

Abstract
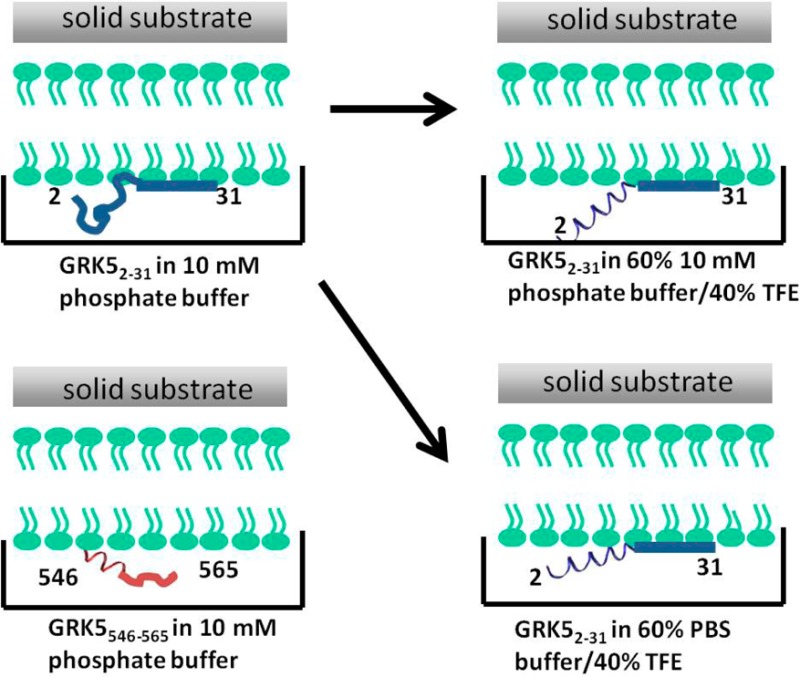
G protein-coupled receptor kinase 5 (GRK5) is thought to associate with membranes in part via N- and C-terminal segments that are typically disordered in available high-resolution crystal structures. Herein we investigate the interactions of these regions with model cell membrane using combined sum frequency generation (SFG) vibrational spectroscopy and attenuated total reflectance–Fourier transform infrared (ATR-FTIR) spectroscopy. It was found that both regions associate with POPC lipid bilayers but adopt different structures when doing so: GRK5 residues 2–31 (GRK52–31) was in random coil whereas GRK5546–565 was partially helical. When the subphase for the GRK52–31 peptide was changed to 40% TFE/60% 10 mM phosphate pH 7.4 buffer, a large change in the SFG amide I signal indicated that GRK52–31 became partially helical. By inspecting the membrane behavior of two different segments of GRK52–31, namely, GRK52–24 and GRK525–31, we found that residues 25–31 are responsible for membrane binding, whereas the helical character is imparted by residues 2–24. With SFG, we deduced that the orientation angle of the helical segment of GRK52–31 is 46 ± 1° relative to the surface normal in 40% TFE/60% 10 mM phosphate pH = 7.4 buffer but increases to 78 ± 11° with higher ionic strength. We also investigated the effect of PIP2 in the model membrane and concluded that the POPC:PIP2 (9:1) lipid bilayer did not change the behavior of either peptide compared to a pure POPC lipid bilayer. With ATR-FTIR, we also found that Ca2+·calmodulin is able to extract both peptides from the POPC lipid bilayer, consistent with the role of this protein in disrupting GRK5 interactions with the plasma membrane in cells.
Introduction
G protein-coupled receptors (GPCRs) are integral membrane proteins that transduce extracellular signals such as light, hormones, and chemoattractants to downstream signal pathways.1 Activated GPCRs are phosphorylated by a family of serine/threonine kinases named G protein-coupled receptor kinases (GRKs), a process that initiates their desensitization. The ability to interact with membranes in which GPCRs are found is essential for GRK function.2 The various GRKs have different ways of associating with lipid bilayers.3 GRK1 and GRK7 are post-translationally modified by prenyl groups at their C-termini. GRK2 and GRK3, on the other hand, bind to membranes by virtue of their C-terminal pleckstrin homology (PH) domains, which bind to acidic phospholipids and interact with heterotrimeric Gβγ subunits, which are prenylated. GRK4, GRK5, and GRK6 constitute a subfamily of GRKs that have 2–3 membrane-binding motifs. The first is a basic segment near the N-terminus that is believed to be responsible for binding phosphatidylinositol-4,5-bisphosphate (PIP2). The second is an amphipathic helix located at the extreme C-terminus that is believed to interact with anionic lipid bilayers. GRK4 and GRK6 are in addition palmitoylated on cysteines immediately C-terminal to this helix. The N-terminal ∼18 amino acids of all GRKs, however, are highly conserved, predicted to have helical propensity, and are essential for phosphorylation of activated GPCRs. Proposed roles for these residues include either direct interaction with activated receptors4 or with the phospholipid bilayer,5,6 either of which is proposed to induce helical character in this region and promote the formation of an activated form of the kinase domain.
Over the past several years, crystallographic studies have yielded new insights into the molecular mechanism for regulation of GRKs by their interactions with receptors and membranes.4 However, crystallographic analysis requires the removal of protein complexes from their native membrane environment and cannot provide direct information on how these molecules are arranged on the membrane surface in situ. Sum frequency generation (SFG) vibrational spectroscopy is a powerful tool to examine peptides and proteins at biointerfaces7−14 such as lipid bilayers.15−21 For example, orientations of peptides with different secondary structures, such as linear α-helices,22,23 bent α-helices,24,25 β-sheets,26 and 310-helices27 associated with solid substrate supported lipid bilayers have been deduced using polarized SFG studies. SFG has also been applied to investigate the membrane orientations of Gβγ, the Gβγ–GRK2 complex, and Gαβγ heterotrimers in situ.28,29 Recently, we showed that the use of both SFG and attenuated total reflectance–Fourier transform infrared (ATR-FTIR) spectroscopy can determine orientations of complex proteins with greater certainty.30 In this research, we used SFG and ATR-FTIR to study the membrane interactions of the N- and C-terminal segments of GRK5 to gain insight into which regions were most important for membrane binding and what structure and orientation they adopt while interacting with membranes.
GRK5 residues 2–31 (GRK52–31) are highly conserved in the GRK4 subfamily of GRKs (Figure 1), which includes GRK4, GRK5, and GRK6. In previous research, it was suggested that residues 22–29, which include basic amino acids Lys22, Arg23, Lys24, Lys26, Lys28, and Lys29, bind to PIP2.31 An overlapping region (residues 20–39) has also been implicated in binding to calmodulin·Ca2+ (CaM·Ca2+).32 The structure of GRK6 (a close homologue of GRK5) determined by X-ray crystallography suggests that the N-terminal portion of the peptide (residues 2–23) is disordered when the enzyme is in an inactive state,33 but residues 2–18 become ordered when the enzyme assumes a more active, presumably receptor-bound conformation.4 However, it is not known if this region forms a platform for binding to lipid membranes or activated GPCRs. Therefore, elucidating the ability of different segments of the GRK5 N-terminus to interact with the membrane is key to understanding how the membrane influences GRK5 function.
Figure 1.

Sequences of the human GRK5 N-terminal and C-terminal peptides used in this study. Residues highlighted in gray adopt an α-helical conformation in the structure of the GRK6·sangivamycin complex.5
The C-terminal residues 552–562 of GRK5 are believed to be another region that interacts with phospholipids. Deletion of these residues results in a 100-fold loss in membrane binding affinity.34 Residues 549–557 are predicted to form an amphipathic helix when bound to membranes.35 In the active conformation of the GRK6 crystal structure, residues 548–557 form an amphipathic helix that docks to the core of the enzyme but is far removed from the predicted membrane surface and the N-terminal segment believed to bind PIP2.4 Thus, either this structural element does not bind to membranes, or it only binds to membranes when GRK6 is in a more inactive state, or the structure represents a soluble form of the enzyme, such as when it translocates to the nucleus to phosphorylate transcription factors.36
By combining data from two complementary optical spectroscopic techniques, SFG and ATR-FTIR, we are seeking to answer the following questions. First, do peptides representing the N-terminal (GRK52–31) and C-terminal (GRK5546–565) regions bind to membranes on their own, and, if so, what structure do they adopt? Second, does PIP2 affect the binding properties of these two peptides? Finally, is CaM·Ca2+ able to dissociate these GRK5 peptides from the membrane, as proposed to be required for nuclear translocation?
Experimental Section
Materials
Peptides GRK52–31, GRK52–24, and GRK5546–565 (Figure 1) were synthesized by the following procedure. Protected amino acids and N-methylpyrrolidone (NMP), 1-hydroxybenzotriazole (HOBt), and O-benzotriazole-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU) were purchased from Creosalus. Acetonitrile, HPLC grade water, trifluoroacetic acid (TFA), diethyl ether, and phenol were from Fisher Scientific. Piperidine, N,N-diisopropylethylamine (DIPEA), dimethylformamide (DMF), thioanisole, triisopropylsilane (TIPS), and calmodulin were from Sigma-Aldrich. Solid-phase synthesis resin NovaPEG Rink Amide Resin (0.5 mmol/g) was purchased from Novabiochem. Analytical HPLC analysis was done using an Alliance system with 250 × 5 mm C18 3 μm column (Vydac). Mass spectrometry analysis was done using a 6130 Quadrupole LC/MS (Agilent Technologies). Semipreparative HPLC purification was performed using a Delta 600 system (Waters) with 150 × 19 mm XBridge Prep C18 10 μm OBD column (Waters). HPLC analysis and purification were done using solvent system 0.1% TFA in water and 0.1% TFA in acetonitrile. Peptides GRK52–24 and GRK52–31 were synthesized using 9-fluorenylmethoxycarbonyl (FMOC) chemistry. The syntheses of C-terminal sequences up to Ala15 were carried out on a CS336X automated synthesizer (C.S. Bio Co.), and the syntheses were then continued on a Discover SPS single mode manual microwave synthesizer (CEM Corp.) (power = 20 W, 5 min per coupling and power 20 W, 1.5 min per deprotection; temperature 70–75 °C). The synthesis scale was 0.2 mmol. The general protocol included double coupling and double deprotection as well as acetylation of the unreacted amino groups. Coupling cycles were performed using 4 equiv of incoming amino acid, HOBt/HBTU in DMF and DIPEA in NMP. Fmoc deprotection was accomplished using 20% piperidine solution in NMP. Cleavage of the peptide from the resin and side-chain deprotection was performed using 10 mL of the mixture DI water:phenol:thioanisole:TIPS:TFA (0.5 mL:0.7 g:0.5 mL:0.25 mL:8.75 mL). The reaction was left running at room temperature for 2 h. After filtration of the resin, crude peptide was precipitated with cold ethyl ether. The resulting crude peptides were purified by semipreparative HPLC, as described above. The purity of the final peptide was analyzed using HPLC and molecular weight confirmed by MS. Peptide GRK525–31 was synthesized by Peptide 2.0 Inc. by a similar approach. POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine) and PIP2 were purchased from Avanti Polar Lipids.
Bilayer Construction
Supported POPC/POPC lipid bilayers were constructed on CaF2 prisms by the Langmuir–Blodgett/Langmuir–Schaefer method, as reported in detail previously.37,38 The first POPC layer is deposited on one of the square faces of the right-angle CaF2 prism with A KSV2000 LB system: The plasma-cleaned prism was first immersed in the LB trough. Then a certain amount of POPC chloroform solution, typically 5 drops of 10 mg/mL, was spread on the water surface until the surface tension reaches ∼5 mN/m. Two barrier arms were suppressed so that the surface tension remained 34 mN/m, while the CaF2 prism was lifted from the subphase. A layer of POPC lipids was deposited on the face perpendicular to the water surface in this way. After aligning the laser beams to find the monolayer signal, a 2 mL reservoir filled with water was placed beneath the prism. POPC lipids were added to the surface of the water in the reservoir so that the surface tension was around 34 mN/m. The reservoir was elevated so that the the lipid monolayer on the water surface contacted with the first layer deposited on the prism to form a lipid bilayer.
SFG Experiments
SFG theory,39−43 our experimental design, and data analysis method23,44 have been reported before. The concentration of each of the four peptides was 3.8 μM, and the peptides were dissolved in 10 mM potassium phosphate buffer (pH 7.4). Because CaF2 prisms were used as substrates to prepare the lipid bilayers, small amounts of Ca2+ may be dissolved in the subphase. 2 mM EDTA was added to the above buffer solution to minimize any influence of the Ca2+ released from the CaF2 substrates. For each of the three N-terminal peptides studied here, we added the peptide into the subphase in contact with the substrate supported bilayer and after equilibration recorded the SFG signal in the water O–H stretching frequency range as well as in the peptide amide I frequency region. For all peptides we studied, the adsorption time on the POPC lipid bilayer in either 10 mM phosphate buffer or PBS buffer was less than 200 s (see the time-dependent SFG signals detected in the CH and OH stretching frequency range in the Supporting Information). For the second step, we substituted the peptide solution subphase with potassium phosphate buffer (∼6 mL in total) to wash off loosely associated peptides and recorded the SFG signal in the water O–H stretching frequency range again. For the last step, we substituted the phosphate buffer subphase with a solution of buffer containing 40% TFE and again collected SFG spectra in the water O–H stretching frequency range and the amide I frequency range. For the C-terminal peptide, we only performed the first two steps of the above procedure. PIP2 experiments were performed in the same way as POPC experiments except that when making bilayers, lipids with a 9:1 molar ratio of POPC:PIP2 were used. Because peptides were used at the same concentration in these experiments yet likely have different affinities and because water signals are also strongly affected by net charge as well as charge distribution in the peptides (and other effects), we defined peptides as weakly membrane associated if the water signal recovered after the buffer wash, as opposed to direct comparison of changes in the SFG signals from water O–H stretching after addition of peptide.
ATR-FTIR Spectroscopy
ATR-FTIR experiments were performed on a Nicolet Magna 550 FTIR spectrometer. Lipid bilayers were deposited on a ZnSe crystal (Specac Ltd. RI, U.K.) using the vesicle fusion method. 1 mL of POPC toluene solution (5 mg/mL) was dried with nitrogen flow and then in vacuum for 2 h. The POPC power was then dissolved in 10 mM phosphate D2O buffer pH 7.4, and the mixture was vortexed for 5 min before addition to the surface of the detachable ZnSe crystal to form bilayers. After 30 min, the vesicles floating in the subphase were washed away using excess buffer. GRK5 peptides were then injected into the subphase (1.6 mL) to achieve a concentration of 11.4 μM. After the system reached equilibrium, spectra before and after extensive wash with D2O buffer were recorded. For GRK5546–565, s- and p-polarized spectra were taken so that data analysis on the peptide orientation could be performed. In the CaM·Ca2+ experiments, after peptides were associated with the lipid bilayers equimolar CaM (11.4 μM) and 50 μM CaCl2 solution were added to the subphase.
Results
SFG Studies on N-Terminal Peptides
We first investigated molecular interactions between the GRK5 N-terminal peptides GRK52–31, GRK52–24, and GRK525–31 and a POPC/POPC lipid bilayer. The POPC/POPC bilayer is zwitterionic, and the electrostatic potential across the bilayer induces the water dipoles to orient near the bilayer surface.45,46 The water region (detected between 2700 and 3700 cm–1)47,48 monitored by SFG spectroscopy can be used to determine the binding properties of ions49,50 or peptides.25 In our experiments, we observed two broad water O–H stretching peaks centered at ∼3200 and 3400 cm–1 in the SFG spectrum from the lipid bilayer/potassium phosphate buffer interface (Figure 2a). Peptides were then added into the subphase, and the system was allowed to reach equilibrium. The water OH stretching signal decreased upon addition of the GRK52–31 or GRK525–31 peptides to the subphase, consistent with their interaction with the POPC/POPC bilayers (Figures 2a and 3a). SFG spectra were also collected after extensive washing, but no substantial changes were observed, suggesting that both GRK52–31 and GRK525–31 peptides are strongly associated with the bilayer. However, the SFG water O–H stretching signal only decreased slightly after the addition of the GRK52–24 peptide to the subphase, and the SFG water signal recovered after washing the interface with buffer (Figure 3c), consistent with GRK52–24 only being weakly associated with the POPC/POPC bilayer. Thus, the highly charged residues spanning residues 25–31 are primarily responsible for membrane binding in these peptides.
Figure 2.
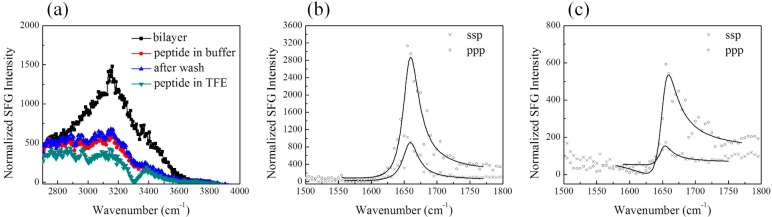
SFG signals from GRK52–31 indicate strong association with model membranes and helical character in a more hydrophobic environment. (a) Spectra in the C–H and O–H stretching frequency region detected from the interface between the POPC/POPC bilayer and buffer alone (black), GRK52–31 in 10 mM phosphate buffer pH 7.4 (red), after washing (blue), and in a mixture of 60% buffer/40% TFE (dark cyan). (b) SFG spectra of GRK52–31 associated with a POPC/POPC bilayer in contact with peptide solution in 60% 10 mM phosphate buffer pH 7.4/40% TFE in the amide I frequency region. (c) SFG spectra of GRK52–31 associated with a POPC/POPC bilayer in contact with 60% PBS/40% TFE.
Figure 3.

SFG ppp signals detected from the GRK525–31 and GRK52–24 peptides indicate that the latter peptide only weakly associates with model membranes. (a) SFG spectra in C–H and O–H stretching frequency region from the interface between the POPC/POPC bilayer and buffer alone (black), GRK525–31 in 10 mM phosphate buffer pH 7.4 (red), after washing (blue), and in a mixture of 60% buffer/40% TFE (dark cyan). (b) SFG spectra in the amide I frequency region from GRK525–31 associated with a POPC/POPC bilayer in 60% 10 mM phosphate buffer pH 7.4/40% TFE. (c) SFG spectra in C–H and O–H stretching frequency region from the interface between the POPC/POPC bilayer and buffer alone (black), GRK52–24 in 10 mM phosphate buffer pH 7.4 (red), after washing (blue), and in a mixture of 60% buffer/40% TFE (dark cyan). (d) SFG spectra in the amide I frequency region from GRK52–24 associated with a POPC/POPC bilayer in contact with peptide solution in 60% 10 mM phosphate buffer pH 7.4/40% TFE.
For all three GRK5 N-terminal peptides, no discernible SFG amide I signal could be detected from the lipid bilayer interfaces after their addition. This suggests that the membrane associated peptides form either ordered structures but with random orientations or essentially random structures. After replacing the subphase with a 40% TFE solution, a strong SFG amide I signal was detected from the GRK52–31 peptide (Figure 2b), but not from GRK525–31 or GRK52–24 (Figures 3b and 3d), consistent with only GRK52–31 forming α-helical structure when the subphase becomes more hydrophobic. This conclusion is also consistent with spectral features detected in the water O–H stretching frequency range after the subphase buffer was replaced by the TFE mixture. Figure 2a shows that only for GRK52–31, a negative peak at ∼3300 cm –1 appeared, originating from the interference between the N–H stretching signals of well-ordered α-helices and the broad water background. This N–H stretch signal can be attributed to the backbone N–H stretch or/and the side chains such as Lys NH3+.51 Although the predicted helical propensity of GRK52–24 is the same as that of GRK52–31, no changes in the spectra upon addition of TFE were detected likely because the peptide was not strongly associated with the membrane and washed off in the previous step.
Orientation Analysis of the α-Helical Segment in GRK52–31
After substituting the subphase with 40% TFE, a prominent α-helical signal centered at ∼1655 cm–1 arises from GRK52–31. This could be interpreted as residues 2–18 adopting an α-helical conformation, consistent with a prior crystal structure of GRK64 and secondary structure predictions. This phenomenon also highlights that SFG, as a second-order nonlinear spectroscopy, is much more sensitive to ordered structure (such as α-helices) than disordered molecules (such as random coils), which is not the case for linear vibrational spectroscopy such as ATR-FTIR.
SFG spectra collected from amide I modes of peptides and proteins using different polarization combinations can be used to determine membrane orientations of peptides and proteins, as shown in a previous publication.22 Using the measured signal strength ratio of the α-helical contribution in the ppp and ssp spectra, we deduced that in 40% TFE, the orientation angle of the helical segment (presumed to be residues 2–18) of GRK52–31 is ∼46 ± 1° relative to the membrane surface normal (with χppp/χssp = 2.08 ± 0.01) if we assume the peptides adopt a single orientation distribution. Interestingly, this orientation angle increases to ∼78 ± 11° (with χppp/χssp = 2.43 ± 0.06) when the ionic strength of the subphase is increased by use of PBS instead of phosphate buffer (Figure 2c). Details of the orientation analysis can be found in the Supporting Information. This result suggests that the increase in ionic strength does not change the conformation of the GRK52–31 but rather changes the charge distribution on the peptide surface and thus facilitates the interaction of helical elements of the peptide with the lipid head groups.
SFG Studies on the C-Terminal Peptide
The SFG spectrum of GRK5546–565 (Figure 4) is similar to that of GRK52–31, in that the two broad peaks at 3200 and 3400 cm–1 decreased and remained so even after extensive washing, indicating strong interaction of GRK5546–565 with the lipid bilayer. However, two new peaks centered at 2876 and 2940 cm–1 appeared. These were also observed for GRK52–31 but were not as significant. These two peaks could be attributed to amino acid side chains,51 disruption of the lipid bilayer,52 or both. The SFG amide I spectra of GRK5546–565, however, is very different from those of the N-terminal peptides. Without changing the subphase into 40% TFE, an amide I signal was readily detected. In the spectra, the peak at 1655 cm–1 is attributed to α-helical structure and the shoulder at ∼1600 cm–1 is likely from amide groups of side chains.53 The peak at 1720 cm–1 is from carbonyl groups in the disrupted lipid bilayer. This agrees with the CH stretching signal change mentioned above, supporting the hypothesis that the lipid bilayer is disrupted. Because the intensity is not as high as that of GRK52–31 in 40% TFE with 10 mM phosphate buffer, no discernible NH peak (∼3300 cm–1) in the water range (3000–4000 cm–1) was detected. Orientation analysis was not performed here due to the low signal-to-noise ratio of the SFG spectra and because there are multiple contributions to the spectra. In summary, the main difference between GRK5546–565 and GRK52–31 is that the former is partially α-helical when associated with lipid bilayers without need for TFE to induce helical structure.
Figure 4.
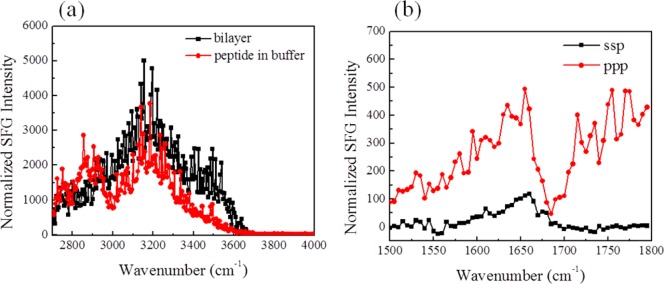
SFG ppp signals detected from GRK5546–565 indicate strong binding to model membranes and helical character. (a) SFG spectra in the C–H and O–H stretching frequency region from the interface between the POPC/POPC bilayer and buffer alone (black) and GRK5546–565 associated in 10 mM phosphate buffer pH 7.4 (red). (b) SFG spectra in the amide I frequency region from GRK5546–565 associated with a POPC/POPC bilayer in 10 mM phosphate buffer pH 7.4.
SFG Studies on the Effect of PIP2
PIP2 is known to enhance the GRK5-mediated phosphorylation of GPCRs.31 In order to test whether this enhancement is related to the membrane binding of the peptides we are studying herein, we constructed (9:1) POPC:PIP2 lipid bilayers and studied its interaction with GRK52–31, GRK52–24, and GRK5546–565. These results (see Supporting Information Figure S2) were then compared to those obtained when using a pure POPC lipid bilayer. The SFG intensities and signal strength ratios of the amide I signals detected in the amide I frequency range using different polarization combinations of the GRK52–31 associated with the two types of bilayers exposed to the solution with 40% TFE were observed to be similar, indicating that PIP2 did not enhance the adsorption of GRK52–31 to the lipid bilayer. The interactions of GRK52–24 and GRK5546–565 with (9:1) POPC:PIP2 bilayers were also similar to those with the pure POPC system. This is reminiscent of protein MARCKS: neither the native protein nor a peptide representing its positive charged cluster requires PIP2 for binding to the membrane. However, PIP2 is laterally sequestered in the presence of MARCKS and the peptide.54 How PIP2 can increase the autophosphorylation of GRK5 and phosphorylation of activated GPCRs calls for further investigation. However, it should be noted that residues 24–31 are well ordered in both available crystal structures of GRK64,33 and that formation of a high affinity site for PIP2 may require the assumption of tertiary structure by this polypeptide, as mandated by the fold of the enzyme. The study on the effect of PIP2 suggests that the conclusions on peptide affinity drawn from Figure 2–4 do not require the existence of PIP2.
ATR-FTIR Studies
Because SFG is sensitive to ordered structures, the signals generated from ordered α-helices are normally much stronger than those detected from random coil. On the other hand, ATR-FTIR spectroscopy detects amide I (1600–1700 cm–1) signals with similar sensitivities from different secondary structural motifs, such as α-helices, random coils, and β-sheets from peptides and proteins.55−59 We used ATR-FTIR spectra to confirm the peptide adsorption behavior detected by SFG. For the ATR-FTIR experiments, the concentrations of all peptides used were 11.4 μM in 10 mM phosphate D2O buffer (pD 7.1). For all the N-terminal peptides, the amide I peak center is around 1642 cm–1 (Figure 5), indicating that the peptides are most likely random coils. For GRK52–31 and GRK525–31, the amide I peak intensities did not change after washing with buffer, but for GRK52–24 the amide I signal decreased to about half, again suggesting a weaker interaction between GRK52–24 and the lipid bilayer, as suggested by the SFG studies. The reason that membrane associated GRK52–24 did not completely disappear after washing, as observed in SFG, is likely because the peptide concentration is 3 times higher than that used in SFG measurements.
Figure 5.

ATR-FTIR spectra of GRK5 N-terminal peptides confirm weak binding of GRK52–24. Spectra of (a) GRK52–31, (b) GRK52–24, and (c) GKR525–31 associated with a POPC/POPC lipid bilayer in the presence of 10 mM phosphate buffer pH 7.4 before (black) and after (red) buffer wash.
By taking secondary derivatives of the ATR-FTIR spectra for GRK5546–565 (Figure 6), we found two peaks centered at 1646 and 1653 cm–1, respectively. The average band position in D2O is reported to be ∼1652 cm–1 for α-helix and ∼1645 cm–1 for disordered secondary structure.57 Therefore, the peak centered at 1646 cm–1 is attributed by random coil and the other at 1653 cm–1 is attributed to α-helices, consistent with SFG results indicating that the GRK5 C-terminal peptide forms an α-helical structure. After extensive washing, the ATR-FTIR signal remained, suggesting a strong interaction with the lipid bilayer, also compatible with the SFG data.
Figure 6.

ATR-FTIR spectra of GRK5546–565 associated with a POPC/POPC lipid bilayer in contact with 10 mM phosphate buffer pH 7.4.
ATR-FTIR Studies of CaM·Ca2+ Interactions with N-Terminal and C-Terminal Peptides
ATR-FTIR was further used to investigate the molecular interactions of GRK52–31 with calmodulin. CaM·Ca2+ itself has very weak binding with the membrane (Supporting Information). As shown in Figure 7a, the addition of equimolar amounts of CaM·Ca2+ and GRK52–31 to the subphase decreased the ATR-FTIR amide I signal by about 50%. Further extensive washing with buffer led to a more substantial decline of the random coil ATR-FTIR signal. This clearly shows that CaM·Ca2+ facilitates the extraction of GRK52–31 from the lipid bilayer. However, CaM·Ca2+ could not extract GRK525–31 from the membrane (Supporting Information), suggesting that the helix formed by residues 2–24 is important for high affinity binding to CaM·Ca2+.60 CaM·Ca2+ also was able to extract GRK5546–565 from our model membranes (Figure 7b). The initial increase in the signal after addition of CaM·Ca2+ to GRK5546–565 was unexpected. However, this may simply reflect that when CaM·Ca2+ forms a complex with this peptide, it remains associated with the membrane to a greater extent than when in complex with the GRK52–31 peptide. Notably, in either case, the subsequent buffer wash eliminates binding, indicating weak binding.
Figure 7.
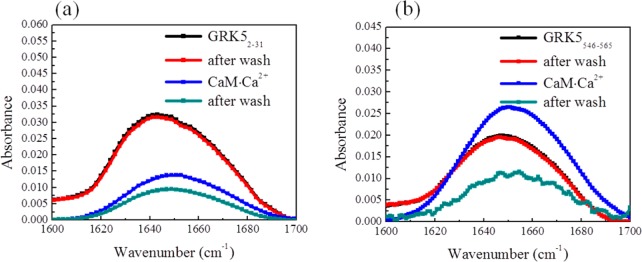
CaM·Ca2+ decreases the association of GRK5 N and C-terminal peptides. ATR-FTIR signals detected before and after the addition of equimolar CaM·Ca2+ to the subphase for peptides (a) GRK52–31 and (b) GRK5546–565. The spectra correspond to before (black), and after (red) washing, to the addition of CaM·Ca2+ to the subphase (blue), and after subsequent washing (dark cyan).
Discussion
Our study is a clear example of how SFG and ATR-FTIR spectroscopies complement each other as methods for interrogating the structure of proteins/peptides at membrane surfaces. Because SFG is a second-order nonlinear optical technique, under the electric dipole approximation, it only detects signal where inversion symmetry is broken. Thus, SFG can minimize the interfering effects of proteins in bulk solution. For example, in our studies, we measured well-defined amide signals using SFG from the GRK52–31 peptide associated with lipid bilayers in contact with solutions with 40% TFE, which generates a high background signal in ATR-FTIR spectroscopy (Supporting Information). Another advantage of SFG is that measurements do not require D2O, which is used in ATR-FTIR to minimize interference by H2O absorption at ∼1650 cm–1. SFG is also more sensitive to ordered secondary structures than disordered ones. We observed a drastic change of the amide I signal of GRK52–31 when its subphase was altered to contain 40% TFE. This change was more subtle in the ATR-FTIR spectra because random coils and α-helices have peak centers close to each other (∼1647 and ∼1653 cm–1, respectively) and usually make similar contributions to the spectra. For large proteins (e.g., GRK5), sometimes the switch from the active state to the inactive state is accompanied by conformational changes. The unique ability of SFG to distinguish random coils from α-helices might shed light on the mechanisms of these processes, which may not be easily distinguishable using ATR-FTIR spectra. On the other hand, ATR-FTIR can directly monitor the adsorption of unstructured peptides and proteins simply by inspecting the amide I signals. Because unstructured domains (e.g., random coils) cannot be readily detected by SFG spectroscopy, the adsorption of such molecules cannot be directly assessed using the SFG amide I signal. However, this goal can be achieved indirectly by monitoring the ordered water signal change in SFG spectra.
Table 1. Fitting Results for SFG Spectra Shown in Figure 2a.
| subphase | polarization | peak center (cm–1) | peak width (cm–1) | χeff | ratio | tilt angle (deg) |
|---|---|---|---|---|---|---|
| 60% phosphate buffer/40% TFE | ssp | 1657 | 14.2 | 14.8 | 2.09 ± 0.01 | 46 ± 1 |
| ppp | 1657 | 14.7 | 51.1 | |||
| PBS buffer/40% TFE | ssp | 1650 | 11.0 | 8.8 | 2.43 ± 0.06 | 78 ± 11 |
| ppp | 1652 | 14.0 | 17.6 |
Errors represent standard deviations of four replicates obtained in each of two individual experiments.
In this work we combined SFG and ATR-FTIR spectroscopies to study the in situ membrane binding potential of two regions of GRK5 previously implicated in binding to phospholipid bilayers. The uniform orientation of water molecules near the bilayer surface was exploited first, as the disappearance of the SFG water signal suggests their displacement by peptide molecules. Whether or not the water signal would resume after washing the system with buffer was used to determine if the peptide molecules are weakly or strongly adsorbed. It was shown that of the three N-terminal peptides, only GRK52–24 binds weakly to the lipid bilayer, suggesting that GRK52–24 alone does not play a significant role in GRK5 membrane binding and that residues 25–31 of the GRK52–31 peptide, which are exceptionally basic and include a tryptophan residue, are primarily responsible for membrane binding in this region. This conclusion is also supported by monitoring the changes in the amide I signal from the peptides before and after washing with buffer using ATR-FTIR. From the amide I SFG signals we found that the segment containing amino acid residues 2–24 of peptide GRK52–31 undergoes a conformational change from a random coil into a well-ordered α-helix when the hydrophobicity of the environment increases (in our experiment by substituting the buffer subphase with a solution containing 40% TFE). It is possible that TFE emulates what happens when this region encounters either an activated GPCR or its own activated kinase domain. Furthermore, the interaction of this region, or of an adjacent region (i.e., residues 25–31), with membranes is not enough to induce order in this segment. Our results are consistent with those reported previously that residues 2–18 play an important role in protein–protein interactions, such as those with activated GPCRs or with the catalytic core of the enzyme to stabilize a more active state.4,2 Polarization-dependent SFG measurements were used to determine the angle of the helical segment of this peptide to the surface normal. This angle was found to increase substantially upon an increase in ionic strength of the surrounding buffer solution. With a similar approach, both SFG and ATR-FTIR results showed that GRK5546–565 was partially helical on POPC lipid bilayers, even in the absence of a helix-inducing agent such as TFE. A model summarizing the membrane interactions of the peptides is shown in Figure 8.
Figure 8.

Schematic showing proposed membrane interaction mechanisms of the GRK5 N-terminal peptide GRK52–31 and the GRK5 C-terminal peptide GRK5546–565.
Therefore, both N- and C-terminal peptide segments of GRK5 contribute to bilayer binding and likely account for the constitutive localization of GRK5 on cell membranes, even though it lacks the palmitoylation found in the closely related enzymes GRK4 and GRK6. Both residues 25–31 and 546–565 bind strongly to membranes, as evidenced by their persistence even after exhaustive washing. However, residues 2–24 at the extreme N-terminus do not represent a strong membrane binding determinant. Instead, our results are most consistent with this highly conserved region only becoming ordered when it forms protein–protein interfaces, such as when in complex with an activated GPCR or when it interacts with the small lobe of the GRK kinase domain. Unexpectedly, PIP2 does not affect the binding properties of the peptides we studied. It is possible that the N-terminal peptide does not fully recapitulate the binding site for this lipid because the peptide is unstructured when bound to membranes, as opposed to the analogous peptide in the context of the full-length enzyme, where its structure is imposed by the fold of the enzyme. The membrane interaction mechanisms of the N-terminal and C-terminal peptides are, however, different. Previous biochemical studies showed that the C-terminal peptide likely forms an amphipathic helix that enhances GRK5 membrane binding.35 The analogous C-terminal region has only been observed in one structure of GRK6 in a relatively active state,4 wherein it forms the expected amphipathic helix. However, the hydrophobic residues bind to the catalytic core of the enzyme, and the helix seems too far from the predicted membrane surface to directly engage lipids. As phospholipids are not present in this crystal structure, the C-terminal structure could represent a crystallographic artifact. Alternatively, because the interactions of the C-terminus of GRK6 with the core of the enzyme are extensive (buried accessible surface area of 2400 Å2), the packing of this helix could represent the situation when GRK6 is in a cytoplasmic and/or autoinhibited state. Because PIP2 is believed to bind to the 25–31 region in the N-terminal region and this site is a structured part of the catalytic core in prior crystal structures, this interaction may be more important for achieving proper orientation of the enzyme at the membrane, whereas the C-terminal amphipathic helix, which is connected to the rest of the enzyme by a 21-amino acid linker, is merely important for maintaining its association at the membrane. CaM·Ca2+ is able to dissociate GRK52–31 and GRK5546–565 peptides from the membrane, consistent with the ability of this protein to drive GRK5 off the membrane of cells and consequently to the nucleus, where it is believed to phosphorylate transcription factors controlling hypertrophic genes.36
Acknowledgments
The work is supported by the National Institute of Health grants GM081655 (to Z.C.) and HL071818 (to J.J.G.T.). B.D. acknowledges the financial support of Barbour Fellowship from the University of Michigan. We genuinely thank Dr. Pei Yang for technical support.
Supporting Information Available
SFG experimental setup, PIP2 related spectra, time-dependent spectra, and SFG data analysis. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Neves S. R.; Ram P. T.; Iyengar R. G protein pathways. Science 2002, 296, 1636–1639. [DOI] [PubMed] [Google Scholar]
- Homan K. T.; Glukhova A.; Tesmer J. J. G. Regulation of G protein-coupled receptor kinases by phospholipids. Curr. Med. Chem. 2013, 20, 39–46. [PubMed] [Google Scholar]
- Gurevich E. V.; Tesmer J. J. G.; Mushegian A.; Gurevich V. V. G protein-coupled receptor kinases: more than just kinases and not only for GPCRs. Pharmacol. Therapeut. 2012, 133, 40–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boguth C. A.; Singh P.; Huang C.; Tesmer J. J. G. Molecular basis for activation of G protein-coupled receptor kinases. EMBO J. 2010, 29, 3249–3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble B.; Kallal L. A.; Pausch M. H.; Benovic J. L. Development of a yeast bioassay to characterize G protein-coupled receptor kinases. Identification of an NH2-terminal region essential for receptor phosphorylation. J. Biol. Chem. 2003, 278, 47466–47476. [DOI] [PubMed] [Google Scholar]
- Pao C. S.; Barker B. L.; Benovic J. L. Role of the amino terminus of G protein-coupled receptor kinase 2 in receptor phosphorylation. Biochemistry 2009, 48, 7325–7333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips D. C.; York R. L.; Mermut O.; McCrea K. R.; Ward R. S.; Somorjai G. A. Side chain, chain length, and sequence effects on amphiphilic peptide adsorption at hydrophobic and hydrophilic surfaces studied by sum-frequency generation vibrational spectroscopy and quartz crystal microbalance. J. Phys. Chem. C 2007, 111, 255–261. [Google Scholar]
- York R. L.; Holinga G. J.; Somorjai G. A. An investigation of the influence of chain length on the interfacial ordering of L-lysine and L-proline and their homopeptides at hydrophobic and hydrophilic interfaces studied by sum frequency generation and quartz crystal microbalance. Langmuir 2009, 25, 9369–9374. [DOI] [PubMed] [Google Scholar]
- Weidner T.; Apte J. S.; Gamble L. J.; Castner D. G. Probing the orientation and conformation of α-helix and β-strand model peptides on self-assembled monolayers using sum frequency generation and NEXAFS Spectroscopy. Langmuir 2010, 26, 3433–3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidner T.; Castner D. G. SFG analysis of surface bound proteins: a route towards structure determination. Phys. Chem. Chem. Phys. 2013, 15, 12516–12524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breen N. F.; Weidner T.; Li K.; Castner D. G.; Drobny G. P. Direct observation of phenylalanine orientations in statherin bound to hydroxyapatite surfaces. J. Am. Chem. Soc. 2009, 131, 14148–14189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidner T.; Breen N. F.; Li K.; Drobny G. P.; Castner D. G. Sum frequency generation and solid-state NMR study of the structure, orientation, and dynamics of polystyrene-adsorbed peptides. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 13288–13293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S.-Y.; Lim S.-M.; Albertorio F.; Kim G.; Gurau M. C.; Yang R. D.; Holden M. A.; Cremer P. S. The Vroman effect: a molecular level description of fibrinogen displacement. J. Am. Chem. Soc. 2003, 125, 12782–12786. [DOI] [PubMed] [Google Scholar]
- Campen R. K.; Ngo T. T. M.; Sovago M.; Ruysschaert J. M.; Bonn M. Molecular restructuring of water and lipids upon the interaction of DNA with lipid monolayers. J. Am. Chem. Soc. 2010, 132, 8037–8047. [DOI] [PubMed] [Google Scholar]
- Engel M. F. M.; vandenAkker C. C.; Schleeger M.; Velikov K. P.; Koenderink G. H.; Bonn M. The polyphenol EGCG inhibits amyloid formation less efficiently at phospholipid interfaces than in bulk solution. J. Am. Chem. Soc. 2012, 134, 14781–14788. [DOI] [PubMed] [Google Scholar]
- Tong Y.; Li N.; Liu H.; Ge A.; Osawa M.; Ye S. Mechanistic studies by sum-frequency generation spectroscopy: hydrolysis of a supported phospholipid bilayer by phospholipase A2. Angew. Chem., Int. Ed. 2010, 49, 2319–2323. [DOI] [PubMed] [Google Scholar]
- Fu L.; Liu J.; Yan E. C. Y. Chiral sum frequency generation spectroscopy for characterizing protein secondary structures at interfaces. J. Am. Chem. Soc. 2011, 133, 8094–8097. [DOI] [PubMed] [Google Scholar]
- Fu L.; Ma G.; Yan E. C. Y. In situ misfolding of human islet amyloid polypeptide at interfaces probed by vibrational sum frequency generation. J. Am. Chem. Soc. 2010, 132, 5405–5412. [DOI] [PubMed] [Google Scholar]
- Diaz A. J.; Albertorio F.; Daniel S.; Cremer P. S. Double cushions preserve transmembrane protein mobility in supported bilayer systems. Langmuir 2008, 24, 6820–6826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkov V.; Bonn M. Structural Properties of gp41 Fusion Peptide at a Model Membrane Interface. J. Phys. Chem. B 2013, 117, 15527–15535. [DOI] [PubMed] [Google Scholar]
- Roeters S. J.; Dijk C. N.; van Torres-Knoop A.; Backus E. H. G.; Campen R. K.; Bonn M.; Woutersen S. Determining in situ protein conformation and orientation from the amide-I sum-frequency generation spectrum: theory and experiment. J. Phys. Chem. A 2013, 117, 6311–6322. [DOI] [PubMed] [Google Scholar]
- Nguyen K. T.; Le Clair S. V.; Ye S.; Chen Z. In situ molecular level studies on membrane related peptides and proteins in real time using sum frequency generation vibrational spectroscopy. J. Phys. Chem. B 2009, 113, 12169–12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding B.; Chen Z. Molecular interactions between cell penetrating peptide Pep-1 and model cell membranes. J. Phys. Chem. B 2012, 116, 2545–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Wang J.; Boughton A. P.; Kristalyn C. B.; Chen Z. Multiple orientation of melittin inside a single lipid bilayer determined by combined vibrational spectroscopic studies. J. Am. Chem. Soc. 2007, 129, 1420–1427. [DOI] [PubMed] [Google Scholar]
- Ding B.; Soblosky L.; Nguyen K.; Geng J.; Yu X.; Ramamoorthy A.; Chen Z. Physiologically-relevant modes of membrane interactions by the human antimicrobial peptide, LL-37, revealed by SFG experiments. Sci. Rep. 2013, 3, 1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao D.; Fu L.; Liu J.; Batista V. S.; Yan E. C. Y. Amphiphilic adsorption of human islet amyloid polypeptide aggregates to lipid/aqueous interfaces. J. Mol. Biol. 2012, 421, 537–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye S.; Nguyen K. T.; Chen Z. Interactions of alamethicin with model cell membranes investigated using sum frequency generation vibrational spectroscopy in real time in situ. J. Phys. Chem. B 2010, 114, 3334–3340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boughton A. P.; Yang P.; Tesmer V. M.; Ding B.; Tesmer J. J. G.; Chen Z. Membrane Orientation of G Gαiβ1γ2 and Gβ1γ2 determined via combined vibrational spectroscopic studies. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, E667–E673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Boughton A. P.; Tesmer J. J. G.; Chen Z. In situ investigation of heterotrimeric G protein γ subunit binding and orientation on membrane bilayers. J. Am. Chem. Soc. 2007, 129, 12658–12659. [DOI] [PubMed] [Google Scholar]
- Yang P.; Boughton A.; Homan K. T.; Tesmer J. J. G.; Chen Z. Membrane orientation of G Gαiβ1γ2 and Gβ1γ2 determined via combined vibrational spectroscopic studies. J. Am. Chem. Soc. 2013, 135, 5044–5051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitcher J. A.; Fredericks Z. L.; Stone W. C.; Premont R. T.; Stoffel R. H.; Koch W. J.; Lefkowitz R. J. Phosphatidylinositol 4,5-bisphosphate (PIP2)-enhanced G protein-coupled receptor kinase (GRK) activity. J. Biol. Chem. 1996, 271, 24907–24913. [DOI] [PubMed] [Google Scholar]
- Pronin A. N.; Satpaev D. K.; Slepak V. Z.; Benovic J. L. Regulation of G protein-coupled receptor kinases by calmodulin and localization of the calmodulin binding domain. J. Biol. Chem. 1997, 272, 18273–18280. [DOI] [PubMed] [Google Scholar]
- Lodowski D. T.; Tesmer V. M.; Benovic J. L.; Tesmer J. J. G. The structure of G protein-coupled receptor kinase (GRK)-6 defines a second lineage of GRKs. J. Biol. Chem. 2006, 281, 16785–16793. [DOI] [PubMed] [Google Scholar]
- Pronin A. N.; Carman C. V.; Benovic J. L. Structure-function analysis of G protein-coupled receptor kinase-5. Biochemistry 1998, 273, 31510–31518. [DOI] [PubMed] [Google Scholar]
- Thiyagarajan M. M.; Stracquatanio R. P.; Pronin A. N.; Evanko D. S.; Benovic J. L.; Wedegaertner P. B. A predicted amphipathic helix mediates plasma membrane localization of GRK5. J. Biol. Chem. 2004, 279, 17989–17995. [DOI] [PubMed] [Google Scholar]
- Gold J. I.; Martini J. S.; Hullmann J.; Gao E.; Chuprun J. K.; Lee L.; Tilley D. G.; Rabinowitz J. E.; Bossuyt J.; Bers D. M.; Koch W. J. Nuclear translocation of cardiac G protein-Coupled Receptor kinase 5 downstream of select Gq-activating hypertrophic ligands is a calmodulin-dependent process. PLoS One 2013, 8, e57324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye S.; Nguyen K. T.; Le Clair S. V.; Chen Z. In situ molecular level studies on membrane related peptides and proteins in real time using sum frequency generation vibrational spectroscopy. J. Struct. Biol. 2009, 168, 61–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Conboy J. C. Direct measurement of the transbilayer movement of phospholipids by sum-frequency vibrational spectroscopy. J. Am. Chem. Soc. 2004, 126, 8376–8377. [DOI] [PubMed] [Google Scholar]
- Gan W.; Wu D.; Zhang Z.; Feng R.; Wang H. Polarization and experimental configuration analyses of sum frequency generation vibrational spectra, structure, and orientational motion of the air/water interface. J. Chem. Phys. 2006, 124, 114705. [DOI] [PubMed] [Google Scholar]
- Chen Z.; Shen Y. R.; Somorjai G. A. Studies of polymer surfaces by sum frequency generation vibrational spectroscopy. Annu. Rev. Phys. Chem. 2002, 53, 437–465. [DOI] [PubMed] [Google Scholar]
- Zhuang X.; Miranda P.; Kim D.; Shen Y. Mapping molecular orientation and conformation at interfaces by surface nonlinear optics. Phys. Rev. B 1999, 59, 12632–12640. [Google Scholar]
- Moad A. J.; Simpson G. J. A unified treatment of selection rules and symmetry relations for sum-frequency and second harmonic spectroscopies. J. Phys. Chem. B 2004, 108, 3548–3562. [Google Scholar]
- Moad A. J.; Moad C. W.; Perry J. M.; Wampler R. D.; Goeken G. S.; Begue N. J.; Shen T.; Heiland R.; Simpson G. J. NLOPredict: Visualization and Data Analysis Software for Nonlinear Optics. J. Comput. Chem. 2007, 28, 1996–2002. [DOI] [PubMed] [Google Scholar]
- Nguyen K. T.; Le Clair S. V.; Ye S.; Chen Z. Molecular interactions between magainin 2 and model membranes in situ. J. Phys. Chem. B 2009, 113, 12358–12363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böckmann R. A.; Hac A.; Heimburg T.; Grubmüller H. Effect of sodium chloride on a lipid bilayer. Biophys. J. 2003, 85, 1647–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurtovenko A. A.; Vattulainen I. Membrane potential and electrostatics of phospholipid bilayers with asymmetric transmembrane distribution of anionic lipids. J. Phys. Chem. B 2008, 112, 1953–1962. [DOI] [PubMed] [Google Scholar]
- Jena K. C.; Hore D. K. Variation of ionic strength reveals the interfacial water structure at a charged mineral surface. J. Phys. Chem. C 2009, 113, 15364–15372. [Google Scholar]
- Jena K. C.; Hore D. K. Water structure at solid surfaces and its implications for biomolecule adsorption. Phys. Chem. Chem. Phys. 2010, 12, 14383–14404. [DOI] [PubMed] [Google Scholar]
- Okur H. I.; Kherb J.; Cremer P. S. Cations bind only weakly to amides in aqueous solutions. J. Am. Chem. Soc. 2013, 135, 5062–5067. [DOI] [PubMed] [Google Scholar]
- Yang Z.; Li Q.; Chou K. C. Structures of water molecules at the interfaces of aqueous salt solutions and silica: cation effects. J. Phys. Chem. C 2009, 8201–8205. [Google Scholar]
- Mermut O.; Phillips D. C.; York R. L.; McCrea K. R.; Ward R. S.; Somorjai G. A. In situ adsorption studies of a 14-amino acid leucine-lysine peptide onto hydrophobic polystyrene and hydrophilic silica surfaces using quartz crystal microbalance, atomic force microscopy, and sum frequency generation vibrational spectroscopy. J. Am. Chem. Soc. 2006, 128, 3598–3607. [DOI] [PubMed] [Google Scholar]
- Ge A.; Wu H.; Darwish T. A.; James M.; Osawa M.; Ye S. Structure and lateral interaction in mixed monolayers of dioctadecyldimethylammonium chloride (DOAC) and stearyl alcohol. Langmuir 2013, 29, 5407–5417. [DOI] [PubMed] [Google Scholar]
- Tamm L. K.; Tatulian S. A. Secondary structure, orientation, oligomerization, and lipid interactions of the transmembrane domain of influenza hemagglutinin. Q. Rev. Biophys. 1997, 30, 365–429. [DOI] [PubMed] [Google Scholar]
- McLaughlin S.; Murray D. Plasma membrane phosphoinositide organization by protein electrostatics. Nature 2005, 438, 605–611. [DOI] [PubMed] [Google Scholar]
- Frey S.; Tamm L. K. Orientation of melittin in phospholipid bilayers. Biophys. J. 1991, 60, 922–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamm L. K.; Tatulian S. A. Infrared spectroscopy of proteins and peptides in lipid bilayers. Q. Rev. Biophys. 1997, 30, 365–429. [DOI] [PubMed] [Google Scholar]
- Barth A.; Zscherp C. What vibrations tell us about proteins. Q. Rev. Biophys. 2002, 35, 369–430. [DOI] [PubMed] [Google Scholar]
- Heimburg T.; Schunemann J.; Weber K.; Geisler N. FTIR-Spectroscopy of multistranded coiled coil proteins. Biochemistry 1999, 38, 12727–12734. [DOI] [PubMed] [Google Scholar]
- Wang J.; Chen J.; Hochstrasser R. M. Local structure of β-hairpin isotopomers by FTIR, 2D IR, and Ab initio theory. J. Phys. Chem. B 2006, 110, 7545–7555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhoads R.; Friedburg F. Sequence motifs for calmodulin recognition. FASEB J. 1997, 11, 331–340. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.