

Abstract
Protein arginine methylation is a posttranslational modification critical for a variety of biological processes. Misregulation of protein arginine methyltransferases (PRMTs) has been linked to many pathological conditions. Most current PRMT inhibitors display limited specificity and selectivity, indiscriminately targeting many methyltransferase enzymes that use S-adenosyl-l-methionine as a cofactor. Here we report diamidine compounds for specific inhibition of PRMT1, the primary type I enzyme. Docking, molecular dynamics, and MM/PBSA analysis together with biochemical assays were conducted to understand the binding modes of these inhibitors and the molecular basis of selective inhibition for PRMT1. Our data suggest that 2,5-bis(4-amidinophenyl)furan (1, furamidine, DB75), one leading inhibitor, targets the enzyme active site and is primarily competitive with the substrate and noncompetitive toward the cofactor. Furthermore, cellular studies revealed that 1 is cell membrane permeable and effectively inhibits intracellular PRMT1 activity and blocks cell proliferation in leukemia cell lines with different genetic lesions.
Introduction
Arginine methylation is a posttranslational modification catalyzed by protein arginine N-methyltransferases (PRMTs). This family of enzymes transfers the methyl group from S-adenosyl-l-methionine (AdoMet, SAM) to the guanidino group of specific arginine residues, leading to mono- or dimethylated arginine residues and releasing S-adenosyl-l-homocysteine (AdoHcy, SAH) as a co-product. This posttranslational modification regulates a wide diversity of biological processes, from chromatin remodeling, signal transduction, RNA splicing, and DNA repair to cell proliferation and differentiation.1−4 In humans, nine PRMTs have been identified so far and are classified into two major groups, type I (PRMT1, -3, -4, -6, and -8) and type II (PRMT5 and -9) according to the specificity and stereochemistry of the methylated product. Type I PRMT enzymes catalyze the formation of asymmetric ω-NG,NG-dimethylarginine (ADMA) residues, whereas the formation of symmetric ω-NG,NG-dimethylarginine (SDMA) residues is maintained by type II enzymes. PRMT1, the predominant mammalian type I enzyme, is identified by yeast two-hybrid screening.5 PRMT1 is ubiquitously expressed and responsible for over 85% of the arginine methylation in mammalian cells.6 PRMT1 has been demonstrated to impact a number of disease pathways. For instance, PRMT1 is an essential element in the oncogenic MLL fusion complexes and confers an aberrant transcriptional activation property critical for the induction of leukemia.7 PRMT1 is also overexpressed in breast cancer and has altered substrate specificity.8 PRMT1-variant 2 is a marker of unfavorable prognosis in colon cancer patients.9 Importantly, given the close correlation of PRMT1 activity with the up-regulation of serum ω-NG,NG-asymmetric dimethyarginine amino acid, which is an endogenous nitric oxide synthase (NOS) inhibitor, PRMT1 has causal relationship with broad cardiovascular implications and inflammatory responses such as diabetes and hypertension.10−12
PRMTs belong to a highly conserved family of proteins in eukaryotes with a conserved catalytic methyltransferase domain. The three-dimensional structures of a few type I PRMTs have been determined.13,14 The structure of the mammalian PRMT1 revealed a two-domain architecture composed of a common AdoMet binding domain and a barrel-like domain with the active site situated between them. Given that the amino acid sequences constituting the AdoMet binding region in PRMTs are largely invariant, specificity in the binding of target proteins likely plays a major role in the substrate selective methylation performed by these enzymes. The selective inhibition of specific PRMTs over others is highly desirable and could yield potential new therapeutics with minimum off-target toxicity. Also, isoform-selective inhibitors are powerful chemical genetic tools for dissecting the enzymatic functions of PRMT1 with spatial and temporal resolution in selected disease pathways.15
A steady progress has been witnessed in the past years in developing PRMT chemical modulators. Substrate-based chemical modalities provide a facile way to PRMT inhibition. AdoMet analogues such as sinefungin and methylthioadenosine were used in the early stage as chemical tools for studying PRMT function, regardless of their pan-antimethylation activity. Interestingly, recent work showed that some compounds containing the structural scaffold of AdoMet linked to arginine motifs can lead to inhibitors with certain isoform selectivity.16−18 Substrate-based inhibitors have particular value for obtaining cocrystal structures of the enzyme–substrate complex, thus being useful molecular tools for understanding substrate recognition mechanism. On the other hand, a number of small molecule PRMT inhibitors have been reported such as AMI-1,19 stilbamidine and allantodapsone,20 RM65,21 pyrazoles,22−25 benzo[d]imidazoles,26 NS-1,27 among others.15,28−33 It should be cautioned that some of these compounds, e.g., AMI-1, NS-1, and A36, most likely target the histone substrate rather than the PRMT1 enzyme.27,34 Notwithstanding recent progress, considerable challenges remain as to how to achieve high potency and selectivity. More diverse chemical structures are waiting to be discovered or designed in order to meet the need of basic biology research and therapeutic development.
Herein we report diamidine-type compounds for selective inhibition of PRMT1 over other PRMTs (CARM1, PRMT5, and PRMT6). The activity of diamidine compounds as therapeutic agents is previously known: inhibition of tachyzoite proliferation against Neospora caninum and Toxoplasma gondii,35 interaction with nuclear DNA topoisomerase II,36 ERG/DNA complex modulation which is important during gene expression,37 and the use as antileishmanial agent.38 2,5-Bis(4-amidinophenyl)furan (furamidine, compound 1) has been shown to concentrate in the cell nucleus and delay parasite maturation,39 serving as an anti-Plasmodium vivax agent6 or potent antiparasitic agent in vitro or in vivo.40 Because of its ability to disrupt mitochondrial membranes, 1 can be used as an antimicrobial agent too.41 Despite such wide applicability, the use of diamidines for PRMT inhibition has not been reported previously except for stilbamidine. We studied a number of diamidine compounds for PRMT inhibition, and several showed clear selectivity for PRMT1. Kinetic data and computer modeling suggest that compound 1 targets the active site pockets of PRMT1.
Results and Discussion
Searching for New PRMT1 Inhibitors
In this work, we attempted to develop PRMT-selective inhibitors containing diamidine groups. Spannhoff and co-workers previously reported that a diamidine compound, stilbamidine, inhibited PRMT1 activity at the micromolar level.20 Since then, however, no further reports have been invested on diamidine-based PRMT inhibitors. Our rationale in considering diamidines for PRMT inhibition was the close resemblance of the amidine group to the guanidine moiety of the substrate arginine. To test this hypothesis, we assessed a series of diamidine compounds for PRMT1 inhibition (Figure 1). In addition to inhibiting PRMT1, the representative member of type I arginine methyltransferases, we also tested the effect of the compounds on the activity of type II methyltransferase PRMT5, with the purpose of gaining type I and/or type II selective inhibitors. We used the typical radiometric P81 filter binding assay to measure the effect of these diamidines on the activity of PRMT1 and PRMT5. In the assay, [3H]-labeled AdoMet and a histone 20-aa H4 peptide from the N-terminal tail of histone H4 (denoted H4-20) were used as substrates. The initial screening for both PRMT1 and PRMT5 inhibition was performed at 20 μM of each compound, and the results are summarized in Table S1. It is clear that different compounds showed varied degrees of inhibitory activity toward PRMT1 and PRMT5. For instance, while 21 showed only very weak activity, 2 was found to block more than 85% of the activity for both PRMT1 and PRMT5.
Figure 1.

Structures of tested amidine compounds.
In consideration of the significance of isoform-selective inhibitors, we are particularly interested in those hits that selectively inhibited PRMT1 or PRMT5 activity. In this regard, compound 1 (furamidine, also known as DB7542) showed more than 75% inhibition of PRMT1 while it had only 11% inhibition against PRMT5, which demonstrates that 1 likely is a selective inhibitor of PRMT1. Indeed, the IC50 of 1 was determined to be 9.4 μM for PRMT1 and 166 μM for PRMT5 (Table 1). Thus, compound 1 exhibited selective inhibition for PRMT1 over PRMT5. Also, the analogue 5 showed comparable potency and selectivity to 1, with IC50 of 7.2 μM for PRMT1 and 186 μM for PRMT5. Stilbamidine, previously reported as a PRMT1 inhibitor, was tested for comparison and showed IC50 values of 15.2 μM for PRMT1 and 44.1 μM for PRMT5. Thus, the selectivity of stilbamidine proved to be inferior to both compound 1 and compound 5.
Table 1. Inhibition of PRMTs by Selected Compoundsa.
| IC50, μM |
||||
|---|---|---|---|---|
| inhibitor | PRMT1 | PRMT5 | CARM1 | PRMT6 |
| 1 | 9.4 ± 1.1 | 166 ± 2 | >400 | 283 ± 37 |
| 5 | 7.2 ± 2.4 | 186 ± 3 | >400 | 211 ± 107 |
| stilbamidine | 15.2 ± 2.3 | 44.1 ± 5.9 | ∼400 | 173 ± 63 |
IC50 values of different diamidine compounds were tested by filter-binding assay with 1 μM H4(1–20) peptide, 0.5 μM [3H]AdoMet, 0.04 μM PRMT1, and PRMT5, and incubation was with varying concentrations of each compound at 30 °C for 8 min. For assay on CARM1 and PRMT6, 1 μM histone H3.1 was used instead of 1 μM H4(1–20) peptide, and the reaction time was 1 h.
Selectivity of Compound 1
PRMTs are largely categorized into two classes, type I and type II, with PRMT1 being representative of type I and PRMT5 of type II.43 Compound 1 showed clear type I selectivity based on the IC50 data for PRMT1 and PRMT5. In an effort to determine the selectivity profile of the diamidine compounds on other type I PRMTs, coactivator-associated arginine methyltransferase 1 (CARM1, PRMT4) and PRMT6 were examined (Table 1). All the experiments were accomplished with the same radioactive P81 filter binding methylation assay. It is an interesting observation that the diamidine compounds tested here, 1 and 5 and stilbamidine, showed stronger inhibition for PRMT1 over the other PRMTs. In particular, the activities of these diamidine compounds were much weaker toward CARM1 and PRMT6. Together, the data in Table 1 demonstrated that 1 and 5 are PRMT1-selective inhibitors.
Structure–Activity Relationship (SAR) of the Diamidine Compounds in PRMT Inhibition
The data shown in Table S1 offer some interesting insights about the SAR of the diamidine compounds in PRMT1 and PRMT5 inhibition. First, the activities of the diamidine compounds are sensitive to alkylation of the terminal amidine moiety. Compounds 9, 12, 17, 18, and 22 having alkyl and 26 having phenyl substituents on the amidine all showed reduced activity in comparison with 1. Likely, these hydrophobic substituents decrease the hydrogen bond donor effect of the diamidine that is important for PRMT1 binding. Steric hindrance is also a factor: the bulkiness on the diamidine termini clearly reduces activities. For example, compound 18 (with the cyclohexyl substituent) showed a weaker activity than 9 and 12, which contained cyclopentyl and cyclopropyl substituents, respectively. Phenyl substitution was even more detrimental than alkylation (e.g., 26 versus 18). Both of the amidine groups seem to be required for efficient inhibition of PRMT1. This can be seen from the activity differences between 25 and 1, 14 and 4, and 24 and 5, the first of which lost one amidine group and showed decreased activity. It is possible that either the hydrogen bond donating ability or the positive charge of the amidine group plays a critical role in binding to PRMT1.
The replacement of the oxygen heteroatom in the furan ring of compound 1 with S or Se atom (compounds 4 and 3) had almost no effect on activity, probably because these three atoms have a similar stereoelectronic property. However, replacement with NH (6) caused a bigger loss in activity, as NH has an additional hydrogen atom. Compound 28, with a phenyl substituent on the pyrrole ring, almost lost all activity, likely because of increased steric bulkiness. The anti-PRMT1 activities of the diamidine compounds are also sensitive to the changes on the two benzene rings. The ortho substitution with a nitrogen atom especially decreased activity (e.g., 7 and 10 in comparison with 1). On the other hand, the meta substitution was less sensitive, as seen in 5 that was equally potent as 1.
The SAR property of the diamidine compounds for PRMT5 inhibition is different from that for PRMT1 inhibition. The most striking feature is that the amidine group (at least one of the two) seems not to be essential for PRMT5 inhibition. Compounds 8 and 14 lost one amidine group compared with 4, but they exhibited a stronger potency for PRMT5. The same phenomenon was seen in 24 versus 5. These results may suggest that monoadimine compounds might be more favorable than diamidine for PRMT5 inhibition. Furthermore, alkylation of the diamidine termini offered a positive contribution for PRMT5 inhibition. Compounds 9, 12, 17, 18, and 26 had an alkyl or phenyl group on the diamidine, and all of them showed better activities than 1 that contained unmodified diamidines. This phenomenon may be an indication that the hydrogen bonding effect of the amidine moiety in PRMT5 binding is not as important as the hydrophobic interaction inferred. Also, compound 28 is more hydrophobic and bigger in size than 1 and showed stronger activity for PRMT5, suggesting that stronger hydrophobicity and bigger size at the central five-member-ring position might be a favorable factor for PRMT5 inhibition. Overall, our biochemical data suggest that PRMT5 prefers to bind those (di)amidine molecules with higher hydrophobicity and more bulkiness. As seen vide infra, our computational analysis shows that PRMT1 has a more compact cavity than PRMT5, and this difference could be an explanation for the selective inhibition of PRMT1 by compounds 1 and 5.
Kinetic Analysis of PRMT1 Inhibition by Compound 1
Since 1 is structurally simple and is one of the best hits among the tested diamidine compounds for PRMT1, we focused on this compound for further mechanistic characterization. To elucidate the inhibition mechanism of compound 1, steady-state kinetic characterization was conducted. In this study, the initial velocities of PRMT1 were measured at several selected concentrations of the inhibitor over a range of varied concentrations of the H4-20 peptide while fixing the concentration of [3H]SAM. The data were plotted with velocities versus H4-20 concentration in Figure 2A. Double-reciprocal plot was also shown with 1/velocity versus 1/[H4-20] (Figure 2B). For a more quantitative analysis, the Michaelis–Menten kinetic data were fit to the following equation:
| 1 |
where [S] is the concentration of the H4-20 peptide. From the fitting we obtained a Kis of 2.6 μM and a Kii of 38 μM. The Kis value is about 14-fold smaller than Kii value, which implies that 1 has a strong nature of competitive inhibition with respect to the peptide substrate. In the double-reciprocal plot, a series of straight lines were intersected closely in the second quadrant, also supporting a pseudo-competitive pattern of inhibition.
Figure 2.

Kinetic analysis of PRMT1 inhibition by compound 1. (A) and (C) are Michaelis–Menten plots, and (B) and (D) are double-reciprocal plot of initial velocities versus varied concentrations of H4-20 or [3H]SAM. Concentration of 1 was selected at 0 μM (▲), 10 μM (■), 20 μM (●), 30 μM (×), and 40 μM (○). In (A) and (B), the concentration of [3H]AdoMet was fixed at 3 μM, and in (C) and (D), the concentration of H4-20 was fixed at 15 μM.
A similar experiment was done in which the initial velocities were checked at several selected concentrations of 1 over a range of varied [3H]SAM concentration when fixing H4-20 concentration (Figure 2C). After the data were processed in an analogous way, a Kis of 16 μM and a Kii of 26 μM were obtained by fitting with eq 1. The resemblance of the Kis and Kii values suggests that compound 1 is classic noncompetitive inhibitor with respect to SAM. It is possible that compound 1 has the capacity to bind both free and SAM-bound PRMT1. In the double-reciprocal plot, a series of straight lines show a pattern of intersection close to the X-axis, further supporting the classic noncompetitive nature of inhibition (Figure 2D).
To further validate that compound 1 is primarily a competitive inhibitor of PRMT1 with respect to the substrate H4-20, we conducted a competitive fluorescence anisotropy binding assay in which a fluorescein-labeled H4 peptide (H4-FL) was used as a substrate ligand for PRMT1 binding. Previously we have shown that anisotropy of H4-FL increases upon binding to PRMT1.27 We introduced varied concentrations of 1 into a mixture containing a fixed concentration of PRMT1 and H4-FL to examine the competition between 1 and H4-FL. As shown in Figure 3, when concentration of 1 was increased, the anisotropy value of the mixture decreased accordingly and reached a plateau of about 0.055. These data clearly illustrated that compound 1 indeed competes with H4-FL for binding to PRMT1, which is in accordance with the results of radiometric steady-state kinetic characterization. Further study showed that 1 did not target the substrate peptide (Figure S-1 in the Supporting Information), excluding the possibility of ligand–substrate interaction.
Figure 3.

Competitive binding measurement with fluorescence anisotropy: fluorescence anisotropy (524 nm) of H4-FL and PRMT1 complex at different concentrations of compound 1. The concentrations of H4-FL and PRMT1 were kept constant at 0.2 and 2.0 μM, respectively.
Molecular Modeling Studies
To date, no crystal structures of human PRMT1 (hPRMT1) have been solved. However, the highly conserved Rattus norvegicus PRMT3 (rPRMT3, PDB code 1F3L)44 and human PRMT3 (hPRMT3, PDB code 3SMQ(45)) structures are available. The rat-PRMT1 X-ray structures (PDB codes 1OR8, 1ORI, 1ORH)46 are not well suited as templates for homology modeling because the crystals were obtained at a nonphysiological pH of 4.7 and an important helical segment near the binding pocket was not resolved (residues 1–40). Thus, we generated a homology model for the active form of hPRMT1 on the basis of the rPRMT3 and hPRMT3 X-ray structures. The sequence identity between the individual enzymes is sufficiently high for this approach; the residues within the binding pocket especially are highly conserved (hPRMT1 and hPRMT3, 47% overall sequence identity; hPRMT1 and rPRMT3, 49% overall sequence identity for the conserved core containing the SAM-binding site and the C-terminal barrel-like domain).
To better understand the mechanism underlying the selective binding of compounds 1 and 5 to PRMT1 versus other PRMTs (e.g., PRMT5), we carried out docking calculations with AutoDock4.247 using as targets the hPRMT1 homology model and the X-ray structure of hPRMT5, respectively. In these calculations, the region encompassing the SAM-binding site and substrate arginine site was included in the bounding box for docking. Subsequently, the energy profile and stability of the predicted structures for the docked complexes were assessed through extensive molecular dynamics (MD) simulations and molecular mechanics/Poisson–Boltzmann solvent-accessible surface area (MM-PBSA) calculations.48,49 The steady-state kinetic analysis and fluorescence anisotropy binding assay showed that compound 1 acted as a primarily competitive inhibitor with respect to H4-20, while noncompetitive versus [3H]SAM MD simulations and MM-PBSA calculation of the hPRMT1 and hPRMT5 in the presence of SAH were also performed to rationalize the differences in inhibition patterns. Table 2 and Table S2 showed the averaged binding free energies for protein–ligand complexes along with corresponding decomposition by energy terms.50 The estimated binding free energy ΔGb was −27.0 kcal mol–1 for the hPRMT1·1 complex and −12.4 kcal mol–1 for the hPRMT5·1 complex. Similarly, ΔGb of the hPRMT1·5 complex was lower than that of the corresponding hPRMT5·5 complex by 22.5 kcal mol–1 (Table S2). This trend agrees with our experimental observation that compounds 1 and 5 bind more favorably to hPRMT1. We further analyzed the free energy components to determine the dominant interactions responsible for the observed binding specificity. According to the components of the binding free energy (Table 2 and Table S2), both the intermolecular (gas phase) van der Waals and electrostatic interactions favor binding. The electrostatic solvation (ΔGpolar) disfavors binding because of desolvation penalty for the ligand and PRMT. Nonpolar solvation, which corresponds to the burial of solvent-accessible surface area (SASA) upon binding, gives a slightly favorable contribution.
Table 2. Free Energy Analysis (kcal mol–1) for the Binding of Compound 1 and SAH to PRMT1 and PRMT5a.
| PRMT1 |
PRMT5 |
|||
|---|---|---|---|---|
| contribution | 1 | SAH | 1 | SAH |
| ΔEele | –511.8 | –138.8 | –500.3 | –112.4 |
| (22.7) | (10.8) | (18.8) | (11.7) | |
| ΔEvdw | –34.5 | –45.8 | –39.6 | –48.2 |
| (5.8) | (4.5) | (3.8) | (4.2) | |
| ΔGnonpol | –5.3 | –6.2 | –5.9 | –6.2 |
| (0.1) | (0.2) | (0.2) | (0.1) | |
| ΔGpolar | 511.3 | 145.6 | 513.6 | 132.3 |
| (20.4) | (6.3) | (15.1) | (10.1) | |
| ΔGsol b | 506.0 | 139.4 | 507.7 | 126.1 |
| (20.4) | (6.3) | (15.1) | (10.0) | |
| ΔGele c | –0.5 | 6.7 | 3.3 | 19.8 |
| (6.81) | (8.7) | (10.3) | (8.3) | |
| ΔHb | –40.4 | –45.1 | –32.2 | –34.5 |
| (6.8) | (8.7) | (9.2) | (6.7) | |
| TΔS | –13.4 | –16.0 | –18.8 | –15.4 |
| (8.01) | (1.0) | (0.6) | (1.0) | |
| ΔGb | –27.0 | –29.1 | –12.4 | –19.1 |
| IC50 (μM)d | 14.2 | 118 | ||
Standard deviation values are shown in parentheses.
Polar/nonpolar (ΔGsol = ΔGpolar + ΔGnonpolar) contributions.
Electrostatic (ΔGele = ΔEele + ΔGpolar) contributions.
IC50 for rat PRMT.
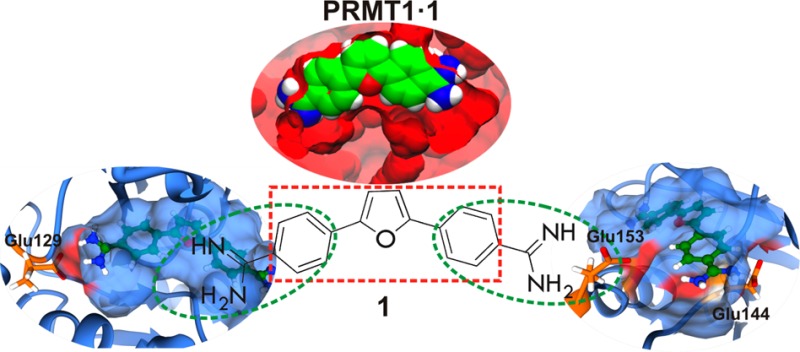
In order to identify the binding mode and the detailed interactions responsible for stabilizing compound 1 and compound 5 in the hPMRT1 and hPRMT5 structures, the binding free energy was decomposed into individual residue contributions and key residues for binding to hPRMT1 and hPRMT5 are shown in Figure 4 and Figure S2. In agreement with the available experimental data, compound 1 and compound 5 bind the hPRMT1 active site and were found to be the stronger inhibitors of hPRMT1 when compared to hPRMT5. A common feature of compound 1 and compound 5 binding to hPRMT1 is the interaction (via hydrogen bonding) of amidine groups with the acidic residues Glu129, Glu144, and Glu153 of hPRMT1 (Figure 4 and Figure S2, left panels). One of the amidine groups extends into the channel, which accommodates the substrate arginine during catalysis. This amidine group is well placed to gain optimal interactions with glutamic acid residues Glu144 and Glu153, reported to be essential for binding and catalysis.13 Notably, the compound 1 and compound 5 scaffold stretches out partially into the SAM adenine binding site, slightly overlapping with the cofactor methyl donor group and forming hydrogen bonds between the second amidine group of the ligand and residue Glu129 of hPRMT1. This binding mode explains why compound 1 is mainly a peptide substrate-competitive inhibitor and partially competes with [3H]SAM from the steady-state kinetic analysis and fluorescence anisotropy binding assay, since compound 1 and compound 5 interact with Glu144 and Glu153 by disrupting the binding with the substrate. The calculations also show that SAH binds slightly more favorably to PRMT compared to compound 1 and compound 5 (Table 2 and Table S2). Several hydrophobic (Met146, Met155, and Thr158) and π–π (Try35, Phe36, and Tyr39, YFXY motif) interactions are detected between the compound 1 and compound 5 ligands and hPRMT1. The YFXY motif in the N-terminal helix αX is invariant among all known PRMTs. In rPRMT3, F218 (YFXY) forms edge to face hydrophobic interaction with adenine groups of AdoHcy, and Tyr217 and Tyr221 (YFXY), whose hydroxyl groups point to the active site residue Glu335. Previously it was determined that deleting helix αX indeed reduced cofactor cross-linking and abolished enzyme activity in PRMT1, suggesting important roles of helix αX both for cofactor binding and for catalysis.13 Compound 5 differs from compound 1 by a single nitrogen atom (amidinophenyl group) replacing carbon and thus shares similar orientation to the latter compound in the hPMRT1 active site.
Figure 4.

Predicted binding modes of compound 1 in PRMT1 and PRMT5 from docking (AutoDock 4.2) and molecular dynamics simulation (NAMD 2.8). Ligand–residue interaction energies from MM/PBSA energy decomposition for (A) PRMT1 and (B) PRMT5. (C, D) Binding modes of compound 1 with (C) PRMT1 and (D) PRMT5. The best docking pose obtained from AutoDock for 1 in complex with the hPRMT1 homology model (based on 1F3L(44) and 3SMQ(45)) and X-ray hPRMT5 (4GQB53) was selected for MD simulation. Dominant structures for the hPRMT1·1 and hPRMT5·1 complexes from the last 20 ns of MD trajectory clustering analysis were used for visualization. PRMT residues engaging the ligand are explicitly shown in ball and stick representation. The protein (in cartoon representation) is colored according to the residue contribution values in the free energy decomposition from red (negative) to blue (positive).
Figure 4 and Figure S2 (right panel) showed the interaction modes of compound 1 and compound 5 with hPMRT5. Both compounds showed a similar type of interaction with the residues of the binding pocket. One amidine group of compound 1 makes electrostatic and hydrogen bond interactions with Glu435 and Glu444, whereas the second guanidine group is involved in hydrogen bonding to Asp419. In the case of compound 5, Glu435 and Glu444 form hydrogen bonds and electrostatic interactions with one guanidine group. In addition, several hydrophobic interactions were formed between the ligands and residues Leu315, Leu316, Leu319, Leu436, Leu437, Met420, and Pro314 in the hPRMT5 active site. However, the energy contributions of these residues to ligand binding in hPRMT5 are below 5 kcal/mol (from free energy decomposition) (Figure 4 and Figure S2, right panels). Thus, electrostatic interactions between the amidine of compound 1 and compound 5 and the carboxylate of Glu in the hPRMT1 structure may facilitate inhibition of PRMT1.
The differences in the binding modes for diamidines (compounds 1 and 5) to hPRMT1 and hPRMT5 are directly linked to the differences in computed binding free energy ΔGb and to the experimentally observed selectivity. Both compounds 1 and 5 have rigid planar scaffolds. The curvature associated with the planar ligand structure enables diamidines to partially occupy the cofactor site and also span the substrate arginine binding site. Thus, the shape of the diamidine inhibitor is compatible with the shape of the cavity formed by the adjacent substrate/cofactor sites in PRMTs. For the same reason compound 1 has also been known to favorably bind DNA in the minor groove (because of compatible curvature). Next, we evaluated the selectivity of compounds 1 and 5 in terms of electrostatic and shape complementarity (Figure 5 and Figure S3). Both binding pockets in PRMT1 and PRMT5 were found to be electrostatically highly complementary to the ligands (Figure 5A and Figure 5B; Figure S3A and S3B). With both pockets negatively charged, electrostatics alone is insufficient to explain selectivity. The difference in ΔGb (and correspondingly in selectivity) arises from the better shape complementarity of compounds 1 and 5 for the PRMT1 pocket (Figure 5C and Figure 5D; Figure S3C and S3D). In PRMT1, the tighter fit of the inhibitors to the binding pocket correlates with the larger computed affinity ΔGb. By contrast, PRMT5 exhibits a larger, partially solvent exposed pocket and binds the ligands less tightly in agreement with the smaller computed affinity ΔGb. Therefore, the shape and rigidity of the diamidine ligands could be tuned to exploit differences in the binding cavities among PRMTs, thus providing an avenue to design more selective PRMT inhibitors.
Figure 5.

Electrostatic and shape complementarity in diamidine binding to PRTM1 and PRMT5: (A) electrostatic potential surface for the binding pocket of PRMT1 with compound 1; (B) electrostatic potential surface for the binding pocket of PRMT5 with compound 1; (C) shape of the binding cavity of PRMT1 (red) with compound 1 (blue); (D) shape of the binding cavity of PRMT5 (red) with compound 1 (blue). The best docking pose obtained from AutoDock for 1 in complex with the hPRMT1 homology model (based on 1F3L(44) and 3SMQ(45)) and X-ray hPRMT5 (4GQB53) was selected for MD simulation. Dominant structures for the hPRMT1·1 and hPRMT5·1 complexes from the last 20 ns of MD trajectory clustering analysis were used for visualization, the same as for Figure 4C,D. The charges of proteins were assigned using PDB2PQR server69 and electrostatic potential was calculated using APBS.68 The electrostatic potential varied from −10KBT/e to +10KBT/e and was depicted using Chimera71 in panels A and B from red to blue, respectively. The ligand in panels A and B is color-coded by AM1BCC charge from red (negative) to blue (positive). The surface was visualized in panels C and D using the program VMD.70
Compound 1 Inhibits Cell Proliferation in Leukemia Cell Lines with Different Genetic Lesions
PRMT1 is highly expressed in many different kinds of tumors as shown from gene expression data from the Oncomine Web site. We rationalize that inhibiting PRMT1 could induce tumor cell death. First, we wanted to verify whether compound 1 inhibited the PRMT1 enzymatic activity in cells. It is known that the ALY protein (also called Yra in yeast) is heavily methylated on the N terminal region and C terminal region flanking the RNA binding domain from the literature51 and from our unpublished data. We used ASYM24 antibody which has been used widely as a generic antibody for methylarginine recognition to detect the protein methylation status of GFP-ALY fusion protein in 293T cells treated with 1 for 15 h. GFP-ALY was immunoprecipitated with GFP antibody (Allele Biotech). In a comparison of lane 1 with lane 2 with and without compound 1 treatment (Figure 6A), it was shown that the expression level of the methylated GFP-ALY protein was significantly reduced when the cells were treated with 20 μM compound 1 when the equal amount of total GFP-ALY protein was loaded onto both lanes (with or without compound 1 treatment). Therefore, we confirmed that the drug is permeable to cell membrane and inhibits cellular PRMT1 activity.
Figure 6.

Compound 1 inhibits proliferation of leukemia cell lines. (A) 1 inhibits GFP-ALY methylation in 293T cells. 20 μM 1 was added to 293T cells for 15 h before harvest. Then the GFP-ALY fusion protein was purified via GFP antibody beads. ASYM24 (Millipore) was used to detect the methylated ALY protein. (B) Compound 1 inhibits leukemic cell growth on day 3. 20 μM 1 was added to the cell culture for 3 days before harvesting for cell viability assay. As a control, the cells were treated with the same amount of DMSO as that added in drug treated samples. The y-axis is the percentage of viable cells in drug treated group by viable cells in control group as denominator. Meg-01 cells and K562 cells have BCR-ABL translocation. HL-60 cells and NB4 cells have PML-RAR α translocation. MOLM13 cells are with MLL-AF9 translocation. HEL cells are with JAK2 V617F mutation. CMK cell, CMY cell, CMS cell, and CHRF cells are with trisomy 21. Jurkat cells derived from T cell leukemia patients had very complicated mutation. (C) Growth curves of CHRF cells. (D) Growth curves of MOLM13 cells. (E) Growth curves of HEL cells.
We have investigated the cell viability with 10 different leukemia cell lines treated with 20 μM compound 1. We measured the number of viable cells in culture every day in 3 consecutive days. We found that compound 1 inhibited cell growth for most of the leukemia cell lines except HEL cells which have JAK2V617F mutations. These results agree with the expected role of PRMT1 in cell proliferation. Interestingly, we found cell lines derived from Down’s syndrome patients and MLL-AF9 patient (such as CMY, CHRF-288-11, and MOLM-13 cells) are more sensitive to PRMT1 inhibitor 1 than cell lines from other mutation backgrounds (such as HEL, Jurkat, and HL-60 cells). The detailed mechanisms of the hypersensitivity would be a subject of further investigation.
Conclusion
We reported a set of diamidine compounds that showed micromolar PRMT inhibition. Among these compounds, 1 and 5 showed potent selective inhibition for PRMT1 compared with the other PRMTs such as CARM1, PRMT5, and PRMT6. Compound 1 is cell membrane permeable and can effectively inhibit PRMT1 activity intracellularly. Compound 1 also inhibited cell proliferation in a panel of leukemia cell lines with different genetic lesions. Interestingly, compound 1 is a well-established inhibitor in a number of previous applications which were described in the Introduction. However, the function of 1 as a selective PRMT inhibitor had not been previously established. The mechanism for the higher affinity of compound 1 for PRMT1 rather than the other PRMTs such as PRMT5 was explored by examining the detailed ligand–protein interaction modules in the active site of the enzyme. Our combined computational and experimental results support that these rigid crescent-shaped compounds span the adjacent substrate and cofactor binding sites. The positively charged amidine functional group serves as an anchor point to ensure binding to the peptide site of PRMT. This dual mode of inhibition was confirmed by kinetic experiments wherein diamidines showed a primarily competitive mode of inhibition for the substrate and a classical noncompetitive (i.e., partially competitive) inhibition toward the cofactor. Further work would be of value to optimize this class of diamidine compounds by rational design to improve their selectivity and/or potency in PRMT inhibition. The disclosed diamidine PRMT inhibitors will be useful chemical probes to investigate new functions of PRMTs in biology.
Experimental Procedures
Compounds
H4-20 and H4-FL were synthesized and described as previously reported.27 The diamidine compounds were provided from Dr. David Boykin’s group at Georgia State University, with >95% purity based on CHN elemental analysis, 1H and 13C NMR, and mass spectrometry.
Protein Expression and Purification
His6x-tagged PRMT1 was expressed in recombinant pET28b vector transformed Escherichia coli BL21(DE3) and purified on Ni-NTA beads. GST-tagged CARM1 and His6x-tagged PRMT6 were expressed and purified as previously described.27
To obtain HA-tagged PRMT5, freshly growing human embryonic kidney 293T (HEK 293T) cells were co-transfected with recombinant plasmid pcDNA3-HA-PRMT5 and plasmid pCMV-Sport6-WDR77 (a gift from Dr. Steve Nilmer). The co-transfected cells were made into lysate by using cold M-PER mammalian protein extraction reagent. HA-PRMT5 inside the lysate was purified on HA-peptide agarose beads by eluting with 2 mg/mL HA-peptide in 1× TBS before protein concentration using ultrafiltration. All the protein concentrations were determined with the Bradford assay.
P81 Filter Binding Assay
Filter binding assay was carried out in 0.65 mL plastic tubes with a 30 μL reaction volume at 30 °C. The reaction buffer was composed of 50 mM HEPES (pH 8.0), 50 mM NaCl, 1 mM EDTA, and 0.5 mM DTT. 0.5 μM tritium labeled S-adenosyl-l-methionine ([3H]SAM, PerkinElmer) was used as methyl donor, and 1 μM histone H4(1–20) was used as the methyl acceptor. Reactions were catalyzed by one of the PRMTs at the designed concentration. Typically, 6 μL of varied concentrations of each candidate inhibitor in reaction buffer was added into the wells prior to the addition of 18 μL of mixture composed of [3H]SAM and a certain kind of PRMT in the same reaction buffer. The 24 μL mixture was incubated at room temperature for 5 min before the reaction was started by adding 6 μL of H4(1–20) dissolved in reaction buffer. Reactions without candidate inhibitor were used as positive control. Reactions with neither H4(1–20) nor candidate inhibitor was used as negative control. After incubation for an appropriate period of time, 20 μL of the reaction mixture was aspirated and spread onto anionic P81 filter paper disks (Whatman). The paper disks loaded with reaction mixture were dried in air for 2 h and then washed with 1 L of 50 mM NaHCO3 (pH 9.0) solution for 15 min three times. Then the paper disks were dried in air overnight before transfer of the disks into 3.5 mL vials full of scintillation oil, and the amount of methylation was quantified by scanning the vials on a scintillation counter (Beckman Coulter, Brea, CA) or a MicroBeta2 (PerkinElmer). The Kcat and Km of PRMT1 for H4(1–20) was obtained by measuring the initial velocity of reaction at different concentrations of H4(1–20) and fitting the kinetic data with Michaelis–Menten equation. The Ki for compound 1 was calculated by using the equation Ki = IC50/(1 + [S]/Km).
Fluorescence Binding Assay
Fluorescence anisotropy of fluorescein-labeled peptides was measured on a Fluoromax-4 spectrofluorometer (Horiba Jobin Yvon). The buffer was the same as that for the P81 filter binding assay. The excitation wavelength and emission wavelength were 498 and 524 nm, respectively. The competitive binding of 5 to PRMT1-H4(1–20)FL solution was measured using the fluorescence anisotropy mode in a similar manner as described previously.52 0.2 μM H4(1–20)FL and 2 μM PRMT1 were mixed, and increasing concentrations of 5 stock were added until the fluorescence anisotropy signals leveled off. The anisotropy values at 524 nm from several scans were plotted as a function of inhibitor concentration.
Homology Modeling
Sequences and structures of Rattus norvegicus protein arginine methyltransferase 3 (rPRMT3), the human PRMT3 (hPRMT3), and the human PRMT5 (hPRMT5) were downloaded from the Protein Data Bank (PDB codes 1F3L,443SMQ,45 and 4GQB(53)). The sequence of the hPRMT1 (GenBank accession number NP_938074.2) was aligned against the sequence of hPRMT3 and rPRMT3 with the ClustalW alignment server (http://www.ebi.ac.uk/Tools/msa/clustalw2/). Then the homology model of the hPRMT1 was built using MODELER 9V10 software based on the alignment with the hPRMT3 and rPRMT3 as templates.54,55
Molecular Docking
Docking was carried out with AutoDock 4.2.47 The molecular structures of compounds 1 and 5 were generated by Omega 2.4.3 program (OpenEye Scientific Software).56 The atomic coordinates of hPRMT1 (homology model) and hPRMT5 (crystal structure) were used as the receptor model for docking. The initial blind docking used a grid box of 120 × 120 × 120 points in three dimensions with a spacing of 0.375 Å centered on the whole AdoMet-binding domain and β-barrel domain and indicated that the major interacting region was located between the two domains. Accordingly, further docking was carried out centered at the AdoMet-binding site and substrate Arg site with a grid box of 64 × 64 × 64 points in three dimensions with a spacing of 0.375 Å.
Molecular Dynamics Simulation
The best docking pose obtained from AutoDock for compound 1 and compound 5 in complex with the hPRMT1 and hPRMT5 was selected for molecular dynamics (MD) study. The steady-state kinetic analysis and fluorescence anisotropy binding assay indicate that compound 1 is mainly a substrate-competitive inhibitor of PRMT1 for H4(1–20) versus [3H]SAM. Therefore, to ensure completeness of the MM/PBSA analysis, we also modeled SAH bound to PRMT1 and PRMT5 in the MD simulations. All energy minimizations and molecular dynamics simulations were performed with NAMD 2.857 using ff99SB force field parameters for the protein58 and gaff parameters59 for the ligands59,60 (compound 1 and compound 5) in explicit solvent (TIP3P water).61 The systems were then minimized for 5000 steps with backbone atoms fixed followed by 5000 steps of minimization with harmonic restraints to remove unfavorable contacts. The systems were then gradually brought up to 300 K and run for 50 ps in the NVT ensemble while keeping the protein backbone restrained. The equilibration was continued for another 2 ns in the NPT ensemble, and the harmonic restraints were gradually released. The 30 ns production simulation was performed in the NPT ensemble (1 atm and 300 K) without constraints. A short-range cutoff of 10 Å was used for the short-range nonbonded interactions with a switching function at 8.5 Å. The long-range electrostatic interactions were treated with a smooth particle mesh Ewald method.62 The r-RESPA multiple time step method63 was employed with a 2 fs integration time step for bonded, 2 fs for short-range nonbonded interactions, and 4 fs for long-range electrostatic interactions. Bonds between hydrogen atoms and heavy atoms of the protein were fixed. Snapshots from the MD trajectories were collected at an interval of 2.0 ps. The free energy of binding for ligands (1, 5, and SAH) to hPRMT1 and hPRMT5 was estimated using the MM-PBSA method in AMBER 9.064 as the average over the last 20 ns (2000 frames) from the trajectories. The MM-PBSA method combines molecular mechanics, Poisson–Boltzmann electrostatics for polar solvation free energy, nonpolar solvation energy based on solvent-accessible surface area, and normal-mode analyses for entropy to calculate the binding free energy for the protein complexes.65−67 The interaction energies were decomposed50 into contributions from the ligands and hPRMT1 or hPRMT5 residue pairs. The same dynamics trajectories utilized in the MM-PBSA calculations were used for the energy decomposition. Electrostatic potentials were calculated using APBS68 and mapped onto the molecular surface corresponding to the PRMT1 and PRMT5 binding. Charges were assigned using the PDB2PQR server.69 PTRAJ module of AMBER TOOLS 12 and VMD70 were used for the analysis of trajectories and structural visualization.
Cellular Study
Cell viability was measured by CellTiter-Glo viability kit (Promega, Madison WI). 1000–1500 cells were seeded in individual wells of a 96-well plate, with 100 μL culture volume per well. All these leukemia cell lines were grown in RPMI medium plus 10% fetal bovine serum. 20 μM 1 or the same amount of DMSO was added to the culture. At 0, 24, 48, 72 h after drug treatment, 100 μL of CellTiter-Glo reagent was added to each well. Luminescence signals, which are proportional to cell viability in each well, were measured by microplate reader (Biotek, Winooski, VT)
GFP Immunoprecipitation Assay
We made a stable cell line from 293T cells expressing GFP-ALY growing in DMEM medium plus 10% fetal bovine serum. With this cell line, we made whole cell extract with H lysis buffer (150 mM NaCl, 20 mM Tris, pH 7.8, 0.5 mM ETDA, 0.2 mM PMSF, 1 mM NaF, 1 mM sodium orthovanadate, 1% Np-40, 1 mM DTT). The cells in H lysis buffer were incubated on ice for 30 min before being cleaned by spinning at 12000g for 20 min. Conjugated GFP antibody beads (Allele Biotech Inc., San Diego, CA) were used to pull down GFP fusion protein. The samples were dissolved in SDS–PAGE gels and detected by ASYM24 antibody (Millipore) in Western blot assay (chemiluminescence reagents).
Acknowledgments
This work was supported by NIH Grant R01GM086717 (Y.G.Z.) and by Georgia State University start-up funds (I.I.) and an NSF CAREER award (Grant MCB-1149521 to I.I.). Computational resources were provided in part by an NSF XSEDE allocation (Grant CHE110042) and through an allocation at National Energy Research Scientific Computing Center (NERSC) supported by the DOE Office of Science (Contract DE-AC02-05CH11231). We are greatly thankful to Prof. David Boykin who kindly provided the diamidine compounds for this study.
Glossary
Abbreviations Used
- PRMT
protein arginine methyltransferase
- AdoMet/SAM
S-adenosyl-l-methionine
- SAH
S-adenosylhomocysteine
- DTT
dithiothreitol
- MM/PBSA
molecular mechanics/Poisson–Boltzmann surface area method
- MD
molecular dynamics
- SAR
structure–activity relationship
Supporting Information Available
Additional experimental and computational data. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
⊥ L.Y.: College of Life Sciences, Hebei University, Baoding, Hebei 071002, China.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Spannhoff A.; Sippl W.; Jung M. Cancer Treatment of the Future: Inhibitors of Histone Methyltransferases. Int. J. Biochem. Cell Biol. 2009, 41, 4–11. [DOI] [PubMed] [Google Scholar]
- Krause C. D.; Yang Z.; Kim Y. S.; Lee J. H.; Cook J. R.; Pestka S. Protein Arginine Methyltransferases: Evolution and Assessment of Their Pharmacological and Therapeutic Potential. Pharmacol. Ther. 2007, 113, 50–87. [DOI] [PubMed] [Google Scholar]
- Di Lorenzo A B. M. Histone Arginine Methylation. FEBS Lett. 2011, 585, 2024–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael C. Y.; Bachand F.; McBride A. E.; Komili S.; Casolari J. M.; Silver P. A. Arginine Methyltransferase Affects Interactions and Recruitment of Mrna Processing and Export Factors. Genes Dev. 2004, 18, 2024–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W.-J.; Gary J. D.; Yang M. C.; Clarke S.; Herschman H. R. The Mammalian Immediate-Early Tis21 Protein and the Leukemia-Associated Btg1 Protein Interact with a Protein-Arginine N-Methyltransferase. J. Biol. Chem. 1996, 271, 15034–15044. [DOI] [PubMed] [Google Scholar]
- Bedford M. T.; Clarke S. G. Protein Arginine Methylation in Mammals: Who, What, and Why. Mol. Cell 2009, 33, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung N.; Chan L. C.; Thompson A.; Cleary M. L.; So C. W. Protein Arginine-Methyltransferase-Dependent Oncogenesis. Nat. Cell Biol. 2007, 9, 1208–1215. [DOI] [PubMed] [Google Scholar]
- Goulet I.; Gauvin G.; Boisvenue S.; Cote J. Alternative Splicing Yields Protein Arginine Methyltransferase 1 Isoforms with Distinct Activity, Substrate Specificity, and Subcellular Localization. J. Biol. Chem. 2007, 282, 33009–33021. [DOI] [PubMed] [Google Scholar]
- Papadokostopoulou A.; Mathioudaki K.; Scorilas A.; Xynopoulos D.; Ardavanis A.; Kouroumalis E.; Talieri M. Colon Cancer and Protein Arginine Methyltransferase 1 Gene Expression. Anticancer Res. 2009, 29, 1361–1366. [PubMed] [Google Scholar]
- Bouras G.; Deftereos S.; Tousoulis D.; Giannopoulos G.; Chatzis G.; Tsounis D.; W Cleman M.; Stefanadis C. Asymmetric Dimethylarginine (ADMA): A Promising Biomarker for Cardiovascular Disease?. Curr. Top. Med. Chem. 2013, 13, 180–200. [DOI] [PubMed] [Google Scholar]
- Chen X.; Niroomand F.; Liu Z.; Zankl A.; Katus H.; Jahn L.; Tiefenbacher C. Expression of Nitric Oxide Related Enzymes in Coronary Heart Disease. Basic Res. Cardiol. 2006, 101, 346–353. [DOI] [PubMed] [Google Scholar]
- Sasser J. M.; Moningka N. C.; Cunningham M. W. Jr.; Croker B.; Baylis C. Asymmetric Dimethylarginine in Angiotensin II-Induced Hypertension. Am. J. Physiol.: Regul., Integr. Comp. Physiol. 2010, 298, R740–R746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Cheng X. Structure of the Predominant Protein Arginine Methyltransferase Prmt1 and Analysis of Its Binding to Substrate Peptides. Structure 2003, 11, 509–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui S.; Wooderchak-Donahue W. L.; Zang T.; Chen D.; Daly M. P.; Zhou Z. S.; Hevel J. M. Substrate-Induced Control of Product Formation by Protein Arginine Methyltransferase 1. Biochemistry 2013, 52, 199–209. [DOI] [PubMed] [Google Scholar]
- Zheng Y. G.; Wu J.; Chen Z.; Goodman M. Chemical Regulation of Epigenetic Modifications: Opportunities for New Cancer Therapy. Med. Res. Rev. 2008, 28, 645–687. [DOI] [PubMed] [Google Scholar]
- Dowden J.; Pike R. A.; Parry R. V.; Hong W.; Muhsen U. A.; Ward S. G. Small Molecule Inhibitors That Discriminate between Protein Arginine N-Methyltransferases Prmt1 and Carm1. Org. Biomol. Chem. 2011, 9, 7814–7821. [DOI] [PubMed] [Google Scholar]
- Osborne T.; Weller Roska R. L.; Rajski S. R.; Thompson P. R. In Situ Generation of a Bisubstrate Analogue for Protein Arginine Methyltransferase 1. J. Am. Chem. Soc. 2008, 130, 4574–4575. [DOI] [PubMed] [Google Scholar]
- t Hart P.; Lakowski T. M.; Thomas D.; Frankel A.; Martin N. I. Peptidic Partial Bisubstrates as Inhibitors of the Protein Arginine N-Methyltransferases. ChemBioChem 2011, 12, 1427–1432. [DOI] [PubMed] [Google Scholar]
- Cheng D.; Yadav N.; King R. W.; Swanson M. S.; Weinstein E. J.; Bedford M. T. Small Molecule Regulators of Protein Arginine Methyltransferases. J. Biol. Chem. 2004, 279, 23892–23899. [DOI] [PubMed] [Google Scholar]
- Spannhoff A.; Heinke R.; Bauer I.; Trojer P.; Metzger E.; Gust R.; Schüle R.; Brosch G.; Sippl W.; Jung M. Target-Based Approach to Inhibitors of Histone Arginine Methyltransferases. J. Med. Chem. 2007, 50, 2319–2325. [DOI] [PubMed] [Google Scholar]
- Spannhoff A.; Machmur R.; Heinke R.; Trojer P.; Bauer I.; Brosch G.; Schule R.; Hanefeld W.; Sippl W.; Jung M. A Novel Arginine Methyltransferase Inhibitor with Cellular Activity. Bioorg. Med. Chem. Lett. 2007, 17, 4150–4153. [DOI] [PubMed] [Google Scholar]
- Purandare A. V.; Chen Z.; Huynh T.; Pang S.; Geng J.; Vaccaro W.; Poss M. A.; Oconnell J.; Nowak K.; Jayaraman L. Pyrazole Inhibitors of Coactivator Associated Arginine Methyltransferase 1 (CARM1). Bioorg. Med. Chem. Lett. 2008, 18, 4438–4441. [DOI] [PubMed] [Google Scholar]
- Huynh T.; Chen Z.; Pang S.; Geng J.; Bandiera T.; Bindi S.; Vianello P.; Roletto F.; Thieffine S.; Galvani A. Optimization of Pyrazole Inhibitors of Coactivator Associated Arginine Methyltransferase 1 (CARM1). Bioorg. Med. Chem. Lett. 2009, 19, 2924–2927. [DOI] [PubMed] [Google Scholar]
- Allan M.; Manku S.; Therrien E.; Nguyen N.; Styhler S.; Robert M.-F.; Goulet A.-C.; Petschner A. J.; Rahil G.; Robert Macleod A. N-Benzyl-1-heteroaryl-3-(trifluoromethyl)-1H-pyrazole-5-carboxamides as Inhibitors of Co-Activator Associated Arginine Methyltransferase 1 (CARM1). Bioorg. Med. Chem. Lett. 2009, 19, 1218–1223. [DOI] [PubMed] [Google Scholar]
- Therrien E.; Larouche G.; Manku S.; Allan M.; Nguyen N.; Styhler S.; Robert M.-F.; Goulet A.-C.; Besterman J. M.; Nguyen H. 1,2-Diamines as Inhibitors of Co-Activator Associated Arginine Methyltransferase 1 (CARM1). Bioorg. Med. Chem. Lett. 2009, 19, 6725–6732. [DOI] [PubMed] [Google Scholar]
- Wan H.; Huynh T.; Pang S.; Geng J.; Vaccaro W.; Poss M. A.; Trainor G. L.; Lorenzi M. V.; Gottardis M.; Jayaraman L. Benzo[d]imidazole Inhibitors of Coactivator Associated Arginine Methyltransferase 1 (CARM1)—Hit to Lead Studies. Bioorg. Med. Chem. Lett. 2009, 19, 5063–5066. [DOI] [PubMed] [Google Scholar]
- Feng Y.; Li M.; Wang B.; Zheng Y. G. Discovery and Mechanistic Study of a Class of Protein Arginine Methylation Inhibitors. J. Med. Chem. 2010, 53, 6028–6039. [DOI] [PubMed] [Google Scholar]
- Heinke R.; Spannhoff A.; Meier R.; Trojer P.; Bauer I.; Jung M.; Sippl W. Virtual Screening and Biological Characterization of Novel Histone Arginine Methyltransferase Prmt1 Inhibitors. ChemMedChem 2009, 4, 69–77. [DOI] [PubMed] [Google Scholar]
- Mai A.; Cheng D.; Bedford M. T.; Valente S.; Nebbioso A.; Perrone A.; Brosch G.; Sbardella G.; De Bellis F.; Miceli M. Epigenetic Multiple Ligands: Mixed Histone/Protein Methyltransferase, Acetyltransferase, and Class III Deacetylase (Sirtuin) Inhibitors. J. Med. Chem. 2008, 51, 2279–2290. [DOI] [PubMed] [Google Scholar]
- Ragno R.; Simeoni S.; Castellano S.; Vicidomini C.; Mai A.; Caroli A.; Tramontano A.; Bonaccini C.; Trojer P.; Bauer I. Small Molecule Inhibitors of Histone Arginine Methyltransferases: Homology Modeling, Molecular Docking, Binding Mode Analysis, and Biological Evaluations. J. Med. Chem. 2007, 50, 1241–1253. [DOI] [PubMed] [Google Scholar]
- Mai A.; Valente S.; Cheng D.; Perrone A.; Ragno R.; Simeoni S.; Sbardella G.; Brosch G.; Nebbioso A.; Conte M.; Altucci L.; Bedford M. T. Synthesis and Biological Validation of Novel Synthetic Histone/Protein Methyltransferase Inhibitors. ChemMedChem 2007, 2, 987–991. [DOI] [PubMed] [Google Scholar]
- Bissinger E.-M.; Heinke R.; Spannhoff A.; Eberlin A.; Metzger E.; Cura V.; Hassenboehler P.; Cavarelli J.; Schüle R.; Bedford M. T. Acyl Derivatives of P-Aminosulfonamides and Dapsone as New Inhibitors of the Arginine Methyltransferase Hprmt1. Bioorg. Med. Chem. 2011, 19, 3717–3731. [DOI] [PubMed] [Google Scholar]
- Dillon M. B.; Bachovchin D. A.; Brown S. J.; Finn M.; Rosen H.; Cravatt B. F.; Mowen K. A. Novel Inhibitors for Prmt1 Discovered by High-Throughput Screening Using Activity-Based Fluorescence Polarization. ACS Chem. Biol. 2012, 7, 1198–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Chen L.; Sinha S. H.; Liang Z.; Chai H.; Muniyan S.; Chou Y.-W.; Yang C.; Yan L.; Feng Y. Pharmacophore-Based Virtual Screening and Biological Evaluation of Small Molecule Inhibitors for Protein Arginine Methylation. J. Med. Chem. 2012, 55, 7978–7987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leepin A.; Stüdli A.; Brun R.; Stephens C. E.; Boykin D. W.; Hemphill A. Host Cells Participate in the in Vitro Effects of Novel Diamidine Analogues against Tachyzoites of the Intracellular Apicomplexan Parasites Neospora caninum and Toxoplasma gondii. Antimicrob. Agents Chemother. 2008, 52, 1999–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortázar T. M.; Coombs G. H.; Walker J. Leishmania Panamensis: Comparative Inhibition of Nuclear DNA Topoisomerase II Enzymes from Promastigotes and Human Macrophages Reveals Anti-Parasite Selectivity of Fluoroquinolones, Flavonoids and Pentamidine. Exp. Parasitol. 2007, 116, 475–482. [DOI] [PubMed] [Google Scholar]
- Nhili R.; Peixoto P.; Depauw S.; Flajollet S.; Dezitter X.; Munde M. M.; Ismail M. A.; Kumar A.; Farahat A. A.; Stephens C. E. Targeting the DNA-Binding Activity of the Human Erg Transcription Factor Using New Heterocyclic Dithiophene Diamidines. Nucleic Acids Res. 2013, 41, 125–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macharia J. C.; Bourdichon A. J.; Gicheru M. M. Efficacy of Trypan: A Diminazene Based Drug as Antileishmanial Agent. Acta Trop. 2004, 92, 267–272. [DOI] [PubMed] [Google Scholar]
- Purfield A. E.; Tidwell R. R.; Meshnick S. R. The Diamidine Db75 Targets the Nucleus of Plasmodium Falciparum. Malar. J. 2009, 8, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M. Z.; Saulter J. Y.; Usuki E.; Cheung Y.-L.; Hall M.; Bridges A. S.; Loewen G.; Parkinson O. T.; Stephens C. E.; Allen J. L. Cyp4f Enzymes Are the Major Enzymes in Human Liver Microsomes That Catalyze the O-Demethylation of the Antiparasitic Prodrug Db289 [2,5-Bis(4-amidinophenyl)furan-bis-O-methylamidoxime]. Drug Metab. Dispos. 2006, 34, 1985–1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F.; Jin W.; Feng J.; Chen A.; Ma Z.; Zhang X. Propamidine Decreas Mitochondrial Complex III Activity of Botrytis Cinerea. BMB Rep. 2010, 43, 614–621. [DOI] [PubMed] [Google Scholar]
- Mathis A. M.; Holman J. L.; Sturk L. M.; Ismail M. A.; Boykin D. W.; Tidwell R. R.; Hall J. E. Accumulation and Intracellular Distribution of Antitrypanosomal Diamidine Compounds Db75 and Db820 in African Trypanosomes. Antimicrob. Agents Chemother. 2006, 50, 2185–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedford M. T.; Richard S. Arginine Methylation an Emerging Regulator of Protein Function. Mol. Cell 2005, 18, 263–272. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Zhou L.; Cheng X. D. Crystal Structure of the Conserved Core of Protein Arginine Methyltransferase Prmt3. EMBO J. 2000, 19, 3509–3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siarheyeva A.; Senisterra G.; Allali-Hassani A.; Dong A.; Dobrovetsky E.; Wasney G. A.; Chau I.; Marcellus R.; Hajian T.; Liu F.; Korboukh I.; Smil D.; Bolshan Y.; Min J.; Wu H.; Zeng H.; Loppnau P.; Poda G.; Griffin C.; Aman A.; Brown P. J.; Jin J.; Al-Awar R.; Arrowsmith C. H.; Schapira M.; Vedadi M. An Allosteric Inhibitor of Protein Arginine Methyltransferase 3. Structure 2012, 20, 1425–1435. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Cheng X. D. Structure of the Predominant Protein Arginine Methyltransferase Prmt1 and Analysis of Its Binding to Substrate Peptides. Structure 2003, 11, 509–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. Autodock4 and Autodocktools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brice A. R.; Dominy B. N. Analyzing the Robustness of the Mm/Pbsa Free Energy Calculation Method: Application to DNA Conformational Transitions. J. Comput. Chem. 2011, 32, 1431–1440. [DOI] [PubMed] [Google Scholar]
- Kollman P. A.; Massova I.; Reyes C.; Kuhn B.; Huo S.; Chong L.; Lee M.; Lee T.; Duan Y.; Wang W. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Acc. Chem. Res. 2000, 33, 889–897. [DOI] [PubMed] [Google Scholar]
- Gohlke H.; Kiel C.; Case D. A. Insights into Protein–Protein Binding by Binding Free Energy Calculation and Free Energy Decomposition for the Ras–Raf and Ras–Ralgds Complexes. J. Mol. Biol. 2003, 330, 891–913. [DOI] [PubMed] [Google Scholar]
- Hung M.-L.; Hautbergue G. M.; Snijders A. P.; Dickman M. J.; Wilson S. A. Arginine Methylation of Ref/Aly Promotes Efficient Handover of Mrna to Tap/Nxf1. Nucleic Acids Res. 2010, 38, 3351–3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y.; Xie N.; Wu J.; Yang C.; Zheng Y. G. Inhibitory Study of Protein Arginine Methyltransferase 1 Using a Fluorescent Approach. Biochem. Biophys. Res. Commun. 2009, 379, 567–572. [DOI] [PubMed] [Google Scholar]
- Antonysamy S.; Bonday Z.; Campbell R. M.; Doyle B.; Druzina Z.; Gheyi T.; Han B.; Jungheim L. N.; Qian Y.; Rauch C. Crystal Structure of the Human Prmt5: Mep50 Complex. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 17960–17965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A. Comparative Protein Modeling by Satisfaction of Spatial Restraints. Mol. Med. Today 1995, 1, 270–277. [DOI] [PubMed] [Google Scholar]
- Martí-Renom M. A.; Stuart A. C.; Fiser A.; Sánchez R.; Melo F.; Šali A. Comparative Protein Structure Modeling of Genes and Genomes. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 291–325. [DOI] [PubMed] [Google Scholar]
- Hawkins P. C.; Skillman A. G.; Warren G. L.; Ellingson B. A.; Stahl M. T. Conformer Generation with Omega: Algorithm and Validation Using High Quality Structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalé L.; Skeel R.; Bhandarkar M.; Brunner R.; Gursoy A.; Krawetz N.; Phillips J.; Shinozaki A.; Varadarajan K.; Schulten K. Namd2: Greater Scalability for Parallel Molecular Dynamics. J. Comput. Phys. 1999, 151, 283–312. [Google Scholar]
- Hornak V.; Abel R.; Okur A.; Strockbine B.; Roitberg A.; Simmerling C. Comparison of Multiple Amber Force Fields and Development of Improved Protein Backbone Parameters. Proteins: Struct., Funct., Bioinf. 2006, 65, 712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Wolf R. M.; Caldwell J. W.; Kollman P. A.; Case D. A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [DOI] [PubMed] [Google Scholar]
- Wang J.; Wang W.; Kollman P. A.; Case D. A. Automatic Atom Type and Bond Type Perception in Molecular Mechanical Calculations. J. Mol. Graphics Modell. 2006, 25, 247–260. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; Darden T.; Lee H.; Pedersen L. G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar]
- Tuckerman M.; Berne B. J.; Martyna G. J. Reversible Multiple Time Scale Molecular Dynamics. J. Chem. Phys. 1992, 97, 1990–2001. [Google Scholar]
- Case D. A.; Cheatham T. E.; Darden T.; Gohlke H.; Luo R.; Merz K. M.; Onufriev A.; Simmerling C.; Wang B.; Woods R. J. The Amber Biomolecular Simulation Programs. J. Comput. Chem. 2005, 26, 1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Morin P.; Wang W.; Kollman P. A. Use of MM-PBSA in Reproducing the Binding Free Energies to HIV-1 RT of TIBO Derivatives and Predicting the Binding Mode to HIV-1 RT of Efavirenz by Docking and MM-PBSA. J. Am. Chem. Soc. 2001, 123, 5221–5230. [DOI] [PubMed] [Google Scholar]
- Yan C.; Kaoud T.; Lee S.; Dalby K. N.; Ren P. Understanding the Specificity of a Docking Interaction between Jnk1 and the Scaffolding Protein Jip1. J. Phys. Chem. B 2011, 115, 1491–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan C.; Xiu Z.; Li X.; Li S.; Hao C.; Teng H. Comparative Molecular Dynamics Simulations of Histone Deacetylase-like Protein: Binding Modes and Free Energy Analysis to Hydroxamic Acid Inhibitors. Proteins: Struct., Funct., Bioinf. 2008, 73, 134–149. [DOI] [PubMed] [Google Scholar]
- Baker N. A.; Sept D.; Joseph S.; Holst M. J.; McCammon J. A. Electrostatics of Nanosystems: Application to Microtubules and the Ribosome. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolinsky T. J.; Nielsen J. E.; McCammon J. A.; Baker N. A. Pdb2pqr: An Automated Pipeline for the Setup of Poisson–Boltzmann Electrostatics Calculations. Nucleic Acids Res. 2004, 32, W665–W667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual Molecular Dynamics. J. Mol. Graphics 1996, 14, 33–38. [DOI] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.