

Abstract
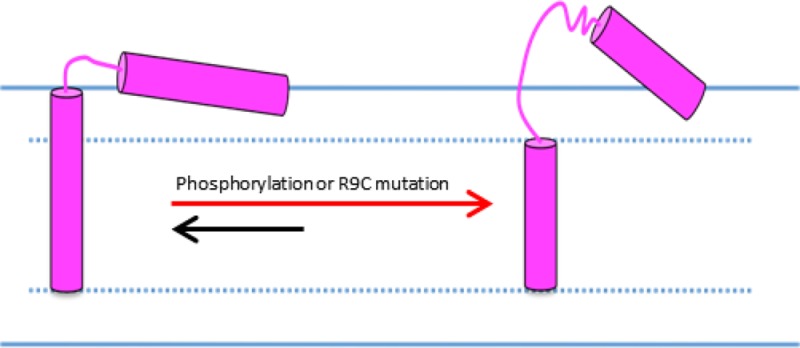
Phospholamban (PLB) is a membrane protein that regulates heart muscle relaxation rates via interactions with the sarcoplasmic reticulum Ca2+ ATPase (SERCA). When PLB is phosphorylated or Arg9Cys (R9C) is mutated, inhibition of SERCA is relieved. 13C and 15N solid-state NMR spectroscopy is utilized to investigate conformational changes of PLB upon phosphorylation and R9C mutation. 13C=O NMR spectra of the cytoplasmic domain reveal two α-helical structural components with population changes upon phosphorylation and R9C mutation. The appearance of an unstructured component is observed on domain Ib. 15N NMR spectra indicate an increase in backbone dynamics of the cytoplasmic domain. Wild-type PLB (WT-PLB), Ser16-phosphorylated PLB (P-PLB), and R9C-mutated PLB (R9C-PLB) all have a very dynamic domain Ib, and the transmembrane domain has an immobile component. 15N NMR spectra indicate that the cytoplasmic domain of R9C-PLB adopts an orientation similar to P-PLB and shifts away from the membrane surface. Domain Ib (Leu28) of P-PLB and R9C-PLB loses the alignment. The R9C-PLB adopts a conformation similar to P-PLB with a population shift to a more extended and disordered state. The NMR data suggest the more extended and disordered forms of PLB may relate to inhibition relief.
Introduction
Phospholamban (PLB) is a 52 amino acid membrane protein that is located in the sarcoplasmic reticulum (SR) membrane of heart muscle cells.1−3 Despite its small size, PLB plays an important role in regulating heart muscle relaxation rate by interacting with sarcoplasmic reticulum Ca2+ ATPase (SERCA).1 In the absence of PLB, SERCA acts as a Ca2+ ion channel that transports Ca2+ ions from the cytosol into the SR and triggers the heart muscle relaxation process.1,2 PLB interacts with SERCA and inhibits SERCA function by decreasing SERCA-Ca2+ affinity.1 The inhibition effect is removed when PLB is phosphorylated at Ser16 by protein kinase A (PKA) or when the cytoplasmic Ca2+ ion concentration is elevated.4,5 PLB inhibition and inhibition relief effects on SERCA regulate SERCA function, which as a result controls the heart muscle relaxation rate.1
PLB has four domains: cytoplasmic domain (domain Ia; residues 1–16), loop region (residues 17–22), domain Ib (residues 23–30), and the transmembrane domain (residues 31–52).6 The bellflower model has been established with the cytoplasmic domain sticking out into the cytosol as an α-helix or as an unstructured coil.7,8 Recent studies have shown that the inconsistency of the cytoplasmic domain conformation results from dynamic changes.9−11 Instead of adopting a fixed conformation in the membrane, it consists of a mixture of two or more conformation states.9 In vivo, PLB forms a pentamer with the cytoplasmic domain as an α-helix laying flat on the membrane surface.12 The flexibility of the cytoplasmic domain enables it to act as the regulatory region of PLB function via shifting its conformation states with the interplay of SERCA, PKA, and the membrane environment.13−15
Domain Ib is less well studied and located on the top of the hydrophobic region of the membrane as an α-helix.6,12 EPR studies have indicated that this segment of PLB is much more dynamic than the cytoplasmic domain.15,16 Two conformation states are identified in domain Ib, which are folded and unfolded.14 Recent studies have suggested that domain Ib is also involved in regulating PLB function.14 The transmembrane domain has abundant leucines and isoleucines forming a Leu-zipper and the three cysteines forming disulfide bonds to help stabilize the PLB pentamer.12 Mutagenesis and protein–protein interaction studies suggest that one face of the transmembrane domain binds to SERCA, while the other face stabilizes the PLB pentamer form by interacting between the monomers.2,17−19
In living cells, ∼78% of PLB are in the pentamer form and the rest are in the monomer form.6 It is the monomer form that interacts with and inhibits SERCA, while the pentamer form acts only as an inactive storage form.1 However, the monomer form alone cannot regulate heart muscle relaxation.20 When PLB is phosphorylated, there is a slightly increase in pentameric form population.21
PLB can adopt four different conformational states (T, T′, R, and R′). The low-energy level T state has the cytoplasmic domain forming an α-helix on top of the membrane surface and domain Ib folded.9 The high energy level R state has the cytoplasmic domain unfolded and extended into the cytosol, and domain Ib is unfolded.9 The two conformation states (folded and unfolded) of domain Ib are independent of the cytoplasmic domain.13 Equilibrium is established between the T and R states with the T state as a dominant population in vivo.12 When phosphorylated, PLB population shifts from the T state to an intermediate T′ state, which gives rise to the more disordered and extended R state.14,21,22 The T′ state has an extended cytoplasmic domain partially attached to the membrane surface.14 The intermediate R′ state has a completely unfolded cytoplasmic domain that is attached to the membrane surface.14 The conformational states are suggested to be well-related to the function of PLB, that is, each state or each dominant state may correspond to a function such as inhibition.13 PLB conformational studies are extremely important to relate structural investigations to the functional studies.
Interplay between PLB, SERCA, and the membrane controls the regulation of PLB on SERCA.13,23−25 However, the mechanism of this regulation is not known. It is believed that the membrane is not only acting as an environment to stabilize or localize SERCA and PLB but is also involved actively in regulating function.13,23,26
PLB mutants are divided into two types: gain-of-function mutants and loss-of-function mutants.18,19,27 Gain-of-function mutants have a superinhibition effect on SERCA when compared with wild-type PLB (WT-PLB), whereas loss-of-function mutants have inhibitory function relief.18,19 Mutation sites are not confined to certain domains according to their effects, which suggest that PLB function is not controlled by certain sites on only one domain.18,19 Several factors may be involved including: PLB oligomerization states, structural conformations, and its interaction with the membrane and SERCA.
This study is focused on the Arg9Cys (R9C)-mutated PLB (R9C-PLB) loss-of-function mutant. R9C-PLB has been identified in individuals who have heart chamber enlargement, which can lead to heart failure in 5 to 10 years.28 Oligomerization has been suggested to partially contribute to R9C-PLB functionality loss.29 Structural conformations between WT-PLB, Ser16-phosphorylated PLB (P-PLB), and R9C-PLB are compared for the first time with NMR spectroscopy.
Solid-state NMR spectroscopy is a robust method to study the structural and dynamic properties of membrane proteins.30−38 Because of the large size, hydrophobicity, and sensitivity to the environment of membrane proteins, traditional structural biology methods are not appropriate to elucidate valid structural information.39−42 The local secondary structure can be investigated by analyzing the 13C NMR spectrum of a 13C=O group on a specific residue.43−49 Static 15N NMR powder pattern spectra reveal information on the backbone dynamics of a specific residue.50−52 With mechanically aligned glass plate samples, membrane proteins are aligned in lipid bilayers with the bilayer normal oriented parallel to the static magnetic field.53−5615N labels at specific sites yield 15N chemical shift resonances to resolve the structural topology.53−55,57−60 The cytoplasmic domain, domain Ib, and transmembrane domain are studied with solid-state NMR spectroscopy to elucidate the secondary structure, backbone dynamics, and topology changes of PLB upon R9C mutation and phosphorylation.
Materials and Methods
Materials
1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) were purchased from Avanti Polar Lipids (Alabaster, AL). The phospholipids were dissolved in chloroform and stored at −20 °C before use. Trifluoroethanol (TFE) and N-[2-hydroxyethyl]piperazine-N′-2-ethane sulfonic acid (HEPES) were purchased from Sigma-Aldrich (St. Louis, MO). Ethylenediaminetetraacetic acid (EDTA) and sodium chloride (NaCl) were purchased from Fisher Scientific (Pittsburgh, PA). Prephosphorylated Fmoc-serine and Fmoc amino acids were obtained from Applied Biosystems (Carlsbad, CA). 13C=O labeled Fmoc-alanine, 13C=O labeled Fmoc-leucine, 15N labeled Fmoc-alanine, and 15N labeled Fmoc-leucine were purchased from Isotec TM/Sigma-Aldrich (Miamisburg, OH). All other peptide synthesis and HPLC reagents and solvents were acquired from VWR (San Dimas, CA). NMR glass sample cells were purchased from New Era Enterprise (Vineland, NJ). Glass plates were purchased from Superior Marienfeld Laboratory Glassware (Lauda-Konigshofen, Germany).
Synthesis, Purification, and Characterization of WT-PLB and R9C-PLB
Specific 13C- or 15N-labeled WT-PLB, P-PLB, and R9C-PLB were synthesized via Fmoc-based solid-phase peptide synthesis on a CEM solid-phase peptide synthesizer. 13C labels were placed on Ala15, Ala24, and Leu39 residues. 15N labels were placed at positions Ala11, Ala24, Leu28, and Leu42. To synthesize the specific labeled P-PLB, we used prephosphorylated Fmoc-serine at Ser16. Crude peptides were obtained after cleavage using a cleavage mixture. A General Electric (GE) AKTA purifier HPLC was utilized to purify peptides by reverse-phase chromatography on a C18 column. The purified peptides were lyophilized and characterized by matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry. The peptides were at least 95% pure.
NMR Sample Preparation
MLVs were prepared according to a protocol established by Rigby and coworkers.61 Lyophilized specific labeled WT-PLB, P-PLB, and R9C-PLB were dissolved in a minimal amount of TFE. POPC (35 mg) was mixed with dissolved peptides (4 mol % with respect to lipid) in a 12 × 75 mm test tube. The solvent was removed by a steady stream of N2 gas and placed in a vacuum desiccator overnight. The lipid peptide mixture was rehydrated with 95 μL of HEPES buffer (5 mM EDTA, 20 mM NaCl, and 30 mM HEPES at pH 7.0). Vesicles were formed in a warm water bath at 45 °C for ∼30 min using a vortexer and bath sonication. The sample was transferred into a 4 mm NMR rotor for solid-state NMR experiments.
Mechanically aligned glass plate samples were prepared as previously described.53−55,57,58 Lyophilized specifically labeled WT-PLB, P-PLB, and R9C-PLB were dissolved in a minimal amount of TFE. Next, PLB (1 mol % with respect to the lipid) was mixed with 60 mg of DOPC/DOPE (molar ratio 4:1). Then, the organic solvent was removed by a steady stream of N2 gas. A thin layer of the mixture was applied to 35–40 glass plates and dried in a vacuum desiccator overnight. 2H-depleted water (4 μL) was added to the sample on each glass plate, and the glass plates were stacked on top of each other. The sample was rehydrated in a hydration chamber with saturated ammonium phosphate (humidity ∼93%) for 24 h at 42 °C. The sample was then transferred to a rectangular glass sample cell and sealed for NMR measurements.
Solid-State NMR Spectroscopy
All NMR spectra were acquired using a 500 MHz WB Bruker Avance solid-state NMR spectrometer. 13C and 15N NMR experiments using MLV samples were performed with a Bruker 4 mm triple resonance CPMAS probe. 13C CPMAS experiments were conducted with a spinning speed at 5 kHz at the magic angle. 1H 90° pulse length was set to 4.5 μs; contact time 3 ms, recycle delay 4 s. Eight k scans were averaged, and 70 Hz line broadening was used. TMS was used as the external reference for the 13C NMR spectra. All 13C NMR spectra were measured at −25 °C. Static 15N NMR experiments using MLV samples were performed using a ramp-CP pulse sequence with 1H decoupling. 1H 90° pulse length was set to 4.7 μs; contact time was 1.5 ms, and recycle delay was 4 s. 120 k scans were averaged, and 300 Hz line broadening was used. Simulations of both the 13C and 15N NMR spectra were performed using DMFIT software.62
Static 15N NMR experiments using mechanically aligned glass plate samples were performed with a flat coil 1H-X Low-E NMR probe.55 The alignment of the samples was checked by observing the static 31P solid-state NMR spectra from the membrane phospholipids. A ramp-CP pulse sequence with 1H decoupling was used to acquire the 15N NMR spectra. 1H 90° pulse length was set to 5 μs. The contact time was 1.5 ms, and the recycle delay was 4 s. 120 k scans were averaged, and 300 Hz line broadening was used. 15NH4Cl was used as the external reference for the 15N NMR spectra. All 15N NMR spectra were acquired at 25 °C.
Results and Discussion
R9C is a loss-of-function mutation of PLB; therefore, R9C-PLB cannot inhibit SERCA as efficiently as WT-PLB.28,63 However, not all PLB mutants abolish its inhibition function. Some mutants increase the inhibition effect and are called superinhibition.52,64 It is not clear why different mutants have different effects. A few properties of these mutants can be investigated to relate to their function variation. PLB affinity to SERCA, interactions with the lipid bilayer, oligomerization states, and conformations are the major factors. Previous studies have indicated that WT-PLB, P-PLB, and a few type of PLB mutants have similar apparent affinity to SERCA.19,65,66 Thus, PLB affinity to SERCA is distinct from PLB inhibitory function regulation. WT-PLB interacts actively with the lipid bilayers by increasing the dynamics of both the membrane surface and hydrophobic region.49 Both P-PLB and R9C-PLB have significantly less interaction with the membrane.26,49 Therefore, interaction with lipids may be involved in regulating PLB function. WT-PLB is dominated by the pentamer form.12 P-PLB and R9C-PLB as well as a few loss-of-function mutants have slightly increased pentamer form population, while some of the gain-of-function mutants have a dominant monomer form.18,19,29 Oligomerization may partially contribute to PLB mutant function regulation. Structural conformations of WT-PLB, P-PLB, and R9C-PLB are compared in this project.
Membrane protein topology, dynamics, and secondary structure are important aspects to investigate in structure biology.67−70 For the membrane protein PLB studies, these investigations can give insights into its structure–function relationship. Veglia and coworkers have studied the conformational states of PLB and the function of corresponding conformations.13 A linear correlation is established.13 It is suggested that the ordered folded T state relates to the inhibition function of PLB, while the disordered and extended T′ state and R state relate to the inhibition relief.13 This helps to explain the loss of inhibition upon phosphorylation on PLB. Phosphorylation shifts PLB conformation states from the dominant T state to the T′ state that are readily exchangeable with the R state.13 It is very likely that the T′ state with the cytoplasmic domain partially unfolded and dissociated from the membrane surface relieves inhibition by PLB. The R9C-PLB is a loss-of-function mutant that also relieves inhibition of SERCA similar to P-PLB.28 Conformation state shifting from the T state to the T′ state or the R state upon R9C mutation would be coupled to functionality loss and make the R9C-PLB conformation studies extremely significant.
Secondary Structure
13C CPMAS solid-state NMR spectroscopy provides secondary structure information by studying the 13C=O signal of a specific residue on a membrane protein.4513C CPMAS NMR spectroscopy of membrane proteins yields well-resolved 13C=O NMR peaks in the region of 170 to 180 ppm.71 A 13C NMR signal with a chemical shift of ∼176 ppm corresponds to an α-helix for alanine and leucine residues, while a peak at ∼173 ppm corresponds to an unstructured coil for an alanine residue.43−46,49
Ala15 is located toward the end of the cytoplasmic domain and is the neighbor of a phosphorylation site (Ser16), thus, it would be sensitive to changes induced by phosphorylation. Secondary structure of the cytoplasmic domain of WT-PLB, P-PLB, and R9C-PLB was examined by 13C CPMAS NMR spectroscopy with a 13C=O label on Ala15. The corresponding 13C solid-state NMR spectra are shown in Figure 1. WT-PLB (Figure 1A) has two peaks at 176.5 and 175.4 ppm, indicating that the cytoplasmic domain of WT-PLB consists of two α-helical structural conformations. Both P-PLB and R9C-PLB (Figure 1B,C) have peaks centered at 176.3 and 174.9 ppm, which are very close to the WT-PLB signals, but the intensity of the 174.9 ppm signal is larger than the peak at 176.3 ppm. This suggests that phosphorylation and R9C mutation increases the population of one of the two α-helical components. Either phosphorylation or R9C mutation of PLB does not induce significant secondary structure changes to the cytoplasmic domain but only involves a population shift.
Figure 1.

13C CPMAS solid-state NMR spectra of POPC MLVs acquired at −25 °C with 13C=O labeled on the Ala15 residue in the cytoplasmic domain of WT-PLB (A), P-PLB (B), and R9C-PLB(C).
Domain Ib was investigated with a 13C=O label at Ala24, which is located near the beginning of domain Ib. Figure 2 shows 13C NMR spectra of the domain Ib of WT-PLB, P-PLB, and R9C-PLB. The WT-PLB sample (Figure 2A) has a major peak at 175.8 ppm with a broad shoulder, while P-PLB and R9C-PLB (Figure 2B,C) have two peaks with one centered at 173.2 ppm with a larger intensity and the other at 175.8 ppm. Domain Ib of WT-PLB forms an α-helix. Interestingly, upon phosphorylation or R9C mutation, a significant population of unstructured coil appears. The data indicate an unfolding or partially unfolding process in domain Ib upon phosphorylation or R9C mutation.
Figure 2.

13C CPMAS solid-state NMR spectra of POPC MLVs acquired at −25 °C with 13C=O labeled on the Ala24 residue in domain Ib of WT-PLB (A), P-PLB (B), and R9C-PLB (C).
The transmembrane domain was probed with a 13C=O label placed at Leu39. The corresponding 13C NMR spectra are shown in Figure 3. WT-PLB and R9C-PLB yield a single well-resolved peak at 176.3 and 176.7 ppm corresponding to a single α-helical component. The data suggest that the transmembrane domain secondary structure is not affected by R9C mutation. Previous studies on P-PLB also indicate an undisturbed transmembrane domain upon phosphorylation.9,13,14
Figure 3.

13C CPMAS solid-state NMR spectra of POPC MLVs acquired at −25 °C with 13C=O labeled on the Leu39 residue in the transmembrane domain of WT-PLB (A) and R9C-PLB (B).
Backbone Dynamics
Backbone dynamics of PLB were investigated by 15N solid-state NMR spectroscopy with 15N labels placed on specific residues. 15N-labeled amide proteins in MLVs adopt random orientations, revealing a 15N powder pattern line shape.72 The chemical shift anisotropy (CSA) width of the powder pattern spectra reflects the backbone dynamics of the specific labeled residue.50,59
Figure 4 compares the 15N NMR spectra of WT-PLB, P-PLB, and R9C-PLB with the 15N label on the Ala11 residue of the cytoplasmic domain. 15N NMR spectra of all three samples consisted of a broad powder pattern line shape that is typical for unoriented solid-state 15N NMR membrane protein samples and an isotropic component at 121–122 ppm. The CSA widths of the broad components of WT-PLB, P-PLB, and R9C-PLB are 171, 168, and 163 ppm. The 15N NMR data indicate that the cytoplasmic domain is a mixture of a dominant ordered component and a small mobile population. The data are consistent with WT-PLB cytoplasmic domain studies that have an equilibrium between the T and R states, in which the T state is immobile and more populated and the R state is more dynamic.12 Upon phosphorylation and R9C mutation, the intensity of the isotropic signal increases and suggests a population shift from the T state to the disordered T′ or R state. Notably, the CSA width of the immobile component of R9C-PLB is reduced by 8 ppm when compared with WT-PLB, while P-PLB induces a 3 ppm CSA width reduction. The 15N NMR data indicate a slightly increased motion of the immobile component of residue Ala11 in the R9C-PLB cytoplasmic domain.
Figure 4.

15N solid-state NMR powder pattern spectra of POPC MLVs acquired at 25 °C with 15N labeled on the Ala11 residue in the cytoplasmic domain of WT-PLB (A), P-PLB (B) and R9C-PLB (C).
Backbone dynamics were investigated in domain Ib with an 15N label on the Ala24 residue. The 15N NMR spectra of all the WT-PLB, P-PLB and R9C-PLB are shown in Figure 5. The 15N NMR spectrum of WT-PLB labeled on Ala24 of domain Ib yields a peak with an undistinguishable isotropic component and a broad powder pattern component. Unlike the two distinct components observed in the cytoplasmic domain, the undistinguishable static 15N NMR peak of domain Ib could rise from the fast exchange between a folded and unfolded state in microseconds.9 The exchange between the states in the cytoplasmic domain is on the 15N NMR spectroscopy time scale (milliseconds),9 and thus both states are observed. The data are consistent with the EPR studies that show two states of domain Ib due to the fast EPR time scale.13 Upon phosphorylation or R9C mutation, there is no significant change in the 15N NMR spectra of domain Ib, indicating the fast exchange between folded and unfolded states.
Figure 5.

15N solid-state NMR powder pattern spectra of POPC MLVs acquired at 25 °C with 15N labeled on the Ala24 residue in the domain Ib of WT-PLB (A), P-PLB (B), and R9C-PLB (C).
For comparison, Figure 6 shows the 15N NMR spectra of WT-PLB and R9C-PLB with the 15N label placed on Leu42 of the transmembrane domain. Characteristic powder pattern line shape spectra with similar CSA width (173 ppm) are observed for both WT-PLB and R9C-PLB. The broad single component 15N spectrum indicates that the transmembrane domain of both forms of PLB is immobile in the membrane. The results are similar to that of the P-PLB form according to previous studies.9,13,14
Figure 6.

15N solid-state NMR powder pattern spectra of POPC MLVs acquired at 25 °C with 15N labeled on the Leu42 residue in the cytoplasmic domain of WT-PLB (A) and R9C-PLB (B).
Structural Topology
The structural topology of PLB with respect to the membrane was studied using 15N solid-state NMR spectroscopy of PLB incorporated into mechanically aligned glass plate samples. In these oriented samples, the direction of the bilayer normal is aligned parallel to the static magnetic field.54 An 15N NMR resonant peak near σ⊥ indicates that the labeled residue is located perpendicular with the bilayer normal, while a 15N NMR peak near σ// indicates the labeled residue is located parallel with the bilayer normal.53−55
Figure 7 compares the 15N NMR spectra of WT-PLB, P-PLB, and R9C-PLB with the 15N label on Ala11 of the cytoplasmic domain. WT-PLB (Figure 7A) has a peak at 71 ppm, which is very close to σ⊥, indicating that WT-PLB cytoplasmic domain lies nearly flat on the surface of the lipid bilayer, which is consistent with previous WT-PLB studies.12,57 P-PLB and R9C-PLB (Figure 7B,C) reveal peaks at 92 and 86 ppm. The shift of the 15N NMR peak indicates a dynamic shift due to backbone motion, a change in cytoplasmic domain topology, or a secondary structure change.
Figure 7.

15N CP solid-state NMR spectra of mechanically aligned DOPC/DOPE (4:1 ratio) lipid bilayers acquired at 25 °C with 15N labeled on the Ala11 residue in the cytoplasmic domain of WT-PLB (A), P-PLB (B), and R9C-PLB (C).
If the 15 ppm downfield shift of R9C-PLB is only due to the backbone dynamic increase, a similar CSA width reduction can be expected when comparing the static 15N NMR spectra of 15N-Ala11 WT-PLB with those of R9C-PLB in MLVs. MLVs are more hydrated than glass plate samples, so a CSA width reduction of ∼15 ppm or larger can be expected. However, R9C-PLB induced an 8 ppm decrease in CSA width when compared with WT-PLB. So, beside the backbone dynamic increase, topology and/or secondary structure change also contributes to the downfield shift of R9C-PLB. No significant secondary structure change is observed at residue Ala15 in the cytoplasmic domain when R9C is mutated. More likely, Ala11 topology changes by shifting away from the membrane surface, as observed in previous P-PLB studies.58 The hypothesis is compatible with the 31P NMR data revealing a significantly less disturbance of R9C-PLB on the lipid bilayer surface when compared with WT-PLB.26 NMR data further confirm the shift of population from a membrane-attached T state toward partially membrane detached and more disordered T′ state or R state upon phosphorylation or R9C mutation.
Aligned 15N NMR spectra of WT-PLB, P-PLB, and R9C-PLB with 15N-label on the Leu28 residue of the domain Ib are shown in Figure 8. Ala24 is not used here because WT-PLB shows no alignment (data not shown). It is not surprising because Ala24 is at the beginning of domain Ib, which is close to the loop region and the water–lipid interface. It is very possible that it does not have a fixed orientation in the lipid bilayer. A 15N NMR resonant peak at 210 ppm is observed for the WT-PLB (Figure 8A). It is near σ// and the 15N NMR peak of WT-PLB with a 15N label on the transmembrane domain.57 Thus, domain Ib of the WT-PLB aligns nearly parallel with the membrane bilayer normal and the transmembrane domain. Interestingly, because all samples were well-aligned with the bilayer normal parallel to the static magnetic field according to the 31P NMR data, upon phosphorylation or R9C mutation, the alignment of domain Ib is lost with the appearance of a broad powder pattern line shape in addition to a 208 ppm peak (Figure 8B,C). The spectra indicate a mixture of an unoriented component and an oriented component parallel to the bilayer normal.
Figure 8.

15N CP solid-state NMR spectra of mechanically aligned DOPC/DOPE (4:1 ratio) lipid bilayers acquired at 25 °C with 15N labeled on the Leu28 residue in the domain Ib of WT-PLB (A), P-PLB (B), and R9C-PLB (C).
The unoriented broad component may be due to backbone dynamic changes, topology, and/or secondary structure changes. If it is not due to the secondary structure change, Leu28 should adopt random orientations as an α-helix in the lipid bilayer. A dramatic disturbance in the lipid acyl chain near the membrane surface is expected. However, 2H NMR spectra investigating lipid acyl chain dynamics do not show any effect upon R9C-PLB or P-PLB addition.26 More likely, the secondary structure change occurs upon R9C mutation or phosphorylation. Unwinding is observed at Ala24 in domain Ib, suggesting a population shift from the dominant folded state toward an unfolded state.
The transmembrane domain topology was investigated with the 15N label on the Leu42 residue (Figure 9). Both WT-PLB and R9C-PLB (Figure 9A,B) show a peak at 208 and 211 ppm, which is near σ//. The 15N NMR spectra indicate that the transmembrane domain of both types of PLB aligns nearly parallel with the bilayer normal, which is also similar to P-PLB according to previous studies.9,13,14
Figure 9.

15N CP solid-state NMR spectra of mechanically aligned DOPC/DOPE (4:1 ratio) lipid bilayers acquired at 25 °C with 15N labeled on the Leu42 residue in the transmembrane domain of WT-PLB (A) and R9C-PLB (B).
Taking all of the data together, when R9C is mutated, the PLB cytoplasmic domain undergoes a possible dissociation process from the membrane surface with increased population of fast backbone motion like the P-PLB. Domain Ib experiences a population shift from a folded to an unfolded state. The transmembrane domain is mostly unaffected. If the T′ state is identified as dominated in P-PLB, a more disordered cytoplasmic domain dissociated from the membrane matches perfectly with the conformation that is observed here for both P-PLB and R9C-PLB. The R state has a completely unfolded cytoplasmic domain and domain Ib. Thus, when phosphorylated or R9C-mutated, PLB conformation probably shifts from a dominant T state to T′ state or even R state. This population shift may induce PLB inhibition function loss. Studies of PLB N27A superinhibition mutant concluded that there is no significant difference between the backbone dynamics of the WT-PLB and N27A-PLB for both the cytoplasmic domain and transmembrane domain.52 The data suggest that N27A-PLB adopts a dominant ordered T state, similar to the WT-PLB, and that this conformation retains or even elevates the inhibition function. Both phosphorylation and R9C mutation appear to shift PLB conformation from the T state to the T′ and/or R state. This more disordered and extended conformation may facilitate the PLB inhibition function relief. The data indicate the correlation between conformational state and function.
The mechanism of structural conformation change by the R9C mutation is unknown. WT-PLB cytoplasmic domain absorbed on the membrane surface could be partially stabilized by the three positively charged Arg residues electrostatically interacting with the negatively charged bilayer surface. When Arg9 is mutated into a neutral Cys, the electrostatic interaction would be weakened, which may initiate the dissociation of the cytoplasmic domain from the membrane surface.
The studies on the interplay between SERCA, PLB, and the membrane indicate that both the transmembrane domain and domain Ib interact with SERCA and that the membrane can bind to SERCA and activate it.13,23,25 Loose binding of the transmembrane domain to SERCA is not crucial for inhibition but only to recruit SERCA.2,23 Thus, it is not surprising to observe the undisturbed transmembrane domain by either phosphorylation or R9C mutation in this study. It is more likely that domain Ib carries out inhibition. Recent studies show that the interaction sites of SERCA with PLB and the membrane may overlap.2,13,23 It is possible that the membrane and PLB domain Ib compete for binding to SERCA, with the PLB inhibiting SERCA while the membrane activates SERCA.2,23 When phosphorylated or R9C-mutated, unwinding of domain Ib may expose the interaction site with SERCA. The dissociation of PLB cytoplasmic domain from the membrane surface may facilitate the lipid to bind to and activate SERCA, which could induce the inhibition relief process. The mechanism of SERCA inhibition relief by P-PLB and R9C-PLB and its mediation by the membrane requires further studies.
Conclusions
On the basis of the solid-state NMR studies in this paper, the R9C mutation of PLB induces the cytoplasmic domain to undergo a change in the population of the α-helical component and partially dissociation from the membrane surface with increased backbone dynamics. Similar effects are noted for the phosphorylated PLB. Domain Ib of the WT-PLB forms a mobile α-helical component. When phosphorylated or R9C-mutated, the population shifts from a folded to an unfolded state in domain Ib are observed. The transmembrane domain is unaffected. The data indicate that phosphorylation or R9C mutation induce a population shift of PLB conformation from a dominant T state to a more extended and disordered conformation, which may correspond to a mixture of T′ state and R state. This conformation change correlates to the T′ state and R state with the PLB inhibition function loss.
Acknowledgments
This research project was supported by NIGMS/NIH (GM108026), R01 GM080542, NSF (CHE-1011909), and NSF MRI-0722403. We thank Dr. Sergey Maltsev for technical assistance and Dr. Shadi Abu-Baker and Dan Mayo for helpful discussion.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- MacLennan D. H.; Kranias E. G. Phospholamban: A Crucial Regulator of Cardiac Contractility. Nat. Rev. Mol. Cell Biol. 2003, 4, 566–577. [DOI] [PubMed] [Google Scholar]
- Toyoshima C.; Asahi M.; Sugita Y.; Khanna R.; Tsuda T.; MacLennan D. H. Modeling of the Inhibitory Interaction of Phospholamban with the Ca2+ ATPase. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller B.; Karim C. B.; Negrashov I. V.; Kutchai H.; Thomas D. D. Direct Detection of Phospholamban and Sarcoplasmic Reticulum Ca-ATPase Interaction in Membranes Using Fluorescence Resonance Energy Transfers. Biochemistry 2004, 43, 8754–8765. [DOI] [PubMed] [Google Scholar]
- Chen Z.; Akin B. L.; Jones L. R. Ca(2+) Binding to Site I of the Cardiac Ca(2+) Pump Is Sufficient to Dissociate Phospholamban. J. Biol. Chem. 2010, 285, 3253–3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catalucci D.; Latronico M. V. G.; Ceci M.; Rusconi F.; Young H. S.; Gallo P.; Santonastasi M.; Bellacosa A.; Brown J. H.; Condorelli G. At Increases Sarcoplasmic Reticulum Ca(2+) Cycling by Direct Phosphorylation of Phospholamban at Thr(17). J. Biol. Chem. 2009, 284, 28180–28187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traaseth N. J.; Shi L.; Verardi R.; Mullen D. G.; Barany G.; Veglia G. Structure and Topology of Monomeric Phospholamban in Lipid Membranes Determined by a Hybrid Solution and Solid-State NMR Approach. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 10165–10170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxenoid K.; Chou J. J. The Structure of Phospholamban Pentamer Reveals a Channel-Like Architecture in Membranes. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 10870–10875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andronesi O. C.; Becker S.; Seidel K.; Heise H.; Young H. S.; Baldus M. Determination of Membrane Protein Structure and Dynamics by Magic-Angle-Spinning Solid-State NMR Spectroscopy. J. Am. Chem. Soc. 2005, 127, 12965–12974. [DOI] [PubMed] [Google Scholar]
- Traaseth N. J.; Veglia G. Probing Excited States and Activation Energy for the Integral Membrane Protein Phospholamban by NMR Cpmg Relaxation Dispersion Experiments. Biochim. Biophys. Acta 2010, 1798, 77–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian P.; Wei D.-Q.; Wang J.-F.; Chou K.-C. An Allosteric Mechanism Inferred from Molecular Dynamics Simulations on Phospholamban Pentamer in Lipid Membranes. PLOS One 2011, 6, e18587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidel K.; Andronesi O. C.; Krebs J.; Griesinger C.; Young H. S.; Becker S.; Baldus M. Structural Characterization of Ca2+-ATPase-Bound Phospholarnban in Lipid Bilayers by Solid-State Nuclear Magnetic Resonance (NMR) Spectroscopy. Biochemistry 2008, 47, 4369–4376. [DOI] [PubMed] [Google Scholar]
- Verardi R.; Shi L.; Traaseth N. J.; Walsh N.; Veglia G. Structural Topology of Phospholamban Pentamer in Lipid Bilayers by a Hybrid Solution and Solid-State NMR Method. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 9101–9106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustavsson M.; Traaseth N. J.; Karim C. B.; Lockamy E. L.; Thomas D. D.; Veglia G. Lipid-Mediated Folding/Unfolding of Phospholamban as a Regulatory Mechanism for the Sarcoplasmic Reticulum Ca(2+)-ATPase. J. Mol. Biol. 2011, 408, 755–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masterson L. R.; Yu T.; Shi L.; Wang Y.; Gustavsson M.; Mueller M. M.; Veglia G. Camp-Dependent Protein Kinase a Selects the Excited State of the Membrane Substrate Phospholamban. J. Mol. Biol. 2011, 412, 155–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim C. B.; Zhang Z. W.; Howard E. C.; Torgersen K. D.; Thomas D. D. Phosphorylation-Dependent Conformational Switch in Spin-Labeled Phospholamban Bound to SERCA. J. Mol. Biol. 2006, 358, 1032–1040. [DOI] [PubMed] [Google Scholar]
- Ghimire H.; Abu-Baker S.; Sahu I. D.; Zhou A. D.; Mayo D. J.; Lee R. T.; Lorigan G. A. Probing the Helical Tilt and Dynamic Properties of Membrane-Bound Phospholamban in Magnetically Aligned Bicelles Using Electron Paramagnetic Resonance Spectroscopy. Biochim. Biophys. Acta, Biomembr. 2012, 1818, 645–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamoon J.; Nitu F.; Karim C.; Thomas D. D.; Veglia G. Mapping the Interaction Surface of a Membrane Protein: Unveiling the Conformational Switch of Phospholamban in Calcium Pump Regulation. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 4747–4752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornea R. L.; Jones L. R.; Autry J. M.; Thomas D. D. Mutation and Phosphorylation Change the Oligomeric Structure of Phospholamban in Lipid Bilayers. Biochemistry 1997, 36, 2960–2967. [DOI] [PubMed] [Google Scholar]
- Lockamy E. L.; Cornea R. L.; Karim C. B.; Thomas D. D. Functional and Physical Competition between Phospholamban and Its Mutants Provides Insight into the Molecular Mechanism of Gene Therapy for Heart Failure. Biochem. Biophys. Res. Commun. 2011, 408, 388–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu G. X.; Li L.; Sato Y.; Harrer J. M.; Kadambi V. J.; Hoit B. D.; Bers D. M.; Kranias E. G. Pentameric Assembly of Phospholamban Facilitates Inhibition of Cardiac Function in Vivo. J. Biol. Chem. 1998, 273, 33674–33680. [DOI] [PubMed] [Google Scholar]
- Paterlini M. G.; Thomas D. D. The Alpha-Helical Propensity of the Cytoplasmic Domain of Phospholamban: A Molecular Dynamics Simulation of the Effect of Phosphorylation and Mutation. Biophys. J. 2005, 88, 3243–3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayadi M.; Feig M. Role of Conformational Sampling of Ser16 and Thr17-Phosphorylated Phospholamban in Interactions with SERCA. Biochim. Biophys. Acta, Biomembr. 2013, 1828, 577–585. [DOI] [PubMed] [Google Scholar]
- Gustavsson M.; Traaseth N. J.; Veglia G. Activating and Deactivating Roles of Lipid Bilayers on the Ca(2+)-ATPase/Phospholamban Complex. Biochemistry 2011, 50, 10367–10374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustavsson M.; Traaseth N. J.; Veglia G. Probing Ground and Excited States of Phospholamban in Model and Native Lipid Membranes by Magic Angle Spinning NMR Spectroscopy. Biochim. Biophys. Acta, Biomembr. 2012, 1818, 146–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akin B. L.; Jones L. R. Characterizing Phospholamban to Sarco(Endo) Plasmic Reticulum Ca2+-ATPase 2a (SERCA2a) Protein Binding Interactions in Human Cardiac Sarcoplasmic Reticulum Vesicles Using Chemical Cross-Linking. J. Biol. Chem. 2012, 287, 7582–7593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X.; Lorigan G. A. Probing the Interaction of Arg9cys Mutated Phospholamban with Phospholipid Bilayers by Solid-State NMR Spectroscopy. Biochim. Biophys. Acta, Biomembr. 2013, 1828, 2444–2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medeiros A.; Biagi D. G.; Sobreira T. J. P.; de Oliveira P. S. L.; Negrao C. E.; Mansur A. J.; Krieger J. E.; Brum P. C.; Pereira A. C. Mutations in the Human Phospholamban Gene in Patients with Heart Failure. Am. Heart J. 2011, 162, 1088–U184. [DOI] [PubMed] [Google Scholar]
- Schmitt J. P.; Kamisago M.; Asahi M.; Li G. H.; Ahmad F.; Mende U.; Kranias E. G.; MacLennan D. H.; Seidman J. G.; Seidman C. E. Dilated Cardiomyopathy and Heart Failure Caused by a Mutation in Phospholamban. Science 2003, 299, 1410–1413. [DOI] [PubMed] [Google Scholar]
- Ha K. N.; Masterson L. R.; Hou Z.; Verardi R.; Walsh N.; Veglia G.; Robia S. L. Lethal Arg9cys Phospholamban Mutation Hinders Ca(2+)-ATPase Regulation and Phosphorylation by Protein Kinase A. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 2735–2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito H.; Naito A. NMR Studies on Fully Hydrated Membrane Proteins, with Emphasis on Bacteriorhodopsin as a Typical and Prototype Membrane Protein. Biochim. Biophys. Acta 2007, 1768, 3145–3161. [DOI] [PubMed] [Google Scholar]
- Brown M. F.; Heyn M. P.; Job C.; Kim S.; Moltke S.; Nakanishi K.; Nevzorov A. A.; Struts A. V.; Salgado G. F. J.; Wallat I. Solid-State H-2 NMR Spectroscopy of Retinal Proteins in Aligned Membranes. Biochim. Biophys. Acta 2007, 1768, 2979–3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahalakshmi R.; Marassi F. M. Orientation of the Escherichia Coli Outer Membrane Protein Ompx in Phospholipid Bilayer Membranes Determined by Solid-State NMR. Biochemistry 2008, 47, 6531–6538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider R.; Ader C.; Lange A.; Giller K.; Hornig S.; Pongs O.; Becker S.; Baldus M. Solid-State NMR Spectroscopy Applied to a Chimeric Potassium Channel in Lipid Bilayers. J. Am. Chem. Soc. 2008, 130, 7427–7435. [DOI] [PubMed] [Google Scholar]
- Opella S. J. NMR and Membrane Proteins. Nat. Struct. Biol. 1997, 4, 845–848. [PubMed] [Google Scholar]
- Opella S. J.; Marassi F. M. Structure Determination of Membrane Proteins by NMR Spectroscopy. Chem. Rev. 2004, 104, 3587–3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong M. Structure, Topology, and Dynamics of Membrane Peptides and Proteins from Solid-State NMR Spectroscopy. J. Phys. Chem. B 2007, 111, 10340–10351. [DOI] [PubMed] [Google Scholar]
- Jesorka A.; Orwar O. Liposomes: Technologies and Analytical Applications. Annu. Rev. Anal. Chem. 2008, 1, 801–832. [DOI] [PubMed] [Google Scholar]
- Lorigan G. A., Magnetic Resonance Spectroscopic Studies of the Integral Membrane Protein Phospholamban. In Modern Magnetic Resonance: Applications in Chemistry, Biological and Marine Sciences; Webb G. A., Ed.; Springer: Dordrecht, The Netherlands, 2008; Vol. 1, pp 313–318. [Google Scholar]
- Kang C.; Li Q. Solution NMR Study of Integral Membrane Proteins. Curr. Opin. Chem. Biol. 2011, 15, 560–569. [DOI] [PubMed] [Google Scholar]
- MacKinnon R. Potassium Channels and the Atomic Basis of Selective Ion Conduction (Nobel Lecture). Angew. Chem., Int. Ed. 2004, 43, 4265–4277. [DOI] [PubMed] [Google Scholar]
- Wlodawer A.; Minor W.; Dauter Z.; Jaskolski M. Protein Crystallography for Non-Crystallographers, or How to Get the Best (but Not More) from Published Macromolecular Structures. FEBS J. 2008, 275, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson M. P.; Friesner R. A.; Xiang Z. X.; Honig B. On the Role of the Crystal Environment in Determining Protein Side-Chain Conformations. J. Mol. Biol. 2002, 320, 597–608. [DOI] [PubMed] [Google Scholar]
- Hirsh D. J.; Hammer J.; Maloy W. L.; Blazyk J.; Schaefer J. Secondary Structure and Location of a Magainin Analogue in Synthetic Phospholipid Bilayers. Biochemistry 1996, 35, 12733–12741. [DOI] [PubMed] [Google Scholar]
- Wildman K. A. H.; Lee D. K.; Ramamoorthy A. Determination of Alpha-Helix and Beta-Sheet Stability in the Solid State: A Solid-State NMR Investigation of Poly (L-Alanine). Biopolymers 2002, 64, 246–254. [DOI] [PubMed] [Google Scholar]
- Tuzi S.; Naito A.; Saito H. A High-Resolution Solid-State C-13-NMR Study on 1-C-13 Ala and 3-C-13 Ala and 1-C-13 Leu and Val-Labeled Bacteriorhodopsin - Conformation and Dynamics of Transmembrane Helices, Loops and Termini, and Hydration-Induced Conformational Change. Eur. J. Biochem. 1993, 218, 837–844. [DOI] [PubMed] [Google Scholar]
- Tuzi S.; Naito A.; Saito H. C-13 NMR-Study on Conformation and Dynamics of the Transmembrane Alpha-Helices, Loops, and C-Terminus of 3-C-13 Ala-Labeled Bacteriorhodopsin. Biochemistry 1994, 33, 15046–15052. [DOI] [PubMed] [Google Scholar]
- Wagner K.; Beck-Sickinger A. G.; Huster D. Structural Investigations of a Human Calcitonin-Derived Carrier Peptide in a Membrane Environment by Solid-State NMR. Biochemistry 2004, 43, 12459–12468. [DOI] [PubMed] [Google Scholar]
- Lee D. K.; Ramamoorthy A. Determination of the Solid-State Conformations of Polyalanine Using Magic-Angle Spinning NMR Spectroscopy. J. Phys. Chem. 1999, 103, 271–275. [Google Scholar]
- Abu-Baker S.; Lorigan G. A. Phospholamban and Its Phosphorylated Form Interact Differently with Lipid Bilayers: A(31)P, (2)H, and (13)C Solid-State NMR Spectroscopic Study. Biochemistry 2006, 45, 13312–13322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colnago L. A.; Valentine K. G.; Opella S. J. Dynamics of Fd-Coat Protein in the Bacteriophage. Biochemistry 1987, 26, 847–854. [DOI] [PubMed] [Google Scholar]
- Abu-Baker S.; Lu J. X.; Chu S. D.; Brinn C. C.; Makaroff C. A.; Lorigan G. A. Side Chain and Backbone Dynamics of Phospholamban in Phospholipid Bilayers Utilizing H-2 and N-15 Solid-State NMR Spectroscopy. Biochemistry 2007, 46, 11695–11706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu S.; Coey A. T.; Lorigan G. A. Solid-State (2)H and (15)N NMR Studies of Side-Chain and Backbone Dynamics of Phospholamban in Lipid Bilayers: Investigation of the N27a Mutation. Biochim. Biophys. Acta 2010, 1798, 210–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs F. A.; Denny J. K.; Song Z.; Quine J. R.; Cross T. A. Helix Tilt of the M2 Transmembrane Peptide from Influenza a Virus: An Intrinsic Property. J. Mol. Biol. 2000, 295, 117–125. [DOI] [PubMed] [Google Scholar]
- Cross T. A.; Opella S. J. Solid-State NMR Structural Studies of Peptides and Proteins in Membranes. Curr. Opin. Struct. Biol. 1994, 4, 574–581. [Google Scholar]
- Gor’kov P. L.; Chekmenev E. Y.; Li C.; Cotten M.; Buffy J. J.; Traaseth N. J.; Veglia G.; Brey W. W. Using Low-E Resonators to Reduce Rf Heating in Biological Samples for Static Solid-State NMR up to 900 MHz. J. Magn. Reson. 2007, 185, 77–93. [DOI] [PubMed] [Google Scholar]
- Sanders C. R.; Hare B. J.; Howard K. P.; Prestegard J. H. Magnetically-Oriented Phospholipid Micelles as a Tool for the Study of Membrane-Associated Molecules. Prog. Nucl. Magn. Reson. Spectrosc. 1994, 26, 421–444. [Google Scholar]
- Abu-Baker S.; Lu J.-X.; Chu S.; Shetty K. K.; Gor’kov P. L.; Lorigan G. A. The Structural Topology of Wild-Type Phospholamban in Oriented Lipid Bilayers Using N-15 Solid-State NMR Spectroscopy. Protein Sci. 2007, 16, 2345–2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu S.; Abu-Baker S.; Lu J.; Lorigan G. A. (15)N Solid-State NMR Spectroscopic Studies on Phospholamban at Its Phosphorylated Form at Ser-16 in Aligned Phospholipid Bilayers. Biochim. Biophys. Acta 2010, 1798, 312–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiburu E. K.; Karp E. S.; Dave P. C.; Damodaran K.; Lorigan G. A. Investigating the Dynamic Properties of the Transmembrane Segment of Phospholamban Incorporated into Phospholipid Bilayers Utilizing H-2 and N-15 Solid-State NMR Spectroscopy. Biochemistry 2004, 43, 13899–13909. [DOI] [PubMed] [Google Scholar]
- Lorigan G. A.; Dave P. C.; Tiburu E. K.; Damodaran K.; Abu-Baker S.; Karp E. S.; Gibbons W. J.; Minto R. E. Solid-State NMR Spectroscopic Studies of an Integral Membrane Protein Inserted into Aligned Phospholipid Bilayer Nanotube Arrays. J. Am. Chem. Soc. 2004, 126, 9504–9505. [DOI] [PubMed] [Google Scholar]
- Rigby A. C.; Barber K. R.; Shaw G. S.; Grant C. W. M. Transmembrane Region of the Epidermal Growth Factor Receptor: Behavior and Interactions Via H-2 NMR. Biochemistry 1996, 35, 12591–12601. [DOI] [PubMed] [Google Scholar]
- Massiot D.; Fayon F.; Capron M.; King I.; Le Calve S.; Alonso B.; Durand J. O.; Bujoli B.; Gan Z. H.; Hoatson G. Modelling One- and Two-Dimensional Solid-State NMR Spectra. Magn. Reson. Chem. 2002, 40, 70–76. [Google Scholar]
- Ceholski D. K.; Trieber C. A.; Holmes C. F. B.; Young H. S. Lethal, Hereditary Mutants of Phospholamban Elude Phosphorylation by Protein Kinase A. J. Biol. Chem. 2012, 287, 26596–26605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akin B. L.; Chen Z.; Jones L. R. Superinhibitory Phospholamban Mutants Compete with Ca(2+) for Binding to SERCA2a by Stabilizing a Unique Nucleotide-Dependent Conformational State. J. Biol. Chem. 2010, 285, 28540–28552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceholski D. K.; Trieber C. A.; Young H. S. Hydrophobic Imbalance in the Cytoplasmic Domain of Phospholamban Is a Determinant for Lethal Dilated Cardiomyopathy. J. Biol. Chem. 2012, 287, 16521–16529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber S. J.; Haydon S.; Thomas D. D. Phospholamban Mutants Compete with Wild Type for SERCA Binding in Living Cells. Biochem. Biophys. Res. Commun. 2012, 420, 236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong M.; Zhang Y.; Hu F.. Membrane Protein Structure and Dynamics from NMR Spectroscopy. Annu. Rev. Phys. Chem. 2012, 63, forthcoming. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diller A.; Loudet C.; Aussenac F.; Raffard G.; Fournier S.; Laguerre M.; Grelard A.; Opella S. J.; Marassi F. M.; Dufourc E. J. Bicelles: A Natural ’Molecular Goniometer’ for Structural, Dynamical and Topological Studies of Molecules in Membranes. Biochimie 2009, 91, 744–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opella S. J.; Zeri A. C.; Park S. H. Structure, Dynamics, and Assembly of Filamentous Bacteriophages by Nuclear Magnetic Resonance Spectroscopy. Annu. Rev. Phys. Chem. 2008, 59, 635–657. [DOI] [PubMed] [Google Scholar]
- Lacapere J.-J.; Pebay-Peyroula E.; Neumann J.-M.; Etchebest C. Determining Membrane Protein Structures: Still a Challenge!. Trends Biochem. Sci. 2007, 32, 259–270. [DOI] [PubMed] [Google Scholar]
- Schaefer J.; Stejskal E. O. C-13 Nuclear Magnetic-Resonance of Polymers Spinning at Magic Angle. J. Am. Chem. Soc. 1976, 98, 1031–1032. [Google Scholar]
- Stockton G. W.; Polnaszek C. F.; Tulloch A. P.; Hasan F.; Smith I. C. P. Molecular-Motion and Order in Single-Bilayer Vesicles and Multilamellar Dispersions of Egg Lecithin and Lecithin-Cholesterol Mixtures-Deuterium Nuclear Magnetic-Resonance Study of Specifically Labeled Lipids. Biochemistry 1976, 15, 954–966. [DOI] [PubMed] [Google Scholar]