

Abstract

Integrase (IN) inhibitors are the newest class of antiretroviral agents developed for the treatment of HIV-1 infections. Merck’s Raltegravir (RAL) (October 2007) and Gilead’s Elvitegravir (EVG) (August 2012), which act as IN strand transfer inhibitors (INSTIs), were the first anti-IN drugs to be approved by the FDA. However, the virus develops resistance to both RAL and EVG, and there is extensive cross-resistance to these two drugs. New “2nd-generation” INSTIs are needed that will have greater efficacy against RAL- and EVG-resistant strains of IN. The FDA has recently approved the first second generation INSTI, GSK’s Dolutegravir (DTG) (August 2013). Our current article describes the design, synthesis, and evaluation of a series of 1,8-dihydroxy-2-oxo-1,2-dihydroquinoline-3-carboxamides, 1,4-dihydroxy-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxamides, and 1-hydroxy-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxamides. This resulted in the identification of noncytotoxic inhibitors that exhibited single digit nanomolar EC50 values against HIV-1 vectors harboring wild-type IN in cell-based assays. Importantly, some of these new inhibitors retain greater antiviral efficacy compared to that of RAL when tested against a panel of IN mutants that included Y143R, N155H, G140S/Q148H, G118R, and E138K/Q148K.
Introduction
After 30 years of intensive research, approximately 30 drugs have been approved for the treatment of acquired immunodeficiency syndrome (AIDS).1,2 Of these, integrase (IN) inhibitors are the newest drug class2 with Merck’s Raltegravir (RAL, 1) (October 2007)3 and Gilead’s Elvitegravir (EVG, 2) (August 2012)4 being the first IN inhibitors to be approved by the FDA (Figure 1). These agents selectively block the strand transfer step (ST) of the integration reaction as compared with the 3′-processing step (3′-P). For this reason, these drugs are called IN strand transfer inhibitors (INSTIs).5 INSTIs, including RAL and EVG, share key structural features. These include a coplanar arrangement of three heteroatoms, whose function is to chelate the two catalytically important Mg2+ ions at the IN active site, which are held in place by a conserved DDE motif (D64, D116, and E152 in HIV-1 IN).6 Additionally, a halobenzyl ring is present that interacts with the penultimate cytosine base near the 3′-end of the viral DNA. This displaces the dA at the very 3′-end of the viral DNA and in so doing prevents the insertion of the viral DNA into the host genome.7 Treatment with RAL and EVG can lead to the development of resistance, and there is extensive shared cross-resistance.8−10 Therefore, “2nd-generation” INSTIs are being developed that have greater efficacies against RAL and EVG-resistant strains of IN.11 GSK’s Dolutegravir (DTG, 3, Figure 1) is a second-generation INSTI that has recently received FDA approval for the treatment of AIDS.1,12,13 However, the finding that DTG, like all anti-HIV drugs, selects for resistant viruses,12 emphasizes the continuing need to develop INSTIs that can effectively inhibit HIV strains that carry the common/extant resistance mutations.
Figure 1.
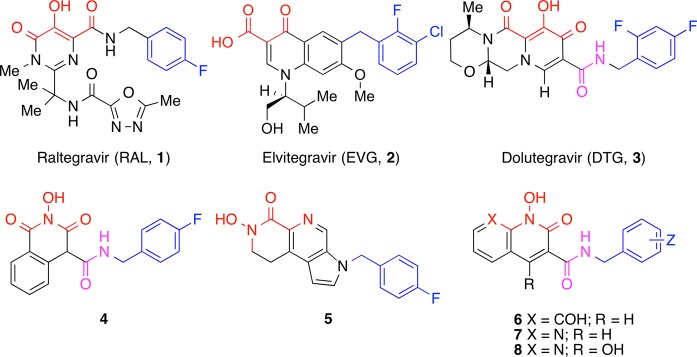
Structures of HIV-1 integrase inhibitors discussed in the text. Mg2+-chelating heteroatoms are shown in red with the halogen-substituted aromatic functionality shown in blue. The amide linkers are shown in magenta.
An attractive property of DTG is its ability to maintain high potencies against mutant strains of HIV that are resistant to RAL and EVG.14 Although DTG contains key structural features that are found in other INSTIs (outlined above and highlighted in Figure 1), it differs in having its halobenzyl group appended via an amide carbonyl that is proximal to but not part of the triad of metal chelating heteroatoms. This is in contrast to RAL and several other INSTIs, in which the halobenzyl amide carbonyl serves as one of the metal-chelating elements in the heteroatom triad. As a result, the halobenzyl linker moiety of DTG has greater flexibility than RAL structurally related INSTIs. This flexibility may contribute to DTG’s ability to bind tightly to both wild-type (WT) and mutant IN–DNA complexes.15−18 While differing in their halobenzyl linker arrangements, both RAL and DTG have a hydroxyl group as the central component of their metal-chelating triad of heteroatoms. We noted that hydroxyl amides can function as high affinity metal-chelating groups.19 In fact, hydroxylamide-containing INSTIs (for example 4) have been reported, which combine a centrally located hydroxylamide metal chelating functionality with flexible halobenzylamide group like the one in DTG.20−24 Alternatively, inhibitors such as 5 employ a hydroxylamide group as the terminal member of the metal-chelating triad, and the halobenzyl group is appended through a 1H-pyrrole ring (Figure 1).25 Our current report describes new INSTIs (6–8, Figure 1) that have a hydroxylamide group as the central component of a triad of metal-chelating heteroatoms, which originate from within bicyclic frameworks. Importantly, the halobenzylamide moieties of many of these inhibitors are appended in a fashion that may not require participation of their amide carbonyls in metal chelation. These inhibitors exhibit high potencies against viral vectors that carry WT IN and the major RAL-resistant IN mutants.
Results and Discussion
Inhibitor Design
In designing the current series of inhibitors, we examined two classes of bicyclic platforms that differed in their presentation of the “left” terminal member of the metal-chelating heteroatom triad. Both of these platforms utilized a 1-hydroxypyridin-2(1H)-one moiety as the central and “right” members of the heteroatom triad. However, one class of inhibitor utilized a phenolic hydroxyl as the “left” terminal metal-chelating heteroatom [1,8-dihydroxyquinolin-2(1H)-ones 6], while the second class employed a ring-embedded nitrogen to give 1-hydroxy-1,8-naphthyridine-2(1H)-ones (7). The latter class of inhibitors was also varied by the introduction of a 4-hydroxyl substituent [1,4-dihydroxy-1,8-naphthyridine-2(1H)-ones 8] (Figure 1). Three different halogen-substituted benzylamides were examined at the 3-position of the “right” 1-hydroxypyridin-2(1H)-one ring because the nature and pattern of halogen phenyl substitution is known to significantly affect the IN inhibitory potency of INSTIs.26
Synthesis
Modification of a previously reported27 low-yield Knoevenagel condensation of o-nitrobenzaldehyde 9a with diethyl malonate using microwave radiation and a solvent mixture that included benzene provided dimethyl 2-(2-nitrobenzylidene)malonate 10a in quantitative yield (Scheme 1).28 Treatment of 3-methoxy-2-nitrobenzaldehyde 9b with diethyl malonate under similar conditions but without the inclusion of benzene gave the corresponding diethyl 2-(3-methoxy-2-nitrobenzylidene)malonate 10b in 90% yield. Initial hydrogenation of 10a (0.15 equiv of platinum(IV) oxide) using previously reported conditions that did not contain DMSO,27 provided a very low yield of the desired methyl 1-hydroxy-2-oxo-1,2-dihydroquinoline-3-carboxylate 11a, accompanied by significant reduction of the C3–C4 double bond and the N-hydroxyl group. However, when the reduction was performed on 10b with the inclusion of DMSO (1.6 equiv),29,30 the desired ethyl 1-hydroxy-8-methoxy-2-oxo-1,2-dihydroquinoline-3-carboxylate 11b was obtained in 82% yield. In choosing the composition and pattern of halogen substitution on the benzyl amide group, we selected 3′-chloro-4′-fluorobenzyl 13a and 3′,4′-difluorobenzyl 13b, respectively, based on our previous findings that these can provide improved IN inhibitory potencies.26,31 In addition, we prepared the 2′,4′-difluorobenzyl-substituted pattern 12c and 13c found in DTG. Demethylation of 13a–c with boron tribromide gave the desired final products 1,8-dihydroxy-2-oxo-1,2-dihydroquinoline-3-carboxamides, 6a–c (Scheme 1).
Scheme 1. Synthesis of Analogues 6a–c and 12c.

Reagents and conditions: (i) HOAc (4 equiv), CH2(CO2Me)2 or CH2(CO2Et)2 (12 equiv), piperidine (1.2 equiv), microwave, 80 °C; (ii) H2, PtO2 (0.15 equiv), rt; (iii) 3-Cl-4-F-BnNH2 (a), 3,4-diF-BnNH2 (b) or 2,4-diF-BnNH2 (c), 60 °C; iv) BBr3, CH2Cl2, rt.
Because DTG is unsubstituted at the position para to its central metal-chelating hydroxyl group (the 2-position of its 5-hydroxy-4-oxo-1,3-dihydropyridine-3-carboxamide moiety), we prepared a series of 1-hydroxy-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxamides (7), which lack a substituent at the corresponding position. Accordingly, in a fashion similar to reported methology,32 treatment of commercially available methyl 2-fluoronicotinate 14 with benzoxylamine in DMSO followed by acylation of the resulting methyl 2-((benzyloxy)amino)nicotinate with methyl 3-chloro-3-oxopropanoate gave the intermediate methyl 2-(N-(benzyloxy)-3-methoxy-3-oxopropanamido)nicotinate 15 (Scheme 2). This was treated with sodium methoxide in methanol to yield the cyclized product, methyl 1-(benzyloxy)-4-hydroxy-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxylate 16 (66% yield for three steps from 14) (Scheme 2). Removal of the 4-hydroxy group was achieved by conversion of 16 to its triflate 17 followed by reduction, and the resulting methyl 1-(benzyloxy)-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxylate 18 was reacted individually with neat 3-chloro-4-fluorobenzylamine (a), 3,4-difluorobenzylamine (b) and 2,4-difluorobenzylamine (c) to provide the corresponding amides 19a–c (Scheme 2). Finally, hydrogenolytic deprotection of the N-hydroxyl group gave the desired 1-hydroxy-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxamides, (7a–c) (Scheme 2). Alternatively, debenzylation of 16 followed by treatment with halobenzylamines (a, b, and c) in DMF afforded the desired 1,4-dihydroxy-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxamides 8a–c along with over-reduced 4-dihydroxy-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxamides (20a–c) (Scheme 2).
Scheme 2. Synthesis of Analogues 7a–c and 8a–c and Over-reduced Products 20a–c.

Reagents and conditions: (i) BnONH2, DMSO, 140 °C; (ii) ClCOCH2CO2CH3, NEt3, CH2Cl2, rt; (iii) NaOMe, MeOH, rt; (iv) Tf2O, 0 °C; (v) PdCl2(PPh3)2, TIS, NEt3, 85 °C; (vi) (a) 3-Cl-4-F-BnNH2, (b) 3,4-diF-BnNH2, or (c) 2,4-diF-BnNH2; (vii) H2, Pd·C.
Evaluation in in Vitro IN Biochemical Assays
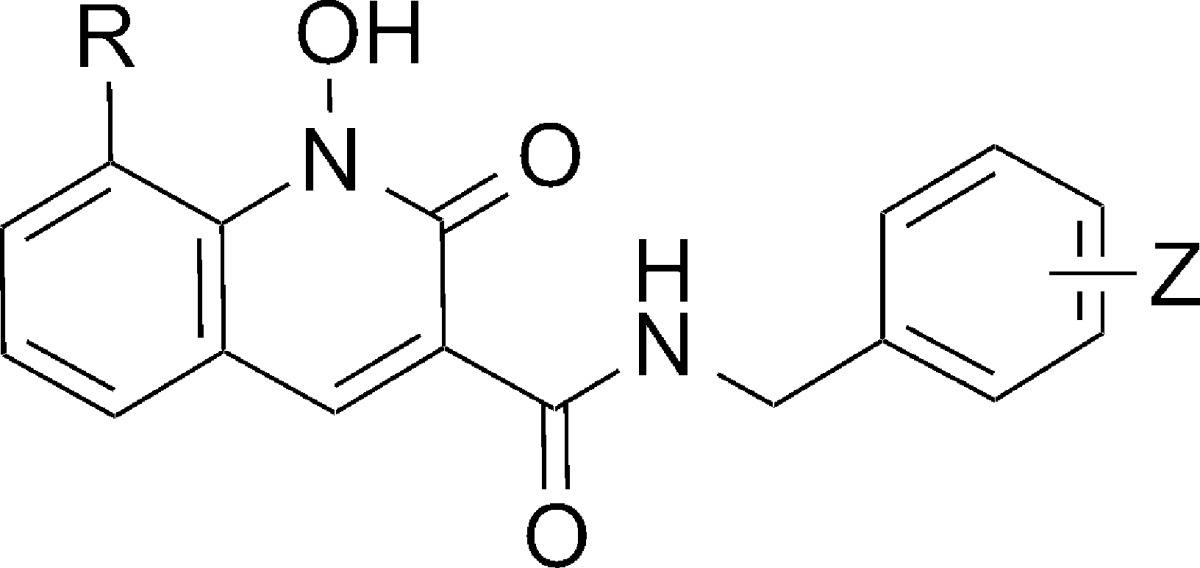
Compounds were evaluated in an IN biochemical assay using radiolabeled oligonucleotides to measure the ability of the compounds to inhibit the 3′-P and ST reactions.33 For the 1,8-dihydroxyquinolin-2(1H)-ones (6), 3′-P reactions were inhibited with IC50 values in the micromolar range, with the 3′-Cl-4′-F and 3′,4′-diF-substituted amides (6a, 3′-P IC50 = 17 μM; and 6b, 3′-P IC50 = 24 μM) being slightly more potent than the 2′,4′-disubstituted analogue 6c (3′-P IC50 = 43 μM) (Table 1). These results are consistent with our previous reports that among the diverse halogen substituent patterns we tested, 3-chloro-4-fluoro benzylamides show the best potencies in in vitro assays.26,31 Selectivity for the ST reaction relative to the 3′-P reaction is a defining characteristic of INSTIs, and in the case of 6a and 6b, selectivity was approximately 30-fold (ST IC50 values of 0.53 μM and 0.78 μM, respectively; Table 1). In contrast, for 6c the ST inhibitory potency (ST IC50 = 18 μM) was very similar to its 3′-P inhibitory potency. There was approximately a 20-fold loss of ST inhibitory potency in going from the 3′,4′-diF to the 2′,4′-diF benzylamide substituent pattern. With the latter pattern of halogen substituents, removal of the 8-hydroxy group abrogated 3′-P inhibitory potency (12c, 3′-P IC50 >333 μM) but had little effect on ST inhibitory potency (12c ST IC50 = 19.7 μM). Insertion of a bulky methoxyl group at the 8-position (13c) also abrogated both 3′-P and ST inhibitory potencies (Table 1).
Table 1. Inhibitory Potencies of 1,8-Dihydroxy-2-oxo-1,2-dihydroquinoline-3-carboxamides 6(a–c) and 1-Hydroxy-2-oxo-1,2-dihydroquinoline-3-carboxamides 12c and 13c Using an in Vitro IN Assay.
| IC50 values (μM) |
||||
|---|---|---|---|---|
| no. | R | Z | 3′-processing | strand transfer |
| 6a | OH | 3′-Cl-4′-F | 17 ± 1 | 0.53 ± 0.13 |
| 6b | OH | 3′, 4′-diF | 24 ± 3 | 0.78 ± 0.22 |
| 6c | OH | 2′, 4′-diF | 43 ± 5 | 18 ± 7 |
| 12c | H | 2′, 4′-diF | >333 | 19.7 ± 2.7 |
| 13c | OCH3 | 2′, 4′-diF | >333 | >333 |
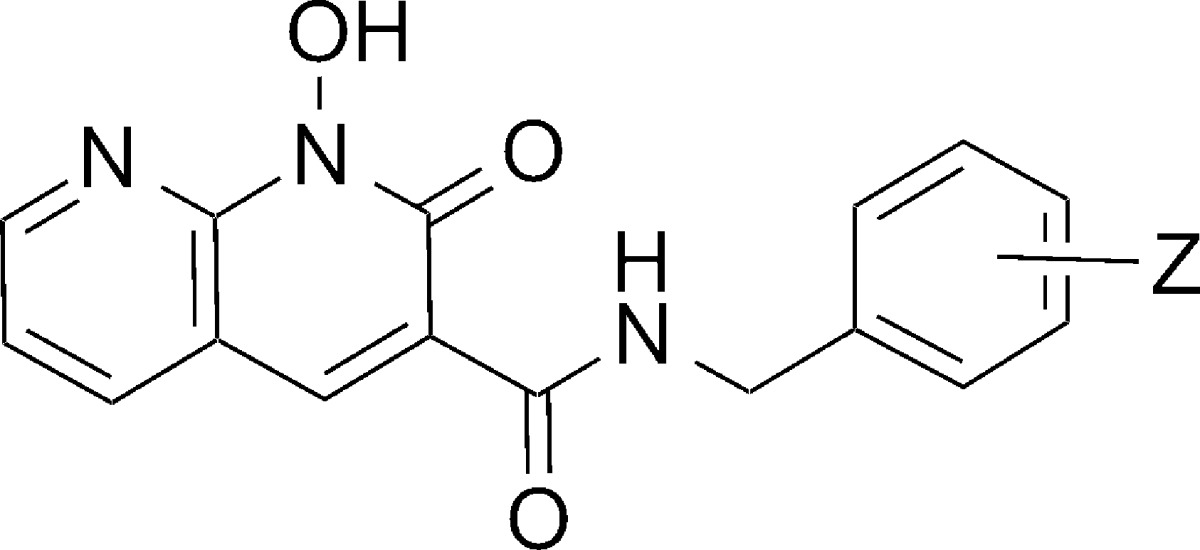
Using the in vitro IN catalytic assay described above, we showed that compounds 7a and 7b exhibited potent ST inhibition, with IC50 values (14 nM and 17 nM, respectively, Table 2). These were approximately 40-fold lower than the IC50 values measured for the corresponding 8-dihydroxyquinolin-2(1H)-ones (6a and 6b, respectively, Table 1); however, high selectivity for inhibiting the ST reaction versus the 3′-P reaction was maintained. Unexpectedly, compound 7c, which contains the benzyl 2′,4′-diF pattern found with DTG, while being slightly less potent (2-fold) than either the 3′-Cl-4′-F or 3′,4′-diF containing compounds (7a and 7b, respectively), exhibited a greater than 400-fold enhancement of ST inhibitory potency (IC50 ≈ 40 nM) relative to the corresponding 8-dihydroxyquinolin-2(1H)-one (6c).
Table 2. Inhibitory Potencies of 1-Hydroxy-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxamides 7(a–c) Using an in Vitro IN Assay.
| IC50 values (μM) |
|||
|---|---|---|---|
| no. | Z | 3′-processing | strand transfer |
| 7a | 3′-Cl-4′-F | 9.4 ± 1.5 | 0.014 ± 0.003 |
| 7b | 3′, 4′-diF | 13.6 ± 1.8 | 0.017 ± 0.003 |
| 7c | 2′, 4′-diF | 6.1 ± 0.8 | 0.041 ± 0.012 |
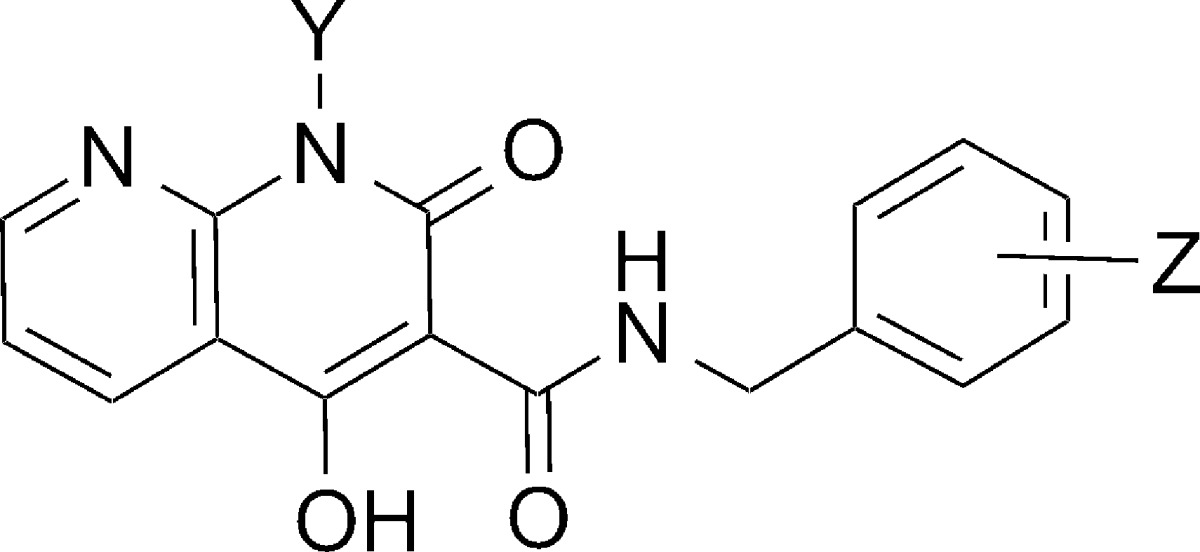
Given the potentially important role of the carboxamide linker arrangement in DTG’s ability to maintain efficacy against RAL- and EVG-resistant strains of mutant IN,15−17 we inserted a hydroxyl group at the 4-position of the 1-hydroxy-1,8-naphthyridine-2(1H)-one ring system to ask whether this could potentially influence the inhibitory profiles by hydrogen bonding with the 3-carboxamide carbonyl. However, this modification resulted in a slight loss of ST inhibitory potencies (8a–c, Table 3). We also showed that removing the hydroxylamide group resulted in a more than two-orders of magnitude loss of ST inhibitory potency (20a–c, Table 3). This is consistent with the important role this hydroxyl may play as the central component of the metal-chelating triad.
Table 3. Inhibitory Potencies of 1,4-Dihydroxy-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxamides 8(a–c) and 4-Dihydroxy-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxamides 20(a–c) Using an in Vitro IN Assay.
| IC50 values (μM) |
||||
|---|---|---|---|---|
| no. | Y | Z | 3′-processing | strand transfer |
| 8a | OH | 3′-Cl-4′-F | 4.7 ± 0.7 | 0.027 ± 0.006 |
| 8b | OH | 3′, 4′-diF | 3.6 ± 0.6 | 0.040 ± 0.010 |
| 8c | OH | 2′, 4′-diF | 1.2 ± 0.2 | 0.055 ± 0.008 |
| 20a | H | 3′-Cl-4′-F | 125 ± 15 | 7.8 ± 1.1 |
| 20b | H | 3′, 4′-diF | 93 ± 8 | 8.1 ± 1.8 |
| 20c | H | 2′, 4′-diF | >333 | 7.1 ± 1.8 |
Evaluation of the Compounds in Cell-Based Antiviral Assays
A primary objective in developing second-generation IN inhibitors is to overcome mutations that are associated with resistance to first-generation inhibitors, such as RAL. To determine the abilities of our current inhibitors to retain efficacy against resistant variants, we evaluated the antiviral potencies in cells infected with one-round HIV-1 vectors that carry mutations that cause resistance to RAL (Table 4).10,34,35 In these assays, RAL (1) exhibited an EC50 value (effective concentration resulting in 50% reduction of luciferase reporter signal) of 4 nM in cells infected with virus containing WT IN. Cells infected with viruses containing either the Y143R or the N155H mutant forms of IN, showed significantly elevated EC50 values (162 nM and 154 nM, respectively). An even greater loss of efficacy (EC50 = 1900 nM) was observed with viruses containing the G140S/Q148H double mutant form of IN (Table 4). In similar assays, treatment of WT-infected cells with inhibitors 7a–c and 8a–c gave EC50 values in the low nanomolar range, with analogues 7c and 8c having the 2′,4′-diF substituted benzyl group (EC50 values of 5 nM and 6 nM, respectively) showing greater efficacy by several fold relative to analogues having either 3′-Cl-4′-F (7a and 8a) or 3′,4′-diF (7b and 8b) substituted benzyl groups. This is in contrast to in vitro assays, where analogues bearing the 2′,4′-diF-substituted benzyl group were slightly less effective ST inhibition (Tables 2 and 3).
Table 4. Antiviral Potencies of Compounds 7(a–c) and 8(a–c) in Cells Infected with HIV-1 Vectors Containing Wild-Type (WT) or Mutant IN.
| EC50 (nM, IN mutantsc) |
||||||
|---|---|---|---|---|---|---|
| no. | CC50 (μM)a | EC50 (nM) WTb | Y143R | N155H | G140S/Q148H | SId |
| 1 | >100 | 4 ± 2 | 162 ± 16 | 154 ± 33 | 1900 ± 300 | >25,000 |
| 7a | >250 | 38 ± 15 | 34 ± 6 | 90 ± 6 | N/Ae | >6,579 |
| 7b | >250 | 62 ± 14 | 40 ± 13 | 2200 ± 61 | N/Ae | >4,032 |
| 7c | >250 | 5.1 ± 1.9 | 4.9 ± 0.8 | 134 ± 23 | 438 ± 121 | >49,020 |
| 8a | 102 ± 18 | 35 ± 12 | 54 ± 9 | 148 ± 8 | 489 ± 62 | 2,914 |
| 8b | 192 ± 19 | 20 ± 6 | 45 ± 12 | 189 ± 70 | 507 ± 125 | 9,600 |
| 8c | 137 ± 20 | 6.2 ± 2.9 | 11 ± 2 | 31 ± 8 | 308 ± 125 | 22,097 |
Cytotoxic concentration resulting in 50% reduction in the level of ATP in human osteosarcoma (HOS) cells.
Values obtained from cells infected with the lentiviral vector harboring WT IN.
Cells were infected with viral vectors carrying IN mutations and indicated values in EC50.
Selectivity index calculated as the ratio of CC50 to EC50.
Not available.
An important feature of the new inhibitors is their ability to maintain high efficacies against RAL-resistant strains. For example, compound 7c exhibits single digit nanomolar antiviral potencies against both WT and the Y143R mutant, while showing a lower potency against the G140S/Q148H mutant (EC50 = 438 nM). However, it is much improved when compared to RAL (1) (EC50 = 1900 nM). Compound 8c also retains good efficacy against the Y143R mutant (EC50 = 11 nM) as well as both the N155H mutant (EC50 = 31 nM; RAL has EC50 = 154 nM) and the G140S/Q148H mutant (EC50 = 308 nM) (Table 4).
While showing nanomolar EC50 values against cells infected with HIV-1 vectors carrying WT and efficacy against some RAL-resistant mutants, analogues 7a–c exhibited very little cytotoxicity, with CC50 values (cytotoxic concentrations were measured as the level of the compound that reduced cellular ATP levels by 50%) greater than 250 μM. In particular, for the 2′,4′-difluorobenzyl amide 7c with an EC50 value of 5.1 nM against WT, this gives a selectivity index (SI = CC50/EC50) greater than 49,000 (Table 4). Analogues 8a–c showed CC50 values between approximately 100 μM (8a) and 200 μM (8b). In the case of 8c, which showed a WT EC50 value of 6.2 nM, a SI value greater than 20,000 was achieved.
Because compounds 7c and 8c show antiviral potencies similar to those of RAL (1) against HIV-1 vectors carrying WT IN, it is important to compare the relative effectiveness of our inhibitors against mutant strains of IN in terms of fold-loss relative to WT. Table 5 contains data showing the efficacy of compounds against the G118R and E138K/Q148K mutants, which have recently been identified through in vitro selection studies with second-generation inhibitors.36 Of particular note is the ability of 8c to maintain efficacy equivalent to that of RAL against the G118R mutant while showing approximately one order of magnitude greater efficacy than RAL against the remaining mutants in the table.
Table 5. Fold Change (FC) of Amides 7c and 8c Compared with That of RAL (1) in Cells Infected with HIV-1 Constructs Carrying WT or Mutant IN.
| EC50 (FC, IN mutantsb) |
||||||
|---|---|---|---|---|---|---|
| no. | EC50 (nM) WTa | Y143R | N155H | G140S/Q148H | G118R | E138K/Q148K |
| 1 | 4 ± 2 | 41× | 38× | 475× | 9× | 375× |
| 7c | 5.1 ± 1.9 | 1× | 26× | 86× | N/Ac | N/Ac |
| 8c | 6.2 ± 2.9 | 2× | 5× | 50× | 6× | 32× |
Values obtained from cells infected with a lentiviral vector harboring WT IN.
Cells were infected with viral constructs carrying IN mutations, and the indicated values correspond to the fold-change (FC) in EC50 relative to WT.
Not available.
Conclusions
A series of bicyclic inhibitors were prepared that employed 1-hydroxy-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxamide (7) and 1,8-dihydroxy-2-oxo-1,2-dihydroquinoline-3-carboxamide (8) ring systems. Key features of these inhibitors include the use of a hydroxylamide group as a metal-chelating component and the inclusion of halobenzylamide functionality that is appended through a linker whose carboxamide carbonyl is not an obligatory component of the key metal-chelating heteroatom triad. Among these IN inhibitors, amides 7c and 8c have single digital nanomolar antiviral EC50 potencies against HIV-1 vectors carrying WT IN. Several compounds have selectivity indices of greater than 20,000, and certain of these inhibitors have greater antiviral efficacies than RAL against a panel of IN mutants that included Y143R, N155H, G118R, and the double mutants G140S/Q148H and E138K/Q148K. Compounds 7c and 8c represent potentially useful platforms for further structural variations intended to find compounds that are more broadly effective against additional resistant strains of the virus.
Experimental Section
General Synthesis
1H and 13C NMR data were obtained on a Varian 400 MHz spectrometer or a Varian 500 MHz spectrometer and are reported in ppm relative to TMS and referenced to the solvent in which the spectra were collected. The solvent was removed by rotary evaporation under reduced pressure, and anhydrous solvents were obtained commercially and used without further drying. Purification by silica gel chromatography was performed using CombiFlash Rf 200i with EtOAc–hexanes solvent systems. Preparative high pressure liquid chromatography (HPLC) was conducted using a Waters Prep LC4000 system having photodiode array detection and Phenomenex C18 columns (Cat. No. 00G-4436-P0-AX, 250 mm × 21.2 mm, 10 μm particle size, 110 Å pore) at a flow rate of 10 mL/min. Binary solvent systems consisting of A = 0.1% aqueous TFA and B = 0.1% TFA in acetonitrile were employed with gradients as indicated. Products were obtained as amorphous solids following lyophilization. Electrospray ionization-mass spectra (ESI-MS) were acquired with an Agilent LC/MSD system equipped with a multimode ion source. Purities of samples subjected to biological testing were assessed using this system and shown to be ≥95%. High-resolution mass spectra (HRMS) were acquired by LC/MS-ESI using an LTQ-Orbitrap-XL at 30K resolution.
General Procedure A for the Synthesis of Diethyl 2-(2-(Nitrobenzylidene)malonates (10a and 10b)
Commercially available 2-nitrobenzaldehydes (9a and 9b) (5 mmol) were added to a solution of dimethyl malonate or diethyl malonate (60 mmol), acetic acid (20 mmol), and piperidine (6 mmol). The mixture was irradiated with microwave radiation with stirring (80 °C, 15 h). The mixture was partitioned between EtOAc (60 mL) and aqueous NaHCO3 (30 mL), and the organic phase was dried (Na2SO4) and filtered, and the filtrate was concentrated. The resulting residue was purified by Combiflash silica gel chromatography (hexanes and EtOAc) to yield either dimethyl (10a) or diethyl 2-(2-nitrobenzylidene)malonate (10b).
Dimethyl 2-(2-Nitrobenzylidene)malonate (10a)
Reaction of commercially available 2-nitrobenzaldehyde (9a) and dimethyl malonate in benzene (8 mL) as described in general procedure A with the inclusion of benzene (8.0 mL) provided 10a as a colorless oil in 100% yield. 1H NMR (500 MHz, CDCl3) δ 8.19 (t, J = 4.1 Hz, 2H), 7.64 (t, J = 7.6 Hz, 1H), 7.56 (t, J = 7.8 Hz, 1H), 7.39 (d, J = 7.6 Hz, 1H), 3.85 (s, 3H), 3.59 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 165.24, 163.68, 147.04, 141.73, 133.88, 130.36, 130.24, 129.92, 128.38, 125.06, 52.83, 52.42. ESI-MS m/z: 266.0 (M+H+).
Diethyl 2-(3-Methoxy-2-nitrobenzylidene)malonate (10b)
Reaction of commercially available 3-methoxy-2-nitrobenzaldehyde (9b) and diethyl malonate as described in general procedure A, provided 10b as a colorless oil in 90% yield. 1H NMR (400 MHz, CDCl3) δ 7.58 (s, 1H), 7.36 (t, J = 8.2 Hz, 1H), 7.05 (d, J = 8.4 Hz, 1H), 6.99 (d, J = 7.9 Hz, 1H), 4.24 (q, J = 7.2 Hz, 2H), 4.16 (q, J = 7.1 Hz, 2H), 3.87 (s, 3H), 1.27 (t, J = 7.1 Hz, 3H), 1.12 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 164.94, 162.88, 151.25, 140.28, 135.29, 131.38 (2C), 127.75, 120.06, 114.03, 62.00, 61.73, 56.55, 13.98, 13.72. ESI-MS m/z: 324.1 (M+H+).
General Procedure B for the Synthesis of Methyl and Ethyl 1-Hydroxy-2-oxo-1,2-dihydroquinoline-3-carboxylates (11a and 11b)
Compound 10a or 10b (0.5 mmol) was dissolved in acetic acid (3 mL) with or without added DMSO (0.8 mmol), followed by the addition of platinum(IV) oxide (0.075 mmol), and the mixture was stirred at room temperature under H2 (22 h). The mixture was filtered and washed by MeOH, and the filtrate was concentrated and purified by Combiflash silica gel chromatography (hexanes and EtOAc) to yield products 11a or 11b.
Methyl 1-Hydroxy-2-oxo-1,2-dihydroquinoline-3-carboxylate (11a)
Treatment of 10a as outlined in general procedure B, without the inclusion of DMSO, afforded 11a as a white solid in 0.7% yield. 1H NMR (500 MHz, CDCl3) δ 8.63 (s, 1H), 7.80–7.77 (m, 3H), 7.39–7.36 (m, 1H), 4.01 (s, 3H). ESI-MS m/z: 220.0 (M+H+).
Ethyl 1-Hydroxy-8-methoxy-2-oxo-1,2-dihydroquinoline-3-carboxylate (11b)
Treatment of 10b as outlined in general procedure B with the inclusion of DMSO afforded 11b as a yellow oil in 82% yield. 1H NMR (400 MHz, CDCl3) δ 8.47 (s, 1H), 7.31–7.29 (m, 1H), 7.23–7.22 (m, 1H), 7.21 (d, J = 1.6 Hz, 1H), 4.41 (q, J = 7.1 Hz, 2H), 3.96 (s, 3H), 1.39 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 163.68, 154.45, 146.70, 141.82, 127.52, 123.81, 122.68, 119.21, 118.55, 116.62, 61.81, 57.64, 14.26. ESI-MS m/z: 264.1 (M+H+).
General Procedure C for the Synthesis of N-(Benzyl)-1-hydroxy-2-oxo-1,2-dihydroquinoline-3-carboxamides (12c and 13a–13c)
A mixture of 11a or 11b (0.4 mmol) with benzylamine (3 mL) was heated with stirring (60 °C, 14 h), and the resulting product mixture was purified by reverse-phase HPLC to provide 12c or 13a–13c.
N-(2,4-Difluorobenzyl)-1-hydroxy-2-oxo-1,2-dihydroquinoline-3-carboxamide (12c)
Treatment of 11a with 2,4-difluorobenzylamine as outlined in general procedure C with purification by preparative reverse-phase HPLC (linear gradient of 30% B to 60% B over 30 min; retention time = 28.1 min) provided 12c as a white solid. 1H NMR (500 MHz, DMSO-d6) δ 11.82 (s, 1H), 10.06 (t, J = 6.2 Hz, 1H), 8.84 (s, 1H), 8.05 (d, J = 8.0 Hz, 1H), 7.82–7.76 (m, 2H), 7.49–7.38 (m, 2H), 7.26 (t, J = 10.0 Hz, 1H), 7.08 (t, J = 8.5 Hz, 1H), 4.61 (d, J = 5.8 Hz, 2H). ESI-MS m/z: 331.1 (M+H+). HRMS calcd for C17H13F2N2O3 [MH+], 331.0889; found, 331.0884.
N-(3-Chloro-4-fluorobenzyl)-1-hydroxy-8-methoxy-2-oxo-1,2-dihydroquinoline-3-carboxamide (13a)
Reaction of 11b and 3-chloro-4-fluorobenzylamine as outlined in general procedure C with purification by preparative reverse-phase HPLC (linear gradient of 30% B to 65% B over 30 min; retention time =27.6 min) provided 13a as a white solid in 62% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.18 (s, 1H), 10.01 (t, J = 6.0 Hz, 1H), 8.70 (s, 1H), 7.57 (dd, J = 8.0, 1.2 Hz, 1H), 7.53–7.51 (m, 1H), 7.34 (ddd, J = 7.2, 6.8, 1.1 Hz, 3H), 7.26 (t, J = 7.9 Hz, 1H), 4.52 (d, J = 6.0 Hz, 2H), 3.83 (s, 3H). ESI-MS m/z: 377.0 (M+H+).
N-(3,4-Difluorobenzyl)-1-hydroxy-8-methoxy-2-oxo-1,2-dihydroquinoline-3-carboxamide (13b)
Reaction of 11b and 3,4-difluorobenzylamine as outlined in general procedure C with purification by preparative reverse-phase HPLC (linear gradient of 30% B to 65% B over 30 min; retention time = 24.8 min) provided 13b as a white solid in 78% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.08 (t, J = 6.0 Hz, 1H), 8.66 (s, 1H), 7.55 (dd, J = 8.0, 1.3 Hz, 1H), 7.38–7.21 (m, 3H), 7.17–7.14 (m, 1H), 7.12–7.08 (m, 1H), 4.51 (d, J = 5.9 Hz, 2H), 3.80 (s, 3H). ESI-MS m/z: 361.1 (M+H+).
N-(2,4-Difluorobenzyl)-1-hydroxy-8-methoxy-2-oxo-1,2-dihydroquinoline-3-carboxamide (13c)
Reaction of 11b and 2,4-difluorobenzylamine as outlined in general procedure C with purification by preparative reverse-phase HPLC (linear gradient of 30% B to 65% B over 30 min; retention time = 27.0 min) provided 13c as a white solid in 74% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.24 (s, 1H), 10.04 (t, J = 6.1 Hz, 1H), 8.75 (s, 1H), 7.62 (dd, J = 7.9, 1.3 Hz, 1H), 7.47 (dd, J = 15.3, 8.7 Hz, 1H), 7.42–7.40 (m, 1H), 7.32 (t, J = 7.9 Hz, 1H), 7.29–7.23 (m, 1H), 7.08 (dd, J = 11.1, 8.6 Hz, 1H), 4.59 (d, J = 5.9 Hz, 2H), 3.88 (s, 3H). ESI-MS m/z: 361.1 (M+H+). HRMS calcd for C18H15F2N2O4 [MH+], 361.0994; found, 361.0990.
General Procedure D for the Synthesis of 1,8-Dihydroxy-2-oxo-1,2-dihydroquinoline-3-carboxamides (6a–6c)
HPLC-purified compounds 13a–13c (0.1 mmol) were stirred at room temperature with boron tribromide (0.8 mmol, 2.0 M in CH2Cl2) (overnight). The reaction was quenched by the addition of MeOH, and the product mixture was purified by reverse-phase HPLC to provide 6a–6c.
N-(3-Chloro-4-fluorobenzyl)-1,8-dihydroxy-2-oxo-1,2-dihydroquinoline-3-carboxamide (6a)
Treatment of 13a as outlined in general procedure D with purification by preparative reverse-phase HPLC (linear gradient of 30% B to 65% B over 30 min; retention time =27.5 min) provided 6a as a white solid in 36% yield.1H NMR (400 MHz, DMSO-d6) δ 10.14 (t, J = 6.0 Hz, 1H), 8.63 (s, 1H), 7.42 (dd, J = 7.4, 1.7 Hz, 1H), 7.39 (dd, J = 8.1, 1.0 Hz, 1H), 7.25–7.22 (m, 3H), 7.17 (t, J = 7.9 Hz, 1H), 7.06 (dd, J = 7.8, 1.1 Hz, 1H), 4.41 (dd, J = 17.7, 6.0 Hz, 2H). ESI-MS m/z: 363.0 (M+H+). HRMS calcd C17H13FClN2O4 [MH+], 363.0542; found, 363.0551.
N-(3,4-Difluorobenzyl)-1,8-dihydroxy-2-oxo-1,2-dihydroquinoline-3-carboxamide (6b)
Treatment of 13b as outlined in general procedure D with purification by preparative reverse-phase HPLC (linear gradient of 30% B to 65% B over 30 min; retention time = 24.9 min) provided 6b as a white solid in 42% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.14 (t, J = 6.1 Hz, 1H), 8.63 (s, 1H), 7.39 (t, J = 6.4 Hz, 1H), 7.29–7.23 (m, 2H), 7.16 (t, J = 7.9 Hz, 2H), 7.06 (dd, J = 7.8, 1.2 Hz, 2H), 4.42 (dd, J = 24.5, 5.8 Hz, 2H). ESI-MS m/z: 347.1 (M+H+). HRMS calcd C17H13F2N2O4 [MH+], 347.0838; found, 347.0847.
N-(2,4-Difluorobenzyl)-1,8-dihydroxy-2-oxo-1,2-dihydroquinoline-3-carboxamide (6c)
Treatment of 13c as outlined in general procedure D with purification by preparative reverse-phase HPLC (linear gradient of 30% B to 65% B over 30 min; retention time = 24.6 min) provided 6c as a white solid in 38% yield. 1H NMR (400 MHz, DMSO-d6) δ 9.99 (t, J = 5.6 Hz, 1H), 8.67 (s, 1H), 7.41 (dd, J = 15.7, 8.0 Hz, 2H), 7.23–7.11 (m, 3H), 7.04 (dd, J = 19.0, 8.2 Hz, 2H), 4.54 (d, J = 5.9 Hz, 2H). ESI-MS m/z: 347.1 (M+H+). HRMS calcd for C17H13F2N2O4 [MH+], 347.0838; found, 347.0835.
Methyl 2-(N-(Benzyloxy)-3-methoxy-3-oxopropanamido)nicotinate (15)
A solution of commercially available methyl 2-fluoronicotinate (14) (1 mmol), O-benzylhydroxylamine (3 mmol), and N-ethyl-N-isopropylpropan-2-amine (3 mmol) in DMSO was subjected to microwave irradiation with stirring (140 °C, 10 h). The resulting mixture was extracted (EtOAc), the organic phase was washed with brine and dried (Na2SO4), the crude product was filtered, and the filtrate was concentrated. Purification by Combiflash silica gel chromatography (hexanes and EtOAc) provided intermediate methyl 2-((benzyloxy)amino)nicotinate as a yellow oil in 86% yield. 1H NMR (400 MHz, CDCl3) δ 10.03 (s, 1H), 8.49 (dd, J = 4.8, 1.9 Hz, 1H), 8.15 (dd, J = 7.8, 1.9 Hz, 1H), 7.50–7.47 (m, 2H), 7.41–7.34 (m, 3H), 6.79 (dd, J = 7.8, 4.8 Hz, 1H), 5.08 (s, 2H), 3.83 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 166.62, 159.66, 153.68 (2C), 139.85, 136.38, 128.86, 128.44, 128.26, 114.58, 114.55, 107.03, 78.05, 52.15. ESI-MS m/z: 259.1 (M+H+). To a solution of this material (2 mmol) and triethylamine (4 mmol) in CH2Cl2 (10 mL) was added methyl 3-chloro-3-oxopropanoate (4 mmol) dropwise, and the mixture was stirred at room temperature (2 h). The crude product was filtered, the filtrate was concentrated, and the residue was purified by Combiflash silica gel chromatography (hexanes and EtOAc) to provide 15 as a yellow oil in 83% yield. 1H NMR (400 MHz, CDCl3) δ 8.64 (s, 1H), 8.18 (d, J = 7.6 Hz, 1H), 7.39–7.38 (m, 2H), 7.35–7.32 (m, 4H), 5.03 (s, 2H), 3.88 (s, 3H), 3.72 (s, 3H), 3.61 (s, 2H). 13C NMR (100 MHz, CDCl3) δ 167.09 (2C), 165.37, 151.13, 139.33, 133.89, 129.59 (3C), 128.94, 128.45 (2C), 123.83, 122.73, 78.18, 52.70, 52.32, 41.13. ESI-MS m/z: 359.1(M+H+).
Methyl 1-(Benzyloxy)-4-hydroxy-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxylate (16)
To a solution of 15 (1.7 mmol) in MeOH (30 mL) was added sodium methanolate (4.2 mmol) (25% in MeOH) at room temperature, and the resulting suspension was stirred at room temperature (overnight). Acidification to pH 4 by the addition of aqueous 2 N HCl gave a precipitate, which was collected and dried to provide 16 as a white solid in 93% yield. 1H NMR (400 MHz, CDCl3) δ 8.79 (dd, J = 4.7, 1.8 Hz, 1H), 8.46 (dd, J = 7.9, 1.8 Hz, 1H), 7.73–7.71 (m, 2H), 7.41–7.36 (m, 3H), 7.28–7.25 (m, 1H), 5.28 (s, 2H), 4.08 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 172.35, 170.14, 156.40, 154.73, 149.54, 134.67, 134.08, 130.00 (2C), 128.94, 128.34 (2C), 118.70, 109.53, 98.85, 78.12, 53.29. ESI-MS m/z: 327.1 (M+H+).
General Procedure E for the Synthesis of 1,4-Dihydroxy-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxamides (8a–c) and 4-Hydroxy-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxamides (20a–20c)
To a degassed solution of 16 (0.2 mmol) and MeOH (15 mL) with EtOAc (5 mL) was added Pd·C (10%, 0.2 mmol), and the mixture was stirred at room temperature under H2 (1 h). The mixture was filtered, and the filtrate was concentrated and dissolved in DMF (1.0 mL). To this was added the appropriate halobenzylamine (2.0 mmol), and the solution was subjected to microwave irradiation with stirring (140 °C, 2 h). Purification of the reaction mixture by reverse-phase HPLC provided the desired final products 8a–c as well as the over-reduced products 20a–20c.
N-(3-Chloro-4-fluorobenzyl)-1,4-dihydroxy-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxamide (8a) and N-(3-Chloro-4-fluorobenzyl)-4-hydroxy-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxamide (20a)
Treatment of 16 as outlined in general procedure E using 3-chloro-4-fluorobenzylamine with purification by preparative reverse-phase HPLC (linear gradient of 20% B to 80% B over 30 min) provided a 43% yield of 8a and 20a as white solids in a 2:1 ratio. For 8a: retention time = 26.4 min. 1H NMR (400 MHz, DMSO-d6) δ 11.01 (bs, 1H), 10.52 (bs, 1H), 8.84 (dd, J = 4.6, 1.7 Hz, 1H), 8.47 (dd, J = 7.9, 1.7 Hz, 1H), 7.60 (d, J = 7.6 Hz, 1H), 7.48–7.42 (m, 1H), 7.42 (d, J = 7.7 Hz, 2H), 4.61 (d, J = 6.2 Hz, 2H). ESI-MS m/z: 364.0 (M+H+). HRMS calcd C16H12ClFN3O4 [MH+], 364.0495; found, 364.0490. For 20a: retention time = 30.7 min. 1H NMR (400 MHz, DMSO-d6) δ 12.31 (s, 1H), 10.59 (t, J = 5.8 Hz, 1H), 8.70 (d, J = 4.6 Hz, 1H), 8.36 (d, J = 7.6 Hz, 1H), 7.60 (d, J = 7.0 Hz, 1H), 7.41 (dd, J = 9.8, 7.0 Hz, 2H), 7.36 (dd, J = 7.9, 4.9 Hz, 1H), 4.59 (d, J = 6.3 Hz, 2H). ESI-MS m/z: 348.0 (M+H+). HRMS calcd C16H12ClFN3O3 [MH+], 348.0546; found, 348.0541.
N-(3,4-Difluorobenzyl)-1,4-dihydroxy-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxamide (8b) and N-(3,4-Difluorobenzyl)-4-hydroxy-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxamide (20b)
Treatment of 16 as outlined in general procedure E using 3,4-difluorobenzylamine with purification by preparative reverse-phase HPLC (linear gradient of 20% B to 80% B over 30 min) provided a 54% yield of 8b and 20b as white solids in a 1:1 ratio. For 8b: retention time = 22.2 min. 1H NMR (400 MHz, DMSO-d6) δ 11.02 (s, 1H), 10.52 (s, 1H), 8.84 (dd, J = 4.6, 1.8 Hz, 1H), 8.47 (dd, J = 7.9, 1.8 Hz, 1H), 7.51–7.37 (m, 3H), 7.24 (s, 1H), 4.60 (d, J = 6.2 Hz, 2H). ESI-MS m/z: 348.0 (M+H+). HRMS calcd C16H12F2N3O4 [MH+], 348.0790; found, 348.0784. For 20b: retention time = 27.4 min. 1H NMR (400 MHz, DMSO-d6) δ 12.33 (s, 1H), 10.59 (s, 1H), 8.70 (d, J = 3.1 Hz, 1H), 8.37 (d, J = 7.8 Hz, 1H), 7.43 (dd, J = 14.0, 5.4 Hz, 2H), 7.42–7.34 (m, 1H), 7.23 (bs, 1H), 4.59 (d, J = 6.1 Hz, 2H). ESI-MS m/z: 332.1 (M+H+). HRMS calcd C16H12F2N3O3 [MH+], 332.0841; found, 332.0835.
N-(2,4-Difluorobenzyl)-1,4-dihydroxy-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxamide (8c) and N-(2,4-Difluorobenzyl)-4-hydroxy-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxamide (20c)
Treatment of 16 as outlined in general procedure E using 2,4-difluorobenzylamine with purification by preparative reverse-phase HPLC (linear gradient of 20% B to 80% B over 30 min) provided a 45% yield of 8c and 20c as white solids in a 1:1 ratio. For 8c: retention time = 24.7 min. 1H NMR (500 MHz, DMSO-d6) δ 10.95 (s, 1H), 10.42 (s, 1H), 8.77 (d, J = 4.6 Hz, 1H), 8.40 (d, J = 7.9 Hz, 1H), 7.49–7.41 (m, 1H), 7.41–7.34 (m, 1H), 7.21 (t, J = 10.1 Hz, 1H), 7.03 (t, J = 8.2 Hz, 1H), 4.57 (d, J = 5.9 Hz, 2H). ESI-MS m/z: 348.0 (M+H+). HRMS calcd C16H12F2N3O4 [MH+], 348.0790; found, 348.0787. For 20c: retention time = 29.0 min. 1H NMR (500 MHz, DMSO-d6) δ 12.32 (s, 1H), 10.57 (s, 1H), 8.70 (s, 1H), 8.36 (d, J = 7.4 Hz, 1H), 7.48 (d, J = 7.5 Hz, 1H), 7.36 (s, 1H), 7.28 (t, J = 9.6 Hz, 1H), 7.10 (s, 1H), 4.62 (d, J = 5.9 Hz, 2H). ESI-MS m/z: 332.1 (M+H+). HRMS calcd C16H12F2N3O3 [MH+], 332.0841; found, 332.0839.
Methyl 1-(Benzyloxy)-2-oxo-4-(((trifluoromethyl)sulfonyl)oxy)-1,2-dihydro-1,8-naphthyridine-3-carboxylate (17)
To a solution of 16 (268 mg, 0.82 mmol) and triethylamine (0.27 mL, 1.97 mmol) in CH2Cl2 (15 mL) was added trifluoromethanesulfonic anhydride (0.17 mL, 0.98 mmol) at 0 °C, and the solution was stirred at 0 °C (0.5 h). The mixture was concentrated and purified by Combiflash silica gel chromatography to provide 17 as a white solid (301 mg, 80% yield). 1H NMR (400 MHz, CDCl3) δ 8.86 (dd, J = 4.7, 1.7 Hz, 1H), 8.21 (dd, J = 8.1, 1.7 Hz, 1H), 7.70–7.67 (m, 2H), 7.43–7.38 (m, 4H), 5.34 (s, 2H), 4.01 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 160.87, 155.20, 154.02, 153.98, 148.70, 148.09, 133.32, 133.23, 130.12, 129.33, 128.48, 120.84, 119.91, 119.86, 116.67, 109.73, 78.72, 53.50. ESI-MS m/z: 459.0 (M+H+).
Methyl 1-(Benzyloxy)-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxylate (18)
To a mixture of 17 (34 mg, 0.074 mmol) and PdCl2(PPh3)2 (5 mg, 7.37 μmol) in DMF (1 mL), triethylamine (0.030 mL, 0.22 mmol) and triisopropylsilane (17 mg, 0.15 mmol) were added. The mixture was heated to 85 °C (24 h). The resulting crude mixture was purified by Combiflash silica gel chromatography (hexanes and EtOAc) to provide 18 as a white solid (12 mg, 52% yield). 1H NMR (400 MHz, CDCl3) δ 8.74 (dd, J = 4.7, 1.8 Hz, 1H), 8.41 (s, 1H), 7.99 (dd, J = 7.8, 1.7 Hz, 1H), 7.69 (dd, J = 7.6, 1.8 Hz, 2H), 7.38–7.30 (m, 3H), 7.26–7.23 (m, 1H), 5.29 (s, 2H), 3.96 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 164.47, 153.41, 141.78, 138.27, 133.86, 132.08, 131.99, 130.10 (2C), 129.05, 128.50, 128.37 (2C), 119.32, 113.30, 78.21, 52.90. ESI-MS m/z: 311.1 (M+H+).
General Procedure F for the Synthesis of 1-(Benzyloxy)-N-(halobenzyl)-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxamides (19a–19c)
A solution of 18 (0.6 mmol) and halobenzylamine (1 mL) was heated with stirring (60 °C, 14 h). The mixture was then extracted (CHCl3) and washed sequentially with aqueous 1 N HCl and brine and dried (Na2SO4). The organic phase was filtered and concentrated, and the crude residue was purified by Combiflash silica gel chromatograpy (hexanes and EtOAc) to provide amides 19(a–c).
1-(Benzyloxy)-N-(3-chloro-4-fluorobenzyl)-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxamide (19a)
Treatment of 18 with 3-chloro-4-difluorobenzylamine as outlined in general procedure F provided 19a as a white solid in 31% yield. 1H NMR (400 MHz, CDCl3) δ 9.90 (t, J = 5.7 Hz, 1H), 8.81 (s, 1H), 8.75 (dd, J = 4.7, 1.8 Hz, 1H), 8.06 (dd, J = 7.8, 1.8 Hz, 1H), 7.61–7.60 (m, 1H), 7.59 (d, J = 1.7 Hz, 1H), 7.36–7.32 (m, 2H), 7.31–7.27 (m, 3H), 7.19–7.15 (m, 1H), 7.03 (t, J = 8.7 Hz, 1H), 5.27 (s, 2H), 4.56 (d, J = 6.0 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 162.45, 159.38, 157.35 (d, J = 248.3 Hz), 153.33, 148.97, 141.41, 138.72, 135.31 (d, J = 3.9 Hz), 133.56, 129.94 (2C), 129.87, 129.24, 128.49 (2C), 128.38 (d, J = 15.3 Hz), 127.43 (d, J = 7.3 Hz), 123.36, 120.00, 116.61 (d, J = 21.2 Hz), 114.12, 78.57, 42.58. ESI-MS m/z: 438.1 (M+H+).
1-(Benzyloxy)-N-(3,4-difluorobenzyl)-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxamide (19b)
Treatment of 18 with 3,4-difluorobenzylamine as outlined in general procedure F provided 19b as a white solid in 51% yield. 1H NMR (400 MHz, CDCl3) δ 9.95 (t, J = 5.8 Hz, 1H), 8.85 (s, 1H), 8.78 (dd, J = 4.7, 1.8 Hz, 1H), 8.09 (dd, J = 7.8, 1.7 Hz, 1H), 7.65–7.64 (m, 1H), 7.62 (d, J = 1.7 Hz, 1H), 7.37–7.30 (m, 4H), 7.20–7.13 (m, 1H), 7.09–7.03 (m, 1H), 6.98–6.94 (m, 1H), 5.29 (s, 2H), 4.59 (d, J = 6.0 Hz, 2H). ESI-MS m/z: 422.1 (M+H+).
1-(Benzyloxy)-N-(2,4-difluorobenzyl)-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxamide (19c)
Treatment of 18 with 2,4-difluorobenzylamine as outlined in general procedure F provided 19c as a white solid in 22% yield. 1H NMR (400 MHz, CDCl3) δ 9.92 (d, J = 5.4 Hz, 1H), 8.87 (s, 1H), 8.82 (ddd, J = 4.7, 1.7, 0.6 Hz, 1H), 8.13 (dd, J = 7.8, 1.7 Hz, 1H), 7.69 (d, J = 2.4 Hz, 1H), 7.68 (dd, J = 3.3, 1.3 Hz, 1H), 7.42–7.34 (m, 5H), 6.88–6.76 (m, 2H), 5.34 (s, 2H), 4.69 (d, J = 5.9 Hz, 2H). ESI-MS m/z: 422.1 (M+H+).
General Procedure G for the Synthesis of 1-(Benzyloxy)-N-(halobenzyl)-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxamides 7(a–c)
To a solution of amide (19a–19c) (0.2 mmol) in a solution of MeOH (10 mL) and EtOAc (3 mL) was added Pd·C (10%, 20 mg), and the mixture was degassed and stirred at room temperature under H2 (1 h). The mixture was then filtered through a small pad of silica gel, the filtrate was concentrated, and the residue was purified by preparative reverse-phase HPLC to provide the target amides 7a–c.
N-(3-Chloro-4-fluorobenzyl)-1-hydroxy-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxamide (7a)
Treatment of 19a as outlined in general procedure G and purification by preparative reverse-phase HPLC (linear gradient of 30% B to 50% B over 30 min; retention time = 22.5 min) provided 7a as a white solid in 41% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.42 (bs, 1H), 9.96 (t, J = 5.9 Hz, 1H), 8.85 (s, 1H), 8.82 (dd, J = 4.7, 1.8 Hz, 1H), 8.51 (dd, J = 7.9, 1.8 Hz, 1H), 7.58 (d, J = 7.8 Hz, 1H), 7.47 (dd, J = 7.8, 4.7 Hz, 1H), 7.40–7.37 (m, 2H), 4.58 (d, J = 6.0 Hz, 2H). ESI-MS m/z: 348.0 (M+H+). HRMS calcd C16H12ClFN3O3 [MH+], 348.0546; found, 348.0542.
N-(3,4-Difluorobenzyl)-1-hydroxy-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxamide (7b)
Treatment of 19b as outlined in general procedure G and purification by preparative reverse-phase HPLC (linear gradient of 20% B to 50% B over 30 min; retention time = 26.1 min) provided 7b as a white solid in 22% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.42 (bs, 1H), 9.96 (t, J = 6.0 Hz, 1H), 8.85 (s, 1H), 8.82 (dd, J = 4.7, 1.8 Hz, 1H), 8.52 (dd, J = 7.8, 1.8 Hz, 1H), 7.47 (dd, J = 7.8, 4.7 Hz, 1H), 7.45–7.37 (m, 2H), 7.23–7.22 (m, 1H), 4.58 (d, J = 6.1 Hz, 2H). ESI-MS m/z: 332.1 (M+H+). HRMS calcd C18H12F2N3O3 [MH+], 332.0841; found, 332.0828.
N-(2,4-Difluorobenzyl)-1-hydroxy-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxamide (7c)
Treatment of 19c as outlined in general procedure G and purification by preparative reverse-phase HPLC (linear gradient of 20% B to 80% B over 30 min; retention time = 19.2 min) provided 7c as a white solid in 52% yield. 1H NMR (400 MHz, DMSO-d6) δ 9.88 (t, J = 5.9 Hz, 1H), 8.78 (s, 1H), 8.75 (dd, J = 4.7, 1.8 Hz, 1H), 8.45 (dd, J = 7.9, 1.8 Hz, 1H), 7.46–7.34 (m, 2H), 7.19 (ddd, J = 10.6, 9.4, 2.6 Hz, 1H), 7.03–6.98 (m, 1H), 4.54 (d, J = 5.9 Hz, 2H). ESI-MS m/z: 332.1 (M+H+). HRMS calcd C16H12F2N3O3 [MH+], 332.0841; found, 332.0834.
Integrase Assays
IN reactions were performed as previously described.35 Briefly, reactions were carried out by adding compounds or an equivalent volume of 100% DMSO (used as solvent for the compounds) to a mixture of 20 nM [γ-32P]-labeled DNA and 400 nM IN in a buffer containing 50 mM morpholinepropanesulfonic acid (pH 7.2), 7.5 mM MgCl2, and 14 mM 2-mercaptoethanol. [γ-32P]-labeled full length oligonucleotide 21T (GTGTGGAAAATCTCTAGCAGT) or precleaved oligonucleotide 19T (GTGTGGAAAATCTCTAGCA), both annealed to the complementary oligonucleotide 21B (ACTGCTAGAGATTTTCCACAC), were used to measure both 3′-processing and strand transfer, respectively.35 Reactions were performed at 37 °C for 2 h and quenched by the addition of an equal volume of loading buffer (formamide containing 1% sodium dodecyl sulfate, 0.25% bromophenol blue, and xylene cyanol). Reaction products were separated in 16% polyacrylamide-denaturing sequencing gels. Dried gels were visualized using Typhoon 8600 (GE Healthcare, Piscataway, NJ). Densitometric analyses were performed using ImageQuant 5.1 software from GE Healthcare. The data analyses (linear regression, 50% inhibitory concentration [IC50] determination, and standard deviation [SD]) were performed from at least 3 independent determinations using Prism 5.0c software from GraphPad.
Cellular Cytotoxicity Assays
The human osteosarcoma cell line, HOS, was obtained from Dr. Richard Schwartz (Michigan State University, East Lansing, MI) and grown in Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA) supplemented with 5% (v/v) fetal bovine serum, 5% newborn calf serum, and penicillin (50 units/mL) plus streptomycin (50 μg/mL; Quality Biological, Gaithersburg, MD). On the day prior to the screen, HOS cells were seeded in a 96-well luminescence cell culture plate at a density of 4000 cells in 100 μL per well. On the day of the screen, cells were treated with compounds in the appropriate concentration range chosen and incubated at 37 °C for 48 h. Cytotoxicity was measured by monitoring ATP levels via a luciferase reporter assay. Cells were lysed in 50 μL of cell lysis buffer (PerkinElmer, Waltham, MA) and shaken at 700 rpm at room temperature for 5 min. After the addition of 50 μL of ATPlite buffer (PerkinElmer) directly onto the lysed cells and shaking at 700 rpm at room temperature (5 min), ATP levels were monitored by measuring luciferase activity using a microplate reader. Activity was normalized to cytotoxicity in the absence of target compounds. KaleidaGraph (Synergy Software, Reading, PA) was used to perform regression analysis on the data. CC50 values were determined from the fit model.
Single-Round HIV-1 Infectivity Assays
Human embryonic kidney cell line 293T was transfected with the pNLNgoMIVR-ΔLUC vector, which was made from pNLNgoMIVR-ΔEnv·HSA by removing the HSA reporter gene and replacing it with a luciferase reporter gene between the NotI and XhoI restriction sites.37 VSV-g-pseudotyped HIV was produced by transfections of 293T cells as described previously.38 On the day prior to transfection, 293T cells were plated on 100-mm-diameter dishes at a density of 1.5 × 106 cells per plate. 293T cells were transfected with 16 μg of pNLNgoMIVR-ΔLUC and 4 μg of pHCMV-g (obtained from Dr. Jane Burns, University of California, San Diego) using the calcium phosphate method. At approximately 6 h after the calcium phosphate precipitate was added, the 293T cells were washed twice with phosphate-buffered saline (PBS) and incubated with fresh media (48 h). The virus-containing supernatants were then harvested, clarified by low-speed centrifugation, filtered, and diluted for preparation in infection assays. On the day prior to the screen, HOS cells were seeded in a 96-well luminescence cell culture plate at a density of 4000 cells in 100 μL per well. On the day of the screen, cells were treated with the compounds from a concentration range of 10 μM to 0.0005 μM using 11 serial dilutions and then incubated at 37 °C (3 h). After this incubation, 100 μL of virus-stock diluted to achieve a maximum luciferase signal between 0.2 and 1.5 RLUs was added to each well, and the plates were incubated at 37 °C (48 h). Infectivity was measured by using the Steady-lite plus luminescence reporter gene assay system (PerkinElmer, Waltham, MA). Luciferase activity was measured by adding 100 μL of Steady-lite plus buffer (PerkinElmer) to the cells, incubating at room temperature (20 min), and measuring luminescence using a microplate reader. Activity was normalized to infectivity in the absence of target compounds. KaleidaGraph (Synergy Software, Reading, PA) was used to perform regression analysis on the data. EC50 values were determined from the fit model.
Vector Constructs
pNLNgoMIVR-ΔEnv·LUC has been described previously.37 The IN coding region was removed from pNLNgoMIVR-ΔEnv·LUC (between KpnI and SalI sites) and inserted between the KpnI and SalI sites of pBluescript II KS+. Using that construct as the wild-type template, we prepared the following IN-resistant mutants via the QuikChange II XL (Stratagene, La Jolla, CA) site-directed mutagenesis protocol: G118R, Y143R, Q148H, Q148K, N155H, G140S + Q148H, G140A + Q148K, and E138K + Q148K. The following sense with cognate antisense (not shown) oligonucleotides (Integrated DNA Technologies, Coralville, IA) was used in the mutagenesis: G118R, 5′-GTACATACAGACAATCGCAGCAATTTCACCAGTAC-3′; E138K, 5′-GGCGGGGATCAAGCAGAAATTTGGCATTCCCTA-3′; G140A, 5′-GGGGATCAAGCAGGAATTTGCCATTCCCTACAATC-3′; G140S, 5′-GGGGATCAAGCAGGAATTTAGCATTCCCTACAATC-3′; Y143R, 5′-GCAGGAATTTGGCATTCCCCGCAATCCCCAAAGTCAAGGA-3′; Q148H, 5′-CATTCCCTACAATCCCCAAAGTCATGGAGTAATAGAATCTA-3′; Q148K, 5′-CATTCCCTACAATCCCCAAAGTAAAGGAGTAATAGAATCTATGAA-3′; and N155H, 5′-CCAAAGTCAAGGAGTAATAGAATCTATGCATAAAGAATTAAAGAAAATTATAGGACA-3′. The double mutant G140S + Q148H was constructed by using the previously generated Q148H mutant and the appropriate oligonucleotide to introduce the second mutation, G140S. The double mutant G140A + Q148K was made by using the Q148K mutant and the appropriate oligonucleotide to introduce the second mutation, G140A. The double mutant E138K + Q148K was made by using the Q148K mutant and the appropriate oligonucleotide to introduce the second mutation, E138K. The DNA sequence of each construct was verified independently by DNA sequencing. The mutant IN coding sequences from pBluescript II KS+ were then subcloned into pNLNgoMIVR-ΔEnv·LUC (between the KpnI and SalI sites) to produce the full-length mutant HIV-1 IN constructs. These DNA sequences were additionally checked independently by DNA sequencing.
Acknowledgments
This work was supported in part by the Intramural Research Program of the NIH, Center for Cancer Research, Frederick National Laboratory for Cancer Research and the National Cancer Institute, National Institutes of Health and the Joint Science and Technology Office of the Department of Defense, and by funds from the Intramural AIDS Targeted Antiviral Program.
Glossary
Abbreviations Used
- HIV-1
human immunodeficiency virus type 1
- AIDS
acquired immune deficiency syndrome
- FDA
Food and Drug Administration
- IN
integrase
- RAL
Raltegravir
- EVG
Elvitegravir
- DTG
Dolutegravir
- 3′-P
3′-processing
- ST
strand transfer
- INSTIs
integrase strand transfer inhibitors
- DNA
deoxyribonucleic acid
- IC50
half-maximum inhibitory concentration
- EC50
half maximal effective concentration
- WT
wild type
- DMSO
dimethyl sulfoxide
- DMF
dimethylformamide
- HPLC
high-pressure liquid chromatography
- HRMS
high-resolution mass spectrometry
Author Present Address
∥ Laboratoire MFP, CNRS - UMR 5234, Université de Bordeaux, Bordeaux Cedex 33076, France
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Metifiot M.; Marchand C.; Pommier Y. HIV integrase inhibitors: 20-Year landmark and challenges. Adv. Pharmacol. 2013, 67, 75–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Santo R.Inhibiting the HIV integration process: past, present, and the future. J. Med. Chem. 2013, DOI: 10.1021/jm400674a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croxtall J. D.; Scott L. J. Raltegravir: In treatment-naive patients with HIV-1 infection. Drugs 2010, 70, 631–642. [DOI] [PubMed] [Google Scholar]
- Wills T.; Vega V. Elvitegravir: A once-daily inhibitor of HIV-1 integrase. Expert Opin. Invest. Drugs 2012, 21, 395–401. [DOI] [PubMed] [Google Scholar]
- Marchand C.; Maddali K.; Metifiot M.; Pommier Y. HIV-1 IN inhibitors: 2010 Update and perspectives. Curr. Top. Med. Chem. 2009, 9, 1016–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metifiot M.; Marchand C.; Maddali K.; Pommier Y. Resistance to integrase inhibitors. Viruses 2010, 2, 1347–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherepanov P.; Maertens G. N.; Hare S. Structural insights into the retroviral DNA integration apparatus. Curr. Opin. Struct. Biol. 2011, 21, 249–256. [DOI] [PubMed] [Google Scholar]
- Perryman A. L.; Forli S.; Morris G. M.; Burt C.; Cheng Y.; Palmer M. J.; Whitby K.; McCammon J. A.; Phillips C.; Olson A. J. A dynamic model of HIV integrase inhibition and drug resistance. J. Mol. Biol. 2010, 397, 600–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geretti A. M.; Armenia D.; Ceccherini-Silberstein F. Emerging patterns and implications of HIV-1 integrase inhibitor resistance. Curr. Opin. Infect. Dis. 2012, 25, 677–686. [DOI] [PubMed] [Google Scholar]
- Delelis O.; Thierry S.; Subra F.; Simon F.; Malet I.; Alloui C.; Sayon S.; Calvez V.; Deprez E.; Marcelin A.-G.; Tchertanov L.; Mouscadet J.-F. Impact of Y143 HIV-1 integrase mutations on resistance to raltegravir in vitro and in vivo. Antimicrob. Agents Chemother. 2009, 54, 491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quashie P. K.; Sloan R. D.; Wainberg M. A.. Novel therapeutic strategies targeting HIV integrase. BMC Med. 2012, 10 (34), DOI: 10.1186/1741-7015-10-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katlama C.; Murphy R. Dolutegravir for the treatment of HIV. Expert Opin. Invest. Drugs 2012, 21, 523–530. [DOI] [PubMed] [Google Scholar]
- Ballantyne A. D.; Perry C. M. Dolutegravir: first global approval. Drugs 2013, 73, 1627–1637. [DOI] [PubMed] [Google Scholar]
- Vandeckerckhove L. GSK-1349572, a novel integrase inhibitor for the treatment of HIV infection. Curr. Opin. Invest. Drugs 2010, 11, 203–212. [PubMed] [Google Scholar]
- Kawasuji T.; Johns B. A.; Yoshida H.; Taishi T.; Taoda Y.; Murai H.; Kiyama R.; Fuji M.; Yoshinaga T.; Seki T.; Kobayashi M.; Sato A.; Fujiwara T. Carbamoyl pyridone HIV-1 integrase inhibitors. 1. Molecular design and establishment of an advanced two-metal binding pharmacophore. J. Med. Chem. 2012, 55, 8735–8744. [DOI] [PubMed] [Google Scholar]
- Kawasuji T.; Johns B. A.; Yoshida H.; Weatherhead J. G.; Akiyama T.; Taishi T.; Taoda Y.; Mikamiyama-Iwata M.; Murai H.; Kiyama R.; Fuji M.; Tanimoto N.; Yoshinaga T.; Seki T.; Kobayashi M.; Sato A.; Garvey E. P.; Fujiwara T. Carbamoyl pyridone HIV-1 integrase inhibitors. 2. Bi- and tricyclic derivatives result in superior antiviral and pharmacokinetic profiles. J. Med. Chem. 2013, 56, 1124–1135. [DOI] [PubMed] [Google Scholar]
- Johns B. A.; Kawasuji T.; Weatherhead J. G.; Taishi T.; Temelkoff D. P.; Yoshida H.; Akiyama T.; Taoda Y.; Murai H.; Kiyama R.; Fuji M.; Tanimoto N.; Jeffrey J.; Foster S. A.; Yoshinaga T.; Seki T.; Kobayashi M.; Sato A.; Johnson M. N.; Garvey E. P.; Fujiwara T. Carbamoyl pyridone HIV-1 integrase inhibitors 3. A diastereomeric approach to chiral nonracemic tricyclic ring systems and the discovery of dolutegravir (S/GSK1349572) and (S/GSK1265744). J. Med. Chem. 2013, 56, 5901–5916. [DOI] [PubMed] [Google Scholar]
- Metifiot M.; Maddali K.; Johnson B. C.; Hare S.; Smith S. J.; Zhao X. Z.; Marchand C.; Burke T. R. Jr.; Hughes S. H.; Cherepanov P.; Pommier Y. Activities, crystal structures and molecular dynamics of dihydro-1H-isoindole derivatives, inhibitors of HIV-1 integrase. ACS Chem. Biol. 2013, 8, 209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangeetha B.; Muthukumaran R.; Amutha R. Pharmacophore modelling and electronic feature analysis of hydroxamic acid derivatives, the HIV integrase inhibitors. SAR QSAR Environ. Res. 2013, 24, 753–771. [DOI] [PubMed] [Google Scholar]
- Plewe M. B.; Butler S. L.; R D. K.; Hu Q.; Johnson T. W.; Kuehler J. E.; Kuki A.; Lam H.; Liu W.; Nowlin D.; Peng Q.; Rahavendran S. V.; Tanis S. P.; Tran K. T.; Wang H.; Yang A.; Zhang J. Azaindole hydroxamic acids are potent HIV-1 integrase inhibitors. J. Med. Chem. 2009, 52, 7211–7219. [DOI] [PubMed] [Google Scholar]
- Tang J.; Maddali K.; Metifiot M.; Sham Yuk Y.; Vince R.; Pommier Y.; Wang Z. 3-Hydroxypyrimidine-2,4-diones as an inhibitor scaffold of HIV integrase. J. Med. Chem. 2011, 54, 2282–2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson T. W.; Tanis S. P.; Butler S. L.; Dalvie D.; DeLisle D. M.; Dress K. R.; Flahive E. J.; Hu Q.; Kuehler J. E.; Kuki A.; Liu W.; McClellan G. A.; Peng Q.; Plewe M. B.; Richardson P. F.; Smith G. L.; Solowiej J.; Tran K. T.; Wang H.; Yu X.; Zhang J.; Zhu H. Design and synthesis of novel N-hydroxy-dihydronaphthyridinones as potent and orally bioavailable HIV-1 integrase inhibitors. J. Med. Chem. 2011, 54, 3393–3417. [DOI] [PubMed] [Google Scholar]
- Desimmie B. A.; Demeulemeester J.; Suchaud V.; Taltynov O.; Billamboz M.; Lion C.; Bailly F.; Strelkov S.; Debyser Z.; Cotelle P.; Christ F. 2-Hydroxyisoquinoline-1,3(2H,4H)-diones (HIDs), novel inhibitors of HIV integrase with a high barrier to resistance. ACS Chem. Biol. 2013, 8, 1187–1194. [DOI] [PubMed] [Google Scholar]
- Billamboz M.; Suchaud V.; Bailly F.; Lion C.; Demeulemeester J.; Calmels C.; Andreola M.-L.; Christ F.; Debyser Z.; Cotelle P. 4-Substituted 2-hydroxyisoquinoline-1,3(2H,4H)-diones as a novel class of HIV-1 integrase inhibitors. ACS Med. Chem. Lett. 2013, 4, 606–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryde D. C.; Webster R.; Butler S. L.; Murray E. J.; Whitby K.; Pickford C.; Westby M.; Palmer M. J.; Bull D. J.; Vuong H.; Blakemore D. C.; Stead D.; Ashcroft C.; Gardner I.; Bru C.; Cheung W.-Y.; Roberts I. O.; Morton J.; Bissell R. A. Discovery of an HIV integrase inhibitor with an excellent resistance profile. MedChemComm 2013, 4, 709–719. [Google Scholar]
- Zhao X. Z.; Maddali K.; Vu B. C.; Marchand C.; Hughes Stephen H.; Pommier Y.; Burke Terrence R. Jr. Examination of halogen substituent effects on HIV-1 integrase inhibitors derived from 2,3-dihydro-6,7-dihydroxy-1H-isoindol-1-ones and 4,5-dihydroxy-1H-isoindole-1,3(2H)-diones. Bioorg. Med. Chem. Lett. 2009, 19, 2714–2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flipo M.; Beghyn T.; Leroux V.; Florent I.; Deprez B. P.; Deprez-Poulain R. F. Novel selective inhibitors of the zinc plasmodial aminopeptidase PfA-M1 as potential antimalarial agents. J. Med. Chem. 2007, 50, 1322–1334. [DOI] [PubMed] [Google Scholar]
- Nicolaou K. C.; Xu J. Y.; Kim S.; Ohshima T.; Hosokawa S.; Pfefferkorn J. Synthesis of the tricyclic core of eleutherobin and sarcodictyins and total synthesis of sarcodictyin A. J. Am. Chem. Soc. 1997, 119, 11353–11354. [Google Scholar]
- Rylander P. N.; Karpenko I. M.; Pond G. R. Selectivity of hydrogenation over platinum metal catalysts: nitroaromatics. Ann. N.Y. Acad. Sci. 1970, 172, 266–275. [Google Scholar]
- Takenaka Y.; Kiyosu T.; Choi J.-C.; Sakakura T.; Yasuda H. Selective synthesis of N-aryl hydroxylamines by the hydrogenation of nitroaromatics using supported platinum catalysts. Green Chem. 2009, 11, 1385–1390. [Google Scholar]
- Zhao X. Z.; Maddali K.; Smith S. J.; Metifiot M.; Johnson B.; Hughes S. H.; Pommier Y.; Burke T. R. Jr. 6,7-Dihydroxy-1-oxoisoindoline-4-sulfonamide-containing HIV-1 Integrase Inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 7309–7313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams P. D.; Venkatraman S.; Langford H. M.; Kim B.; Booth T. M.; Grobler J. A.; Staas D.; Ruzek R. D.; Embrey M. W.; Wiscount C. M.; Lyle T. A.. Preparation of 1-Hydroxynaphthyridin-2(1H)-one Derivatives as Anti-HIV Agents. Patent WO2008010964(A1).
- Zhao X. Z.; Semenova E. A.; Vu B. C.; Maddali K.; Marchand C.; Hughes S. H.; Pommier Y.; Burke T. R. Jr. 2,3-Dihydro-6,7-dihydroxy-1H-isoindol-1-one-based HIV-1 integrase inhibitors. J. Med. Chem. 2008, 51, 251–259. [DOI] [PubMed] [Google Scholar]
- Malet I.; Delelis O.; Valantin M.-A.; Montes B.; Soulie C.; Wirden M.; Tchertanov L.; Peytavin G.; Reynes J.; Mouscadet J.-F.; Katlama C.; Calvez V.; Marcelin A.-G. Mutations associated with failure of raltegravir treatment affect integrase sensitivity to the inhibitor in vitro. Antimicrob. Agents Chemother. 2008, 52, 1351–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metifiot M.; Maddali K.; Naumova A.; Zhang X.; Marchand C.; Pommier Y. Biochemical and pharmacological analyses of HIV-1 integrase flexible loop mutants resistant to raltegravir. Biochemistry 2010, 49, 3715–3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesplede T.; Quashie P. K.; Wainberg M. A. Resistance to HIV integrase inhibitors. Curr. Opin. HIV AIDS 2012, 7, 401–408. [DOI] [PubMed] [Google Scholar]
- Zhao X. Z.; Maddali K.; Metifiot M.; Smith S. J.; Vu B. C.; Marchand C.; Hughes S. H.; Pommier Y.; Burke T. R. Jr. Bicyclic hydroxy-1H-pyrrolopyridine-trione containing HIV-1 integrase inhibitors. Chem. Biol. Drug Des. 2012, 79, 157–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachmann A.; Koenig J.; Julius C.; Feldbruegge M. A reverse genetic approach for generating gene replacement mutants in Ustilago maydis. Mol. Genet. Genomics 2004, 272, 216–226. [DOI] [PubMed] [Google Scholar]