

Abstract
Histone deacetylase (HDAC) proteins are promising targets for cancer treatment, as shown by the approval of two HDAC inhibitors for the treatment of cutaneous T-cell lymphoma. HDAC1 in particular has been linked to cell growth and cell cycle regulation and is therefore an attractive target for anticancer drugs. The HDAC1 active site contains a hydrophobic 11 Å active-site channel, with a 14 Å internal cavity at the bottom of the active site. Several computational and biochemical studies have proposed an acetate-escape hypothesis where the acetate byproduct of the deacetylation reaction escapes via the 14 Å internal cavity. Selective HDAC inhibitors that bind to the 14 Å cavity have also been created. To understand the influence of amino acids lining the HDAC1 14 Å cavity in acetate escape and inhibitor binding, we used mutagenesis coupled with acetate competition assays. The results indicate that amino acids lining the 14 Å cavity are critical for catalytic activity and acetate competition, confirming the role of the cavity in acetate escape. In addition, these mutagenesis studies will aid in HDAC1-inhibitor design that exploits the 14 Å cavity.
Introduction
Acetylation of the ε-amino group of specific lysine residues on histone proteins can influence gene expression by modulating chromatin structure.1 Two classes of enzymes, histone acetyltransferase and histone deacetylase (HDAC), mediate the balance between acetylated and deacetylated states. Deacetylase-mediated removal of acetyl groups is generally linked to gene repression.2 Several studies have shown that inhibiting deacetylation in tumor cells leads to cell differentiation and reduced cell growth, making HDAC proteins attractive anticancer targets.2−4 Currently, two HDAC inhibitors, vorinostat5 and romidepsin6 (Figure 1A), are approved as cancer therapeutics. The discovery of additional HDAC inhibitors for cancer therapy is a promising area of research.7
Figure 1.
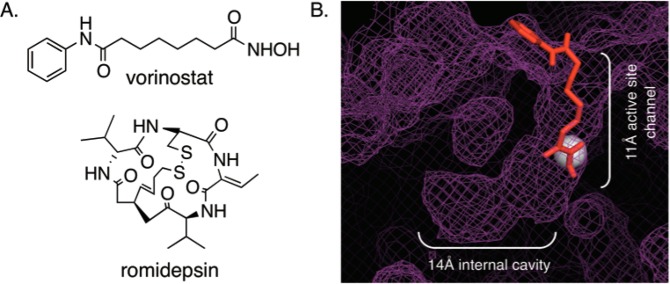
(A) Structures of two HDAC inhibitors, vorinostat and romidepsin. (B) Docked image of vorinostat (red, ball and stick) in the HDAC1 crystal structure (purple mesh; PBD 4BKX). Vorinostat is bound in the 11 Å active-site channel, whereas the 14 Å internal cavity is located adjacent to the catalytic metal (gray sphere).
Phylogenetically, HDAC proteins are grouped into four main classes.8 Class I HDAC proteins are homologous to yeast Rpd3 and include HDAC1, HDAC2, HDAC3, and HDAC8.9−12 HDAC4, HDAC5, HDAC6, HDAC7, and HDAC9 belong to the class II subfamily and have sequence homology to yeast Hda1.13−15 HDAC11 is the only class IV member with similarities to both classes I and II.16,17 With similar metal-dependent catalytic mechanisms, most HDAC inhibitors display broad activity against all isoforms.18 Ultimately, the discovery of isoform-selective inhibitors will be more focused if the involvement of each HDAC isoform in cancer formation is well-characterized.
Among the class I HDAC isoforms, HDAC1 is an important player in cancer. For example, HDAC1-knockout embryonic mouse cells were nonviable, with retarded growth and reduced proliferative rates.19 Mouse fibroblast cells lacking HDAC1 were antiproliferative and exhibited G1 cell cycle arrest accompanied by upregulation of cell cycle regulators p21 and p57.20 Aberrant cell cycle regulation and proliferation are hallmarks of cancer formation, making HDAC1 an exciting target for anticancer drug design. HDAC1 is also overexpressed in a variety of cancer tissues, including ovarian,21,22 prostate,23 pancreas,24 and leukemia.25
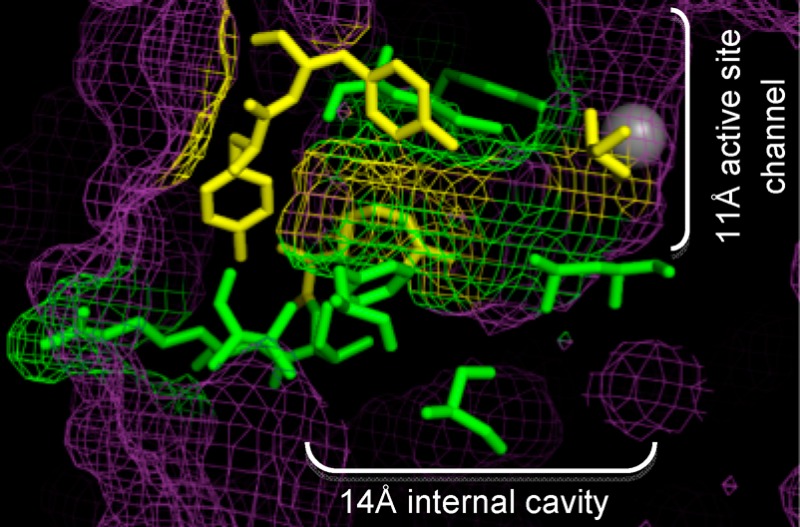
With its significant role in cancer-related events, HDAC1-selective inhibitors would be valuable tools to study cancer cell biology as well as to provide lead compounds for improved anticancer therapies. Efforts to design selective inhibitors have been frustrated by the slow progress in structural characterization. Crystal structures have been reported to date for HDAC2, HDAC3, HDAC4, HDAC7, HDAC8,26−31 and, very recently, HDAC1.32 In addition, homology models of the HDAC isoforms have been helpful in inhibitor design.33,34 The crystal structure of HDAC1 revealed that the active site contains a hydrophobic 11 Å channel with a catalytic metal ion (Figure 1B). HDAC inhibitors have been designed that fit in this long channel by making favorable hydrophobic interactions with amino acids lining the channel along with chelation to the metal ion.
At the bottom of the active site and next to the catalytic metal ion is the 14 Å internal cavity (Figure 1B), which is postulated to allow the acetate byproduct of the deacetylation reaction to escape.29,35,36 Specifically, computational studies using the HDAC1 homology model identified amino acids in the 14 Å cavity that are likely to facilitate acetate escape by forming favorable ionic interactions.35 Acetate-docking studies further revealed important ionic interactions with several 14 Å cavity residues, including R27 and R16 in HDLP.35,37 Interestingly, HDAC inhibitors containing substituents that fit into the 14 Å cavity have shown isoform selectivity.38−40 The implications for these finding is that novel HDAC inhibitors can be designed to exploit the 14 Å cavity.39 Although the 14 Å cavity of HDAC1 may play a critical role in acetate escape and impart selective substrate/inhibitor binding, experimental studies probing the 14 Å cavity of HDAC1 are lacking.
To understand the contribution of residues lining the 14 Å cavity of HDAC1 to catalytic activity and protein associations, the recently published HDAC1 crystal structure was used to identify amino acids residing in the cavity. Alanine single point mutants of 14 Å cavity residues were generated and tested for deacetylase activity. Furthermore, we performed acetate competition assays to determine whether alanine mutants affect HDAC1 binding to acetate. The results suggest that amino acids lining the 14 Å cavity are important to maintain HDAC1 catalytic activity and acetate escape.
Results
Residues in the 14 Å Cavity of HDAC1 Are Critical for Activity
Computational and crystallography studies predicted that HDAC1 Y23 and R34 form a charged pocket in the 14 Å cavity and bind to, and thereby stabilize, the acetate byproduct of the deacetylation reaction (Figure 2A, yellow residues).35 Polar residues Y24 and C151 were also speculated to line the opposite side of the 14 Å cavity to influence acetate binding and to restore the active-site configuration after reaction (Figure 2A, yellow residues). In addition, these four amino acids might influence substrate binding and orientation as a result of their close proximity to the active site. Among these four residues, R34 and C151 are almost strictly conserved among all HDAC isoforms (92 and 100% identity, Figure 2B), whereas Y23 and Y24 are poorly conserved (Figure 2B).
Figure 2.
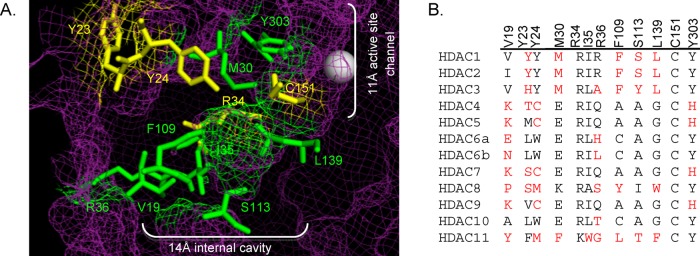
(A) Amino acid residues in the 14 Å cavity of HDAC1 that are mutated in this study (yellow and green, ball and stick structures) are highlighted in the HDAC1 crystal structure (shown as purple mesh, 4BKX). The metal ion required for catalysis is shown as a gray sphere. (B) Catalytic domains of the class I, II, and IV human HDAC proteins were aligned (ClustalW), and residues located in the 14 Å internal cavity are shown. Residues differing from the most highly conserved at each position are highlighted in red. The numbering at the top is for HDAC1. (Genbank accession numbers: HDAC1, Q13547.1; HDAC2, Q92769.2; HDAC3, NP_003874.2; HDAC4, AAD29046.1; HDAC5, AAD29047.1; HDAC6, AAD29048.1; HDAC7, NP_056216.2; HDAC8, CAB90213.1; HDAC9, AAK66821.1; HDAC10, NP_114408.3; and HDAC11, NP_079103.2.)
To determine the influence of Y23, Y24, R34, and C151 on HDAC1 enzymatic activity, each amino acid was individually mutated to alanine, and the mutants were tested for deactylation activity. Briefly, alanine mutants or wild-type HDAC1 were expressed in T-Ag Jurkat cells as FLAG-tagged fusion proteins, immunoprecipitated with α-FLAG agarose beads, and tested for deacetylase activity using an in vitro fluoresence assay, as previously described.41 The catalytically inactive mutant H141A was included for comparison.42 Alanine mutation of all four residues significantly reduced HDAC1 catalytic activity (Figure 3A and Table S1). The highly conserved R34A and C151A mutants demonstrated 31 and 21% remaining activity, respectively, compared to wild type (100%), which was similar to inactive H141A (20%). Mutating Y23 and Y24 to alanine also led to mutants with 35 and 26% catalytic activity remaining, respectively. The HDAC1 homology model predicts that R34 and C151 are closer to the active site, whereas Y23 and Y24 exist closer to the solvent-exposed region of the 14 Å cavity (Figure 2A, yellow residues). Despite their position, the alanine-scan results indicate that Y23, Y24, R34, and C151 are critical to maintain the catalytic activity of HDAC1.
Figure 3.

Y23A, Y24A, R34A, and C151A mutants affect HDAC1 deacetylase activity but not protein association. (A) Wild-type or mutant proteins were expressed in T-Ag Jurkat cells as FLAG-tagged fusion proteins, immunoprecipitated with anti-FLAG-agarose beads, and tested for catalytic activity using an in vitro fluorescence assay (histogram) or separated on SDS-PAGE and probed with anti-FLAG antibody (gel image). The histogram shows the mean percent of at least four independent trials, which were normalized to wild-type deacetylase activity (set to 100%). The standard error is shown as error bars (Table S1). (B). Immunoprecipitates of wild type and alanine mutants in panel A were further probed with anti-FLAG, anti-RbAp48, or anti-mSin3A antibodies to assess protein association.
Mutation of 14 Å Cavity Residues Does Not Influence Protein Associations
The mammalian corepressor proteins mSin3A and RbAp48 tie HDAC1 to promoter-bound transcription factors, leading to transcriptional gene silencing.42−45 The binding of HDAC1 to corepressors not only recruits the deacetylase to promoter regions but also leads to an increase in deacetylase activity.43 To test whether the reduced activity observed with Y23A, Y24A, R34A, and C151A mutants was due to poor binding to associating partners, biochemical purification was performed. HDAC wild type or mutants were immunoprecipitated with FLAG-agarose beads, separated on SDS-PAGE, and probed with mSin3A or RbAp48 antibodies. All four mutants displayed an equal ability to coimmunoprecipitate mSin3A and RbAp48, independent of deacetylase activity (Figure 3B, compare the wild type to mutant lanes). As a control, HDAC1 H141A recovered levels of mSin3A and RbAp48 comparable to the wild type (Figure 3B, compare wild type to H141A lanes), as previously reported.41 The results suggest that the reduced activity of HDAC1 mutants is independent of corepressor binding. In addition, the data suggest that the global structure of the mutants is not significantly disturbed.
Additional Residues in the 14 Å Cavity of HDAC1 Are Also Critical for Activity
In addition to Y23, Y24, R34, and C151, multiple residues comprise the remainder of the 14 Å cavity. The HDAC1 crystal structure was used to identify V19, M30, I35, F109, L139, and Y303 in the 14 Å cavity (Figure 2A, green residues). These amino acids are hydrophobic or polar and are therefore not expected to participate as critically in acetate binding. However, because of their proximity to the active site, these amino acids may influence substrate binding and restoration of the active-site configuration. We were also interested in testing the role of R36 and S113 in HDAC1 activity. Although the crystal structure shows that the guanidinium side chain of R36 is positioned away from the 14 Å cavity (Figure 2B), earlier studies suggested that it resides in the cavity.33 In the case of S113, computational studies with an HDAC1 homology model suggested that the amino acid at position 113 dictates selective binding of HDAC inhibitors into the 14 Å cavity.39 Therefore, mutagenesis studies with residues comprising the 14 Å cavity, including S113, may assist in future inhibitor-design efforts.
To determine the influence of V19, M30, I35, R36, S113, F109, L139, and Y303 on HDAC1 enzymatic activity, each amino acid was individually mutated to alanine, overexpressed in T-Ag Jurkat cells, immunoprecipitated, and tested for activity, as discussed earlier. All mutants displayed reduced HDAC1 catalytic activity (Figure 4A and Table S2). Alanine replacement of strictly conserved Y303 resulted in a mutant displaying 6% remaining activity relative to wild type (100%). The homology model predicts that Y303 is positioned at the junction of the 11 Å active-site channel and the 14 Å cavity (Figure 2A), and the hydroxyl group of Y303 is thought to stabilize the hemiketal intermediate produced during the deacetylation reaction through hydrogen bonding,36 which is consistent with its role in maintaining activity. Likewise, the V19A, M30A, I35A, F109A, and L139A mutants displayed between 6 and 39% deacetylase activities, which are comparable to the inactive H141A mutant (20%) and suggest that all are important for HDAC1 deacetylase activity. Although the crystal structure suggests that the guanidinium side chain of R36 is positioned away from the 14 Å cavity, the R36A mutant displayed only 25% deacetylases activity, suggesting that it plays a supportive role. Finally, the S113A mutant was the most active of this series, maintaining 62% deacetylase activities. Coimmunoprecipitation experiments with the alanine mutants also confirmed that protein associations were unaltered (Figure 4B). The combined mutagenesis data suggests that seven out of eight 14 Å cavity mutants tested are critical for maintaining HDAC1 enzymatic activity.
Figure 4.

Additional 14 Å cavity alanine mutants affect HDAC1 deacetylase activity but have no effect on the protein association. (A) Wild-type or mutant proteins were expressed in T-Ag Jurkat cells as FLAG-tagged fusion proteins, immunoprecipitated with anti-FLAG-agarose beads, and tested for catalytic activity using an in vitro fluorescence assay (histogram) or separated on SDS-PAGE and probed with anti-FLAG antibody (gel image). The histogram shows the mean percent of at least four independent trials normalized to wild-type deacetylase activity (set to 100%). The standard error is shown with error bars (Table S2). (B) Immunoprecipitates from panel A were further probed with anti-FLAG, anti-RbAp48, and anti-mSin3A antibodies to assess protein association.
Substitution Mutants of 14 Å Cavity Residues Are Also Inactive
Consistent with the acetate-escape hypothesis, acetate could form favorable electrostatic interactions with charged or polar residues within the cavity, including Y23, Y24, R34, R36, C151, and Y303. Accordingly, substitution of these residues to the similar charged or polar functional groups could promote favorable interactions with acetate and maintain enzymatic activity. To probe this possibility, several substitution mutants were created. Residues R34 and R36 were replaced with lysine to retain positive charge. Similarly, C151 was replaced by serine to maintain polarity. In the case of Y23, Y24, and Y303, phenylalanine substitution mutants would preserve aromaticity while removing polarity. Alternatively, replacement of Y23 with cysteine, histidine, or arginine would maintain polarity, charge, and/or aromaticity. In total, substitution mutants were created to probe the role of charge, polarity, and aromaticity in the 14 Å cavity on activity.
The Y23F, Y23C, Y23H, Y23R, Y24F, R34K, R36K, C151S, and Y303F single-point HDAC1 mutants were expressed in T-Ag Jurkat cells as FLAG-tagged fusion proteins, immunoprecipitated with FLAG-agarose beads, and tested for deacetylase activity using an in vitro fluoresence assay, as described earlier (Figure 5A and Table S3). The substitution mutants significantly influenced HDAC1 catalytic activity and, in most cases, displayed similar or lower activities compared to the alanine mutants (compare Figures 3A and 5A). In the case of the Y23 mutant series, Y23C maintained similar activity to the alanine mutant (36 versus 35%), whereas the Y23F and Y23H mutants displayed reduced activity (12%). Lysine did not compensate for arginine, as shown by the low activities of the R34K and R36K mutants (17 and 33%, respectively), which is comparable to the activities of the R34A and R36A mutants (31 and 25%, respectively). Likewise, the C151A and C151S mutants demonstrated similar activities (21 and 30%, respectively). Only in two cases did substitution mutants enhance activity compared to the corresponding alanine mutants. In comparison to the 26% activity of Y24A, Y24F displayed 50% activity (a 2-fold increase). More dramatically, Y303F demonstrated 38% activity compared to the 6% activity of Y303A (a 6-fold increase). All mutants were capable of interacting with associated proteins mSin3A and RbAp48 (Figure 5B), indicating that they maintain their globular structure. In total, the mutagenesis data suggests that Y23, R34, R36, and C151 are uniquely suited to promote HDAC1 deacetylase activity, whereas phenylalanine partially substitutes for Y24 and Y303.
Figure 5.

Substitution mutants of the 14 Å cavity residues display reduced activity. (A) Wild-type or mutant proteins were expressed in T-Ag Jurkat cells as FLAG-tagged fusion proteins, immunoprecipitated with anti-FLAG-agarose beads, and tested for catalytic activity using an in vitro fluorescence assay (histogram) or separated on SDS-PAGE and probed with anti-FLAG antibody (gel image). The histogram shows the mean percent of at least four independent trials normalized to wild-type deacetylase activity (set to 100%). The standard error is shown as error bars (Table S3). (B) Coimmunoprecipitates of mutants in panel A were probed with anti-FLAG, anti-RbAp48, and anti-mSin3A antibodies to assess protein association.
Acetate Inhibition Studies
The mutagenesis results indicate that amino acids in the 14 Å cavity promote HDAC1 enzymatic activity. One hypothesis to account for the essential role of the 14 Å cavity in activity is via acetate release after deacetylation. Prior computational work postulated that charged and polar amino acids Y23, Y24, R34, R36, and C151 form favorable electrostatic/hydrogen-bond interactions with acetate.35,42 To assess experimentally the influence of 14 Å cavity residues on acetate binding, acetate inhibition experiments were performed. The expectation was that mutants would show reduced sensitivity to acetate inhibition compared to wild-type HDAC1 if each residue plays a role in acetate binding. As a critical control, the partially active F205Y mutant was included because of its location in the solvent-exposed surface of the 11 Å channel;41 F205 mutation should not influence acetate release and, consequently, should demonstrate identical acetate inhibition sensitivity compared to wild-type HDAC1.
To test the role of 14 Å cavity mutants in acetate release experimentally, wild-type or mutant proteins were immunoprecipitated with FLAG-agarose beads, and the extent of inhibition by acetate was determined using dose–response curves (Table 1, Figure S1, and Tables S4–S10). Mutant proteins displayed reduced sensitivity to acetate inhibition. The Y23A, Y24A, and R34A mutants were the most insensitive to acetate inhibition, with roughly 3-fold reduced IC50 values (Table 1, 19–21 mM) compared to the wild type (7.2 mM). The R36A and C151A mutants also showed a roughly 2-fold reduction in acetate inhibition compared to the wild type (Table 1). As a critical control, the F205Y mutation that is located in the 11 Å channel and not expected to influence acetate release displayed identical acetate inhibition sensitivity as wild-type HDAC1 (Table 1, 7.2 and 8.0 mM). Collectively, the acetate competition data suggest that both charged and polar amino acid residues in the 14 Å cavity influence HDAC1 binding to acetate. These experimental studies are consistent with earlier computational work documenting the involvement of the HDAC1 14 Å cavity in acetate release.46
Table 1. Acetate Inhibition Experiments.
| sample | IC50 (mM) |
|---|---|
| wild type | 7.2 ± 0.54 |
| HDAC1 F205Y | 8.0 ± 1.5 |
| HDAC1 Y23A | 21 ± 1.5 |
| HDAC1 Y24A | 19 ± 1.1 |
| HDAC1 R34A | 21 ± 1.6 |
| HDAC1 R36A | 16 ± 1.3 |
| HDAC1 C151A | 13 ± 1.1 |
IC50 values from at least three independent trials are shown with standard error. Dose-dependent curves and data are shown in Figure S1 and Tables S4–S10.
Inhibitor Selectivity Study
Several inhibitors with selectivity to HDAC1 and HDAC2 have been reported, where isoform preference was attributed to interactions with residues in the 14 Å cavity.38−40,47 In one study, docking experiments suggested that the HDAC1/2-selecitivity was due to variable amino acids present at the position corresponding to S113 in HDAC1 (Figure 2B).39 Specifically, HDAC3 positions a tyrosine at that position, and the docking analysis suggested that the larger size of tyrosine blocks access of the inhibitor to the 14 Å cavity, explaining the >1400-fold selectivity against HDAC3. Interestingly, the relatively small alanine residue is found at the corresponding position in HDAC4–7, yet several inhibitors in the series, including 2-thiophenyl biaryl inhibitor 1 (Figure 6A), also demonstrated >1400-fold selectivity against HDAC4–8. The fact that HDAC4–7 display alanine at the position corresponding to S113 in HDAC1 yet bind 1 poorly is inconsistent with the proposed hypothesis that HDAC1/2-selecitivity is due to the steric size of amino acids at this position.
Figure 6.

Inhibitory potency of 1 against wild-type and S113A HDAC1. (A) Structure of HDAC1/2-selective compound 1. (B) Wild-type (closed circles) or S113A (open circles) proteins were expressed in T-Ag Jurkat cells as FLAG-tagged fusion proteins, immunoprecipitated with anti-FLAG-agarose beads, and tested for catalytic activity using an in vitro chemiluminescence assay in the absence or presences of increasing concentrations of 1. Dose–response curves of from three independent trials with error bars indicating standard error are shown (Tables S11 and 12).
With the availability of the S113A HDAC1 mutant, we tested the influence of S113 on the selectivity of compound 1 for HDAC1. Wild-type or S113A mutant proteins were immunoprecipitated with FLAG-agarose beads, and the extent of inhibition by 1 was determined using dose–response curves (Figure 6B and Tables S11 and S12). Wild-type HDAC1 demonstrated an IC50 value of 40 ± 4 nM, which is similar to prior reports.39 The S113A HDAC1 mutant demonstrated an IC50 value of 204 ± 20 nM, which is a sensitivity difference of roughly 5-fold compared to the wild type. Given the 1400-fold inhibitory selectivity of 1 against the HDAC isoforms containing an alanine at the corresponding S113 position, the 5-fold difference suggests that S113 is only a partial determinant of selectivity. These mutagenesis studies implicate additional 14 Å cavity residues outside of S113 as critical contributors to selective HDAC1 inhibition by 1.
Discussion
The 14 Å cavities of HDAC isoforms have long been hypothesized to act as an exit route for acetate following deacetylation.35−37,42 In one study, docking showed that the protonated and charged forms of both acetate and acetic acid localize next to the catalytic metal ion, R16, and R17 in HDLP. In contrast, similar studies with N-hydroxyacetamide, which is neutral and similar in structure to the HDAC inhibitor vorinostat (Figure 1A), revealed preferred binding in the 11 Å channel but not the 14 Å cavity.35 In addition, amino acids located toward the solvent-exposed regions of the 14 Å cavity demonstrate flexibility, indicating that side-chain movement of amino acids could exchange cavity contents with the bulk water solvent for the next cycle of deacetylation.29,35,36 To date, no experimental data have confirmed the role of the 14 Å cavity in HDAC1 enzymatic activity and acetate binding.
An alanine scan was used to determine the influence of 12 amino acid residues lining the 14 Å cavity of HDAC1 on enzymatic activity and binding to acetate.41,46 Eleven of the alanine mutants (V19A, Y23A, Y24A, M30A, R34A, I35A, R36A, F109A, L139A, C151A, and Y303A) displayed reduced HDAC1 deacetylase activity, with only 6–39% activity compared to wild type (set at 100%, Figures 3A and 4A). Only one mutant, S113A, maintained significant deacetylase activity (61% compared to wild type, Figure 4A). Computational studies previously suggested that S113 of HDAC1 is responsible for the isoform selectivity observed by inhibitors that access the 14 Å cavity;38,39 however, S113 is positioned adjacent to, but not within, the 14 Å cavity in the HDAC1 crystal structure (Figure 2A). In addition, alanine is found at position 113 in six of the 11 HDAC isoforms (Figure 2B, HDAC4, 5, 6, 7, 9, and 10). Therefore, alanine mutation at S113 may preserve HDAC activity because of its distal location and presence in other isoforms. Alanine is also found in other isoforms at position V19 (HDAC10), I35 (HDAC8), R36 (HDAC3), and F109 (HDAC4, 5, 7, and 9); however, alanine is not well conserved among all HDAC isoforms at these positions (Figure 2B) and was not tolerated by HDAC1 (Figure 4A). In total, the results indicate that residues in the 14 Å cavity are critical for HDAC1 activity, with alanine partially substituting for only S113.
Because polar and charged residues lining the 14 Å cavity are implicated in acetate binding, substitution mutants maintaining the polarity, charge, or aromaticity of 14 Å cavity residues were created to probe compensatory activities. Substitution mutants displayed reduced deacetylase activities compared to wild type, with similar or lower activity compared to the alanine mutants (Figure 5A). Y24F was the only substitution mutant that maintained a significant deacetylase activity (50% compared to wild type, Figure 5A). In addition, Y303F maintained 38% activity (Figure 5A), which was 6-fold elevated compared to Y303A. These substitution studies suggest that aromaticity at positions 24 and 303 plays an important role in the 14 Å cavity, although the presence of a polar hydroxyl group is also important. The combined mutagenesis studies suggest that Y23, Y24, R34, R36, C151, and Y303 are each uniquely suited to maintain the full enzymatic activity of HDAC1.
The loss in enzymatic activity of 14 Å cavity mutants can be attributed to two possibilities. First, cavity residues might influence substrate binding or govern the organization of the active-site residues, which ultimately affects activity. Second, cavity residues might influence the release of the acetate byproduct. To distinguish between these possibilities, the influence of mutation on acetate binding was determined. Alanine mutation of both polar and charged residues reduced acetate inhibition by 2- to 3-fold compared to wild type, whereas the 11 Å channel F205Y mutant showed no change. These findings are consistent with previous work with recombinant HDAC8 where acetate inhibition was 160-fold reduced with HDAC8 R37A (equivalent to R34A in HDAC1) compared to wild type (IC50 of 400 mM with HDAC8 R37A versus 2.5 mM with wild type).46 The larger fold difference observed with recombinant HDAC8 compared to mammalian HDAC1 could be a result of the mammalian versus bacterial expression of the proteins. With both HDAC1 and HDAC8, mutation of 14 Å cavity residues removed key contacts required for acetate binding, resulting in reduced acetate inhibitory sensitivities. Combined, the mutagenesis data is consistent with the hypothesis that the 14 Å cavity plays a role in acetate binding and release during the deacetylation reaction.
In addition to acetate binding, the 14 Å cavity has also been implicated to transporting water to the HDAC active site.35 Computational studies with HDAC8 suggested that R37 (R34 in HDAC1) interacts with nearby residues (G303 and G305 in HDAC8, for example) to act as a gatekeeper to acetate/water passage.37,46 Moreover, it was hypothesized that R37 mutation results in the flooding of the active site with water, which inhibits deacetylation.46 R34 in the HDAC1 homology model is within hydrogen-bonding distance of G300; however, the cavity is not gated by this interaction, as with the HDAC8 structure. More recent computational work with an HDAC1 homology model predicted aromatic gating of water away from the 14 Å cavity by Y22, Y24, and F109 (Figure 2A).48 In addition, water accessibility was shown to contribute to the binding of hydroxamate-containing inhibitors, including vorinostat.49 Combined, it is interesting to speculate that mutation of residues in the 14 Å cavity alters the accessibility of water to the active site, perhaps by perturbing an aromatic-gating mechanism. Indeed, alanine mutants of putative gatekeeper residues (Y24A and F109A) demonstrated reduced activity (20–35%, Figures 3A and 4A). In addition, the partial recovery of activity by the Y24F mutants (Figure 5A, 2-fold) is consistent with the possibility that aromatic gating plays a role in maintaining the structure and function of the 14 Å cavity.
HDAC1 has been linked to tumorigenesis, making it an attractive target for cancer therapy. Computational studies have suggested that HDAC1 and HDAC2 can be selectively targeted through the 14 Å cavity.35 Indeed, several inhibitors that access the 14 Å cavity displayed selectivity for HDAC1 and 2 compared to HDAC3–8.38−40,47 In these cases, the HDAC1/2-selecitivity was computationally rationalized because of the presence of S113 in HDAC1 versus tyrosine in HDAC3 (Figure 2B).39 Interestingly, alanine is the most abundant residue at position 113, with 60% conservation (Figure 2B). Despite the relatively small size of the alanine residue, HDAC4, 5, 6, and 7 demonstrate poor binding to HDAC1/2-selective inhibitors, which suggests that the size of the residue at S113 in HDAC1 may only minimally influence selectivity. To assess the influence of the S113 residue on the selectivity of inhibitors that access the 14 Å cavity, the potency of HDAC1/2-selective inhibitor 1 was determined with the HDAC S113A mutant. The data show that the S113A mutant displays only a 5-fold reduced sensitivity to 1 compared to wild-type HDAC1, which is significantly less than the observed 1400-fold selectivity of 1 for HDAC1. Therefore, the data suggest that the amino acid at the S113 position only partially accounts for the observed selectivity. In fact, the recently published crystal structure of HDAC1 positions S113 adjacent to, but not within, the 14 Å cavity (Figure 2A). Combined, the current evidence suggests that structural features in the 14 Å cavity other than the S113 residue likely govern selectivity.
A more recent study reported HDAC8-selective inhibitors that bind the 14 Å cavity.37 In this case, selectivity was attributed to π-stacking interactions between the inhibitor and W141 in HDAC8 (aligned with L139 in HDAC1). Among the isoforms, only HDAC11 also contains an aromatic residue at this position (F141), whereas all others display either glycine (60% conservation) or leucine (25% conservation, Figure 2B). The HDAC1 L139A mutant displayed weak activity (39%, Figure 4A), implicating structural differences in this region of the 14 Å cavity. In total, the mutagenesis studies here are consistent with prior work suggesting that residues at the base of the 14 Å cavity impart unique structural features that can be exploited in isoform selective inhibitor design. Future studies that elucidate critical 14 Å cavity residues involved in inhibitor binding will be helpful to create additional selective HDAC inhibitors.
The acetate competition results presented here also suggest that acetate mimics appended near the metal-binding moiety of HDAC inhibitors may interact with 14 Å cavity residues to enhance binding affinity. To date, inhibitors with aromatic groups adjacent to the metal-binding moiety were designed to access the 14 Å cavity (Figure 6A).38,39 Creation of HDAC inhibitors displaying acetate mimics may enhance binding affinity while also influencing selectivity. Given the wide interest in HDAC inhibitors as pharmacological tools and anticancer drugs, inhibitor design exploiting structural differences and favorable binding interactions in the 14 Å cavity is an exciting future area of development.
Experimental Procedures
Mutagenesis, Mammalian Expression, and Western Blot Analysis of HDAC1 Mutants
HDAC1 single point mutants were cloned into the pBJ5HDAC1-FLAG expression plasmid using NotI and EcoRI restriction sites, as previously described.41 All mutants were confirmed by DNA sequencing (see the Supporting Information for primer sequences). T-Ag Jurkat cells50 were grown in RPMI-1640 media supplemented with 10% FBS and 1% anti-mycotic (GIBCO) at 37 °C in a 5% CO2 environment. Plasmid DNA (20 μg, pBJ5HDAC1-FLAG or mutants) was separately transiently transfected into 40 × 106 cells by electroporation. After a 48 h growth period, the cells were harvested, washed with PBS (137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, and 1.4 mM KH2PO4, pH 7.3), and used immediately or stored at −80 °C until use. Cells were lysed in 1 mL of cold Jurkat lysis buffer (50 mM Tris, pH 8, 150 mM NaCl, 10% glycerol, and 0.5% Triton X-100) containing 1× protease inhibitor cocktail set V (Calbiochem) at 4 °C for 10 min with rotation before centrifugation to collect the soluble lysate. Anti-FLAG agarose beads (10 μL of bead slurry, Sigma) were used to immunoprecipitate HDAC1-FLAG wild-type or mutant proteins from the lysates. Immunoprecipitated proteins were split into two equal portions. One portion was used in the HDAC activity assay described below, whereas the second portion was separated using 10% SDS-PAGE, transferred to a PVDF membrane (Immobilon P, Millipore), and probed with anti-Flag (Sigma), anti-mSin3A (Santa Cruz), or anti-RbAp48 (Sigma) antibodies.
HDAC Activity Assay
The Fluor de Lys fluorescence activity assay (Biomol) was used to determine HDAC activity by following the manufacturer’s protocol. Immunoprecipitated proteins (approximately 20 μg) were incubated with HDAC assay buffer (25 μL) and Fluor de Lys substrate (25 μL of 100 μM) at 37 °C for 45 min with shaking (900 rpm). Then, developer (50 μL of a 1× solution) was added and incubated for 5 min. Fluorescence signal was measured using the GENios Plus plate-reader fluorimeter (Tecan). The fluorescence signal was background-corrected against a reaction containing HDAC assay buffer, substrate, and developer but no HDAC enzyme. The background-corrected signal from each mutant was normalized to the wild type (set at 100%). The mean and standard error from at least three independent trials are shown in Figures 3–5 (Tables S1–S3).
Acetate Inhibition Assays
To perform the acetate inhibition assays, the HDAC activity assay was performed as described51−53 with a few modifications. In this case, HDAC or mutant proteins were immunoprecipitated from a 40 × 106 cell transfection using 40 μL of anti-Flag beads (Sigma) and were then split into five equal aliquots. Each immunoprecipitate aliquot was preincubated with HDAC assay buffer (25 μL) in the absence or presence of sodium acetate for 15 min at 37 °C with shaking (900 rpm). The Fluor de Lys substrate was then added and incubated as described for the fluorescence activity assay. The concentrations of sodium acetate used are shown in Tables S4–S10 and represent the final concentrations after addition of substrate. The deacetylase reaction was developed and analyzed as described above. All reactions in either the presence or absence of sodium acetate were background-corrected to a reaction containing HDAC assay buffer, substrate, and developer but no HDAC enzyme. Percent inhibition was determined by dividing the fluorescence signal at each acetate concentration to the no acetate control for each mutant and then multiplying by 100 (Tables S4–S10). Standard error from at least three independent trials is shown (Tables S4–S10). IC50 values were determined by fitting the data to a sigmoidal curve using Kaleidograph software (Figure S1).
Compound 1 Inhibition Assays
The inhibition studies with compound 1 were performed with the HDAC-Glo chemiluminescence assay (Promega). FLAG-tagged wild-type and S113A mutant proteins were immunoprecipitated and divided into aliquots, as described for the acetate studies. Then, each immunoprecipitated protein aliquot was incubated with HDAC-Glo assay buffer (24 μL) and varying concentrations of 1 (1 μL). The concentrations of 1 used are shown in Tables S11 and S12 and represent the final concentrations after addition of substrate/developer solution. Because of the slow binding kinetics of benzamides, compound 1 was preincubated with the immunoprecipitated enzyme for 2 h at 30 °C with shaking (900 rpm). Then, the substrate/developer solution (2 μL of 100 μM substrate containing a 1000-fold dilution of developer) was added and incubated at 30 °C for 30 min with shaking (900 rpm). Chemiluminescence signal was measured using the GENios Plus plate-reader fluorimeter (Tecan). The reactions were analyzed as described for the acetate inhibition experiments (Tables S11 and S12).
Docking Study
The crystal structure for HDAC1 was downloaded from the RCSB Protein Data Bank (HDAC1 PDB ID: 4BKX). PyMOL (Schrodinger, LLC) was used to delete the MTA1 corepressor chain, acetate, and potassium and sulfate ions in the HDAC1 crystal structure. AutoDockTools-1.5.4 program54,55 was used to add all hydrogen atoms, modify histidine protonation (H140 and H141) by adding only HD1, compute Gasteiger charges, and merge all nonpolar hydrogen followed by generation of the pdbqt output file. The charge of the zinc atom was manually changed from zero to +2. A grid box with a spacing of 0.375 Å, size of 56 × 42 × 38, and coordinates for the center of the grid box (−48.000, 18.000, −3.750) were used. AutoGrid 4.2 was used to precalculate and generate the grid maps files required for the docking calculations. Vorinostat was drawn in ChemBioDraw Ultra, and Chem 3D Pro was used to run MM2 for energy minimization. Then, AutoDockTools-1.5.4 was used to add hydrogens, compute Gasteiger charges, merge nonpolar hydrogens, choose torsions, and generate the pdbqt file. All acyclic bonds were made rotatable except the amide bonds. AutoDock 4.255 was used to perform the docking calculations using a genetic algorithm. The generated pdbqt file for the enzyme was set as a rigid macromolecule, and the genetic algorithm search parameters were set to 100 GA runs for each ligand with a population size of 150, a maximum number of 2.5 × 105 energy evaluations, a maximum number of 2.7 × 104 generations, a mutation rate of 0.2, and a crossover rate of 0.8. The docking parameters were set to default. The output DLG file was converted to pdbqt format, and the results were visualized in PyMOL. The lowest-energy pose consistent with metal binding is shown in Figure 1B.
Acknowledgments
We thank the National Institute of Health (GM079529) and Wayne State University for funding, G. R. Crabtree for the T-Ag Jurkat cells, and G. Padige and M. Embogama for comments on the manuscript.
Glossary
Abbreviations Used
- HDAC
histone deacetylase
- FBS
fetal bovine serum
- PBS
phosphate buffered saline
Supporting Information Available
Primer sequences used in HDAC1 mutagenesis; percent deacetylase activities for all alanine and substitution mutants; and dose-dependent curves and data for acetate and compound 1 inhibition studies. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
All cloning and experiments were performed by M.W. except for the F109A mutant and compound 1 experiments, which were performed by D.N. A.N. performed the docking study to create Figure 1B. M.P. assisted in the design, analysis, and interpretation of the experiments. All authors assisted in the writing of the manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Grunstein M. Histone acetylation in chromatin structure and transcription. Nature 1997, 389, 349–352. [DOI] [PubMed] [Google Scholar]
- Hebbes T. R.; Thorne A. W.; Crane-Robinson C. A direct link between core histone acetylation and transcriptionally active chromatin. EMBO J. 1988, 7, 1395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richon V. M.; Emiliani S.; Verdin E.; Webb Y.; Breslow R.; Rifkind R. A.; Marks P. A. A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 3003–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen L. A.; Amin S.; Marks P. A.; Rifkind R. A.; Desai D.; Richon V. M. Chemoprevention of carcinogen-induced mammary tumorigenesis by the hybrid polar cytodifferentiation agent, suberanilohydroxamic acid (SAHA). Anticancer Res. 1999, 19, 4999–5005. [PubMed] [Google Scholar]
- Mann B. S.; Johnson J. R.; Cohen M. H.; Justice R.; Pazdur R. FDA approval summary: Vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007, 12, 1247–52. [DOI] [PubMed] [Google Scholar]
- Prince H. M.; Dickinson M. Romidepsin for cutaneous T-cell lymphoma. Clin. Cancer Res. 2012, 18, 3509–3515. [DOI] [PubMed] [Google Scholar]
- Glaser K. B. HDAC inhibitors: Clinical update and mechanism-based potential. Biochem. Pharmacol. 2007, 74, 659–671. [DOI] [PubMed] [Google Scholar]
- Gregoretti I. V.; Lee Y. M.; Goodson H. V. Molecular evolution of the histone deacetylase family: Functional implications of phylogenetic analysis. J. Mol. Biol. 2004, 338, 17–31. [DOI] [PubMed] [Google Scholar]
- Taunton J.; Hassig C. A.; Schreiber S. L. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science 1996, 272, 408–411. [DOI] [PubMed] [Google Scholar]
- Yang W. M.; Yao Y. L.; Sun J. M.; Davie J. R.; Seto E. Isolation and characterization of cDNAs corresponding to an additional member of the human histone deacetylase gene family. J. Biol. Chem. 1997, 272, 28001–28007. [DOI] [PubMed] [Google Scholar]
- Hu E.; Chen Z.; Fredrickson T.; Zhu Y.; Kirkpatrick R.; Zhang G. F.; Johanson K.; Sung C. M.; Liu R.; Winkler J. Cloning and characterization of a novel human class I histone deacetylase that functions as a transcription repressor. J. Biol. Chem. 2000, 275, 15254–15264. [DOI] [PubMed] [Google Scholar]
- Van den Wyngaert I.; de Vries W.; Kremer A.; Neefs J.; Verhasselt P.; Luyten W. H.; Kass S. U. Cloning and characterization of human histone deacetylase 8. FEBS Lett. 2000, 478, 77–83. [DOI] [PubMed] [Google Scholar]
- Grozinger C. M.; Hassig C. A.; Schreiber S. L. Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 4868–4873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao H. Y.; Downes M.; Ordentlich P.; Evans R. M. Isolation of a novel histone deacetylase reveals that class I and class II deacetylases promote SMRT-mediated repression. Genes Dev. 2000, 14, 55–66. [PMC free article] [PubMed] [Google Scholar]
- Zhou X.; Marks P. A.; Rifkind R. A.; Richon V. M. Cloning and characterization of a histone deacetylase, HDAC9. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 10572–10577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L.; Cueto M. A.; Asselbergs F.; Atadja P. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J. Biol. Chem. 2002, 277, 25748–25755. [DOI] [PubMed] [Google Scholar]
- Villagra A.; Cheng F.; Wang H. W.; Suarez I.; Glozak M.; Maurin M.; Nguyen D.; Wright K. L.; Atadja P. W.; Bhalla K.; Pinilla-Ibarz J.; Seto E.; Sotomayor E. M. The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance. Nat. Immunol. 2009, 10, 92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan N.; Jeffers M.; Kumar S.; Hackett C.; Boldog F.; Khramtsov N.; Qian X.; Mills E.; Berghs S. C.; Carey N.; Finn P. W.; Collins L. S.; Tumber A.; Ritchie J. W.; Jensen P. B.; Lichenstein H. S.; Sehested M. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem. J. 2008, 409, 581–589. [DOI] [PubMed] [Google Scholar]
- Lagger G.; O’Carroll D.; Rembold M.; Khier H.; Tischler J.; Weitzer G.; Schuettengruber B.; Hauser C.; Brunmeir R.; Jenuwein T.; Seiser C. Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression. EMBO J. 2002, 21, 2672–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi T.; Cubizolles F.; Zhang Y.; Reichert N.; Kohler H.; Seiser C.; Matthias P. Histone deacetylases 1 and 2 act in concert to promote the G1-to-S progression. Genes Dev. 2010, 24, 455–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khabele D.; Son D. S.; Parl A. K.; Goldberg G. L.; Augenlicht L. H.; Mariadason J. M.; Rice V. M. Drug-induced inactivation or gene silencing of class I histone deacetylases suppresses ovarian cancer cell growth: Implications for therapy. Cancer Biol. Ther. 2007, 6, 795–801. [DOI] [PubMed] [Google Scholar]
- Hayashi A.; Horiuchi A.; Kikuchi N.; Hayashi T.; Fuseya C.; Suzuki A.; Konishi I.; Shiozawa T. Type-specific roles of histone deacetylase (HDAC) overexpression in ovarian carcinoma: HDAC1 enhances cell proliferation and HDAC3 stimulates cell migration with downregulation of E-cadherin. Int. J. Cancer 2010, 127, 1332–1346. [DOI] [PubMed] [Google Scholar]
- Song Y.; Shiota M.; Tamiya S.; Kuroiwa K.; Naito S.; Tsuneyoshi M. The significance of strong histone deacetylase 1 expression in the progression of prostate cancer. Histopathology 2011, 58, 773–780. [DOI] [PubMed] [Google Scholar]
- Gao D.-J.; Xu M.; Zhang Y.-Q.; Du Y.-Q.; Gao J.; Gong Y.-F.; Man X.-H.; Wu H.-Y.; Jin J.; Xu G.-M.; Li Z.-S. Upregulated histone deacetylase 1 expression in pancreatic ductal adenocarcinoma and specific siRNA inhibits the growth of cancer cells. Pancreas 2010, 39, 994–1001. [DOI] [PubMed] [Google Scholar]
- Wada T.; Kikuchi J.; Nishimura N.; Shimizu R.; Kitamura T.; Furukawa Y. Expression levels of histone deacetylases determine the cell fate of hematopoietic progenitors. J. Biol. Chem. 2009, 284, 30673–30683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bressi J. C.; Jennings A. J.; Skene R.; Wu Y.; Melkus R.; Jong R. D.; O’Connell S.; Grimshaw C. E.; Navre M.; Gangloff A. R. Exploration of the HDAC2 foot pocket: Synthesis and SAR of substituted N-(2-aminophenyl)benzamides. Bioorg. Med. Chem. Lett. 2010, 20, 3142–3145. [DOI] [PubMed] [Google Scholar]
- Guo L.; Han A.; Bates D. L.; Cao J.; Chen L. Crystal structure of a conserved N-terminal domain of histone deacetylase 4 reveals functional insights into glutamine-rich domains. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 4297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuetz A.; Min J.; Allali-Hassani A.; Schapira M.; Shuen M.; Loppnau P.; Mazitschek R.; Kwiatkowski N. P.; Lewis T. A.; Maglathin R. L.; McLean T. H.; Bochkarev A.; Plotnikov A. N.; Vedadi M.; Arrowsmith C. H. Human HDAC7 harbors a class IIa histone deacetylase-specific zinc binding motif and cryptic deacetylase activity. J. Biol. Chem. 2008, 283, 11355–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannini A.; Volpari C.; Filocamo G.; Casavola E. C.; Brunetti M.; Renzoni D.; Chakravarty P.; Paolini C.; Francesco R. D.; Gallinari P.; Steinkuhler C.; Marco S. D. Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 15064–15069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somoza J. R.; Skene R. J.; Katz B. A.; Mol C.; Ho J. D.; Jennings A. J.; Luong C.; Arvai A.; Buggy J. J.; Chi E.; Tang J.; Sang B.-C.; Verner E.; Wynands R.; Leahy E. M.; Dougan D. R.; Snell G.; Navre M.; Knuth M. W.; Swanson R. V.; McRee D. E.; Tari L. W. Structural snapshots of human HDAC8 provide insights into the class I histone deacetylases. Structure 2004, 12, 1324–1334. [DOI] [PubMed] [Google Scholar]
- Watson P. J.; Fairall L.; Santos G. M.; Schwabe J. W. Structure of HDAC3 bound to co-repressor and inositol tetraphosphate. Nature 2012, 481, 335–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millard C. J.; Watson P. J.; Celardo I.; Gordiyenko Y.; Cowley S. M.; Robinson C. V.; Fairall L.; Schwabe J. W. R. Class I HDACs share a common mechanism of regulation by inositol phosphates. Mol. Cell 2013, 51, 57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D. F.; Helquist P.; Wiech N. L.; Wiest O. Toward selective histone deacetylase inhibitor design: Homology modeling, docking studies, and molecular dynamics simulations of human class I histone deacetylases. J. Med. Chem. 2005, 48, 6936–6947. [DOI] [PubMed] [Google Scholar]
- Ortore G.; Di Colo F.; Martinelli A. Docking of hydroxamic acids into HDAC1 and HDAC8: A rationalization of activity trends and selectivities. J. Chem. Inf. Model. 2009, 49, 2774–2785. [DOI] [PubMed] [Google Scholar]
- Wang D. F.; Wiest O.; Helquist P.; Lan-Hargest H. Y.; Wiech N. L. On the function of the 14Å long internal cavity of histone deacetylase-like protein: Implications for the design of histone deacetylase inhibitors. J. Med. Chem. 2004, 47, 3409–3417. [DOI] [PubMed] [Google Scholar]
- Finnin M. S.; Donigian J. R.; Cohen A.; Richon V. M.; Rifkind R. A.; Marks P. A.; Pavletich N. P. Structure of a histone deacetylase homologue bound to trichostatin A. Nature 1999, 401, 188–193. [DOI] [PubMed] [Google Scholar]
- Whitehead L.; Dobler M. R.; Radetich B.; Zhu Y.; Atadja P. W.; Claiborne T.; Grob J. E.; McRiner A.; Pancost M. R.; Patnaik A.; Shao W.; Shultz M.; Tichkule R.; Tommasi R. A.; Vash B.; Wang P.; Stams T. Human HDAC isoform selectivity achieved via exploitation of the acetate release channel with structurally unique small molecule inhibitors. Bioorg. Med. Chem. 2011, 19, 4626–4634. [DOI] [PubMed] [Google Scholar]
- Moradei O. M.; Mallais T. C.; Frechette S.; Paquin I.; Tessier P. E.; Leit S. M.; Fournel M.; Bonfils C.; Trachy-Bourget M. C.; Liu J. H.; Yan T. P.; Lu A. H.; Rahil J.; Wang J.; Lefebvre S.; Li Z. M.; Vaisburg A. F.; Besterinan J. M. Novel aminophenyl benzamide-type histone deacetylase inhibitors with enhanced potency and selectivity. J. Med. Chem. 2007, 50, 5543–5546. [DOI] [PubMed] [Google Scholar]
- Methot J. L.; Chakravarty P. K.; Chenard M.; Close J.; Cruz J. C.; Dahlberg W. K.; Fleming J.; Hamblett C. L.; Hamill J. E.; Harrington P.; Harsch A.; Heidebrecht R.; Hughes B.; Jung J.; Kenific C. M.; Kral A. M.; Meinke P. T.; Middleton R. E.; Ozerova N.; Sloman D. L.; Stanton M. G.; Szewczak A. A.; Tyagarajan S.; Witter D. J.; Paul Secrist J.; Miller T. A. Exploration of the internal cavity of histone deacetylase (HDAC) with selective HDAC1/HDAC2 inhibitors (SHI-1:2). Bioorg. Med. Chem. Lett. 2008, 18, 973–978. [DOI] [PubMed] [Google Scholar]
- Witter D. J.; Harrington P.; Wilson K. J.; Chenard M.; Fleming J. C.; Haines B.; Kral A. M.; Secrist J. P.; Miller T. A. Optimization of biaryl selective HDAC1&2 inhibitors (SHI-1:2). Bioorg. Med. Chem. Lett. 2008, 18, 726–731. [DOI] [PubMed] [Google Scholar]
- Weerasinghe S. V. W.; Estiu G.; Wiest O.; Pflum M. K. H. Residues in the 11 Å channel of histone deacetylase 1 promote catalytic activity: Implications for designing isoform-selective histone deacetylase inhibitors. J. Med. Chem. 2008, 51, 5542–5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassig C. A.; Tong J. K.; Fleischer T. C.; Owa T.; Grable P. G.; Ayer D. E.; Schreiber S. L. A role for histone deacetylase activity in HDAC1-mediated transcriptional repression. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 3519–3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alland L.; David G.; Shen-Li H.; Potes J.; Muhle R.; Lee H. C.; Hou H. Jr.; Chen K.; DePinho R. A. Identification of mammalian Sds3 as an integral component of the Sin3/histone deacetylase corepressor complex. Mol. Cell. Biol. 2002, 22, 2743–2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laherty C. D.; Yang W. M.; Sun J. M.; Davie J. R.; Seto E.; Eisenman R. N. Histone deacetylases associated with the mSin3 corepressor mediate mad transcriptional repression. Cell 1997, 89, 349–356. [DOI] [PubMed] [Google Scholar]
- Nicolas E.; Morales V.; Magnaghi-Jaulin L.; Harel-Bellan A.; Richard-Foy H.; Trouche D. RbAp48 belongs to the histone deacetylase complex that associates with the retinoblastoma protein. J. Biol. Chem. 2000, 275, 9797–9804. [DOI] [PubMed] [Google Scholar]
- Haider S.; Joseph C. G.; Neidle S.; Fierke C. A.; Fuchter M. J. On the function of the internal cavity of histone deacetylase protein 8: R37 is a crucial residue for catalysis. Bioorg. Med. Chem. Lett. 2011, 21, 2129–2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieliauskas A. V.; Pflum M. K. H. Isoform-selective histone deacetylase inhibitors. Chem. Soc. Rev. 2008, 37, 1402–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyaanamoorthy S.; Chen Y. P. Exploring inhibitor release pathways in histone deacetylases using random acceleration molecular dynamics simulations. J. Chem. Inf. Model. 2012, 52, 589–603. [DOI] [PubMed] [Google Scholar]
- Wu R.; Lu Z.; Cao Z.; Zhang Y. Zinc chelation with hydroxamate in histone deacetylases modulated by water access to the linker binding channel. J. Am. Chem. Soc. 2011, 133, 6110–6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northrop J. P.; Ullman K. S.; Crabtree G. R. Characterization of the nuclear and cytoplasmic components of the lymphoid-specific nuclear factor of activated T cells (NF-AT) complex. J. Biol. Chem. 1993, 268, 2917–2923. [PubMed] [Google Scholar]
- Bieliauskas A.; Weerasinghe S.; Pflum M. H. Structural requirements of HDAC inhibitors: SAHA analogs functionalized adjacent to the hydroxamic acid. Bioorg. Med. Chem. Lett. 2007, 17, 2216–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S. E.; Weerasinghe S. V.; Pflum M. K. The structural requirements of histone deacetylase inhibitors: Suberoylanilide hydroxamic acid analogs modified at the C3 position display isoform selectivity. Bioorg. Med. Chem. Lett. 2011, 21, 6139–6142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S. E.; Pflum M. K. H. The structural requirements of histone deacetylase inhibitors: Suberoylanilide hydroxamic acid analogs modified at the C6 position. Bioorg. Med. Chem. Lett. 2012, 22, 7084–7086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanner M. F. Python: A programming language for software integration and development. J. Mol. Graphics Modell. 1999, 17, 57–61. [PubMed] [Google Scholar]
- Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.