

Abstract
Coronaviruses (CoVs) cause numerous diseases, including Middle East respiratory syndrome and severe acute respiratory syndrome, generating significant health-related and economic consequences. CoVs encode the nucleocapsid (N) protein, a major structural protein that plays multiple roles in the virus replication cycle and forms a ribonucleoprotein complex with the viral RNA through the N protein’s N-terminal domain (N-NTD). Using human CoV-OC43 (HCoV-OC43) as a model for CoV, we present the 3D structure of HCoV-OC43 N-NTD complexed with ribonucleoside 5′-monophosphates to identify a distinct ribonucleotide-binding pocket. By targeting this pocket, we identified and developed a new coronavirus N protein inhibitor, N-(6-oxo-5,6-dihydrophenanthridin-2-yl)(N,N-dimethylamino)acetamide hydrochloride (PJ34), using virtual screening; this inhibitor reduced the N protein’s RNA-binding affinity and hindered viral replication. We also determined the crystal structure of the N-NTD–PJ34 complex. On the basis of these findings, we propose guidelines for developing new N protein-based antiviral agents that target CoVs.
Introduction
Coronaviruses (CoVs) are a large group of RNA viruses with single-stranded RNA genomes that cause various upper and lower respiratory tract infections in both humans and animals.1,2 The human coronavirus strains OC43 and 229E (HCoV-OC43 and HCoV-229E) were identified in the 1960s.3 Between 2003 and 2004, the severe acute respiratory syndrome coronavirus (SARS-CoV) caused a worldwide epidemic and had a significant economic impact in the countries affected by the outbreak.4 In 2004, another alphacoronavirus (HCoV-NL63) was isolated from a 7-month-old child in the Netherlands suffering from bronchiolitis and conjunctivitis.5 In 2005, Woo et al. discovered the novel betacoronavirus HKU1 in patients with respiratory tract infections.6 Recently, the Middle East respiratory syndrome coronavirus (MERS-CoV) was found in patients with severe acute respiratory tract infections in the Middle East. As is true for all coronavirus infections, there is no currently available efficacious therapy.
The CoVs have several conserved structural proteins: the matrix (M), the small envelope (E) proteins, the trimeric spike (S) glycoproteins, and the nucleocapsid (N) proteins.7 Some variants have a third glycoprotein, HE (hemagglutinin esterase), which is present in most betacoronaviruses.8,9 The N protein is a major structural CoV protein that serves multiple purposes, such as packaging the RNA genome into helical ribonucleoproteins, modulating host cell metabolism, and regulating viral RNA synthesis during replication and transcription.10,11 The N protein binds to the viral RNA genome, forming a long helical nucleocapsid structure or ribonucleoprotein (RNP) complex.12 In situ cross-linking and immunological experiments revealed that the RNP formation is critical for maintaining an ordered RNA conformation suitable for replicating and transcribing the viral genome.10,13 Other studies implicate the N protein in the regulation of cellular processes, including actin reorganization, host cell cycle progression, and apoptosis.14−16 The N protein is capable of inducing protective immune responses against CoV and is a key antigen for developing a sensitive diagnostic assay.17
Coronavirus N proteins contain three domains: an N-terminal RNA-binding domain (NTD), a C-terminal dimerization domain (CTD), and a poorly structured central Ser/Arg (SR)-rich linker. Previous studies have revealed that the N- and C-terminal domains of the CoV N proteins are responsible for RNA binding and oligomerization, respectively.18−20 The central region of the N protein has also been shown to contain an RNA-binding region and primary phosphorylation sites.21,22 The crystal structures of several CoV N-NTDs, including those encoded by SARS,23 infectious bronchitis virus (IBV),24,25 HCoV-OC43,20 and mouse hepatitis virus (MHV)26 have been solved. Additionally, several critical residues have been identified for RNA binding and virus infectivity in the N-terminal domain of coronaviral N proteins.23,26−28 However, the structural and mechanistic basis for RNA binding and RNP formation remains largely unknown. Understanding these aspects should facilitate the discovery of agents that specifically block the formation of RNP during CoV genome replication. We report the crystal structures of HCoV-OC43 N-NTD complexed with ribonucleoside 5′-monophosphates as a model for understanding the molecular interactions that govern CoV N-NTD binding to RNA. We also describe the structure of HCoV-OC43 N-NTD complexed with a new N protein inhibitor, N-(6-oxo-5,6-dihydrophenanthridin-2-yl)(N,N-dimethylamino)acetamide hydrochloride (PJ34), and demonstrate the ability of PJ34 to interfere with both the RNA-binding activity of the N protein and virus replication. Our findings will aid in the development of new drugs that interfere with viral N proteins and viral replication in HCoVs.
Results
Cocrystal Structure of the HCoV-OC43 N-NTD with AMP
We were unable to find any previous reports describing the atomic structure of CoV N protein–RNA complexes. To begin to elucidate how RNA and the N protein interact, we determined the crystal structure of HCoV-OC43 N-NTD complexed with AMP. The complete statistics for the data collection and refinement of HCoV-OC43 N-NTD complexed with AMP are summarized in Table 1. The complex contained one ribonucleoside 5′-monophosphate-binding site alongside two β strands (β2 and β3) (Figure 1A). As with apo HCoV-OC43 N-NTD, the HCoV-OC43 N-NTD complexes all contain a core located within amino acids 105–120 that comprises five β strands, one α helix, and a disordered loop that extends away from the core. The flexible loop region lies between β2 and β3 (residues 115–117) and exhibits a low electron density in the initial 2Fo–Fc map. The AMP base was inserted into a hole in the N-NTD that was almost perpendicular to the phosphate moiety (Figure 1B,C). The phosphate group was bound to a basic and conserved 5′-phosphate-binding site that contained the largest positively charged region on the N-NTD surface. The HCoV-OC43 N-NTD has the same folding pattern as is found in the SARS-CoV, IBV, and MHV N-NTD;20 however, the positions of the secondary structural elements and loops vary between the species.
Table 1. Data Collection and Refinement Statistics for HCoV-OC43 N-NTD–AMP and HCoV-OC43 N-NTD–PJ34 Crystals.
| HCoV-OC43 N-NTD–AMP | HCoV-OC43 N-NTD–PJ34 | |
|---|---|---|
| PDB code | 4LI4 | 4KXJ |
| space group | P65 | P65 |
| resolution (Å) | 30–1.71 (1.77–1.71)a | 30–2.65 (2.74–2.65)a |
| wavelength (Å) | 1.00000 | 1.00000 |
| a = b (Å) | 81.919 | 81.684 |
| c (Å) | 42.892 | 42.950 |
| no. of obsd reflns | 124447 | 33506 |
| no. of unique reflns | 17943 | 4854 |
| completeness (%) | 97.8(100.0)a | 99.7(99.8)a |
| Rmerge (%) | 2.6(16.4)a | 8.2(42.8)a |
| I/σ(I) | 55.0(12.64)a | 26.4(5.7)a |
| refinement | ||
| no. of reflns | 17501 | 4235 |
| Rwork (95% data) | 0.22 | 0.18 |
| Rfree (5% data) | 0.25 | 0.22 |
| bond lengths (Å) | 0.008 | 0.013 |
| bond angles (deg) | 1.485 | 2.032 |
| no. of protein atoms | ||
| mean B value (Å2) | 33.54 | 32.35 |
| no. of ligand atoms | ||
| mean B value (Å2) | 20.24 | 24.09 |
| no. of water molecules | ||
| mean B value (Å2) | 37.63 | 29.3 |
| Ramachandran statistics (%) | ||
| most favored region | 94.5 | 91.5 |
| generally allowed region | 2.4 | 6.2 |
| others | 3.1 | 2.3 |
Values in parentheses are for the highest resolution shells.
Figure 1.

Structural overview of the HCoV-OC43 N-NTD–AMP complex. (A) Ribbon representation of HCoV-OC43 N-NTD with AMP depicted as a stick structure. (B) Electrostatic surface of the OC43 N-NTD–AMP complex. Blue denotes positive charge potential, while red indicates negative charge potential. (C) Map of the conserved surfaces of selected CoV N-NTDs (see Figure S1, Supporting Information).
AMP binding to the N-NTD is clearly unambiguously defined in the resulting electron density maps provided in Figure 2A. AMP shows a temperature factor of about 20 Å2, compared with an average overall temperature factor of around 35 Å2. Figure 2B reveals the detailed interactions between the AMP and HCoV-OC43 N-NTD. The amino acid composition of this binding site includes Ser 64, Gly 68, Arg 122, Tyr 124, Tyr 126, and Arg 164. The positively charged group in the Arg 122 side chain provides an ionic interaction with the AMP monophosphate with a distance of 3.8 Å, whereas the Gly 68 backbone forms hydrogen bonds with the monophosphate group in the AMP with a distance of 2.4 Å. Additionally, the carbonyl oxygen and amide nitrogen of the Ser 64 backbone form hydrogen bonds with the ribose 2′-hydroxyl substituents and N7 of the base with distances of 3.0 and 2.7 Å, respectively. Tyr 124 is located on the surface of the N protein in the HCoV-OC43 N-NTD and is directly involved in the interactions with the AMP base through π–π stacking. The phenolic hydroxyl group substituent on Tyr 126 forms hydrogen bonds with the sixth amino groups present in the AMP adenine ring with a distance of 3.1 Å. Hydrogen bonds also form between the 2′-hydroxyl group of the AMP ribose and the Arg 164 side chain. The Arg 122, Tyr 124, Tyr 126, and Arg 164 side chains generate a distinct ribonucleotide-binding pocket and interact with the ribonucleoside 5′-monophosphate via hydrogen bonding, ionic bonding, and π–π stacking forces (Figure 2C). These amino acids are sequentially and structurally conserved in other HCoV N proteins (Figure S2, Supporting Information); therefore, they are likely essential for RNA recognition and interaction in all coronavirus N proteins. In addition, the structure of the N-NTD in the AMP co-complex is essentially identical to the previously published structure of apo HCoV-OC43 N-NTD with a root-mean-square deviation (RMSD) value of 0.19 Å (123 equivalent Cα atoms) (Figure 2D). Only the phenyl group of F57 is displaced backward by 1 Å to prevent steric hindrance at the AMP entrance. We solved the structures of three additional HCoV-OC43 N-NTD complexes (cytosine monophosphate (CMP), guanosine monophosphate (GMP), and uridine monophosphate (UMP)), all featured protein–RNA interactions similar to the interaction of the HCoV-OC43 N-NTD–AMP complex. See Figure S3 in the Supporting Information. A comparison of the amino acid composition of ribonucleoside 5′-monophosphate-binding sites in the HCoV-OC43 N-NTD complexes (Figure S3D) shows that amino acid residues Ser 64, Phe 66, Gly 68, Arg 122, Tyr 124, Tyr 126, and Arg 164 are interactive in more than two HCoV-OC43 N-NTD complex structures, indicating their importance in RNA binding. Compared to AMP, the higher B-factors of CMP, GMP, and UMP indicate the ligand for CMP, GMP, and UMP is quite flexible in the ribonucleotide-binding pocket of HCoV-OC43 N-NTD (Table 1; Table S1, Supporting Information). On the basis of the electron density maps of similar resolution data, it appeared that the structure of the AMP complex showed better defined electron density for all components of the nucleotide (Figure 2A). These results suggest that this pocket is probably specific for AMP. More studies are needed to prove this in the future.
Figure 2.

Crystal structure of HCoV-OC43 N-NTD complexed with AMP. (A) Unbiased differences in the AMP electron density contoured at 2.9σ. (B) Detailed stereoview of the interactions at the AMP-binding site. The AMP molecule binds to this site via Ser 64, Gly 68, Arg 122, Tyr 124, Tyr 126, and Arg 164. The dotted green lines indicate hydrogen bonds. The red dashed lines indicate ionic interactions. (C) Schematic diagram of the AMP bound to the HCoV-OC43 N-NTD. The hydrogen-bonding interactions mediated by the side- and main-chain atoms are displayed as solid and dashed green lines, respectively. The ionic interactions mediated by the side-chain atoms are displayed as dashed red lines. The stacking interactions mediated by the side-chain atoms are indicated by the solid orange lines. (D) Structural superimposition of the native HCoV-OC43 N-NTD (green) with HCoV-OC43 N-NTD–AMP (cyan) at the residues involved in the AMP binding.
RNA-Binding Activity Analyses of Wild-Type and Mutant HCoV-OC43 N Proteins
We replaced amino acid residues Arg 122, Tyr 124, Tyr 126, and Arg 164 with alanine and used surface plasmon resonance (SPR) analysis to determine their interactions in the binding between the full-length HCoV-OC43 NPs and RNA. Depending on the virus strain, there are two to four UCUAA pentanucleotide repeats, with the last repeat being UCUAAAC and termed the intergenic (IG) sequence at the 3′ end of the leader.29,30 Previous studies showed that HCoV N protein has high affinity for the intergenic sequence.11,31 Therefore, the repeated intergenic sequence of HCoV-OC43, 5′-bio(UCUAAAC)4-3′, was used as a probe in our SPR experiments. The association constants, Kd (kd/ka), for the various HCoV-OC43 N protein and RNA complexes were obtained from kinetic analyses of SPR experiments (Table 2). The dissociation constants for RNA binding to R122A, Y124A, Y126A, and R164A range from 3.83 × 10–8 to 8.13 × 10–8 M and are much larger than those for the wild type (WT). Thus, we identified several amino acids in the HCoV-OC43 N protein that are important for RNA binding, especially R122, Y124, and R164. Previously, Keane et al. reported that R127 and Y127 in MHV, which correspond to R122 and Y124 in HCoV-OC43, play key roles in RNA binding.23,26,27 In addition, the alanine substitution of Y94 in the NTD of the IBV N protein, which corresponds to Y126 in HCoV-OC43, led to a significant decrease in its RNA-binding affinity. These results are consistent with our observations.
Table 2. Numerical Kd Valuesa Obtained from the Kinetic Analysis of the SPR Experiments Examining Binding of HCoV-OC43 WT and Mutant N Proteins to RNA.
| Kd (10–8 M) | Kd (10–8 M) | ||
|---|---|---|---|
| wild type | 1.17 ± 0.08 | Y124A | 8.13 ± 0.59 |
| R122A | 7.19 ± 0.57 | Y126A | 3.83 ± 0.25 |
| R164A | 7.09 ± 0.45 |
Kd value obtained from kd divided by ka.
Since R122A, Y124A, and R164A mutants have significant effects on the RNA-binding activity of the N protein, we monitored levels of viral RNA encoding the M protein to determine the effects of R122A, Y124A, and R164A mutants on the viral replication. Because all OC43 genes are transcribed concordantly throughout HCoV-OC43 infection, levels of the M protein gene should reflect virus replication. Therefore, 293T cells were transfected with plasmids encoding the mutant N protein and its WT counterpart followed by infection with HCoV-OC43. As shown in Figure S4 (Supporting Information), in cells transfected with plasmids encoding the mutant N protein and infected with virus, levels of M RNA were significantly decreased compared to those detected in cells transfected with plasmids encoding the WT N protein. These results support the notion that these amino acids of HCoV-OC43 are important for RNA-binding affinity.
Effect of PJ34 on Virus Replication and the RNA-Binding Affinity of the N Protein
Next, a virtual screening was performed, targeting the AMP-binding site of N-NTD. Potential hits with high docking scores (87 compounds) (Table S2, Supporting Information) were further analyzed on the basis of these docking results. We found that nine of the potential hits showed interaction characteristics reminiscent of those between AMP and HCoV-OC43 N-NTD (Table S3, Supporting Information). First, they all contain an aromatic core able to stack onto Y124 of the N-NTD. Second, the aromatic core contains hydrogen-bond-forming moieties to mediate the specific interactions with the N-NTD. Third, the aromatic core contains an attached branching moiety (or moieties) to fit into the ribonucleotide-binding pocket. More importantly, among the 87 potential hits, these 9 compounds were readily available commercially. We further studied the effects of the nine compounds on the RNA-binding capacity of N protein by SPR experiments. Two compounds, O3 and PJ34, decreased the RNA-binding capacity of N protein by more than 10% (Table S3). Because PJ34 and O3 were predicted to bind to the N-NTD ribonucleotide-binding pocket, we next studied PJ34 and O3 in virus replication assays. To enhance virus replication, we transfected N cDNA into cells prior to infection because previous results showed that increased N protein expression enhanced replication.20 In general, a 10 μM concentration of each candidate compound was used in subsequent assays. If the candidate compound at this concentration was effective, we considered the compound worthy of continued development. We monitored M mRNA levels in infected 293T cells in the presence of PJ34 and O3 at 10 μM both with and without exogenous N protein expression. Because the M gene is transcribed throughout the HCoV-OC43 infection, M mRNA levels should reflect viral replication. As predicted from previous studies, M mRNA levels were increased 7-fold in infected cells transfected with plasmid that encodes the WT N protein compared to those that did not receive transfected N cDNA (2.41 and 0.32 in the presence and absence of N protein expression, respectively) (Figure 3A,B). In the PJ34-treated cells, M mRNA levels were 0.65 and 0.11 in the presence and absence of N protein expression, respectively, while in the O3-treated cells, corresponding M mRNA levels were 2.46 and 0.22. Thus, M mRNA levels were reduced both in the presence and in the absence of N protein expression after treatment with PJ34 at 10 μM (Figure 3A,B), whereas O3 treatment was not effective. PJ34 has been shown to have therapeutic efficacy in several noninfectious conditions. In one instance, PJ34 treatment reduced central nervous system inflammation and maintained neurovascular integrity in mice during the onset of experimental allergic encephalomyelitis.32 This compound also exhibited neuroprotective effects in both in vivo and in vitro stroke models.33 To determine whether PJ34 hindered the HCoV-OC43 replication by interfering with the binding of the N protein to RNA, we used SPR to determine the effect of compound PJ34 on the RNA-binding affinity toward HCoV-OC43 N protein. In the presence of PJ34 under saturation conditions, RNA affinity for HCoV-OC43 NP decreased with decreasing resonance unit (RU) values for PJ34 (Figure 3C). The HCoV-OC43 N protein exhibited weaker RNA binding in the presence of PJ34 with a 5-fold increase in the dissociation constant (Figure 3D). Therefore, PJ34 antagonizes the binding activity between HCoV-OC43 N protein and RNA. Consequently, the data indicate that PJ34 interacts with the HCoV-OC43 N protein, decreasing RNA binding and subsequently decreasing viral replication.
Figure 3.

Virus replication is inhibited by PJ34 with (A) and without (B) exogenous wild-type HCoV-OC43 N protein expression. (A) Cells were transfected with a plasmid, pcDNA3.1, encoding the WT N protein or not (B) prior to being infected with HCoV-OC43 as described in the Experimental Section. The samples were subsequently analyzed for their matrix protein (MP) gene transcript levels in the vehicle-treated, PJ34-treated, and O3-treated cells. (B) No transfection. Quantitative data are reported as the means ± SD, n = 3. (C) Sensorgram of the interaction between the immobilized single-stranded RNA and full-length HCoV-OC43 N proteins in the presence of PJ34 at 10 μM. (D) Kinetic analyses expressed as the dissociation constants for HCoV-OC43 N proteins binding to RNA with and without PJ34. The N protein:drug molar ratio was 1:100.
Crystal Structure of HCoV-OC43 N-NTD Complexed with PJ34
To determine the mechanism of PJ34 (Figure 4A) binding to the HCoV-OC43 N protein, N-NTD crystals were soaked in PJ34 under the conditions described in the Experimental Section. We used molecular replacement to resolve the HCoV-OC43 N-NTD–PJ34 complex structure at a 2.65 Å resolution and refined this model to an Rwork:Rfree ratio of 18%:22% (Table 1). The complex revealed additional unbiased density around the ribonucleotide-binding pocket of the N-NTD, suggesting that the affinity of N-NTD for PJ34 was sufficiently high to inhibit the RNA-binding affinity of N protein (Figure 4B). As with the HCoV-OC43 N-NTD–AMP complex, one PJ34-binding site was noted near the β2 and β3 strands (Figure 4C). The complex adopted a U-shaped β platform that contained five β strands across the structure and resembled the N protein NTDs in other CoVs.23−26 On the basis of the surface charge distribution, the polycyclic ring of PJ34 intercalates into the N-NTD hole parallel to the long axis of the protein structure (Figure 4D).
Figure 4.
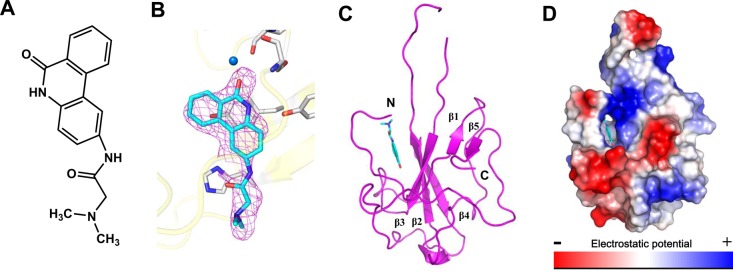
(A) Chemical structure of PJ34. Structural overview of the HCoV-OC43 N-NTD–PJ34 complex. (B) Unbiased difference electron density of PJ34 contoured at 2.9σ. (C) Ribbon representation of the HCoV-OC43 N-NTD with PJ34 depicted as a stick model. (D) Electrostatic surface of the HCoV-OC43 N-NTD–PJ34 complex. Blue denotes positive charge potential, while red indicates negative charge potential.
Parts A and B of Figure 5 reveal the detailed interactions with PJ34. The amino acid composition of this binding site includes Ser 64, Phe 66, Tyr 124, Tyr 126, and His 104. The NH functionalities from the backbone amide groups on Ser 64 are 3.3 Å from the carbonyl group on the PJ34 6-phenanthridinone ring, indicating that a hydrogen bond may form between Ser 64 and PJ34. Hydrogen bonds also form between the 6-phenanthridinone ring and the backbone carbonyl group of Phe 66 via water molecules. The nitrogen atom in the PJ34 6-phenanthridinone ring also forms a single hydrogen bond with the Tyr 126 side chain of the HCoV-OC43 N-NTD with a distance of 2.9 Å. The aromatic ring on the PJ34 6-phenanthridinone participates in stacking interactions with the His 104 and Tyr 124 side chains. A comparison between the N-NTD in both its native and PJ34-complexed forms generated a low RMSD of 0.20 Å, indicating that binding with PJ34 requires no significant conformational change in the N-NTD (Figure 5C). The key PJ34-interactive residues of the native and complexed forms superimpose well; however, the phenyl group on the Phe 57 side chain in the N-NTD–PJ34 complex rotates over 90°, and the imidazole side group of His 104 moves back 0.8 Å to avoid steric hindrances and to accommodate PJ34.
Figure 5.
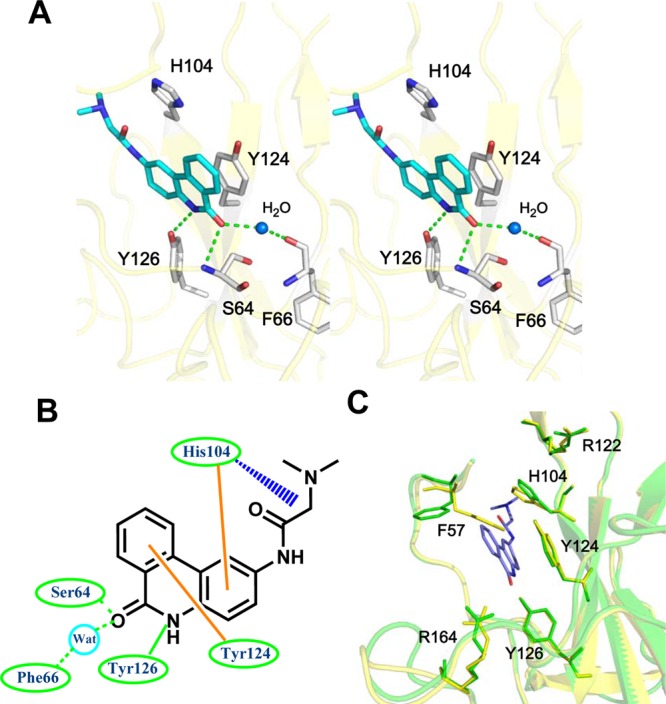
Crystal structure of the HCoV-OC43 N-NTD complexed with PJ34. (A) Detailed stereoview of the interactions at the PJ34-binding site. The PJ34 molecule binds to this site via Ser 64, Phe 66, His 104, Tyr 124, and Tyr 126. The dotted green lines denote hydrogen bonds. The red dashed lines indicate van der Waals interactions. (B) Schematic diagram of PJ34 bound to HCoV-OC43 N-NTD. The hydrogen-bonding interactions mediated by the side- and main-chain atoms are marked as solid and dashed green lines, respectively. The van der Waals interactions mediated by the side-chain atoms are denoted as blue dashed lines. The stacking interactions mediated by the side-chain atoms are marked as solid orange lines. (C) Structural superimposition of the native HCoV-OC43 N-NTD (green) with HCoV-OC43 N-NTD–PJ34 (cyan) at the residues involved in PJ34 binding.
Comparison of Crystal Structures of HCoV-OC43 N-NTD Complexed with PJ34 and AMP
Comparison of the PJ34- and AMP-bound HCoV-OC43 N-NTD crystal structures demonstrates that PJ34 and AMP target the same pocket within HCoV-OC43 N-NTD. Although the HCoV-OC43 N-NTD–PJ34 and HCoV-OC43 N-NTD–AMP crystal structures superimpose with a low RMSD of 0.21 Å, the PJ34-binding orientation differs from that of AMP. PJ34 binds more closely to the N-terminus loop of the HCoV-OC43 N-NTD than does AMP, with the Phe 57 side chain rotating 90° counterclockwise upon PJ34 binding, compared to that of the AMP-bound HCoV-OC43 N-NTD. The branch moiety of PJ34 inserts into an interior core of N-NTD that is opposite the ribose moiety of AMP coming from the inside out. PJ34 also lacks a phosphate group and fails to match AMP’s interactions with the positively charged Arg 122 (Figure S5, Supporting Information). Nevertheless, several of the examined interactions between the N-NTD and PJ34 were similar to those between N-NTD and AMP, particularly those with Ser 64, Tyr 124, and Tyr 126 (Figures 2C and 5B).
Discussion
The N protein is the most abundant viral polypeptide in CoV-infected cells and is responsible for recognizing RNA and forming a filamentous nucleocapsid.18 Because CoVs are significant threats to both humans and domestic animals, understanding the molecular mechanisms governing RNP formation may facilitate better management of CoV infections. Previous X-ray analysis revealed that the folding of the N protein’s N-terminal domain is essentially conserved across various CoV strains20,24,26,34 with a right-handed fist-shaped structure in which the palm and finger are rich in basic residues, while the flexible loops remain ordered around the β-sheet core of the NTD, possibly providing a scaffold for RNA binding. X-ray diffraction analyses of RNA-binding proteins complexed with ribonucleoside monophosphate have been used in several studies to identify the unique ribonucleotide-binding site in the RNA-binding domain.35,36 Here, we report an N-NTD–ribonucleoside 5′-monophosphate complex crystal structure that comprises a pocket for accommodation of ribonucleotide binding. On the basis of the structures of the N-NTD–ribonucleotide complex, two tyrosine residues on HCoV-OC43 NTD (Tyr 124 and Tyr 126) were found to interact with RNA bases via stacking and hydrogen-bonding interactions, respectively. Similar interactions were observed in complexes between vesicular stomatitis virus nucleoprotein and RNA.37 In addition, two arginine residues of HCoV-OC43 NTD, Arg 122 and Arg 164, interact with the phosphate group and ribose though ionic and hydrogen-bonding interactions, respectively. These four residues were conserved in other HCoVs as well, and the results suggest that the ribonucleotide-binding pocket of the HCoV N-NTD exists among different CoVs (Figure S6, Supporting Information). No structural data are available regarding CoV N protein binding to single-stranded RNA. To understand the structural interactions responsible for the RNA recognition by HCoV-OC43 N-NTD, we modeled the structure of HCoV-OC43 N-NTD in an RNA-bound state using the crystal structure of the N-NTD–AMP complex as a template (Figure S7, Supporting Information). Previous studies indicated that the positively charged amino acid, Arg 106, located at the cleft in the HCoV-OC43 N-NTD structure, is conserved in all CoV N proteins and interacts nonspecifically with the RNA phosphate backbone.20 This model indicates that the RNA-binding region of the N-NTD contains Arg 106, Arg 122, Tyr 124, Tyr 126, and Arg 164 and expands from the β-sheet core to the exterior loop region. A previous study showed that other conserved positively charged residues in the positively charged loop of HCoV-OC43 N protein, including R107, K110, and R117, were also involved in RNA binding.20
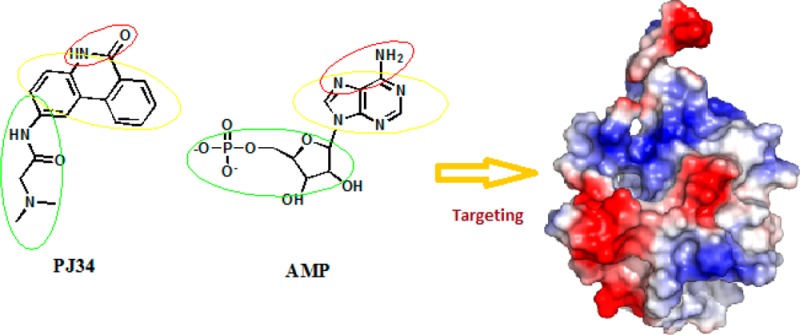
Current antiviral drugs developed to treat CoV infections primarily target the 3C-like (3CL) and papain-like (PLP) proteases.38 However, antiviral protease inhibitors may nonspecifically act on the cellular homologous protease, resulting in host cell toxicity and severe side effects. Therefore, novel antiviral strategies are needed to combat acute respiratory infections caused by CoV. The CoV nucleocapsid protein is a multifunctional RNA-binding protein that is necessary for viral RNA transcription and replication. Recent studies suggest that N proteins in infections caused by coronaviruses and other viruses will be useful antiviral drug targets because they serve many critical functions during the viral life cycle. Two strategies to inhibit oligomeric N protein function have been reported.39 The first strategy is to impair normal N protein function by interfering with monomer–oligomer equilibrium through either enhancement or inhibition of its oligomerization. The second one is to target the RNA-binding site, which contains a number of conserved residues. In one study, nuclozin analogues were shown to inhibit influenza A virus replication by preventing RNP formation during viral particle production.40 Lo et al. identified an antiviral peptide that interferes with the CTD oligomerization of the HCoV N protein and inhibits HCoV.19 The results presented herein provide a detailed, high-resolution picture of the ribonucleotide monophosphate bound to the CoV N-NTD and identify a unique ribonucleotide-binding pocket in the center of the CoV N-NTD. Mutation of RNA-binding residues in the NTD of the coronaviral N protein led to a significant decrease in its RNA-binding affinity and subsequent decrease in viral replication. Therefore, the N-terminal RNA-binding domain of coronaviral N protein would be a validated target for broad-spectrum antiviral drugs through interference with the RNA-binding activity of the N protein. Compounds binding to this site that act as competitive N protein inhibitors may be employed to combat highly pathogenic CoVs. PJ34 has been reported to protect mice against brain ischemia, splanchnic ischemia, reperfusion, and lipopolysaccharide (LPS) toxicity, in addition to various models of local inflammation.41 We also found that the cell viability was not affected by treatment with PJ34 alone up to 20 μM for 24 h in cell lines. Therefore, the efficacy of PJ34 is relatively diverse while its safety is high, making PJ34 an ideal new candidate for antiviral therapy. We found that PJ34 at 10 μM inhibits coronavirus replication and potently interferes with the RNA-binding activity of HCoV OC43 N protein by targeting the N-NTD ribonucleotide-binding pocket. On the other hand, O3 did not abolish HCoV-OC43 viral replication, likely because O3 is closer to the HCoV-OC43 N-NTD disordered loop than is AMP, which hinders any orientation suitable for the formation of a hydrogen bond network with the β1 strand, based on the docking results. Moreover, since O3 is as a potent inhibitor of CDC25 protein phosphatases,42,43 most likely some O3 molecules will bind CDC25 and consequently lose the ability to inhibit HCoV-OC43 N-NTD interactions. On the basis of the mechanisms of action of compounds such as AMP and PJ34 and the chemical features common to these two distinct compound classes, we formulated three general guidelines for developing CoV N-NTD-targeting agents: First, a polycyclic aromatic core is required to enable π–π stacking with the tyrosine residues in the N-NTD. Second, introducing hydrogen-bond-forming moieties to the aromatic core mediates specific interactions with the N-NTD. Third, attaching a branching moiety (or moieties) that fits the ribonucleotide-binding pocket can enhance the drug affinity and specificity (Figure 6).
Figure 6.
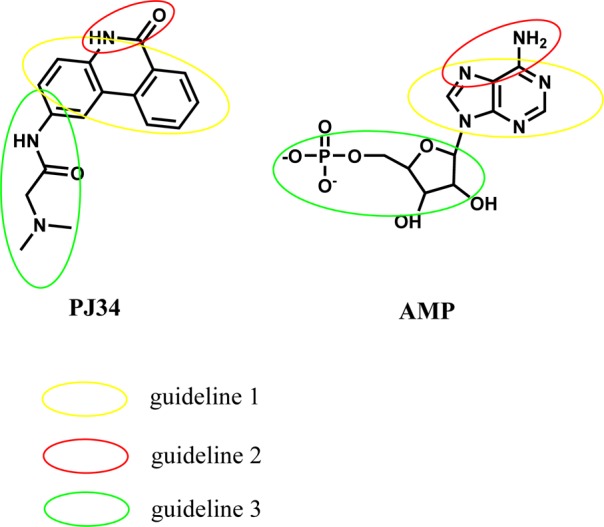
Three general guidelines deduced from the molecular structures of PJ34 and AMP.
In summary, we reported the crystal structures of the CoV N-NTD in complex with five ligands, AMP, CMP, GMP, UMP, and PJ34. These structures not only advance our understanding of the RNA-binding mechanisms of CoV N-NTD and illustrate the conformational landscapes of drug-binding pockets, but will also guide the design of novel antiviral agents useful for treating pathogenic HCoV infections.
Experimental Section
Chemicals
The drugs and reagents supplied in ≥95.0% purity as determined by HPLC were purchased from Sigma Chemical Co. (St. Louis, MO) and used without further purification.
Cloning, Protein Expression, and Purification
The HCoV-OC43 N-NTD gene expression and protein purification were performed according to previously described methods.44 The pET28a/N-NTD construct was transferred into nonauxotrophic Escherichia coli cells capable of BL21 (DE3) protein expression. Protein expression was induced by adding IPTG to 1 mM, followed by incubation at 10 °C for 24 h. After the bacteria were harvested via centrifugation (3500g, 30 min, 4 °C), the bacterial pellets were treated with lysis buffer (50 mM Tris-buffered saline [pH 7.3], 150 mM NaCl, and 15 mM imidazole). The soluble proteins were obtained from the supernatant after centrifugation (15 000 rpm, 30 min, 4 °C). The HCoV-OC43 N-NTD proteins carrying a His6-tag at their N-termini were purified using a Ni–nitrilotriacetic acid (NTA) column (Novagen) with an elution gradient ranging from 15 to 300 mM imidazole. Pure fractions were collected and dialyzed against a low-salt buffer. The purified protein was finally concentrated using a 3 kDa cutoff membrane in Amicon ultra-15 centrifugal filter units (Millipore, MA) and stored at −80 °C. The protein concentrations were determined using the Bradford method with Bio-Rad protein assay reagents.
Crystallization and Data Collection
HCoV-OC43 N-NTD crystals were grown as previously described:44 The crystallization solution (2 μL) was mixed with 1.5 μL of purified protein solution (8 mg mL–1) and 0.5 μL of 40% hexanediol at room temperature (∼298 K) and equilibrated against a 400 μL solution in the well of a Cryschem plate at 293 K. The crystallization conditions required a 0.5 M succinic acid–phosphate–glycine (SPG) buffer at pH 6.0 with 50% PEG 1500. The crystalline HCoV-OC43 N-NTD–AMP, HCoV-OC43 N-NTD–CMP, HCoV-OC43 N-NTD–GMP, and HCoV-OC43 N-NTD–UMP complexes were obtained via cocrystallization with an HCoV-OC43 N-NTD solution (8 mg/mL) preincubated for 30 min with 2 mM AMP, 5 mM CMP, 2 mM GMP, and 5 mM UMP, respectively. Crystals of the HCoV-OC43 N-NTD–PJ34 complex were obtained by soaking a native HCoV-OC43 N-NTD crystal for 1.5 h at 4 °C in a solution containing 5 mM PJ34 in 0.25 M SPG buffer at pH 6.0 and 25% PEG 1500. The crystals were flash-cooled under flowing nitrogen gas at 100 K. The X-ray diffraction data for the HCoV-OC43 N-NTD were collected at the National Synchrotron Radiation Research Centre (NSRRC; Hsinchu, Taiwan), BL13B1. All diffraction images were recorded using an ADSD Q315 charge-coupled device (CCD) detector, and the data were processed and scaled using the HKL2000 software package.45 The data collection statistics of HCoV-OC43 N-NTD complexed with ligand are summarized in Table 1 and Table S1 in the Supporting Information.
Structural Determination and Refinement
The structures of the HCoV-OC43 N-NTD complexes with ribonucleoside 5′-monophosphates or PJ34 were determined using the previously resolved structure of the native HCoV-OC43 N-NTD (3J3K)20 because the new crystals were isomorphic. For each structure, iterative cycles of model building were performed using Mifit and computational refinement via CNS and PHENIX;46,47 5% reflections were set aside for the Rfree calculations (Table 1 and Table S1 in the Supporting Information).48 The stereochemical quality of the structures was assessed using the PROCHECK program.49 The molecular figures were produced using PyMOL (DeLano Scientific, http://www.pymol.org).
Drug Discovery of the N Protein Inhibitor
For drug screening, the HCoV-OC43 N-NTD–AMP complex crystal structure was used as a template, and a large-scale molecular-docking-based library screen was conducted to identify compounds that might bind to the AMP-binding site on the N proteins. Several commercial drug databanks, including Acros Organics, Sigma Aldrich Inc., and Bachem Inc. from the ZINC databases, were screened to obtain compounds that act on the N protein by using the LIBDOCK molecular docking software. The N protein’s binding pocket was represented using a set of spheres, and each compound in the database was docked in the important pocket of the N protein; this pocket included Tyr 124, Tyr 126, Arg 122, and Arg 164 because they are involved in optimal RNA binding. We identified 87 potential compounds with high docking scores. Nine of the potential hits were identified among the 87 hits that include three interaction characters with HCoV-OC43 N-NTD, which are similar to the interactions between AMP and HCoV-OC43 N-NTD.
Site-Directed Mutagenesis
The single mutants were constructed using a QuikChange kit (Stratagene) with a plasmid containing an open reading frame that encodes the full-length HCoV-OC43 N protein as the template for mutagenesis. The PCR reaction used Pfu DNA polymerase, and each cycle involved heating the sample at 95 °C for 30 s, 55 °C for 1 min, and 68 °C for 2 min/kb of plasmid length; this sequence was repeated for a total of 16 cycles. The templates were digested with DpnI and transformed into E. coli XL-1 cells. All mutations were confirmed by automated sequencing in both directions.
SPR Binding Experiments
SPR experiments were performed as previously described.50 The affinity, association, and dissociation between the HCoV-OC43 N proteins and RNA were measured using a BIAcore 3000A SPR instrument (Pharmacia, Uppsala, Sweden) equipped with a SensorChip SA5 from Pharmacia; the refractive index change of the sensor chip surface was monitored. These changes are proportional to the quantity of analyte bound. The change in SPR angle is reported in resonance units. First, the surface was washed three times by injecting 10 μL of a 100 mM NaCl solution with 50 mM NaOH. To control the quantity of RNA (or DNA) bound to the SA chip surface, the biotinylated oligomer was manually immobilized onto the surface of a streptavidin chip. The chip surface was subsequently washed with 10 μL of 10 mM HCl to eliminate nonspecific binding. The N proteins (WT and mutants) were dissolved in 50 mM Tris (pH 7.3) with 150 mM NaCl and 0.1% CHAPS prior to passing over the chip surface for 140 s at 30 μL/min to achieve equilibrium. Next, a blank buffer solution was passed over the chip to initiate the dissociation reaction; this step was continued for an additional 600 s, allowing the reaction to reach completion. After 600 s, the surface was recovered by washing with 10 μL of 0.1% SDS for each single-stranded RNA. The sensorgrams revealing interactions between the RNA and protein were analyzed using BIA evaluation software (version 3) to determine the dissociation constants (kd/ka). To analyze the effect of PJ34 on the interactions between the N proteins and RNA, the N proteins were used with PJ34 in 50 mM Tris (pH 7.5), 150 mM NaCl, and 0.1% CHAPS injected onto the sensor chip.
Viral Infection and Real-Time Polymerase Chain Reaction (RT-PCR)
An RT-PCR was performed as previously described.51 First, the 293T cells were cultured in DMEM culture medium containing 10% fetal bovine serum (FBS; Atlanta Biologicals), 1% nonessential amino acid (NEAA; Invitrogen), and 10 μM β-mercaptoethanol (β-ME). Then 3 × 105 293T cells were seeded into each well of a 12-well plate one day prior to transfection. During the viral replication assay, the cells were transfected or not with pcDNA3.1/NP (WT and mutants) containing FuGENE 6 (Roche). Four days postinfection, the media were removed, the cells were lysed in 1 mL of Trisol (Invitrogen), the RNA was extracted following the manufacturer’s instructions, and 2 μg of the RNA was used as a template for the cDNA synthesis. The cDNA (2 μL) was added to 23 μL of a PCR cocktail containing 2 × SYBR Green Master Mix (ABI, Foster City, CA) and a 0.2 μM concentration of both the sense and antisense primers (IDT DNA, Coralville, IA). The amplification was performed in an ABI Prism 7700 thermocycler (ABI). The specificity of the amplification was confirmed via dissociation curve analysis. The data were collected and recorded using the ABI Prism 7700 software and expressed as a function of the threshold cycle (Ct); the threshold cycle describes the fluorescence intensity in a given reaction tube as it rises above the background level (calculated as 10 times the mean standard deviation of the fluorescence in all wells over the baseline cycles). The specific primers used to assay the expression of OC43 M and the housekeeping gene GAPDH were Fwd-ATGTTAGGCCGATAATTGAGGACTAT, Rev-AATGTAAAGATGGCCGCGTAT and Fwd-CCACTCCTCCACCTTTGA, Rev-ACCCTGTTGCTGTAGCCA, respectively.
HCoV-OC43 N-NTD–ssRNA Complex Modeling
We used the crystal structure of the N-NTD–AMP complex as a template to construct a plausible N-NTD–ssRNA complex using the molecular modeling programs Discovery Studio 2.5 and CNS.46 On the basis of the N-NTD–AMP complex crystal structure, we extended three and one nucleotide(s) from the 5′ and 3′ ends of the AMP, respectively, using the biopolymer module of Discovery Studio 2.5. The complex structure was further refined using CNS. The RNA force field parameters of Parkinson et al. were utilized.52 The quality of the model geometry was evaluated using the RMS derivation of the bond length and bond angle.
Acknowledgments
We thank Dr. Tai-huang Huang and Chung-ke Chang (Academia Sinica) for helpful discussions and guidance. We thank the NSRRC staff for the data collection. This work was supported by an NSC grant (100-2113-M-005-004-MY3) (M.-H.H.) and an NIH grant (NS36592).
Glossary
Abbreviations Used
- CoVs
coronaviruses
- MERS
Middle East respiratory syndrome
- SARS
severe acute respiratory syndrome
- N
nucleocapsid
- RNP
ribonucleoprotein
- NTD
N-terminal domain
- HCoV-OC43
human CoV-OC43
- PJ34
N- (6-oxo-5,6- dihydrophenanthridin-2-yl)(N,N-dimethylamino)acetamide hydrochloride
- M
matrix
- E
envelope
- S
spike
- HE
hemagglutinin esterase
- CTD
C-terminal dimerization domain
- IBV
infectious bronchitis virus
- MHV
mouse hepatitis virus
- AMP
adenosine monophosphate
- CMP
cytosine monophosphate
- GMP
guanosine monophosphate
- UMP
uridine monophosphate
- WT
wide type
- O3
6-chloro-7-((2-morpholin-4-ylethyl)amino)quinoline-5,8-dione
- PDB
Protein Data Bank
Supporting Information Available
Figures S1–S7 and Tables S1–S3. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
The atomic coordinates and structural factors for HCoV-OC43 N-NTD complexed with AMP (4LI4), CMP (4LMC), GMP (4LM9), UMP (4LM7), and PJ34 (4KXJ) were deposited in the wwPDB.
Author Contributions
# S.P. and M.-H.H. contributed equally to this work.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Peiris J. S.; Chu C. M.; Cheng V. C.; Chan K. S.; Hung I. F.; Poon L. L.; Law K. I.; Tang B. S.; Hon T. Y.; Chan C. S.; Chan K. H.; Ng J. S.; Zheng B. J.; Ng W. L.; Lai R. W.; Guan Y.; Yuen K. Y. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: A prospective study. Lancet 2003, 361, 1767–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss S. R.; Leibowitz J. L. Coronavirus pathogenesis. Adv. Virus Res. 2011, 81, 85–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh E. E.; Shin J. H.; Falsey A. R. Clinical impact of human coronaviruses 229E and OC43 infection in diverse adult populations. J. Infect. Dis. 2013, 208, 1634–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skowronski D. M.; Astell C.; Brunham R. C.; Low D. E.; Petric M.; Roper R. L.; Talbot P. J.; Tam T.; Babiuk L. Severe acute respiratory syndrome (SARS): A year in review. Annu. Rev. Med. 2005, 56, 357–81. [DOI] [PubMed] [Google Scholar]
- Vabret A.; Mourez T.; Dina J.; van der Hoek L.; Gouarin S.; Petitjean J.; Brouard J.; Freymuth F. Human coronavirus NL63, France. Emerging Infect. Dis. 2005, 11, 1225–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo P. C.; Lau S. K.; Chu C. M.; Chan K. H.; Tsoi H. W.; Huang Y.; Wong B. H.; Poon R. W.; Cai J. J.; Luk W. K.; Poon L. L.; Wong S. S.; Guan Y.; Peiris J. S.; Yuen K. Y. Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J. Virol. 2005, 79, 884–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brian D. A.; Baric R. S. Coronavirus genome structure and replication. Curr. Top. Microbiol. Immunol. 2005, 287, 1–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogue B. G.; Brian D. A. Structural proteins of human respiratory coronavirus OC43. Virus Res. 1986, 5, 131–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai M. M.; Cavanagh D. The molecular biology of coronaviruses. Adv. Virus Res. 1997, 48, 1–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stohlman S. A.; Baric R. S.; Nelson G. N.; Soe L. H.; Welter L. M.; Deans R. J. Specific interaction between coronavirus leader RNA and nucleocapsid protein. J. Virol. 1988, 62, 4288–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson G. W.; Stohlman S. A.; Tahara S. M. High affinity interaction between nucleocapsid protein and leader/intergenic sequence of mouse hepatitis virus RNA. J. Gen. Virol. 2000, 81, 181–8. [DOI] [PubMed] [Google Scholar]
- Masters P. S.; Sturman L. S. Background paper. Functions of the coronavirus nucleocapsid protein. Adv. Exp. Med. Biol. 1990, 276, 235–8. [DOI] [PubMed] [Google Scholar]
- Tang T. K.; Wu M. P.; Chen S. T.; Hou M. H.; Hong M. H.; Pan F. M.; Yu H. M.; Chen J. H.; Yao C. W.; Wang A. H. Biochemical and immunological studies of nucleocapsid proteins of severe acute respiratory syndrome and 229E human coronaviruses. Proteomics 2005, 5, 925–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh P. K.; Chang S. C.; Huang C. C.; Lee T. T.; Hsiao C. W.; Kou Y. H.; Chen I. Y.; Chang C. K.; Huang T. H.; Chang M. F. Assembly of severe acute respiratory syndrome coronavirus RNA packaging signal into virus-like particles is nucleocapsid dependent. J. Virol. 2005, 79, 13848–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surjit M.; Liu B.; Chow V. T.; Lal S. K. The nucleocapsid protein of severe acute respiratory syndrome-coronavirus inhibits the activity of cyclin-cyclin-dependent kinase complex and blocks S phase progression in mammalian cells. J. Biol. Chem. 2006, 281, 10669–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du L.; Zhao G.; Lin Y.; Chan C.; He Y.; Jiang S.; Wu C.; Jin D. Y.; Yuen K. Y.; Zhou Y.; Zheng B. J. Priming with rAAV encoding RBD of SARS-CoV S protein and boosting with RBD-specific peptides for T cell epitopes elevated humoral and cellular immune responses against SARS-CoV infection. Vaccine 2008, 26, 1644–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang F. Y.; Lin L. C.; Ying T. H.; Yao C. W.; Tang T. K.; Chen Y. W.; Hou M. H. Immunoreactivity characterisation of the three structural regions of the human coronavirus OC43 nucleocapsid protein by Western blot: Implications for the diagnosis of coronavirus infection. J. Virol. Methods 2013, 187, 413–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C. K.; Chen C. M.; Chiang M. H.; Hsu Y. L.; Huang T. H. Transient oligomerization of the SARS-CoV N protein—Implication for virus ribonucleoprotein packaging. PLoS One 2013, 8, e65045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Y. S.; Lin S. Y.; Wang S. M.; Wang C. T.; Chiu Y. L.; Huang T. H.; Hou M. H. Oligomerization of the carboxyl terminal domain of the human coronavirus 229E nucleocapsid protein. FEBS Lett. 2013, 587, 120–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen I. J.; Yuann J. M.; Chang Y. M.; Lin S. Y.; Zhao J.; Perlman S.; Shen Y. Y.; Huang T. H.; Hou M. H. Crystal structure-based exploration of the important role of Arg106 in the RNA-binding domain of human coronavirus OC43 nucleocapsid protein. Biochim. Biophys. Acta, Proteins Proteomics 2013, 1834, 1054–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C. K.; Sue S. C.; Yu T. H.; Hsieh C. M.; Tsai C. K.; Chiang Y. C.; Lee S. J.; Hsiao H. H.; Wu W. J.; Chang W. L.; Lin C. H.; Huang T. H. Modular organization of SARS coronavirus nucleocapsid protein. J. Biomed. Sci. 2006, 13, 59–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wootton S. K.; Rowland R. R.; Yoo D. Phosphorylation of the porcine reproductive and respiratory syndrome virus nucleocapsid protein. J. Virol. 2002, 76, 10569–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saikatendu K. S.; Joseph J. S.; Subramanian V.; Neuman B. W.; Buchmeier M. J.; Stevens R. C.; Kuhn P. Ribonucleocapsid formation of severe acute respiratory syndrome coronavirus through molecular action of the N-terminal domain of N protein. J. Virol. 2007, 81, 3913–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan H.; Ooi A.; Tan Y. W.; Wang S.; Fang S.; Liu D. X.; Lescar J. The nucleocapsid protein of coronavirus infectious bronchitis virus: Crystal structure of its N-terminal domain and multimerization properties. Structure 2005, 13, 1859–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaram H.; Fan H.; Bowman B. R.; Ooi A.; Jayaram J.; Collisson E. W.; Lescar J.; Prasad B. V. X-ray structures of the N- and C-terminal domains of a coronavirus nucleocapsid protein: Implications for nucleocapsid formation. J. Virol. 2006, 80, 6612–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossoehme N. E.; Li L.; Keane S. C.; Liu P.; Dann C. E. 3rd; Leibowitz J. L.; Giedroc D. P. Coronavirus N protein N-terminal domain (NTD) specifically binds the transcriptional regulatory sequence (TRS) and melts TRS-cTRS RNA duplexes. J. Mol. Biol. 2009, 394, 544–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Y. W.; Fang S.; Fan H.; Lescar J.; Liu D. X. Amino acid residues critical for RNA-binding in the N-terminal domain of the nucleocapsid protein are essential determinants for the infectivity of coronavirus in cultured cells. Nucleic Acids Res. 2006, 34, 4816–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane S. C.; Liu P.; Leibowitz J. L.; Giedroc D. P. Functional transcriptional regulatory sequence (TRS) RNA binding and helix destabilizing determinants of murine hepatitis virus (MHV) nucleocapsid (N) protein. J. Biol. Chem. 2012, 287, 7063–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Zhang X. The leader RNA of coronavirus mouse hepatitis virus contains an enhancer-like element for subgenomic mRNA transcription. J. Virol. 2000, 74, 10571–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawicki S. G.; Sawicki D. L.; Siddell S. G. A contemporary view of coronavirus transcription. J. Virol. 2007, 81, 20–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welter L. M.; Stohlman S. A.; Deans R. J. MHV leader RNA secondary structure affects binding to the nucleocapsid protein. Adv. Exp. Med. Biol. 1990, 276, 247–54. [DOI] [PubMed] [Google Scholar]
- Scott G. S.; Kean R. B.; Mikheeva T.; Fabis M. J.; Mabley J. G.; Szabo C.; Hooper D. C. The therapeutic effects of PJ34 [N-(6-oxo-5,6-dihydrophenanthridin-2-yl)-N,N-dimethylacetamide.HCl], a selective inhibitor of poly(ADP-ribose) polymerase, in experimental allergic encephalomyelitis are associated with immunomodulation. J. Pharmacol. Exp. Ther. 2004, 310, 1053–61. [DOI] [PubMed] [Google Scholar]
- Teng F.; Beray-Berthat V.; Coqueran B.; Lesbats C.; Kuntz M.; Palmier B.; Garraud M.; Bedfert C.; Slane N.; Berezowski V.; Szeremeta F.; Hachani J.; Scherman D.; Plotkine M.; Doan B. T.; Marchand-Leroux C.; Margaill I. Prevention of rt-PA induced blood-brain barrier component degradation by the poly(ADP-ribose)polymerase inhibitor PJ34 after ischemic stroke in mice. Exp. Neurol. 2013, 248, 416–28. [DOI] [PubMed] [Google Scholar]
- Ma Y.; Tong X.; Xu X.; Li X.; Lou Z.; Rao Z. Structures of the N- and C-terminal domains of MHV-A59 nucleocapsid protein corroborate a conserved RNA-protein binding mechanism in coronavirus. Protein Cell 2010, 1, 688–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank F.; Sonenberg N.; Nagar B. Structural basis for 5′-nucleotide base-specific recognition of guide RNA by human AGO2. Nature 2010, 465, 818–22. [DOI] [PubMed] [Google Scholar]
- Zhao C.; Lou Z.; Guo Y.; Ma M.; Chen Y.; Liang S.; Zhang L.; Chen S.; Li X.; Liu Y.; Bartlam M.; Rao Z. Nucleoside monophosphate complex structures of the endonuclease domain from the influenza virus polymerase PA subunit reveal the substrate binding site inside the catalytic center. J. Virol. 2009, 83, 9024–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green T. J.; Zhang X.; Wertz G. W.; Luo M. Structure of the vesicular stomatitis virus nucleoprotein-RNA complex. Science 2006, 313, 357–60. [DOI] [PubMed] [Google Scholar]
- Ramajayam R.; Tan K. P.; Liang P. H. Recent development of 3C and 3CL protease inhibitors for anti-coronavirus and anti-picornavirus drug discovery. Biochem. Soc. Trans. 2011, 39, 1371–5. [DOI] [PubMed] [Google Scholar]
- Chenavas S.; Crepin T.; Delmas B.; Ruigrok R. W.; Slama-Schwok A. Influenza virus nucleoprotein: Structure, RNA binding, oligomerization and antiviral drug target. Future Microbiol. 2013, 8, 1537–45. [DOI] [PubMed] [Google Scholar]
- Hung H. C.; Liu C. L.; Hsu J. T.; Horng J. T.; Fang M. Y.; Wu S. Y.; Ueng S. H.; Wang M. Y.; Yaw C. W.; Hou M. H. Development of an anti-influenza drug screening assay targeting nucleoproteins with tryptophan fluorescence quenching. Anal. Chem. 2012, 84, 6391–9. [DOI] [PubMed] [Google Scholar]
- Clark I. A.; Alleva L. M.; Mills A. C.; Cowden W. B. Pathogenesis of malaria and clinically similar conditions. Clin. Microbiol. Rev. 2004, 17, 509–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazo J. S.; Aslan D. C.; Southwick E. C.; Cooley K. A.; Ducruet A. P.; Joo B.; Vogt A.; Wipf P. Discovery and biological evaluation of a new family of potent inhibitors of the dual specificity protein phosphatase Cdc25. J. Med. Chem. 2001, 44, 4042–9. [DOI] [PubMed] [Google Scholar]
- Pu L.; Amoscato A. A.; Bier M. E.; Lazo J. S. Dual G1 and G2 phase inhibition by a novel, selective Cdc25 inhibitor 6-chloro-7-[corrected](2-morpholin-4-ylethylamino)-quinoline-5,8-dione. J. Biol. Chem. 2002, 277, 46877–85. [DOI] [PubMed] [Google Scholar]
- Chen I. J.; Chou C. C.; Liu C. L.; Lee C. C.; Kan L. S.; Hou M. H. Crystallization and preliminary X-ray diffraction analysis of the N-terminal domain of human coronavirus OC43 nucleocapsid protein. Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun. 2010, 66, 815–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor W.; Cymborowski M.; Otwinowski Z.; Chruszcz M. HKL-3000: the integration of data reduction and structure solution—From diffraction images to an initial model in minutes. Acta Crystallogr., D: Biol. Crystallogr. 2006, 62, 859–66. [DOI] [PubMed] [Google Scholar]
- Brunger A. T.; Adams P. D.; Clore G. M.; DeLano W. L.; Gros P.; Grosse-Kunstleve R. W.; Jiang J. S.; Kuszewski J.; Nilges M.; Pannu N. S.; Read R. J.; Rice L. M.; Simonson T.; Warren G. L. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr., D: Biol. Crystallogr. 1998, 54, 905–21. [DOI] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkoczi G.; Chen V. B.; Echols N.; Headd J. J.; Hung L. W.; Jain S.; Kapral G. J.; Grosse Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R. D.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. The Phenix software for automated determination of macromolecular structures. Methods 2011, 55, 94–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger A. T. Assessment of phase accuracy by cross validation: The free R value. Methods and applications. Acta Crystallogr., D: Biol. Crystallogr. 1993, 49, 24–36. [DOI] [PubMed] [Google Scholar]
- Morris A. L.; MacArthur M. W.; Hutchinson E. G.; Thornton J. M. Stereochemical quality of protein structure coordinates. Proteins 1992, 12, 345–64. [DOI] [PubMed] [Google Scholar]
- Lo Y. S.; Tseng W. H.; Chuang C. Y.; Hou M. H. The structural basis of actinomycin D-binding induces nucleotide flipping out, a sharp bend and a left-handed twist in CGG triplet repeats. Nucleic Acids Res. 2013, 41, 4284–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pewe L.; Zhou H.; Netland J.; Tangudu C.; Olivares H.; Shi L.; Look D.; Gallagher T.; Perlman S. A severe acute respiratory syndrome-associated coronavirus-specific protein enhances virulence of an attenuated murine coronavirus. J. Virol. 2005, 79, 11335–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson G.; Vojtechovsky J.; Clowney L.; Brunger A. T.; Berman H. M. New parameters for the refinement of nucleic acid-containing structures. Acta Crystallogr., D: Biol. Crystallogr. 1996, 52, 57–64. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.