

Abstract
A better understanding of protein aggregation is bound to translate into critical advances in several areas, including the treatment of misfolded protein disorders and the development of self-assembling biomaterials for novel commercial applications. Because of its ubiquity and clinical potential, albumin is one of the best-characterized models in protein aggregation research; but its properties in different conditions are not completely understood. Here, we carried out all-atom molecular dynamics simulations of albumin to understand how electrostatics can affect the conformation of a single albumin molecule just prior to self-assembly. We then analyzed the tertiary structure and solvent accessible surface area of albumin after electrostatically triggered partial denaturation. The data obtained from these single protein simulations allowed us to investigate the effect of electrostatic interactions between two proteins. The results of these simulations suggested that hydrophobic attractions and counterion binding may be strong enough to effectively overcome the electrostatic repulsions between the highly charged monomers. This work contributes to our general understanding of protein aggregation mechanisms, the importance of explicit consideration of free ions in protein solutions, provides critical new insights about the equilibrium conformation of albumin in its partially denatured state at low pH, and may spur significant progress in our efforts to develop biocompatible protein hydrogels driven by electrostatic partial denaturation.
1. Introduction
The study of protein aggregation is critically important for understanding the etiology of many misfolded protein disorders such as Alzheimer’s disease, Parkinson’s disease, type 2 diabetes, and sickle cell anemia.1 In addition to a deeper understanding of these conditions, lessons learned from protein aggregation studies have led to the fabrication of several commercially interesting self-assembling biomaterials.1 Not surprisingly, given their intrinsic biocompatibility and similarities to the extracellular matrix of certain tissues,2,3 biological hydrogels are used extensively in medical applications. These biological hydrogels have been synthesized from a variety of biomacromolecules by forming intermolecular cross-links via thermal or chemical methods.2,3 Proteins are one such type of complex biomacromolecules with a well-established hierarchical structure, from the primary sequence of amino acid residues to multiprotein assemblies at the quaternary level. The structural complexity of a single protein’s three-dimensional structure (tertiary level) depends on the delicate interplay between electrostatic, hydrophobic, hydrogen bonding, and other interactions, whose modifications can result in significant conformational changes.4 Evolutionary optimization of these interactions in physiological environments has resulted in protein conformations that are functionally operational.5 However, by altering the electrostatic charges on protein surfaces in a targeted manner, we can unlock the original protein structure leading to the exposure of buried hydrophobic regions. These regions could ideally drive new quaternary assemblies that promote hydrogel formation while preserving some of the original protein functionality in unchanged domains. Earlier studies have utilized similar approaches to probe the mechanisms behind the formation of functional6 and diseased7 amyloid structures.
The simulations in the present study explore the origins of intermolecular aggregation of albumin at the level of atomistic interactions prior to hydrogel formation. The results are likely to shed critical light into the fundamental understanding of the competition between folding and assembly of macromolecules and also for the understanding of their influence in many clinically relevant phenomena, such as amyloid formation and the slow releasing properties of drug carrier systems that exploit albumin’s natural drug binding capacity.8
Albumin is a 66 kDa water-soluble, monomeric protein, and the most abundant protein in blood plasma (40–50 mg/mL). It has three primary domains that are arranged in a heart shape with 17 disulfide bond linkages that stabilize the domains.9 It serves as the primary carrier of various solutes in plasma, including cations, bilirubin, fatty acids, and therapeutic drugs.9 There is extensive literature regarding serum albumin’s affinities to various compounds,10−13 denaturation conditions,4,14−17 gelation mechanisms,18−26 and current or potential medical uses.27−33 Albumin hydrogels formed by thermal or chemical cross-linking (e.g., glutaraldehyde) or by polymer-albumin conjugated methods have been reported.9,25,32,34−36 However, these hydrogel systems typically require either the physical/chemical modification of the albumin or the incorporation of synthetic components into the hydrogel network.
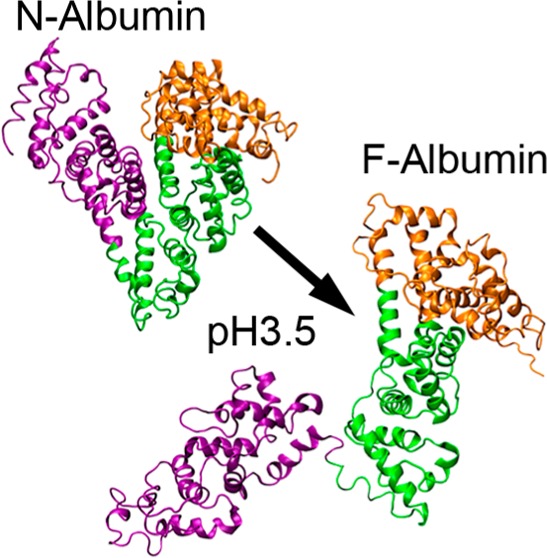
Albumin can reversibly and drastically change its conformation when exposed to changes in solution pH (transitions occurring at pH 2.7, 4.3, 8, and 10).4,9 For example, at pH 7.4, albumin has a normal heartlike structure (N isoform), while at pH 3.5 it has a partially expanded cigarlike shape (F isoform).37 Below its transition point at pH 2.7, albumin denatures into its fully expanded E isoform. During the N–F isoform transition, bovine serum albumin (BSA) passes through its isoelectric point at pH 4.7 and the net charge on the protein changes from −16 at pH 7.4 to +100 at pH 3.5.9 Low solution pH also shifts the denaturation temperature of BSA from 62 °C (at pH 7.4) to 46.8 °C (at pH 3.5).38 In concentrated solutions, we have observed that BSA proteins in the F isoform can self-assemble into a solid hydrogel network within 24 h at room temperature (RT; 25 °C) or in 30 min at 37 °C but do not form networks in the E isoform.39 In contrast, pure N isoform BSA solutions do not exhibit this gelation behavior unless the temperature rises above 62 °C when it triggers thermal denaturation of the N isoform.35 These findings suggest that the self-assembly of albumin hydrogels at RT hinges on the presence of a specific set of physicochemical features that are strongly favored in the F isoform. This raises the important question of what other interactions might be recruited in order to overcome the highly charged nature of the F isoform. To answer this question, we used atomistic molecular dynamics simulations to calculate the conformational changes in BSA due to the changes in pH from the N isoform structure and studied the interaction between two partially denatured proteins.
2. Methods
Atomistic BSA Model Simulations
We performed a series of molecular dynamics (MD) simulations of bovine serum albumin (BSA) to develop a model of the protein at pH 3.5 and then used the model to investigate intermolecular interactions between two proteins. Fully atomistic MD simulations were performed using the GROMACS 4.5.4 simulation package.40−43 The tertiary structure of BSA was first obtained by submitting the BSA primary sequence (GenBank: CAA76847.1) to a protein homology modeling server (CPHmodels 3.0).44,45 CPHmodels identified HSA as the closest existing protein structure to BSA and the result matches well (RMSD = 1.39 Å) with recent crystallographic BSA structures.46 The resulting output file was used as the basis for all subsequent atomistic simulations of BSA. The all-atom optimized potential for liquid simulations (OPLS/AA) force field parameters47 were used to describe interactions among the atoms. FAMBE-pH, a program that calculates the total solvation free energies of proteins as a function of pH, was used to calculate the ionization state of titratable residues (ASP, GLU, HIS, LYS, ARG) on BSA at pH 7.4 and 3.5.48 The 1:1 salt effect is included, indirectly, in the FAMBE-pH method as was done for the salt-dependent generalized Born method.48 Protonation states were fixed to the model for each pH and the model was then energetically stabilized by steepest descent algorithm, followed by an equilibration for at least 1 ns in water at 300K. The protein was then immersed in a 17 × 7 × 7 nm3 box of SPC water molecules49 to allow room for protein expansion along the long axis of the simulation box, and a simulation was run for production for 64 ns in canonical (NVT) ensemble at constant temperature 300 K with Nose-Hoover temperature coupling method.40,47 Analysis of protein secondary structures was performed by the STRIDE webserver.50
Circular Dichroism Experiments
Dilute solutions (0.005 wt %) of essentially fatty acid free bovine serum albumin (A6003, Sigma, St. Louis, MO) in deionized water were titrated to different pH levels near the N–F transition (3.5, 4, 4.5) with HCl. Solutions were loaded into triple rinsed quartz cuvettes and evaluated by Circular Dichroism spectrography (J-815, JASCO Inc., Easton, MD) with a wavelength scan from 190 to 260 nm in triplicate. Internal heating elements in the J-815 were used to thermally denature dilute albumin solutions (0.005 wt %) at pH 7.4 to 60 and 80 °C.
Electrostatic Potential Calculations
A Python script was written to compute the electrostatic potential explicitly (including all water and counterion molecules) at each point along the Connolly surface of the protein with the following equation:
where the sum runs over all atoms i that are within 3 nm from the point of the Connolly surface, Qi is the charge of atom i, and ri is the distance between the charge i and the Connolly surface. The Connolly surface was computed using the built in GROMACS g_sas command with settings identical to those used by the program APBS51 embedded in Chimera52 that was used to generate the Poisson–Boltzmann potential surface. The radius of the solvent probe was 1.4 Å with 20 dots per sphere on the surface. A 3 nm radius cutoff was used in calculating the electrostatic potential contribution of every atom near each mesh point. Visualizations of molecular structures are performed with the VMD 1.9.1 software package.53
3. Results and Discussion
While the atomic structure of human albumin at pH 7.4 has been determined at a 2.5 Å resolution,54 only low-resolution 3D models based on X-ray scattering (SAXS) data exist for the F isoform.37 Therefore, to study protein aggregation of F isoform bovine serum albumin at pH 3.5, we first needed to generate an accurate model of the F isoform albumin. Recent advances in computational power, MD software,41 and theoretical methods to calculate titration states of residues in large proteins48 now enable us to simulate these conformational changes from first principles. Since the size of the simulations required to model pH atomistically in this system remains prohibitively expensive, we utilized a program called FAMBE-pH to calculate the total solvation free energies of proteins as a function of pH.48 Briefly, this program employs a combination of approaches to calculate these free energies and involves (i) solving the Poisson equation with a fast adaptive multigrid boundary element method (FAMBE); (ii) calculating electrostatic free energies of ionizable residues at neutral and charged states; (iii) defining a precise dielectric surface interface; (iv) tessellating the dielectric surface with multisized boundary elements; and (v) including 1:1 salt effects.48 The computation of the free energy of solvation by FAMBE-pH includes the following terms: (1) the free energy of creation of a molecular cavity in the water; (2) the free energy of van der Waals interactions between the protein and the water solvent; (3) the free energy of polarization of the water solvent by the protein; and (4) the free energy of equilibrium titration of protein for a given pH and conformation.55 Since the number of ionizable groups in albumin (198) is more than ∼20–25, the Tanford-Schellman integral was used to calculate the equilibrium proton binding/release.48 With this program, we calculated the ionization state of titratable residues (ASP, GLU, HIS, LYS, ARG) at pH 3.5 (Figure 1a). Residues with carboxylic acid groups that increase in charge state from −1 to 0 (ASP and GLU) between pH 7.4 and pH 3.5 are shown in red while residues with primary and secondary amines (LYS, ARG, and HIS) that increase in charge from 0 to +1 are shown in blue (Figure 1). At pH 7.4, FAMBE-pH correctly predicted the deprotonation of all ASP and GLU residues: the protonation of all ARG and LYS residues, and a balance of protonated and deprotonated HIS residues were consistent with an expected overall net charge of −9 (Figure 1b). At pH 3.5, FAMBE-pH predicted that all ASP and GLU residues become protonated and that the remaining HIS residues also be protonated, while LYS and ARG residues remain unchanged (Figure 1b). The locations of these residues on BSA are distributed uniformly over the tertiary structure (Figure 1b) and represent the ionization state of BSA at pH 3.5 (net charge of +100). While the pH of the protein was effectively set to 3.5, the conformational structure was still that of the N isoform. This predicted net charge was higher than the net charge (+65) and effective charge (+13) for albumin molecules at pH 3.5, as determined by experimental titration and electrophoresis NMR experiments,56,57 but this may be due to the fact that the structure of the protein was not yet in its ideal conformation. This difference can also be explained by the fact that any observable measurement should be computed from an ensemble of structures via a Boltzmann average, however, this is not feasible with the existent computational resources.
Figure 1.
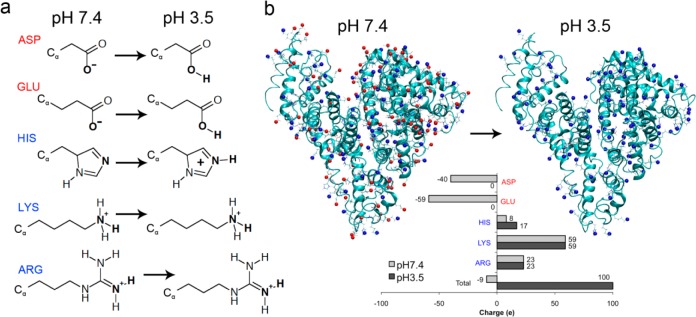
Determination of ionization state of titratable residues for simulations. (a) Chemical structures of titratable residues (ASP, GLU, HIS, LYS, ARG) and the resulting ionized structure at pH 3.5. Red and blue color denotes negatively and positively charged residues, respectively. (b) Localization of ionized residues on albumin at pH 7.4 and pH 3.5. Inset shows the total charge per residue type and for the protein overall at the two pH values. At pH 7.4, the protein has a total charge of −9, while at pH 3.5 the charge is +100 (including the amine terminal group).
To produce the conformational changes induced by the change in the number of charges upon pH change, we added 100 neutralizing counterion charges, a large water box, and ran a large molecular dynamics simulation with ∼300 000 atoms. We observed that the electrostatic repulsions between the three domains in the protein induced a conformational transition from the N isoform to an F-type isoform as shown in the simulation snapshots for the time evolution of this process in the presence of neutralizing counterions (Figure 2a). Within tens of nanoseconds, the distance between domains 1 (orange) and 3 (purple) has increased, with the area between domain 2 (green) and domain 3 acting as a hinge for the expansion as predicted in the literature.58,59 After the initial expansion within this time, the conformation remained stable for up to 64 ns without significant conformational change (Figure 2b). To quantify the simulated expansion, we measured the interdomain distances between center of mass of domain 1 and domain 3 (Figure 2c). Consistent with the simulation snapshots, the initial rate of protein expansion was ∼1.2 nm/ns. Final interdomain spacings of albumin was found to increase from 3.47 ± 0.12 nm (N isoform) to 7.26 ± 0.32 nm (F isoform) (Figure 2c).
Figure 2.

Partial unfolding simulations of albumin with titratable residues set to pH 3.5 ionization states. Orange, green, and purple regions denote domains 1, 2, and 3 respectively. (a) Snapshots of albumin conformations simulation during partial electrostatically triggered denaturation. (b) Final simulation conformations of albumin at pH 3.5. Locations of positive charges and counterions are represented on the right. (c) Distance measured between the center of mass of domain 1 and domain 3 during simulations with counterions (red) in comparison with physiological albumin at pH 7.4 (blue). Insets depict albumin final conformations along each path and are colored with their electrostatic surface potential at the vdW distance (blue is positive, and red is negative).
In addition to tertiary structural changes, the partial denaturation also resulted in a net loss of alpha helical secondary structure, from 62.9% ± 2.9% in the N isoform to 53.2% ± 2.2% in the F isoform (Figure 3a). When resolved by domain, differences in the degree of preservation emerged. Domain 1 was the most preserved with a nonsignificant (p > 0.05) decrease in alpha helical content from 58.6% ± 3.8% in the N isoform to 55.8% ± 2.4% in the F isoform. In contrast, both domains 2 and 3 had significant (p < 0.01) decreases in helical content (domain 2: N = 69.3% ± 3.8% to F = 57.7% ± 3.7% and domain 3: N = 61.0% ± 2.8% to F = 46.6% ± 3.9%). Alpha helical signatures calculated from simulations were consistent with the presence of alpha helical signatures measured experimentally via circular dichroism spectroscopy at different pH values (3.5, 4, 4.5) in the F isoform range (Figure 3b). In contrast, circular dichroism data for thermally denatured albumin near the limit (60 °C) and above (80 °C) albumin’s denaturation temperature, reveals complete or near complete loss of all native secondary structures (Figure 3b). Persistent secondary structural content in pH denatured albumin supports the notion that this partial denaturation pathway does not require disruption of the entire protein as in the case for thermally denatured albumin. The predicted and observed preservation of secondary structures further supports the use of hydrogels formed by the electrostatic triggering method of partial denaturation for drug delivery applications, particularly for drugs that utilize binding sites in domain 1.
Figure 3.
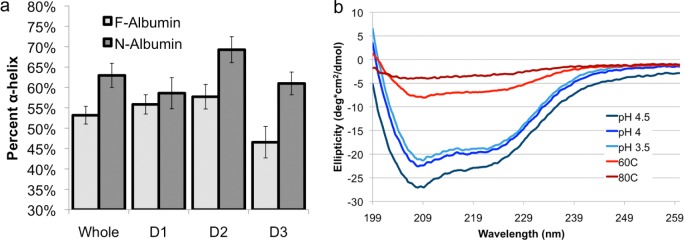
(a) Percentage of helices in each domain for both N and F BSA isoforms. All F domains loose a fraction of their helical content to turn/coil structures during the partial denaturation in comparison to N conformations. (b) Circular dichroism data of dilute solutions of BSA (0.005 wt %) at low pH and high temperature showing the relative degree of secondary structure denaturation. Electrostatically triggered denaturation avoids total loss of secondary structures as observed in thermal denaturation.
Having evaluated both tertiary and secondary structural changes during the N–F conformational transition, we then analyzed the effects of this transition at the individual residue level. Specifically, we focused on the change in solvent exposure of hydrophobic residues to investigate whether hydrophobic attractions might be present that could help explain the observed protein aggregation. We calculated the solvent accessible surface (SAS) area for each residue and categorized all residues as hydrophobic or hydrophilic as determined by the Serada et al. scale (Figure 4).60 We normalized measured SAS areas by the number of atoms contained within each category (domain 1/2/3 and hydrophobic/hydrophilic) for each of the (N and F) isoforms and report the absolute values (Figure 4a). The hydrophobic SAS for domains 1, 2, and 3 in the F isoform was 0.0362 ± 0.0007 nm2/atom, 0.0406 ± 0.0009 nm2/atom, and 0.0388 ± 0.0011 nm2/atom, respectively. In the N isoform, the hydrophobic SAS for domains 1, 2, and 3 was 0.0333 ± 0.0008 nm2/atom, 0.0361 ± 0.0008 nm2/atom, and 0.0355 ± 0.0007 nm2/atom, respectively. The analysis shows that all three domains have a statistically significant (p < 0.0001) increase in the SAS area of hydrophobic residues during the N–F transition. The differences between these absolute SAS area values (domain 1: 0.0028 ± 0.0016 nm2/atom; domain 2: 0.0045 ± 0.0018 nm2/atom; domain 3: 0.0033 ± 0.0019 nm2/atom) during the N–F transition reiterate the increase in hydrophobic SAS area for each domain (Figure 4b). In contrast, the SAS area of hydrophilic residues decreased significantly during the N–F transition. Hydrophilic SAS for domains 1, 2, and 3 in the F isoform was 0.0550 ± 0.0010 nm2/atom, 0.0549 ± 0.0012 nm2/atom, and 0.0524 ± 0.0011 nm2/atom respectively. In the N isoform, hydrophilic SAS for domains 1, 2, and 3 was 0.0576 ± 0.0010 nm2/atom, 0.0548 ± 0.0010 nm2/atom, and 0.0760 ± 0.0009 nm2/atom respectively. From a physical point of view of the entire protein, the hydrophobicity increases by 16% and the hydrophilicity decreases by 13%.
Figure 4.

Solvent accessible surface areas for hydrophobic and hydrophilic moieties in the N and F BSA isoforms represented for (a) each domain individually and (b) the change due to the N–F transition. The SAS increases for hydrophobic moieties and decreases hydrophilic ones in the F isoform.
The total SAS area measurements when both hydrophobic and hydrophilic residues are taken together can be used to infer whether the individual domains are expanding or collapsing (Figure 4a). Although all of the N–F differences were different, the difference in domain 1 was modest (N = 0.0429 ± 0.0007 nm2/atom, F = 0.0436 ± 0.0007 nm2/atom). This small change is consistent with the earlier result that the change in alpha helical content was not significantly different between the two isoforms. However, the domain 2 expanded (N = 0.0435 ± 0.0007 nm2/atom, F = 0.0462 ± 0.0009 nm2/atom) and domain 3 collapsed (N = 0.0524 ± 0.0006 nm2/atom, F = 0.0441 ± 0.0009 nm2/atom) to a greater degree during the transition.
The large decrease in hydrophilic SAS area measured for domain 3 is worth noting. This effect is likely due to several reasons; first is the fact that ASP and GLU residues are protonated at pH 3.5 and thus, less hydrophilic, and second is the greater loss of secondary structure in domain 3. Taken together, these two effects allow ASP and GLU residues to become buried, reducing their SAS area contribution (Figure 5). While ASP and GLU residue SAS areas decrease in every domain, they are disproportionately represented in domain 3, making these effects more noticeable. On the whole, the protein is more hydrophobic in the F isoform than in the N isoform. The increases in hydrophobic SAS area and decreases in hydrophilic SAS area suggest that aggregation of F isoform BSA molecules in high concentrations may be due to intermolecular hydrophobic interactions.
Figure 5.

Solvent accessible surface areas for ASP and GLU residues in the N and F BSA isoforms in the total protein and in for each domain. Normally hydrophilic residues ASP and GLU face the solvent at pH 7.4 but are hydrophobic when protonated at pH 3.5.
To test this hypothesis, we investigated the interactions between two proteins using our new F isoform albumin models. We placed two of these configurations in contact such that their newly exposed hydrophobic surfaces, as determined by the increase in local hydrophobic SAS, were facing each other. With this arrangement, the effective concentration of albumin in water in this simulation was ∼7 mg/mL, substantially lower than the experimentally observed threshold for gelation (15 mg/mL) but sufficient for examining the interaction between two proteins. We run two types of simulations, one with explicit counterions and the other without them. The absence of counterions, while unphysical, results in a tremendous speed up of the simulations and the aim was to check whether physical insightful results could be obtained. However, in the absence of counterions, large electrostatic repulsions between the proteins forced them to move away from each other soon after overcoming the initial contact attraction (Figure 6a), leading to a result that is qualitatively wrong, as shown next, demonstrating the importance of appropriately counting for the explicit counterions.
Figure 6.

Dimerization of two F conformation albumin proteins. (a) Minimum distance measured between two F-isoform BSA structures placed near each other and simulated with and without system neutralizing counterions. Proteins with counterions allowed proteins to stay within 0.25 nm of each other (black line) until they separated after 36 ns. Absence of counterions allowed unscreened repulsive electrostatic interactions to rapidly overcome attractions (red line). (b) Configurations of two proteins from (a) at 10 ns. The top pair (green protein and white protein) corresponds to the no counterion simulation and the bottom pair corresponds to the counterion simulation. Individual surface atoms are colored by the change in free energy due to solvation in water (kcal/mol). Hydrophobic and hydrophilic atoms are colored red and blue, respectively. The blue arrow indicates the point of contact between the two proteins.
In the presence of counterions necessary to maintain system electroneutrality (200 Cl–), the two proteins stayed within 0.25 nm of each other, as indicated by the minimum distance measured between the two proteins (Figure 6a). The persistent point of contact between the two proteins was located in domain 2 but this may be an artifact of the initial protein placement (Figure 6b). Interestingly, after 36 ns, the two proteins separated from each other. This suggests that the attraction observed between the two proteins may be a result of a local minimum in the free energy as a result of the increased hydrophobicity but would need to be corroborated with additional simulations.
Calculation of the electrostatic surface energy potential provides an additional method to evaluate the intermolecular interactions. The usual way to determine electrostatic potentials in proteins is by solving the Poisson–Boltzmann (PB) equation. However, it is not clear how good the mean-field approximation would be in a system with such larger number of charges. Therefore, we performed PB calculations and explicit determination of the electrostatic potentials from the findings of the positions of all the molecules, including the ions, from the simulations. Explicit electrostatic potential calculations that factor the contribution of counterions in the system results in surface potentials that are more negative when compared to the result from PB (Figure 7a). Particularly interesting is that, in the scale shown in Figure 7a, the PB results show an almost constant, relatively high, positive potential that directly reflects the charge on the proteins, that is, the +100 that result from the low pH. In sharp contrast, the explicit calculations demonstrate relatively large, variation of the electrostatic potential across the protein surface, showing that the explicit positions of the counterions plays a dramatic role in determining the structure and interactions of proteins. This is very important since the PB calculations would suggest strong attractive interactions between the protein (anywhere on its surface!) and negatively charged molecules, or surfaces. On the other hand, the full calculations show a much more complex surface that could lead to a variety of possible interactions.
Figure 7.

Explicit counterion and PB calculated electrostatic surface potentials for (a) single proteins and (b) aggregated proteins. Explicit counterion calculations result in a more negative electrostatic potential when compared to PB. Explicit calculations that ignore counterion contributions duplicate the positive electrostatic potentials shown by PB. Potentials are shown at the Connolly and SAS surfaces are shown for all cases. For clarity, aggregated proteins are colored individually (orange and purple) to help differentiate them in the potential surface representation. (c) Residue ARG 208 is an example of a residue which is has a positive potential when calculated with PB and a negative potential when calculated with explicit counterions. A histogram of the distances to the nearest Cl– ion for the charged N+ atom on ARG 208 demonstrates that this residue is typically bound to Cl–. (d) Localization of residues selected for further analysis. ARG 208 is shown in silver. Inset depicts ARG 484 (green), LYS 474 (blue), GLU 478 (red), and LYS 350 (yellow) are all located near the point of contact between the two proteins (orange and purple). (e) Histograms of distances to nearest Cl– ions for the four selected residues near the point of contact in d and magnified representations of charged atoms associated with Cl–. Orange and purple coloring of residue names indicate which protein the residue is from.
While in many cases the PB calculation is sufficient, it misses many important details regarding the effect of individual counterions in highly charged systems. For example, at residue ARG 208 (Figure 7a, green arrow), PB predicts the nitrogen atom to have a positive electrostatic potential. In fact, the explicit calculation indicates the potential is negative due to the attraction of a neighboring Cl– counterion (Figure 7c, right). A histogram of the distances to the nearest Cl– ion for the charged N+ atom on ARG 208 demonstrates that this residue is typically bound to a counterion (Figure 7c, left).
In the case of two proteins interacting with each other, we observe similar effects of the electrostatic surface potential calculation as in the single protein case (Figure 7b). To underscore the important contribution of these counterions on the interpretation of the electrostatic potential, we have additionally computed the explicit electrostatic potential while ignoring the counterions present (Figure 7b center). This results in a relatively high, positive potential similar to the one calculated by PB (Figure 7b right). We also show the potentials calculated at the Connolly surface (0.14 nm) and the SAS surface (1.4 nm) to demonstrate how the potential becomes more negative as we move away from the positive charges on the protein. This detail is largely lost in the PB calculation where the effects of numerous positive surface charges persist for greater distances.
Four additional residues at the point of contact between the two proteins are highlighted for further analysis (Figure 7d). All four (and two in particular, LYS 350 and LYS 474) rarely had any associated counterions in the single protein case. But, when brought in contact with another highly charged protein, all four residues were substantially more likely to have counterions present (Figure 7e). Both LYS 350 and LYS 474 were rarely seen without a counterion present after dimerization. In the case of GLU 478 and LYS 350, a chlorine ion was found close to both proteins (orange and purple). While there are many other positively charged surface residues on both proteins, they do not all recruit counterions to them as in the case of LYS 350 and LYS 474. This is due to the inherent entropic cost of binding every free counterion with every positively charged residue but it becomes more likely when the proteins are dimerized (Figure 7e). The increased likelihood of finding nearby counterions in the dimerized state suggests the attraction of these counterions is necessary to neutralize residue charges and promote protein aggregation.
Thus far, these observations support the hypothesis that hydrophobic interactions from the protein core and counterion association to charged residues at the proteins point of contact drives the self-assembly of the hydrogel network. Importantly, the electrostatically driven denaturation observed in these fully atomistic BSA simulations captures the conformational structures predicted by others in the literature4,37 but with a much greater accuracy.
4. Conclusions
Our results provide insights into what are the interactions necessary to overcome the highly charged nature of the F isoform in forming protein aggregates. When the individual proteins are highly charged, strong intramolecular electrostatic repulsions trigger a partial denaturation of the protein. We used FAMBE-pH to calculate the total solvation free energy of the protein as a function of pH and determined the probability of residue ionization on albumin. Lowering the solution pH to 3.5 and simulating with molecular dynamics enables albumin to make the N to F isoform transition in a manner that is driven by electrostatic repulsions, and that results in the exposure of core hydrophobic regions. These hydrophobic regions are critically involved in the aggregation of the proteins despite the electrostatic repulsions still present between proteins. Interprotein electrostatic repulsions are mitigated by the attraction of counterions to charged residues at the point of contact. Extended simulations after 36 ns showed separation of two proteins, suggesting a local free energy minimum for the aggregated state with two proteins at subthreshold concentrations. Larger simulations with more than four interacting proteins would be necessary to meet the threshold concentration but are computationally demanding to perform. An explicit counterion calculation of electrostatic surface potentials resulted in new insights that were missed by conventional PB calculations. Solving the electrostatic surface potential with explicit consideration of counterions may be a useful approach in other protein and drug binding studies. Analysis of the protein conformation reveals that alpha helical structures in domain 1 are preserved and that the total secondary structural content is more preserved when compared to thermally denatured albumin gels. Future studies will explore whether such preserved structures, particularly in domain 1, can retain the binding capacity of N isoform albumin for use in drug delivery or toxin removal applications. Building on this improved understanding of partially denatured albumin conformations, puts us in a better position to harness these electrostatically triggered hydrophobically self-assembled protein gelation mechanisms to reveal new solutions to longstanding problems in drug delivery and unwanted protein self-assembly, for example, amyloid formation.
Acknowledgments
This research was supported in part by the National Institutes of Health under Ruth L. Kirschtein National Research Service Award 1F31EB014698-01 from the National Institute of Biomedical Imaging and Bioengineering. This research was also supported by a grant from IMASL-CONICET PIP-112-2011-0100030 (J.A.V.) and Project 328402 from the National University of San Luis (J.A.V.), Argentina. Molecular graphics representations were performed with the VMD package. Poisson–Boltzmann molecular graphics and analyses were performed with the UCSF Chimera package. Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Zhang S. G. Fabrication of Novel Biomaterials through Molecular Self-Assembly. Nat. Biotechnol. 2003, 21101171–1178. [DOI] [PubMed] [Google Scholar]
- Peppas N. A.; Hilt J. Z.; Khademhosseini A.; Langer R. Hydrogels in Biology and Medicine: From Molecular Principles to Bionanotechnology. Adv. Mater. 2006, 18111345–1360. [Google Scholar]
- Slaughter B. V.; Khurshid S. S.; Fisher O. Z.; Khademhosseini A.; Peppas N. A. Hydrogels in Regenerative Medicine. Adv. Mater. 2009, 2132–333307–3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter D. C.; Ho J. X. Structure of Serum-Albumin. Adv. Protein Chem. 1994, 45, 153–203. [DOI] [PubMed] [Google Scholar]
- Branden C.; Tooze J.. Introduction to Protein Structure, 2nd ed.; Garland: New York, 1999. [Google Scholar]
- Pfefferkorn C. M.; McGlinchey R. P.; Lee J. C. Effects of pH on Aggregation Kinetics of the Repeat Domain of a Functional Amyloid, Pmel17. Proc. Natl. Acad. Sci. U.S.A. 2010, 1075021447–21452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Es-haghi A.; Shariatizi S.; Ebrahim-Habibi A.; Nemat-Gorgani M. Amyloid Fibrillation in Native and Chemically-Modified Forms of Carbonic Anhydrase II: Role of Surface Hydrophobicity. Biochim. Biophys. Acta, Proteins Proteomics 2012, 18243468–477. [DOI] [PubMed] [Google Scholar]
- Krebs M. R. H.; Domike K. R.; Donald A. M. Protein Aggregation: More than Just Fibrils. Biochem. Soc. Trans. 2009, 37, 682–686. [DOI] [PubMed] [Google Scholar]
- Peters T.All About Albumin; Elsevier: New York, 1995. [Google Scholar]
- Brandt J.; Fredriks M.; Andersson L. O. Coupling of Dyes to Biopolymers by Sensitized Photooxidation - Affinity Labeling of a Binding-Site in Bovine Serum-Albumin. Biochemistry 1974, 13234758–4764. [DOI] [PubMed] [Google Scholar]
- Day Y. S. N.; Myszka D. G. Characterizing a Drug’s Primary Binding Site on Albumin. J. Pharm. Sci. 2003, 922333–343. [DOI] [PubMed] [Google Scholar]
- Hamilton J. A.; Era S.; Bhamidipati S. P.; Reed R. G. Locations of the 3 Primary Binding-Sites for Long-Chain Fatty-Acids on Bovine Serum-Albumin. Proc. Natl. Acad. Sci. U.S.A. 1991, 8862051–2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilger C.; Grigioni F.; De Beaufort C.; Michel G.; Freilinger J.; Hentges F. Differential Binding of IgG and IgA Antibodies to Antigenic Determinants of Bovine Serum Albumin. Clin. Exp. Immunol. 2001, 1233387–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barone G.; Capasso S.; DelVecchio P.; DeSena C.; Fessas D.; Giancola C.; Graziano G.; Tramonti P. Thermal Denaturation of Bovine Serum Albumin and Its Oligomers and Derivatives pH Dependence. J. Therm. Anal. 1995, 4561255–1264. [Google Scholar]
- Chmelik J.; Anzenbacher P.; Chmelikova J.; Matejckova M.; Kalous V. Mechanism of Denaturation of Human-Serum Albumin by Urea. Collect. Czech. Chem. Commun. 1988, 532411–422. [Google Scholar]
- Gianazza E.; Galliano M.; Miller I. Structural Transitions of Human Serum Albumin: An Investigation Using Electrophoretic Techniques. Electrophoresis 1997, 185695–700. [DOI] [PubMed] [Google Scholar]
- Giancola C.; DeSena C.; Fessas D.; Graziano G.; Barone G. DSC Studies on Bovine Serum Albumin Denaturation - Effects of Ionic Strength and SDS Concentration. Int. J. Biol. Macromol. 1997, 203193–204. [DOI] [PubMed] [Google Scholar]
- Baier S. K.; Decker E. A.; McClements D. J. Impact of Glycerol on Thermostability and Heat-Induced Gelation of Bovine Serum Albumin. Food Hydrocolloids 2004, 18191–100. [Google Scholar]
- Baier S. K.; McClements D. J. Combined Influence of NaCl and Sucrose on Heat-Induced Gelation of Bovine Serum Albumin. J. Agric. Food Chem. 2003, 51278107–8112. [DOI] [PubMed] [Google Scholar]
- Clark A. H.; Kavanagh G. M.; Ross-Murphy S. B. Globular Protein Gelation - Theory and Experiment. Food Hydrocolloids 2001, 154–6383–400. [Google Scholar]
- Gosal W. S.; Ross-Murphy S. B. Globular Protein Gelation. Curr. Opin. Colloid Interface Sci. 2000, 53–4188–194. [Google Scholar]
- Haque Z. Z.; Aryana K. J. Effect of Copper, Iron, Zinc and Magnesium Ions on Bovine Serum Albumin Gelation. Food Sci. Technol. Res. 2002, 811–3. [Google Scholar]
- Matsudomi N.; Oshita T.; Kobayashi K.; Kinsella J. E. Alpha-Lactalbumin Enhances the Gelation Properties of Bovine Serum-Albumin. J. Agric. Food Chem. 1993, 4171053–1057. [Google Scholar]
- Matsudomi N.; Rector D.; Kinsella J. E. Gelation of Bovine Serum-Albumin and Beta-Lactoglobulin - Effects of pH, Salts and Thiol Reagents. Food Chem. 1991, 40155–69. [Google Scholar]
- Navarra G.; Giacomazza D.; Leone M.; Librizzi F.; Militello V.; Biagio P. L. S. Thermal Aggregation and Ion-Induced Cold-Gelation of Bovine Serum Albumin. Eur. Biophys. J. 2009, 384437–446. [DOI] [PubMed] [Google Scholar]
- Renard D.; Lefebvre J. Gelation of Globular-Proteins - Effect of Ph and Ionic-Strength on the Critical Concentration for Gel Formation - a Simple-Model and Its Application to Beta-Lactoglobulin Heat-Induced Gelation. Int. J. Biol. Macromol. 1992, 145287–291. [DOI] [PubMed] [Google Scholar]
- Abraxis BioScience. Abraxane for Injectable Suspension (paclitaxel protein-bound particles for injectable suspension); 2005, 1–20; www.drugs.com.
- Christiansen C.; Kryvi H.; Sontum P. C.; Skotland T. Physical and Biochemical-Characterization of Albunex, a New Ultrasound Contrast Agent Consisting of Air-Filled Albumin Microspheres Suspended in a Solution of Human Albumin. Biotechnol. Appl. Biochem. 1994, 19, 307–320. [PubMed] [Google Scholar]
- Christiansen C.; Vebner A. J.; Muan B.; Vik H.; Haider T.; Nicolaysen H.; Skotland T. Lack of an Immune-Response to Albunex(R), a New Ultrasound Contrast Agent Based on Air-Filled Albumin Microspheres. Int. Arch. Allergy Imm 1994, 1044372–378. [DOI] [PubMed] [Google Scholar]
- Iemma F.; Spizzirri U. G.; Puoci F.; Muzzalupo R.; Trombino S.; Picci N. Radical Cross-Linked Albumin Microspheres as Potential Drug Delivery Systems: Preparation and in Vitro Studies. Drug Delivery 2005, 124229–234. [DOI] [PubMed] [Google Scholar]
- Pande S.; Vyas S. P.; Dixit V. K. Localized Rifampicin Albumin Microspheres. J. Microencapsulation 1991, 8187–93. [DOI] [PubMed] [Google Scholar]
- Rubino O. P.; Kowalsky R.; Swarbrick J. Albumin Microspheres as a Drug-Delivery System - Relation among Turbidity Ratio, Degree of Cross-Linking, and Drug-Release. Pharm. Res. 1993, 1071059–1065. [DOI] [PubMed] [Google Scholar]
- Kakinoki S.; Taguchi T. Antitumor Effect of an Injectable in-Situ Forming Drug Delivery System Composed of a Novel Tissue Adhesive Containing Doxorubicin Hydrochloride. Eur. J. Pharm. Biopharm. 2007, 673676–681. [DOI] [PubMed] [Google Scholar]
- Iemma F.; Spizzirri U.; Muzzalupo R.; Puoci F.; Trombino S.; Picci N. Spherical Hydrophilic Microparticles Obtained by the Radical Copolymerisation of Functionalised Bovine Serum Albumin. Colloid Polym. Sci. 2004, 2833250–256. [Google Scholar]
- Murata M.; Tani F.; Higasa T.; Kitabatake N.; Doi E. Heat-Induced Transparent Gel Formation of Bovine Serum-Albumin. Biosci., Biotechnol., Biochem. 1993, 57143–46. [DOI] [PubMed] [Google Scholar]
- Oss-Ronen L.; Seliktar D. Photopolymerizable Hydrogels Made from Polymer-Conjugated Albumin for Affinity-Based Drug Delivery. Adv. Biomater. 2010, 121–245–52. [DOI] [PubMed] [Google Scholar]
- Leggio C.; Galantini L.; Pavel N. V. About the Albumin Structure in Solution: Cigar Expanded Form versus Heart Normal Shape. Phys. Chem. Chem. Phys. 2008, 10456741–6750. [DOI] [PubMed] [Google Scholar]
- Yamasaki M.; Yano H.; Aoki K. Differential Scanning Calorimetric Studies on Bovine Serum-Albumin 0.1. Effects of pH and Ionic-Strength. Int. J. Biol. Macromol. 1990, 124263–268. [DOI] [PubMed] [Google Scholar]
- Baler K.; Carignano M. A.; Szleifer I.; Ameer G. A.. Albumin Hydrogels Formed by Electrostatically Triggered Self-Assembly and Their Drug Delivery Behavior. Unpublished work. [DOI] [PMC free article] [PubMed]
- Berendsen H. J. C.; Vanderspoel D.; Vandrunen R. Gromacs - a Message-Passing Parallel Molecular-Dynamics Implementation. Comput. Phys. Commun. 1995, 911–343–56. [Google Scholar]
- Hess B.; Kutzner C.; van der Spoel D.; Lindahl E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, And Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 43435–447. [DOI] [PubMed] [Google Scholar]
- Lindahl E.; Hess B.; Van Der Spoel D. GROMACS 3.0: A Package for Molecular Simulation and Trajectory Analysis. J. Mol. Model. 2001, 78306–317. [Google Scholar]
- Van der Spoel D.; Lindahl E.; Hess B.; Groenhof G.; Mark A. E.; Berendsen H. J. C. Gromacs: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26161701–1718. [DOI] [PubMed] [Google Scholar]
- Nielsen M., Lundegaard C., Lund O., Petersen T. N., CpHModels-3.0. Remote homology Modeling Using Structure Guided Profile Sequence Alignment and Double-Sided Baseline Corrected Scoring Scheme. Abstract at the CASP8 Conference 2008, 193. [Google Scholar]
- Nielsen M., Lundegaard C., Lund O., Petersen T. N., CPHmodels-3.0 - Remote Homology Modeling Using Structure Guided Sequence Profiles. Nucleic Acids Res. 2010, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bujacz A. Structures of Bovine, Equine and Leporine Serum Albumin. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2012, 68, 1278–1289. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L.; Maxwell D. S.; TiradoRives J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 1184511225–11236. [Google Scholar]
- Vorobjev Y. N.; Vila J. A.; Scheraga H. A. FAMBE-pH: A Fast and Accurate Method to Compute the Total Solvation Free Energies of Proteins. J. Phys. Chem. B 2008, 1123511122–11136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen H. J.; Van Gunsteren W. F.; Zwinderman H. R.; Geurtsen R. G. Simulations of Proteins in Water. Ann. N.Y. Acad. Sci. 1986, 482, 269–86. [DOI] [PubMed] [Google Scholar]
- Heinig M.; Frishman D. STRIDE: A Web Server for Secondary Structure Assignment from Known Atomic Coordinates of Proteins. Nucleic Acids Res. 2004, 32, W500–W502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker N. A.; Sept D.; Joseph S.; Holst M. J.; McCammon J. A. Electrostatics of Nanosystems: Application to Microtubules and the Ribosome. Proc. Natl. Acad. Sci. U.S.A. 2001, 981810037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF Chimera - A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25131605–1612. [DOI] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual Molecular Dynamics. J. Mol. Graphics Modell. 1996, 14133–38. [DOI] [PubMed] [Google Scholar]
- Sugio S.; Kashima A.; Mochizuki S.; Noda M.; Kobayashi K. Crystal Structure of Human Serum Albumin at 2.5 Angstrom Resolution. Protein Eng. 1999, 126439–446. [DOI] [PubMed] [Google Scholar]
- Vorobjev Y. N. Advances in Implicit Models of Water Solvent to Compute Conformational Free Energy and Molecular Dynamics of Proteins at Constant pH. Adv. Protein Chem. Struct. Biol. 2011, 85, 281–322. [DOI] [PubMed] [Google Scholar]
- Bohme U.; Scheler U. Effective Charge of Bovine Serum Albumin Determined by Electrophoresis NMR. Chem. Phys. Lett. 2007, 435, 342–345. [Google Scholar]
- Tanford C.; Buzzell J. G.; Rands D. G.; Swanson S. A. The Reversible Expansion of Bovine Serum Albumin in Acid Solutions. J. Am. Chem. Soc. 1955, 77246421–6428. [Google Scholar]
- Geisow M. J.; Beaven G. H. Physical and Binding Properties of Large Fragments of Human-Serum Albumin. Biochem. J. 1977, 1633477–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M. Y. Direct Evidence for the Involvement of Domain-III in the N-F Transition of Bovine Serum-Albumin. Biochem. J. 1986, 2361307–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sereda T. J.; Mant C. T.; Sonnichsen F. D.; Hodges R. S. Reversed-Phase Chromatography of Synthetic Amphipathic Alpha-Helical Peptides as a Model for Ligand/Receptor Interactions Effect of Changing Hydrophobic Environment on the Relative Hydrophilicity/Hydrophobicity of Amino-Acid Side-Chains. J. Chromatogr. A 1994, 6761139–153. [DOI] [PubMed] [Google Scholar]