

Abstract
A mass spectrometry (MS) method is described here that can reproducibly identify hundreds of peptides across multiple experiments. The method uses intelligent data acquisition to precisely target peptides while simultaneously identifying thousands of other, nontargeted peptides in a single nano-LC–MS/MS experiment. We introduce an online peptide elution order alignment algorithm that targets peptides based on their relative elution order, eliminating the need for retention-time-based scheduling. We have applied this method to target 500 mouse peptides across six technical replicate nano-LC–MS/MS experiments and were able to identify 440 of these in all six, compared with only 256 peptides using data-dependent acquisition (DDA). A total of 3757 other peptides were also identified within the same experiment, illustrating that this hybrid method does not eliminate the novel discovery advantages of DDA. The method was also tested on a set of mice in biological quadruplicate and increased the number of identified target peptides in all four mice by over 80% (826 vs 459) compared with the standard DDA method. We envision real-time data analysis as a powerful tool to improve the quality and reproducibility of proteomic data sets.
Keywords: nano-LC–MS/MS, elution order alignment, data-dependent acquisition, peptide identification, real-time data analysis, discovery, target
Introduction
Large-scale proteomic studies make use of a variety of tools and techniques to achieve depth and wide coverage of proteomes. The most popular method for sequencing proteomes is shotgun sequencing where peptides are digested from extracted proteins, separated with chromatography (HPLC), and then mass-analyzed using mass spectrometry (MS).1,2 Since complex proteomes can encompass thousands of proteins, leading to millions of peptides, deciding how to allocate the limited mass spectrometer bandwidth is key to successful analysis.3 By far the most successful method for this time management is data-dependent acquisition (DDA), where intact peptide precursors are first mass-analyzed (MS1), specific m/z features are then selected to undergo fragmentation, and finally the fragment ions are mass-analyzed again (MS/MS). This process is repeated throughout the LC separation, resulting in a large collection of MS and MS/MS spectra. Peptides are eventually identified from the fragmentation spectra and then assembled into protein groups.4−8 This approach has produced outstanding results in the past decade, but due to a variety of reasons (e.g., large protein dynamic range, speed of MS instrumentation, separation efficiency, etc.) undersampling of proteomes is very common. In other words, not every peptide is identified in every nano-LC–MS/MS experiment. Incomplete data sets limit the questions researchers can answer; in particular, when biological replication is used to increase statistical power, many measurements become worthless if they cannot be measured reproducibly.9 Because proteomics seeks to answer global biological questions, reproducible peptide identification between data sets is mandated.10−12
Many studies have outlined the problem of poor peptide reproducibility.13−17 Aebersold succinctly summarized that irreproducibility is a multifaceted issue, depending on user experience, equipment, and data analysis, among others.18 He outlines that there are two main approaches in tackling irreproducibility. First, exhaustively identify every peptide in a sample—an approach that is becoming more feasible as technology improves.19−21 The more common approach, as many other researchers have embarked on, is to focus on a smaller subset of peptides and to thoroughly identify and quantify those using targeted methods.22 Methods such as selected reaction monitoring (SRM)23 are powerful and reproducible but are low-throughput, targeting a few hundred peptides at most in a single nano-LC–MS/MS experiment.24−27 Targeted methods almost exclusively rely on retention-time-based scheduling to improve identification reproducibility and throughput, segmenting the MS duty cycle among the target peptides. In SRM methods, a series of MS/MS transitions for each targeted peptide is automatically collected at the appropriate retention time (RT), removing the dependence on MS1 detection. This requires precise knowledge of the peptide RT for the LC–MS system and is low-throughput because only one set of transitions is monitored at a given point in time. Recent work on intelligent SRM (iSRM) increases throughput by monitoring only a subset of transitions for each target, switching to normal SRM when these transitions are detected.28 We sought to expand upon the idea of intelligent real-time switching of methods by combining the enhanced reproducibility of targeted scheduled methods with the novel discovery advantages of DDA in a single hybrid method. Our goals were three-fold: first, to develop a method that increases the throughput of targeting; second, to replace retention-time based scheduling and its laborious method development with a more robust and straightforward peptide elution ordering; and last, to maintain the discovery aspect of DDA sampling while simultaneously targeting a subset of peptides.
In the past decades, a few computational approaches have been aimed at solving the problem of poor reproducibility. The concept of accurate mass tags (AMTs) was first introduced by Smith et al. as a means to identify peptides in multiple runs based on accurate mass and RT.29 This concept was further expanded with PepMiner and PEPPeR, tools for clustering features among multiple data sets.30,31 Most notably, Prakash et al. introduced the concept of aligning multiple MS data sets based on peptide relative elution order (EO) into signal maps.32 To date, these and other computational methods33−38 have been performed postacquisition, attempting to improve already collected data. We seek to improve the reproducibility at the source by improving the algorithms the MS uses to select precursors to fragment. We and others have proposed using real-time data analysis and dynamic MS control as a means for improving the quality of acquired spectra.39−41 These methods rely on determining peptide spectrum matches (PSMs) in real time and using those identifications to make informed, dynamic decisions. However, real-time identification has some setbacks: (1) MS/MS spectra are not always identified leaving the data incomplete, (2) wrongly assigned PSMs could negatively affect performance, and (3) a reduction in the instrument duty cycle decreases the number of MS/MS performed. These, and other issues, have lead us to investigate alternative ways for detecting peptides in real-time, primarily through accurate mass measurements. Here we present our findings on combining accurate mass, EOs, and real-time data analysis to improve the sampling reproducibility of the MS.
Experimental Procedures
Yeast Culture
Saccharomyces cerevisiae strain BY4741 was grown in yeast extract peptone dextrose media (YPD) (1% yeast extract, 2% peptone, 2% dextrose). A starter culture was added to 2 L of media and was propagated for ∼12 generations (20 h) to a total OD600 of ∼2. The cells were pelleted with centrifugation at 5000 rpm for 5 min, the supernatant was decanted, and the pellet was resuspended in chilled NanoPure water. Washing with water was repeated twice, and the final pelleting was performed at 5000 rpm for 10 min. The pellet was resuspended in lysis buffer composed of 50 mM Tris pH8, 8 M urea, 75 mM sodium chloride, 100 mM sodium butyrate, protease, and phosphatase inhibitor tablet (Roche). Cell lysing was performed with glass bead milling in a stainless-steel container (Retsch). A 2.5 mL aliquot of resuspended yeast was shaken with 2 mL of acid-washed glass beads at 30 Hz for 4 min, followed by 1 min of rest, for eight cycles.
Mouse Handling and Tissue Isolation
Four male C57BL/B6 mice were bred from in-house colonies and housed in an environmentally controlled facility with free access to water and standard rodent chow (Purina #5008). Mice were kept in accordance to the University of Wisconsin-Madison Research Animals Resource Center and NIH guidelines for care and use of laboratory animals. At 10 weeks of age, mice were sacrificed by decapitation after a 4 h fast. Eight tissues were dissected from the mice (cerebellum, cerebrum, kidney, heart, liver, lung, extensor digitorum longus, and spleen), flash frozen in liquid nitrogen, and stored at −80 °C. Tissues were homogenized in 1 mL of lysis buffer/100 mg tissue (8 M urea, 50 mM Tris, 100 mM NaCl, 1 mM CaCl2, 100 mM sodium butyrate, 5 μM MS-275, 0.2 μM SAHA, Roche protease, and phosphatase inhibitor tablets).
Sample Preparation
Protein was quantified by BCA (Pierce) and reduced with 5 mM dithiothreitol and incubated for 45 min at 55 °C. Alkylation was performed with 15 mM iodoacetamide for 30 min in the dark and quenched with 5 mM dithiothreitol. Urea concentration was diluted to 1.5 M with 50 mM Tris pH 8.0. Proteolytic digestion was performed by the addition of Trypsin (Promega), 1:50 enzyme to protein ratio, and incubated at ambient temperature overnight. For quantitative studies, the resulting peptides were labeled with TMT 8-plex (Pierce) isobaric tag and mixed.42,43 All samples were desalted using C-18 solid-phase extraction (SPE) columns (Waters, Milford, MA) prior to nano-LC–MS/MS analysis.
Nano LC–MS/MS Analysis
Peptides were separated with online reverse-phase chromatography using a nanoACQUITY UPLC system (Waters, Milford, MA). Peptides were first loaded onto a precolumn (75 μm ID, 5 cm Magic C18 particles, Bruker, Michrom) for 10 min at 1 μL/min flow rates. Peptides were then separated on a 30 cm analytical column (75 μm ID, 5 cm Magic C18 particles) for either 100 or 165 min over a linear gradient from 8 to 35% acetonitrile at 300 nL/min. Mass analysis was performed on an LTQ Orbitrap Elite44 mass spectrometer (Thermo Fisher Scientific, San Jose, CA) using 60 000 resolving power (RP) MS1 scans. Peptides selected for MS/MS analysis used a 2 Th isolation width, were fragmented with HCD (NCE = 35), and were analyzed in the Orbitrap at 15 000 RP or 30 000 RP for quantitative experiments. Unless otherwise noted, data-dependent analysis was performed selecting the top 15 most intense m/z features (charge state >1) for MS/MS analysis. Dynamic exclusion settings were enabled for 35 s at ±10 ppm mass window, 1 occurrence with a maximum of 500 exclusions at any given point in time. Automatic gain control (AGC) was enabled, and MS1 targets were set to 1 × 106 and MS/MS targets were set to 5 × 104. Accurate mass inclusion list experiments would prioritize MS/MS sampling from a list of targets at ±10 ppm mass tolerances. Remaining MS/MS events were filled with normal top-N DDA approaches. Intelligent data acquisition control was implemented using the ion trap control language (ITCL, Thermo Fisher Scientific), and the pseudocode of these modifications is included in the Supporting Information. In brief, following MS1 analysis, the spectra were analyzed using algorithms written in ITCL to select targets for MS/MS analysis (described herein). Any remaining MS/MS slots would be filled by the unmodified DDA firmware code. For information on implementing the modified firmware code, please contact Thermo Fisher Scientific. All nanoLC–MS/MS experiments in the Thermo .raw format are located on the Chorus Project Website (https://chorusproject.org/) under the ‘Elution Order Algorithm’ project.
Data Analysis
Thermo .raw files were processed using the Coon OMSSA Proteomic Analysis Software Suite (COMPASS)45 and in-house software. In brief, raw files were converted to the dta file format (DTA Generator) and were searched using the Open Mass Spectrometry Search Algorithm (OMSSA, v 2.1.9).46 Yeast data were searched against a target-decoy47 database of yeast ORFs (www.yeastgenome.com, February 3, 2011) and mouse data from UniProt canonical database. Peptides were generated from a tryptic digestion with up to three missed cleavages, carbamidomethylation of cysteines as fixed modifications, and oxidation of methionines as variable modifications. For quantitative experiments, a fixed modification of 8-plex TMT tag was added to lysines and peptide n-terminus, with a variable modification of 8-plex TMT tag on tyrosines. Precursor mass tolerance was 100 ppm using the multiisotope function (-tem 4 -ti 4), and product ions were searched at 0.015 Da tolerances. Peptide spectral matches (PSMs) were reduced to unique peptide sequences (I/L ambiguity removed) and validated using FDR Optimizer based on q values and precursor mass accuracy (<10 ppm) at a 1% peptide-level false discovery rate (FDR).48−50 Protein groups were constructed from peptide identifications according to the law of parsimony and filtered to a 1% protein-level FDR (Protein Hoarder). For quantitative data sets, peptides were quantified with TagQuant (v1.4) using the generated TMT 8-plex reporter ions, corrected for isotopic impurities, and normalized to total protein abundance. Quantitative significance (p value) was determined by the Student’s t test with Storey correction assuming equal variances.51 Peptide EO determination algorithms were performed by custom software developed in C# with the Microsoft .NET Framework version 4.5. This software is available for download by visiting www.chem.wisc.edu/∼coon/software.php.
Results and Discussion
Irreproducible Peptide Identification
In DDA peptide precursors are selected for fragmentation based on intensity in a MS1 survey scan. This straightforward approach has proven to be a simple and powerful technique. However, it is pestered with inconsistent sampling and therefore irregular peptide identification between experiments. The DDA method is inherently stochastic in nature, depending heavily on the consistency of the input data (MS1) to deliver reproducible peptide identification (MS/MS). Even the slightest change in the chromatography or ionization efficiencies will have repercussions on the collection of the whole data set, as selecting m/z features for MS/MS analysis is often dependent on previous decisions (e.g., dynamic exclusion). To characterize the extent these minor changes have on the reproducibility of peptide identifications, six replicate injections of a tryptic digest of yeast whole cell lysate were analyzed using DDA on the same nano-LC–MS/MS system over a span of 10 days. On average, each experiment identified 13 289 ± 340 unique peptide sequences (I/L ambiguity removed) at a 1% peptide-level FDR, indicating a highly consistent separation and nearly identical instrument performance. Of the 23 919 unique peptides identified in total, only 5404 (22.6%) of those peptide were identified in all six experiments (Figure 1). A significant portion were only identified once (7474, 31.2%), while the remaining peptides were divided between two and five experiments. This clearly demonstrates the irreproducibility of DDA sampling on the same peptide solution. The reproducibility of identified protein groups fares better; 1708 of 3054 (56%) protein groups were identified in every experiment. The higher overlap percentage is because many different peptides can make up one protein group, minimizing the importance of identifying the same peptides in all experiments. However, post-translation modification (PTM) analysis requires identification of the same sites to compare between experiments, demanding the need for high peptide overlap. PTM analysis and quantitation is becoming more prominent in the literature, thus making this a growing problem in the field. Two reasons can be attributed to the poor reproducibility of stochastic DDA sampling. First, precursors having low signal-to-noise (S/N) are affected first by changes in chromatography and ionization. For example, a precursor with a maximal S/N of four may have been sampled and identified in one experiment, but in the next experiment, the S/N may have dropped below the detection threshold and excluded from being sampled. This is evident when 8883 MS1 features from peptides identified in one or all of the six experiments were examined for their maximal S/N (Supplemental Figure 1 in the Supporting Information). For peptides identified once, 2707 (30.5%) had a maximal S/N ≤ 4, while only 814 (9.2%) precursors identified in every experiment had similar maximum S/N. The other reason for inconsistent peptide identification is increased MS1 spectral complexity, specifically its effect on charge-state assignment. In proteomic MS/MS workflows, precursors are often only selected when they exhibit a well-defined charge state—usually where z > 1, as singly charged precursors fragment poorly and usually do not lead to positive identifications. Increases in spectral complexity hinder the charge-state determination algorithms, especially for low S/N precursors. This results in skipping precursors even if its signal-to-noise is above the sampling threshold.
Figure 1.
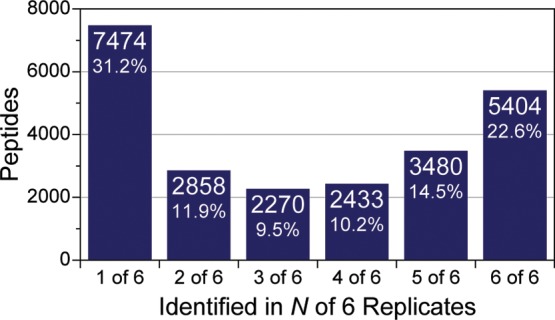
Overlap of peptide identification among the analysis of six technical replicates. Six nano-LC–MS/MS experiments produced 23 919 unique peptide identifications in total, but only one-fifth of the identifications were observed in all six replicates. A large percentage (31.2%) of the peptides were only detected in one of the six experiments.
Retention-Time-Based Targeting
When good peptide identification reproducibility is needed, RT-based targeting, that is, scheduling, has been the method of choice. Here peptides of interest are assigned an expected elution time and MS/MS is triggered, regardless of MS1 detection, during the appropriate time range. This avoids the two issues with DDA sampling previously described and enables much higher reproducibility. However, such methods are laborious to construct and maintain—identical LC and MS parameters must be kept between experiments to minimize any variances in RTs of the peptides.
To assess the degree of variance in peptide RTs that occurs in normal nano-LC–MS/MS experiments, two of the yeast DDA experiments described above, performed 10 days apart, were compared. The first experiment (July 22, D0) produced 13 529 unique peptides, and the second experiment (July 31, D9) identified 13 433 yeast peptides. Together, 7589 peptides were in common and the apex RT of their elution in each experiment is plotted in Figure 2A. The relationship between RTs of matched peptides is highly linear (R2 = 0.9989) but has a nonunity slope and nonzero intercept (m = 1.033; b = −0.647). While the slope is very close to 1, even the slightest deviation (0.033), compounded over time, leads to large RT differences late in the separation (e.g., ∼1.6 min shift at 70 min). On the whole, the average RT deviation was nearly 1 min (μ = −0.805 min) with a broad distribution over a 2 min range (Figure 2B). Typically, the assigned peptide elution times must be corrected to encompass this shift.
Figure 2.
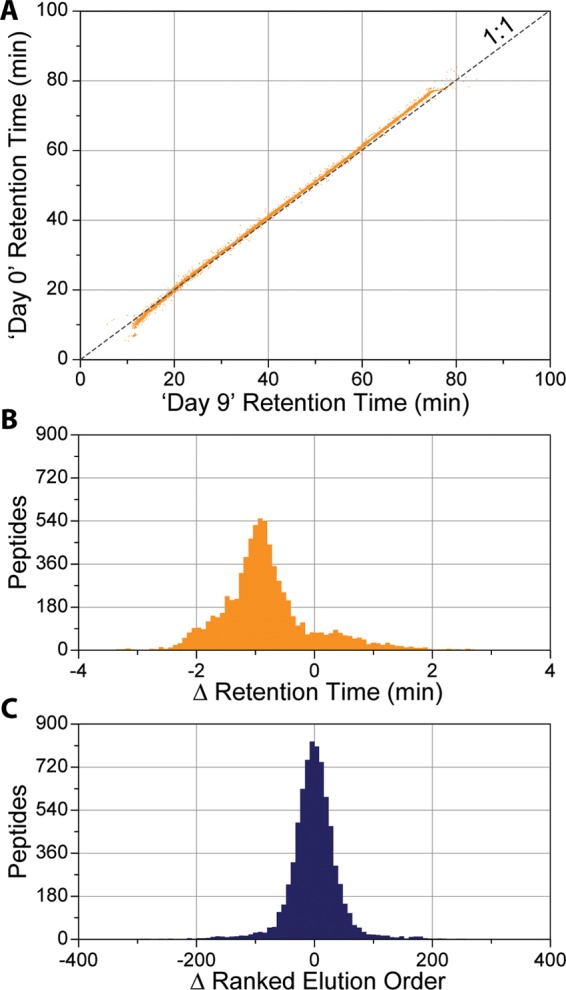
To assess the deviation in retention times for matched samples, we ran two identical nano-LC–MS/MS experiments 10 days apart on the same LC–MS system. (A) The relationship between apex retention times of the 7589 unique peptides common between experiments displays a high degree of linearity (R2 = 0.9989) but a skewed slope and nonzero intercept (m = 1.033; b = −0.647). (B) Average deviation from unity was nearly a minute off (μ = −0.805 min), with a broad distribution over 2 min wide. (C) Peptides ranked by their relative elution order exhibit a normal distribution around zero (μ = −1.097).
We hypothesize that—due to the degree of linearity in peptide RTs—we could avoid these corrections by scheduling peptides based on their relative EO, opposed to their absolute RT. Under similar LC conditions (i.e., same particles, temperature, column length, phase, etc.) peptides elute in the same relative order regardless of separation duration or slope. For example, if peptide ‘A’ elutes before peptide ‘B’ in a 30 min LC gradient, the same ordering is preserved with a 60 min LC gradient, even if the absolute RTs vary greatly. When many peptides’ EOs are taken into account (e.g., thousands of peptides), they provide a simple way to correct for elution variation dynamically. This is evident when we took the 7589 peptides and rank ordered them based on their apex RTs for both the D0 and D9 experiments and plotted the difference between matched peptides (Figure 2C). Here the values are normally distributed around zero (μ = −1.097) with a full width at half-maximum (fwhm) of only ∼100. EO can be useful even under extreme differences under chromatographic conditions as well.
To simulate dynamic chromatographic conditions, we separated yeast peptides under two different LC gradient profiles. The resulting peptide identifications were again matched between the runs, and the RT difference was plotted (Supplemental Figure 2A in the Supporting Information). These data show an average deviation of 10 min between the two gradients (Supplemental Figure 2B in the Supporting Information), but when ranked by their EOs, the two experiments show a linear slope of 1 with a normal distribution of ranked EOs around zero (Supplemental Figure 2C,D in the Supporting Information).
Real-Time Elution Ordering Alignment
We reasoned that using EO could improve the irreproducible sampling of DDA, similarly to scheduled methods, but on a larger scale and more robustly. The question shifts from “What RT is it?” as scheduled methods ask, to “What is the current EO?” By knowing which peptides are currently eluting from the LC, combined with the a priori knowledge of their EO, we predict with high fidelity what peptides are going to subsequently elute.
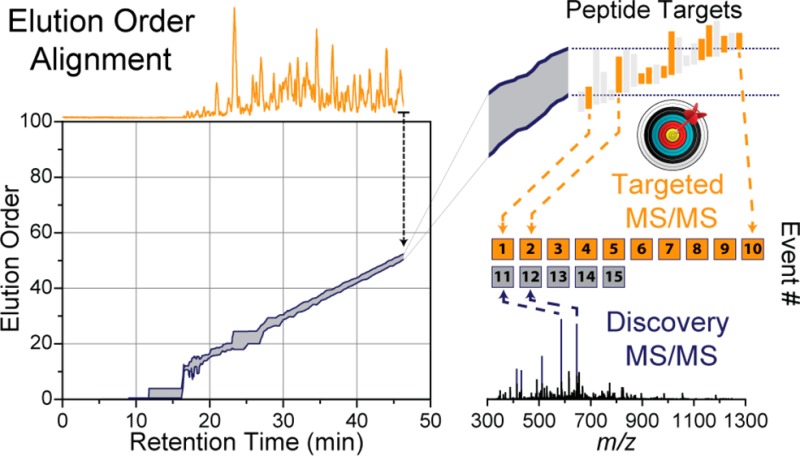
Prior knowledge is needed of the sample to adequately calculate the EOs of the peptides in the sample. With time-based scheduled methods, many cursory experiments are performed to optimize the RTs of the targeted peptides. To reduce variances in RTs, it is vital that these initial experiments are conducted exactly the same as the targeted experiments. In stark contrast, EOs can be determined using a variety of methods. First, much work has been devoted to determining peptide hydrophobicities from theoretical calculations of the amino acid sequence.52−55 A simple list of peptides, ordered by their hydrophobicities, can produce a highly linear elution ordering. Second, previously collected data of the sample can produce an accurate elution ordering as long as the LC conditions are similar enough. This enables the combination of multiple data sets to produce a single EO versus m/z map (elution order map, EOM), regardless of their individual separation durations. This is accomplished by rank ordering all peptide identifications in a given run and normalizing their orderings between 0 and 100 (where 100 represents the last eluting peptide). These normalized values are then matched between experiments and aligned using a simple algorithm to produce the final EOM as shown in Figure 3A. Lastly, the most robust method for determining peptide EOs is to perform a discovery experiment right before the targeted experiment. Regardless of how EO is determined, the final EOM is uploaded onto the instrument and is accessed throughout the course of the subsequent analyses.
Figure 3.
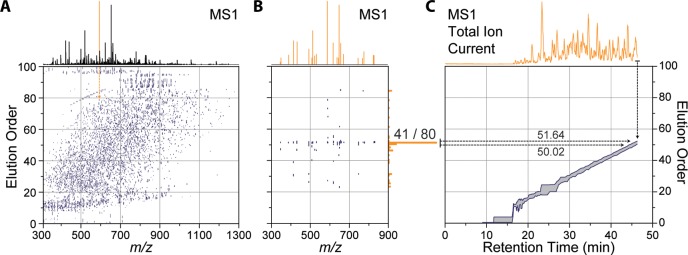
Real-time elution order alignment algorithm. 46.3 min into a nano-LC–MS/MS experiment, an MS1 scan is performed (A) and m/z features are matched to a 2D ion map stored on the instrument. (B) 21 of the peaks match 80 features in the ion map at a 10 ppm tolerance. Of these, over half (41 of 80) were mapped to one elution order bin (51 elution order). (C) A rolling elution order range is continually updated throughout the nano-LC–MS/MS experiment.
Prior to targeted analysis, a list of peptide targets, along with their relative EOs, is also uploaded to the instrument (Figure 4B). Each target is assigned an EO range (first and last appearance) depending on its length of elution in the discovery experiments. (See Figure 4C for zoom in.) Maintaining a dynamic EO range for each peptide is needed as different peptides elute for different amount of time during the separation. During the targeted analysis, instead of relying on absolute RT to trigger targeted MS/MS scans, determining the current EO becomes the main goal of the method. We have designed an online peptide elution order alignment (EOA) algorithm that takes a single MS1 spectrum and computes the current EO therefrom. In brief, following MS1 acquisition, the EOA algorithm takes the most intense m/z feature and extracts all EO values from the uploaded EOM at a narrow m/z tolerance (e.g., 10 ppm) (Figure 3A). Each m/z feature is matched in a similar fashion, and the resulting EO values are stored in a separate array (Figure 3B). In this example MS1, 21 m/z features matched a total of 80 EO values. When binned into 1 EO-wide bins, 41 of these values are contained within a single bin at 50 EO units. This indicates with high confidence that the current EO is somewhere near 50. To determine the EO precisely, the algorithm then calculates the 95% confidence interval around the max EO bin and stores the minimum (50.02) and maximum (51.64) EO. This process is repeated for each MS1, and over time the calculated EO range constructs a rolling average, as shown in Figure 3C. The EOA algorithm is expedient, taking on average 26 ms per MS1 to execute and does not induce a statistically significant change in the total number of MS/MS scans performed (Supplemental Figure 3 in the Supporting Information).
Figure 4.
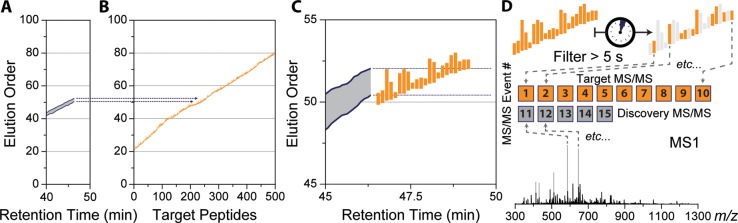
Following determination of the current elution order range (A), target peptides (B) sharing a similar elution order value are selected (C, rectangles represent individual peptides). Peptide targets within the elution order range are filtered based on when they were last sampled for MS/MS (D), leaving only targets that have been waiting the longest (e.g., > 5 s, highlighted rectangles). Those filtered peptides are then immediately sampled by MS/MS, regardless of MS1 detection (D). Unfilled MS/MS events are automatically filled with m/z features picked by the intensity-based DDA algorithm using normal sampling parameters (e.g., dynamic exclusion, intensity threshold, charge state exclusion, etc.).
Once the current EO range is determined, peptides sharing a similar EO are selected for MS/MS analysis. In brief, the current EO range is intersected with the target peptides already uploaded on the instrument (Figure 4B), and peptides whose EO overlaps the current EO range are stored as potential targets (Figure 4C). These peptides have a high probability of imminently eluting because they share very similar EO values with the current overall EO value. To prevent oversampling of any given target, potential targets are filtered based on how long since they were last sampled. Peptides that have been waiting the longest (e.g., >5 s) are automatically triggered for MS/MS analysis regardless of MS1 detection. Unfilled MS/MS events are then populated using normal DDA top-N approaches, excluding any m/z previously selected to be targeted (Figure 4D). This data collection scheme enables repetitive, consistent targeting of multiple peptides over their elution while allowing DDA scans to facilitate discovery. The EOA algorithm is compatible with other quantitative strategies such as parallel reaction monitoring (PRM),56,57 where peptide targets are repeatedly sampled (MS/MS) over their elution, and the resulting fragment ions are extracted to provide quantitative information (Supplemental Figure 4 in the Supporting Information).
Improving Peptide Identification in Multiple Experiments
We reasoned that the EOA algorithm would improve the reproducibility of peptide identification across multiple runs. Additionally, we increased the proteomic complexity by using a mammalian system (mouse) instead of yeast to determine how sample complexity affects the algorithm. Here a male C57BL/B6 mouse was sacrificed at 10 weeks, eight organs were harvested, and peptides from a tryptic digestion of each organ were labeled with a TMT 8-plex tag. First, six DDA top-15 nano-LC–MS/MS experiments were performed on the peptide sample. From the results of these discovery experiments, 500 peptides—identified in only three of the six experiments—were randomly selected to serve as peptide targets. These targets represent peptides that are difficult to identify reproducibly using standard DDA methodology. Additionally, we chose 500 targets because this represents the limit of the number of targets one could target with an inclusion list on the Orbitrap Elite MS in a single nano-LC–MS/MS analysis. Each target peptide’s EO was calculated from the three discovery experiments they were identified in, combined into a single EOM, and then uploaded to the instrument prior to targeting (Figure 4B). The same vial of mouse peptides, kept at 4 °C in an autosampler, used in the discovery experiments was then analyzed using DDA, followed by an accurate mass inclusion list (INC) and last intelligent data acquisition (IDA). This sequence was repeated for six technical replicates. On average, only 256 (51%) of the targeted peptides were identified in each of the DDA experiments (Figure 5A, 1% peptide-level FDR, error bars represent one σ). This is consistent with targeted peptides, as they originated from three of the original six discovery experiments (50%). The accurate mass inclusion list modestly increases identifications to an average of 280 (56%) targets per experiment. The biggest improvement is realized with IDA, where 440 of 500 targets (88%) were identified on average in each nano-LC–MS/MS experiment. When all six experiments for each method were combined and analyzed together, IDA identified 483 target peptides at least once, while INC identified 456 and DDA identified 426 at least once. Notably, 69 of the targets were only identified by IDA and not by DDA, while only 13 unique targets were discovered by DDA and not identified by IDA (Figure 5C and Supplemental Figure 5 in the Supporting Information). These results indicate that both DDA and the inclusion list undersampled the targeted peptides; presumably this is a result of either low S/N or poor charge-state determination of the precursor ions in the MS1. The IDA method avoids both of these issues by sampling regardless of MS1 detection, depending only on the target’s expected elution ordering.
Figure 5.
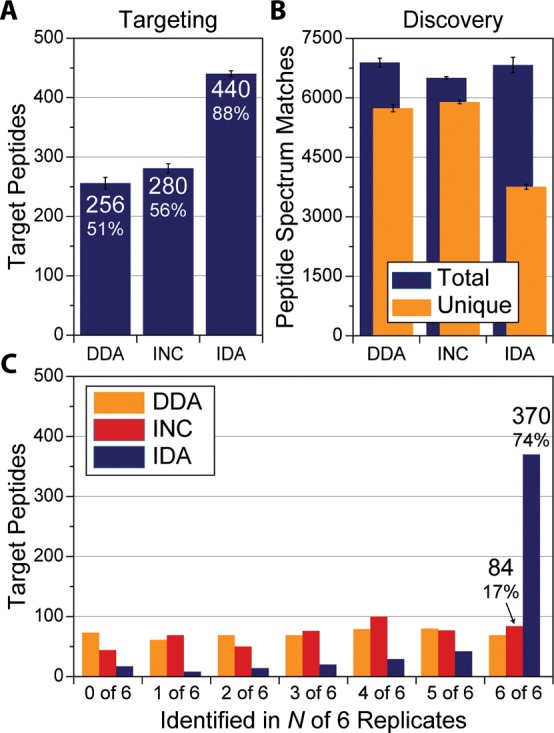
Subset of 500 mouse peptides were targeted with DDA, an accurate mass inclusion list (INC), and our intelligent data acquisition (IDA) method in hexplicate. (A) IDA identified the most target peptides of the three methods (error bars represent the 1 σ). (B) Discovery identifications by three methods show only a slight decline in the total number of peptides identified using IDA. (C) 74% of the targets were observed in all six technical replicates when IDA was used compared with <20% for the inclusion list or data-dependent acquisition.
Since the IDA method enables simultaneous DDA MS/MS sampling, comparisons of the total number of peptide identifications between the three acquisition methods can be made (Figure 5B). Each method produced nearly the same number of PSMs. A difference appears at the unique PSMs level (i.e., peptides), where both DDA and INC produced similar number of identifications (∼5800 peptides) but dropped to ∼3700 using IDA. We attributed this decline primarily to the redundant sampling of target peptides with the IDA method compared with the other methods. IDA identified each target 4.3 times on average, compared with 0.59 and 0.63 for DDA and INC, respectively, a ∼7:1 ratio. This is in agreement with the ratio of dynamic exclusion times between methods; IDA uses 5 s for each target compared with the longer dynamic exclusion time (35 s, 1:7) used in the DDA and INC methods. The oversampling of target peptides in IDA increases the likelihood of identification. We feel that it is an acceptable trade-off between maximizing reproducibility for a subset of peptides and a slight decline in total identified peptides. The increased reproducibility is demonstrated in Figure 5C; the IDA method identified 370 (74%) of the same peptides in all six experiments. The same cannot be said for DDA or INC; they managed to identify only 69 and 84 peptides in all six experiments, respectively. This represents an increase of over 340% in the number of peptide targets that were seen in all replicates.
Improved Reproducibility in Biological Systems
All data previously described have consisted of technical replicates of the same sample, injected with the same HPLC and analyzed using the same MS. These technical replicates are ideal to develop acquisitions methods on, primarily because the same peptides should exist in each injection, which removes sample variability from obfuscating the results. However, biological replication in proteomic studies is becoming more prevalent due to the increase in statistical power it affords. Four male C57BL/B6 mice were sacrificed at 10 weeks, eight organs were harvested, and peptides from a tryptic digestion of each organ were labeled with a TMT 8-plex tag to test whether intelligent data acquisition improves reproducibility in biological systems (Figure 6A,B). The tagged peptides from each mouse were mixed together and separated over a 165 min gradient and sampled using a DDA top-15 method to generate a list of peptide targets. An average of 8683 ± 313 peptide sequences were identified in each mouse for a total of 13 502 unique sequences. Of these, only 3969 (29.4%) peptides were identified in every mouse (Figure 6C). A subset of 1500 peptides was selected from the peptides detected in either two or three of four mice and sorted based on their assigned EOs (Figure 6D). Here peptide targets were chosen to be evenly distributed in the EO dimension to limit the number of coeluting peptides at given point. In subsequent targeting experiments, each mouse sample was analyzed twice, once using DDA and the other IDA, for a total of eight experiments. When the DDA targeting experiments were analyzed, an average of 810 (54%) target peptides were identified (Figure 7A, 1% peptide-level FDR, error bars represent one σ). Using IDA, this number increases to 1072 (71.5%). In total, over half of the targeted peptides (826, 55.1%) were identified in all four mice when using IDA compared with only 30.6% (459) using DDA (Figure 7B). The IDA method represents a nearly 80% improvement over DDA in the number of peptide targets it identifies in all mice. This increase in reproducible identification improves the quantitative results as well. When each tissue is compared with liver, the number of quantified peptides that are statistically significant (p value <0.05, Student’s t test with Storey Correction) is on average 227 greater with IDA compared with DDA (Figure 7C). For example, when the quantitative data for muscle is compared with that for liver (Figure 7D), IDA produced 826 significantly different peptides while only 531 were significant for DDA, a 56% increase. This can be directly attributed to increased reproducibility in identification across biological samples.
Figure 6.
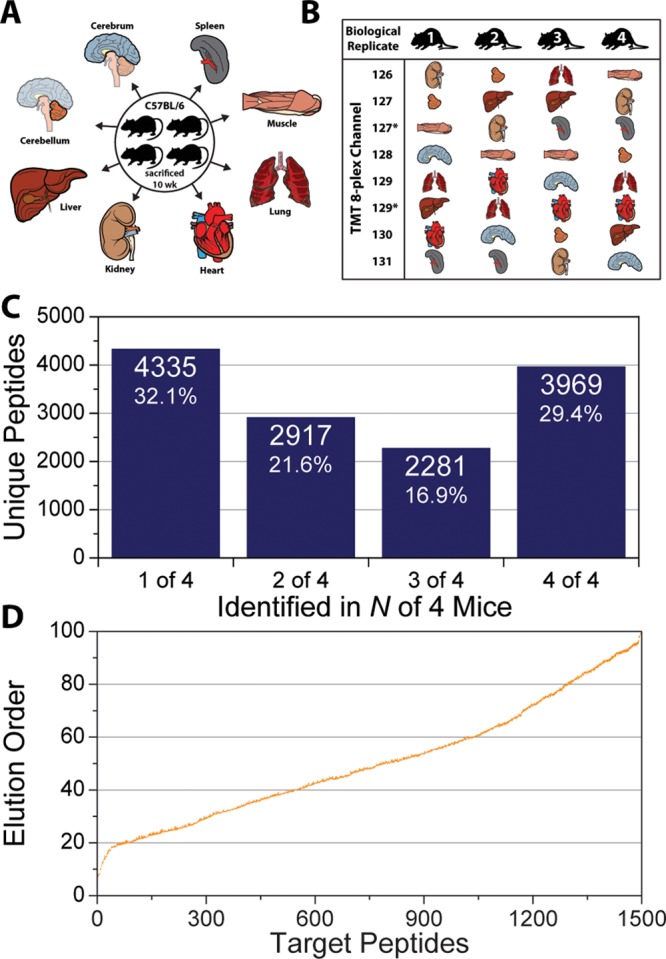
(A) Four C57Bl/6 mice were sacrificed at 10 weeks of age, and eight organs were harvested from each mouse. (B) Peptides resulting from a tryptic digestion of lysates from each organism were labeled with TMT 8-plex tags in a randomized order. (C) 165 min nano-LC–MS/MS experiments using DDA top-15 method identified only 3969 peptides in all four mice. (D) A subset of 1500 peptide targets was selected from peptides detected in only two or three of all four mice.
Figure 7.
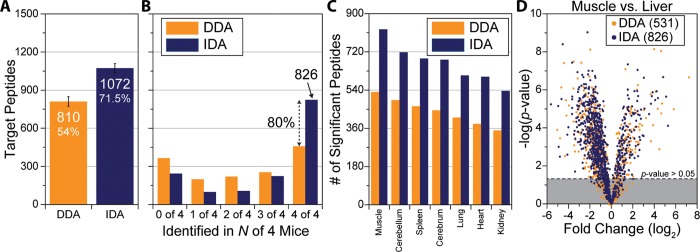
(A) In four subsequent nano-LC–MS/MS experiments, only 810 of 1500 mouse peptide targets were identified with DDA. The identifications improve to 1072 when IDA is used (error bars represent 1 σ). (B) In total, 826 target peptides were identified in all four mice when IDA was used to target. This number falls to only 459 peptides when DDA is used. (C) The number of statistically significant differences (p value <0.05) quantified when each tissue is compared with liver is greater with IDA than DDA. (D) When comparing target peptides identified in the muscle versus the liver, IDA quantified 826 statistically significant peptides compared with only 531 when DDA was used, a 56% increase.
Conclusions
The ability to identify the same peptides in multiple experiments reproducibly is increasingly important in proteomic analysis because increased statistical power is demanded. Historically, the most common acquisition method, DDA, has been used to sample large portions of proteomes, but it lacks adequate peptide identification reproducibility. We expand upon our previous IDA work and introduce the concept of using EO as a way to schedule and target peptides. Here we have described an online EOA algorithm that automatically adjusts to different chromatographic conditions to deliver consistent scheduling and robust reproducibility. The method is capable of targeting large number of peptides (>500) in a single run with minimal upfront preparation and effort. Using this method, we have shown improvements in peptide identification overlap among multiple experiments compared with DDA (88% compared with 50% identification overlap in six experiments). The EOA algorithm is capable of improving reproducibility even for highly variable samples. In four mice, our method was able to identify 806 target peptides compared with only 459 using normal DDA sampling.
We believe that such technologies can now be applied to traditional SRM methods that use triple quadrupole mass spectrometers. Here periodic full MS scans could be performed and analyzed to calculate the current EO and adjust the timing of the SRM transitions. One challenge would be the decreased specificity in determining EO from low-resolution scans. However, using a more adaptable metric for scheduling (elution ordering vs RT) could potentially increase the portability and robustness of SRM methods while reducing development time. Additionally, improved quantitative results could be obtained by deliberately oversampling one particular peptide target during its elution and quantifying with PRM or label-free methods.
Unlike SRM methods, where every MS/MS scan is predetermined, a novel aspect of our method is the flexibility of combining both targeted and discovery analysis in a single nano-LC–MS/MS experiment. The MS intelligently switches between targeted and discovery modes depending on what peptides are currently eluting, without any human intervention. In one experiment, over 3700 unique mouse peptides were discovered while simultaneously targeting 500 other peptides. Such hybrid MS methods enable both a focused and holistic view on the same sample, something that is welcomed when sample-limited.
Until comprehensive proteomic coverage is routinely obtained, targeted methods will be heavily used and developed. We have explored increasing the intelligence of MS methods as a means to improve the throughput and power of peptide targeting without sacrificing the novel discovery aspect of DDA sampling. Future work includes improvements to the determination of EOs, increasing the success rate of target identification, exploring additional quantitative strategies (e.g., PRM, label-free), and maximizing the throughput to target larger portions of the proteome without laborious upfront work.
Acknowledgments
We thank Jae Schwartz, John Syka, and Jens Griep-Raming for their thoughtful discussion and comments. This work was funded by the grants from the National Institutes of Health (NIH) (GM081629 to J.J.C and DK098672 to D.J.P.) and Thermo Fisher Scientific. M.T.M. was supported by NIH National Research Service Award T32 GM07215.
Supporting Information Available
IDA pseudo code. Figure S-1. Peptides that were identified in one of six or six of six DDA top-15 experiments analyzed for their maximal MS1 S/N. Figure S-2. Two technical replicates of a yeast DDA top-15 method using two different LC gradients. Figure S-3. EOA algorithm is expedient and induces only a slight increase in the MS1 duty cycle compared with normal DDA method. The distributions of scan times for IDA are bimodal because the EOA algorithm can be triggered every other MS1 because the current elution order changes only slightly between consecutive MS1 scans. Figure S-4. Example of PRM scan sequences obtainable using the IDA method. Figure S-5. Analysis of 500 mouse peptides that were targeted in six nanoLC–MS/MS experiments using IDA and DDA. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Washburn M. P.; Wolters D.; Yates J. R. 3rd. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 2001, 193242–247. [DOI] [PubMed] [Google Scholar]
- Wolters D. A.; Washburn M. P.; Yates J. R. 3rd. An automated multidimensional protein identification technology for shotgun proteomics. Anal. Chem. 2001, 73235683–5690. [DOI] [PubMed] [Google Scholar]
- Michalski A.; Cox J.; Mann M. More than 100,000 detectable peptide species elute in single shotgun proteomics runs but the majority is inaccessible to data-dependent LC–MS/MS. J. Proteome Res. 2011, 1041785–1793. [DOI] [PubMed] [Google Scholar]
- Eng J. K.; McCormack A. L.; Yates J. R. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994, 511976–989. [DOI] [PubMed] [Google Scholar]
- Sadygov R. G.; Cociorva D.; Yates J. R. 3rd Large-scale database searching using tandem mass spectra: looking up the answer in the back of the book. Nat. Methods 2004, 13195–202. [DOI] [PubMed] [Google Scholar]
- Venable J. D.; Dong M. Q.; Wohlschlegel J.; Dillin A.; Yates J. R. Automated approach for quantitative analysis of complex peptide mixtures from tandem mass spectra. Nat. Methods 2004, 1139–45. [DOI] [PubMed] [Google Scholar]
- Hoopmann M. R.; Merrihew G. E.; von Haller P. D.; MacCoss M. J. Post analysis data acquisition for the iterative MS/MS sampling of proteomics mixtures. J. Proteome Res. 2009, 841870–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesvizhskii A. I.; Aebersold R. Interpretation of shotgun proteomic data: the protein inference problem. Mol. Cell. Proteomics 2005, 4101419–1440. [DOI] [PubMed] [Google Scholar]
- Bantscheff M.; Schirle M.; Sweetman G.; Rick J.; Kuster B. Quantitative mass spectrometry in proteomics: a critical review. Anal. Bioanal. Chem. 2007, 38941017–1031. [DOI] [PubMed] [Google Scholar]
- Ideker T.; Thorsson V.; Ranish J. A.; Christmas R.; Buhler J.; Eng J. K.; Bumgarner R.; Goodlett D. R.; Aebersold R.; Hood L. Integrated genomic and proteomic analyses of a systematically perturbed metabolic network. Science 2001, 2925518929–934. [DOI] [PubMed] [Google Scholar]
- Aebersold R.; Mann M. Mass spectrometry-based proteomics. Nature 2003, 4226928198–207. [DOI] [PubMed] [Google Scholar]
- Molloy M. P.; Brzezinski E. E.; Hang J.; McDowell M. T.; VanBogelen R. A. Overcoming technical variation and biological variation in quantitative proteomics. Proteomics 2003, 3101912–1919. [DOI] [PubMed] [Google Scholar]
- Liu H.; Sadygov R. G.; Yates J. R. 3rd. A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal. Chem. 2004, 76144193–4201. [DOI] [PubMed] [Google Scholar]
- Wolf-Yadlin A.; Hautaniemi S.; Lauffenburger D. A.; White F. M. Multiple reaction monitoring for robust quantitative proteomic analysis of cellular signaling networks. Proc. Natl. Acad. Sci. U. S. A 2007, 104145860–5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabb D. L.; Vega-Montoto L.; Rudnick P. A.; Variyath A. M.; Ham A. J.; Bunk D. M.; Kilpatrick L. E.; Billheimer D. D.; Blackman R. K.; Cardasis H. L.; Carr S. A.; Clauser K. R.; Jaffe J. D.; Kowalski K. A.; Neubert T. A.; Regnier F. E.; Schilling B.; Tegeler T. J.; Wang M.; Wang P.; Whiteaker J. R.; Zimmerman L. J.; Fisher S. J.; Gibson B. W.; Kinsinger C. R.; Mesri M.; Rodriguez H.; Stein S. E.; Tempst P.; Paulovich A. G.; Liebler D. C.; Spiegelman C. Repeatability and reproducibility in proteomic identifications by liquid chromatography-tandem mass spectrometry. J. Proteome Res. 2010, 92761–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson T.; Mann M.; Aebersold R.; Yates J. R. 3rd; Bairoch A.; Bergeron J. J. Mass spectrometry in high-throughput proteomics: ready for the big time. Nat. Methods 2010, 79681–685. [DOI] [PubMed] [Google Scholar]
- Pachl F.; Ruprecht B.; Lemeer S.; Kuster B. Characterization of a high field Orbitrap mass spectrometer for proteome analysis. Proteomics 2013, 13172552–2562. [DOI] [PubMed] [Google Scholar]
- Aebersold R. A stress test for mass spectrometry-based proteomics. Nat. Methods 2009, 66411–412. [DOI] [PubMed] [Google Scholar]
- Thakur S. S.; Geiger T.; Chatterjee B.; Bandilla P.; Frohlich F.; Cox J.; Mann M. Deep and highly sensitive proteome coverage by LC–MS/MS without prefractionation. Mol. Cell. Proteomics 2011, 108M110 003699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraj N.; Kulak N. A.; Cox J.; Neuhauser N.; Mayr K.; Hoerning O.; Vorm O.; Mann M. System-wide perturbation analysis with nearly complete coverage of the yeast proteome by single-shot ultra HPLC runs on a bench top Orbitrap. Mol. Cell. Proteomics 2012, 113M111 013722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert A. S.; Richards A. L.; Bailey D. J.; Ulbrich A.; Coughlin E. E.; Westphall M. S.; Coon J. J. The One Hour Yeast Proteome. Mol. Cell. Proteomics 2014, 13, 339–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitski M. M.; Fischer F.; Mathieson T.; Sweetman G.; Lang M.; Bantscheff M. Targeted data acquisition for improved reproducibility and robustness of proteomic mass spectrometry assays. J. Am. Soc. Mass Spectrom. 2010, 21101668–1679. [DOI] [PubMed] [Google Scholar]
- Lange V.; Picotti P.; Domon B.; Aebersold R. Selected reaction monitoring for quantitative proteomics: a tutorial. Mol. Syst. Biol. 2008, 4, 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picotti P.; Bodenmiller B.; Mueller L. N.; Domon B.; Aebersold R. Full dynamic range proteome analysis of S. cerevisiae by targeted proteomics. Cell 2009, 1384795–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picotti P.; Rinner O.; Stallmach R.; Dautel F.; Farrah T.; Domon B.; Wenschuh H.; Aebersold R. High-throughput generation of selected reaction-monitoring assays for proteins and proteomes. Nat. Methods 2010, 7143–46. [DOI] [PubMed] [Google Scholar]
- Picotti P.; Aebersold R. Selected reaction monitoring-based proteomics: workflows, potential, pitfalls and future directions. Nat. Methods 2012, 96555–566. [DOI] [PubMed] [Google Scholar]
- Sabido E.; Wu Y.; Bautista L.; Porstmann T.; Chang C. Y.; Vitek O.; Stoffel M.; Aebersold R. Targeted proteomics reveals strain-specific changes in the mouse insulin and central metabolic pathways after a sustained high-fat diet. Mol. Syst. Biol. 2013, 9, 681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyonami R.; Schoen A.; Prakash A.; Peterman S.; Zabrouskov V.; Picotti P.; Aebersold R.; Huhmer A.; Domon B. Increased selectivity, analytical precision, and throughput in targeted proteomics. Mol. Cell. Proteomics 2011, 102M110 002931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith R. D.; Anderson G. A.; Lipton M. S.; Pasa-Tolic L.; Shen Y.; Conrads T. P.; Veenstra T. D.; Udseth H. R. An accurate mass tag strategy for quantitative and high-throughput proteome measurements. Proteomics 2002, 25513–523. [DOI] [PubMed] [Google Scholar]
- Beer I.; Barnea E.; Ziv T.; Admon A. Improving large-scale proteomics by clustering of mass spectrometry data. Proteomics 2004, 44950–960. [DOI] [PubMed] [Google Scholar]
- Jaffe J. D.; Mani D. R.; Leptos K. C.; Church G. M.; Gillette M. A.; Carr S. A. PEPPeR, a platform for experimental proteomic pattern recognition. Mol. Cell. Proteomics 2006, 5101927–1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash A.; Mallick P.; Whiteaker J.; Zhang H.; Paulovich A.; Flory M.; Lee H.; Aebersold R.; Schwikowski B. Signal maps for mass spectrometry-based comparative proteomics. Mol. Cell. Proteomics 2006, 53423–432. [DOI] [PubMed] [Google Scholar]
- Radulovic D.; Jelveh S.; Ryu S.; Hamilton T. G.; Foss E.; Mao Y.; Emili A. Informatics platform for global proteomic profiling and biomarker discovery using liquid chromatography-tandem mass spectrometry. Mol. Cell. Proteomics 2004, 310984–997. [DOI] [PubMed] [Google Scholar]
- Listgarten J.; Emili A. Statistical and computational methods for comparative proteomic profiling using liquid chromatography-tandem mass spectrometry. Mol. Cell. Proteomics 2005, 44419–434. [DOI] [PubMed] [Google Scholar]
- Shen Y.; Strittmatter E. F.; Zhang R.; Metz T. O.; Moore R. J.; Li F.; Udseth H. R.; Smith R. D.; Unger K. K.; Kumar D.; Lubda D. Making broad proteome protein measurements in 1–5 min using high-speed RPLC separations and high-accuracy mass measurements. Anal. Chem. 2005, 77237763–7773. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Yi E. C.; Li X. J.; Mallick P.; Kelly-Spratt K. S.; Masselon C. D.; Camp D. G. 2nd; Smith R. D.; Kemp C. J.; Aebersold R. High throughput quantitative analysis of serum proteins using glycopeptide capture and liquid chromatography mass spectrometry. Mol. Cell. Proteomics 2005, 42144–155. [DOI] [PubMed] [Google Scholar]
- Lin H.; He L.; Ma B. A combinatorial approach to the peptide feature matching problem for label-free quantification. Bioinformatics 2013, 29141768–1775. [DOI] [PubMed] [Google Scholar]
- Bateman N. W.; Goulding S. P.; Shulman N.; Gadok A. K.; Szumlinski K. K.; Maccoss M. J.; Wu C. C. Maximizing peptide identification events in proteomic workflows utilizing data-dependent acquisition. Mol. Cell. Proteomics 2014, 13, 329–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey D. J.; Rose C. M.; McAlister G. C.; Brumbaugh J.; Yu P.; Wenger C. D.; Westphall M. S.; Thomson J. A.; Coon J. J. Instant spectral assignment for advanced decision tree-driven mass spectrometry. Proc. Natl. Acad. Sci. U. S. A. 2012, 109228411–8416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graumann J.; Scheltema R. A.; Zhang Y.; Cox J.; Mann M. A framework for intelligent data acquisition and real-time database searching for shotgun proteomics. Mol. Cell. Proteomics 2012, 113M111 013185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webber J. T.; Askenazi M.; Ficarro S. B.; Iglehart M. A.; Marto J. A. Library dependent LC–MS/MS acquisition via mzAPI/Live. Proteomics 2013, 1391412–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner T.; Becher I.; Sweetman G.; Doce C.; Savitski M. M.; Bantscheff M. High-resolution enabled TMT 8-plexing. Anal. Chem. 2012, 84167188–7194. [DOI] [PubMed] [Google Scholar]
- McAlister G. C.; Huttlin E. L.; Haas W.; Ting L.; Jedrychowski M. P.; Rogers J. C.; Kuhn K.; Pike I.; Grothe R. A.; Blethrow J. D.; Gygi S. P. Increasing the multiplexing capacity of TMTs using reporter ion isotopologues with isobaric masses. Anal. Chem. 2012, 84177469–7478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalski A.; Damoc E.; Lange O.; Denisov E.; Nolting D.; Muller M.; Viner R.; Schwartz J.; Remes P.; Belford M.; Dunyach J. J.; Cox J.; Horning S.; Mann M.; Makarov A. Ultra high resolution linear ion trap Orbitrap mass spectrometer (Orbitrap Elite) facilitates top down LC MS/MS and versatile peptide fragmentation modes. Mol. Cell. Proteomics 2012, 113O111 013698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger C. D.; Phanstiel D. H.; Lee M. V.; Bailey D. J.; Coon J. J. COMPASS: a suite of pre- and post-search proteomics software tools for OMSSA. Proteomics 2011, 1161064–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geer L. Y.; Markey S. P.; Kowalak J. A.; Wagner L.; Xu M.; Maynard D. M.; Yang X.; Shi W.; Bryant S. H. Open mass spectrometry search algorithm. J. Proteome Res. 2004, 35958–964. [DOI] [PubMed] [Google Scholar]
- Elias J. E.; Gygi S. P. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 2007, 43207–214. [DOI] [PubMed] [Google Scholar]
- Kall L.; Storey J. D.; MacCoss M. J.; Noble W. S. Posterior error probabilities and false discovery rates: Two sides of the same coin. J. Proteome Res. 2008, 7140–44. [DOI] [PubMed] [Google Scholar]
- Kall L.; Storey J. D.; MacCoss M. J.; Noble W. S. Assigning significance to peptides identified by tandem mass spectrometry using decoy databases. J. Proteome Res. 2008, 7129–34. [DOI] [PubMed] [Google Scholar]
- Hsieh E. J.; Hoopmann M. R.; MacLean B.; MacCoss M. J. Comparison of Database Search Strategies for High Precursor Mass Accuracy MS/MS Data. J. Proteome Res. 2010, 921138–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey J. D.; Tibshirani R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. U.S.A. 2003, 100169440–9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krokhin O. V.; Craig R.; Spicer V.; Ens W.; Standing K. G.; Beavis R. C.; Wilkins J. A. An improved model for prediction of retention times of tryptic peptides in ion pair reversed-phase HPLC: its application to protein peptide mapping by off-line HPLC-MALDI MS. Mol. Cell. Proteomics 2004, 39908–919. [DOI] [PubMed] [Google Scholar]
- Krokhin O. V. Sequence-specific retention calculator. Algorithm for peptide retention prediction in ion-pair RP-HPLC: application to 300- and 100-A pore size C18 sorbents. Anal. Chem. 2006, 78227785–7795. [DOI] [PubMed] [Google Scholar]
- Petritis K.; Kangas L. J.; Yan B.; Monroe M. E.; Strittmatter E. F.; Qian W. J.; Adkins J. N.; Moore R. J.; Xu Y.; Lipton M. S.; Camp D. G. 2nd; Smith R. D. Improved peptide elution time prediction for reversed-phase liquid chromatography-MS by incorporating peptide sequence information. Anal. Chem. 2006, 78145026–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spicer V.; Grigoryan M.; Gotfrid A.; Standing K. G.; Krokhin O. V. Predicting retention time shifts associated with variation of the gradient slope in peptide RP-HPLC. Anal. Chem. 2010, 82239678–9685. [DOI] [PubMed] [Google Scholar]
- Peterson A. C.; Russell J. D.; Bailey D. J.; Westphall M. S.; Coon J. J. Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Mol. Cell. Proteomics 2012, 11111475–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallien S.; Duriez E.; Crone C.; Kellmann M.; Moehring T.; Domon B. Targeted proteomic quantification on quadrupole-orbitrap mass spectrometer. Mol. Cell. Proteomics 2012, 11121709–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.