [Google Scholar]
- Masten AS, Faden VB, Zucker RA, Spear LP. Underage drinking: a developmental framework. Pediatrics. 2008;121 (Suppl 4):S235–S251. doi: 10.1542/peds.2007-2243A. [DOI] [PubMed] [Google Scholar]
- McClain JA, Hayes DM, Morris SA, Nixon K. Adolescent binge alcohol exposure alters hippocampal progenitor cell proliferation in rats: effects on cell cycle kinetics. J Comp Neurol. 2011;519:2697–2710. doi: 10.1002/cne.22647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClain JA, Morris SA, Marshall A, Nixon K. Ectopic hippocampal neurogenesis in adolescent male rats following alcohol dependence. Addiction Biol. 2013 doi: 10.1111/adb.12075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina KL, Schweinsburg AD, Cohen-Zion M, Nagel BJ, Tapert SF. Effects of alcohol and combined marijuana and alcohol use during adolescence on hippocampal volume and asymmetry. Neurotoxicol Teratol. 2007;29:141–152. doi: 10.1016/j.ntt.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris SA, Eaves DW, Smith AR, Nixon K. Alcohol inhibition of neurogenesis: a mechanism of hippocampal neurodegeneration in an adolescent alcohol abuse model. Hippocampus. 2010;20:596–607. doi: 10.1002/hipo.20665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel BJ, Schweinsburg AD, Phan V, Tapert SF. Reduced hippocampal volume among adolescents wth alcohol use disorders without psychiatric comorbidity. Psychiatry Res. 2005;139:181–190. doi: 10.1016/j.pscychresns.2005.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naimi TS, Nelson DE, Brewer RD. The intensity of binge alcohol consumption among U.S. adults. Am J Prev Med. 2003;38:201–207. doi: 10.1016/j.amepre.2009.09.039. [DOI] [PubMed] [Google Scholar]
- Nelson TO, Graf A, Dunlosky J, Marlatt A, Walker D, Luce K. Effect of acute alcohol intoxication on recall and on judgments of learning during the acquisition of new information. In: Mazzoni G, Nelson TO, editors. Metacognition and cognition neuropsychology: Monitoring and control processes. Mahwah, New Jersey: Erlbaum; 1999. pp. 161–180. [Google Scholar]
- Neto FL, Borges G, Torres-Sanchez S, Mico JA, Berrocoso E. Neurotrophins role in depression neurobiology: a review of basic and clinical evidence. Curr Neuropharmacol. 2011;9:530–552. doi: 10.2174/157015911798376262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NIAAA. Binge drinking defined. NIAAA Newslett. 2004;3:3. [Google Scholar]
- Nixon K, Crews FT. Temporally specific burst in cell proliferation increases hippocampal neurogenesis in protracted abstinence from alcohol. J Neurosci. 2004;24:9714–9722. doi: 10.1523/JNEUROSCI.3063-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey SC, Ugale R, Zhang H, Tang L, Prakash A. Brain chromatic remodeling: a novel mechanism of alcoholism. J Neurosci. 2008;28:3729–3737. doi: 10.1523/JNEUROSCI.5731-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey SC, Zhang H, Roy A, Misra K. Central and medial amygdaloid brain-derived neurotrophic factor signaling plays a critical role in alcohol-drinking and anxiety-like behaviors. J Neurosci. 2006 doi: 10.1523/JNEUROSCI.4988-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao U. Links between depression and substance abuse in adolescents. neurobiological mechanism. Am J Prev Med. 2006;31:S161–S174. doi: 10.1016/j.amepre.2006.07.002. [DOI] [PubMed] [Google Scholar]
- Regier DA, Farmer ME, Rae DS, Locke BZ, Keth SJ, Judd LL, Goodwin FK. Comorbidity of mental disorders with alcohol and other drug abuse: results from the Epidemiological Catchment Area (ECA) Study. JAMA. 1990;264:2511–2518. [PubMed] [Google Scholar]
- Rohde P, Lewinsohn M, Seeley JR. Comorbidity of unipolar depression. II. Comorbidity with other mental disorders in adolescents and adults. J Abnorm Psychol. 1991;100:214–222. [PubMed] [Google Scholar]
- Schulteis G, Archer C, Tapert SF, Frank LR. Intermittent binge alcohol exposure during the periadolescent period induces spatial working memory deficits in young rats. Alcohol. 2008;42:459–467. doi: 10.1016/j.alcohol.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweinsburg AD, McQueeny T, Nagel BJ, Eyler LT, Tapert SF. A preliminary study of functional magnetic resonance imaging response during verbal encoding among adolescent binge drinkers. Alcohol. 2010;44:111–117. doi: 10.1016/j.alcohol.2009.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheline YI, Gado MH, Kraemer HC. Untreated depression and hippocampal volume loss. Am J Psychiatry. 2003;160:1516–1518. doi: 10.1176/appi.ajp.160.8.1516. [DOI] [PubMed] [Google Scholar]
- Shen K, Cowan CW. Guidance molecules in synapse formation and plasticity. Cold Spring Harb Perspect Biol. 2010;2:a001842. doi: 10.1101/cshperspect.a001842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spear LP. The adolescent brain and age-related behavioral manifestations. Neurosci Biobehav Rev. 2000;24:417–463. doi: 10.1016/s0149-7634(00)00014-2. [DOI] [PubMed] [Google Scholar]
- Stevenson JR, Schroeder JP, Nixon K, Besheer J, Crews FT, Hodge CW. Abstinence following alcohol drinking produces depression-like behavior and reduced hippocampal neurogenesis in mice. Neuropsychopharmacology. 2009;34:1209–1222. doi: 10.1038/npp.2008.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan LE, Fiellin DA, O’Connor PG. The prevalence and impact of alcohol problems in major depression: a systematic review. Am J Med. 2005;118:330–341. doi: 10.1016/j.amjmed.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Tapia-Arancibia L, Rage F, Givalos L, Dingeon P, Arancibia S, Beauge F. Effects of alcohol on brain-derived neurotrophic factor mRNA expression in discrete regions of the rat hippocampus and hypothalamus. J Neurosci Res. 2001;63:200–208. doi: 10.1002/1097-4547(20010115)63:2<200::AID-JNR1012>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Vicki M, Kuntsche E, Gmel G. Drinking at European universities? a review of students’ alcohol use. Addictive Behaviors. 2010;35:913–924. doi: 10.1016/j.addbeh.2010.06.015. [DOI] [PubMed] [Google Scholar]
- White AM, Swartzwelder HS. Age-related effects of alcohol on memory and memory-related brain function in adolescents and adults. Recent Dev Alcohol. 2005;17:161–176. doi: 10.1007/0-306-48626-1_8. [DOI] [PubMed] [Google Scholar]