

The peripheral immune response is altered in Huntington’s disease, but the underlying mechanisms are unclear. Using RNA interference to lower huntingtin levels in leucocytes from patients, Träger et al. reverse disease-associated phenotypes including cytokine elevation and transcriptional dysregulation, and argue for a direct effect of mutant huntingtin on NFκΒ signalling.
Keywords: Huntington’s disease, immunology, myeloid cells, gene lowering
Abstract
Huntington’s disease is an inherited neurodegenerative disorder caused by a CAG repeat expansion in the huntingtin gene. The peripheral innate immune system contributes to Huntington’s disease pathogenesis and has been targeted successfully to modulate disease progression, but mechanistic understanding relating this to mutant huntingtin expression in immune cells has been lacking. Here we demonstrate that human Huntington’s disease myeloid cells produce excessive inflammatory cytokines as a result of the cell-intrinsic effects of mutant huntingtin expression. A direct effect of mutant huntingtin on the NFκB pathway, whereby it interacts with IKKγ, leads to increased degradation of IκB and subsequent nuclear translocation of RelA. Transcriptional alterations in intracellular immune signalling pathways are also observed. Using a novel method of small interfering RNA delivery to lower huntingtin expression, we show reversal of disease-associated alterations in cellular function–the first time this has been demonstrated in primary human cells. Glucan-encapsulated small interfering RNA particles were used to lower huntingtin levels in human Huntington’s disease monocytes/macrophages, resulting in a reversal of huntingtin-induced elevated cytokine production and transcriptional changes. These findings improve our understanding of the role of innate immunity in neurodegeneration, introduce glucan-encapsulated small interfering RNA particles as tool for studying cellular pathogenesis ex vivo in human cells and raise the prospect of immune cell-directed HTT-lowering as a therapeutic in Huntington’s disease.
Introduction
Huntington’s disease is an incurable, autosomal dominant neurodegenerative disorder caused by a CAG repeat expansion in exon 1 of the huntingtin (HTT) gene leading to an expanded stretch of 36 or more glutamine residues in the N-terminal region of the HTT protein (The Huntington's Disease Collaborative Research Group, 1993). The disease is characterized by progressive cognitive, psychiatric and motor impairments caused by neuronal dysfunction and cell death.
Although primary pathology in Huntington’s disease is believed to arise from basal ganglia degeneration, HTT expression has been found in all tissues studied (Li et al., 1993). Indeed, numerous studies of patients with Huntington’s disease and mouse models have described abnormalities in peripheral tissues, including weight loss, muscle wasting, diabetes and changes in the neuro-endocrine system (van der Burg et al., 2009). Mutant HTT expression in non-neuronal cells in both the brain and the periphery may contribute to Huntington’s disease neuropathology.
HTT is expressed in immune cells (Weiss et al., 2012), and both central and peripheral immune system abnormalities have been shown in patients with Huntington’s disease (Soulet and Cicchetti, 2011). Microglia, the resident immune cells of the brain (Ransohoff and Perry, 2009), are sustained by self-renewal (Ajami et al., 2007); however, disrupting the blood–brain barrier by irradiation has shown that blood monocytes are able to populate the brain (Simard and Rivest, 2004). Microglial activation, seen in post-mortem Huntington’s disease brain tissue (Sapp et al., 2001) and by PET imaging, occurs in Huntington’s disease gene carriers before symptom onset (Tai et al., 2007). We have previously demonstrated peripheral immune system dysfunction in Huntington’s disease, including changes in innate immune proteins in patient plasma (Dalrymple et al., 2007). Moreover, elevated plasma cytokine (Björkqvist et al., 2008) and chemokine (Wild et al., 2011) levels in patients correlate with disease progression and can be detected years before disease onset. We have shown that primary human monocytes are hyper-reactive in response to lipopolysaccharide, producing increased levels of interleukin (IL)-6. This phenotype is replicated in murine mutant HTT expressing macrophages and microglia, demonstrating that peripheral cells could mirror pathology in the CNS in Huntington’s disease (Björkqvist et al., 2008).
Furthermore, several recent studies have suggested that the peripheral immune system can act as a modifier of Huntington’s disease neuropathology. Transplantation of wild-type bone marrow into Huntington’s disease mice partially rescues their motor defects, increases synaptogenesis and reduces elevated plasma cytokine levels (Kwan et al., 2012a). Peripheral administration of a kynurenine 3-monooxygenase (KMO) inhibitor extends lifespan, prevents synaptic loss and decreases microglial activation in Huntington’s disease mice. As the drug cannot cross the blood–brain barrier, the neuroprotective effect is secondary to inhibition of KMO in peripheral immune cells (Zwilling et al., 2011). Furthermore, treatment with a cannabinoid receptor 2 agonist known to dampen immune responses, suppresses motor deficits and CNS inflammation while extending life span in a Huntington’s disease mouse model. This positive effect can be blocked with an antagonist that is restricted to the periphery, demonstrating the importance of peripheral immune cells in modulating pathogenesis (Bouchard et al., 2012). These studies provide strong evidence that the immune system plays a disease-modifying role in Huntington’s disease neuropathogenesis, but the mechanism(s) by which mutant HTT expression in immune cells causes this dysfunction has not yet been established.
Intracellular signalling pathways leading to the activation of the transcription factor NFκB are important regulators of cytokine production and play a key role in inflammation. Events such as the activation of Toll-like receptors (TLRs) lead to signal transduction through adapter proteins MyD88 and IRAK1, leading to the phosphorylation and activation of IKK. This kinase phosphorylates IκB, which is then ubiquitinated and degraded by the proteasome, whereby it dissociates from the NFκB transcription factor subunits (RelA, RelB, cRel, NFκB1, NFκB2) that it sequesters in an inactive state in the cytoplasm. The free NFκB molecules can then translocate into the nucleus and activate gene transcription (Hayden and Ghosh, 2012). The NFκB pathway has previously been implicated in Huntington’s disease, with Khoshnan et al. (2004) having shown in inducible PC12 cells and striatal extracts from R6/2 Huntington’s disease mice that overexpression of mutant HTT exon 1 can activate the NFκB pathway by directly interacting with IKKγ (Khoshnan et al., 2004). Similarly, a recent study has shown enhanced NFκB signalling in astrocytes isolated from R6/2 mice (Hsiao et al., 2013). It remains to be shown that this interaction also occurs in a human system with expression of full-length HTT at normal allelic expression levels.
The present work seeks to identify the mechanism of dysfunction in primary human Huntington’s disease monocytes and macrophages ex vivo. We have characterized immune cell dysfunction by detailed cytokine profiling and study of upstream intracellular signalling pathways, identifying NFκB pathway dysregulation as the cause of immune dysfunction. We have used overexpression studies and a novel small interfering RNA-mediated knock-down technique to investigate the role cell-intrinsic HTT plays in human Huntington’s disease monocyte and macrophage function, demonstrating the feasibility of reversing peripheral immune dysregulation by cell-targeted HTT-lowering.
Materials and methods
Collection and classification of human samples
All human experiments were performed in accordance with the Declaration of Helsinki and approved by University College London (UCL)/UCL Hospitals Joint Research Ethics Committee. All subjects provided informed written consent. Classification of patients is detailed in the Supplementary material. Subjects’ demographic are provided in Supplementary Table 1.
Isolation of human monocytes and macrophages
Cells were isolated from whole blood, as previously described (Björkqvist et al., 2008) and in the Supplementary material. Cells were cultured in RPMI culture medium supplemented with 10% foetal calf serum, 2 mM l-glutamine, 50 U/ml penicillin and 50 µg/ml streptomycin (Invitrogen). Monocytes were allowed to rest for 16 h before experimental use. Culture medium was supplemented with 20 ng/ml granulocyte macrophage-colony stimulating factor (GM-CSF) for 6 days to differentiate monocytes into macrophages.
Mutant HTT expression in U937 cells
U937 cells (Sundström and Nilsson, 1976) were transduced with lentiviral constructs containing human HTT exon 1 sequences with either 29, 71 or 129 CAG repeats, together with GFP, or a control vector containing GFP but no HTT exon 1. For details of vectors, viral production and transduction, see the online Supplementary material. Transduced U937 cells were tested for HTT protein expression using a time resolved fluorescence resonance energy transfer (TR-FRET) immunoassay. HTT exon 1 expressing U937 cells were seeded into 24-well plates at 5 × 105 cells per well and differentiated into mature monocytes using 10 nM phorbol 12-myristate 13-acetate (PMA) for 3 days (Alciato et al., 2010) before cytokine profiling.
HTT silencing
Monocytes and macrophages were incubated with β1,3-d-glucan-encapsulated small interfering RNA particles (GeRPs) for 4 h, after which fresh medium was added to the cultures. GeRP uptake was visualized by seeding 1 × 105 monocytes per 13 mm coverslip, incubating them with empty green fluorescent GeRPs for 12 h and mounting onto slides with 1 µg/ml DAPI. Images were acquired using a Zeiss 510 meta microscope (objective ×63/1.4 oil DIC, 1024 × 1024), overlaying the bright-field image of the cells with the 405 nm and 488 nm fluorescence channels for DAPI and green fluorescence, respectively. Macrophages, which were transfected on Day 3 of the differentiation protocol, were transfected with green fluorescent GeRPs containing no small interfering RNA at various ratios (1:1, 1:3, and 1:10) before uptake rates were measured by flow cytometry. Cells were fixed with 3.7% paraformaldehyde for 10 min, washed with fluorescence-activated cell sorting (FACS) buffer (PBS containing 1% foetal calf serum and 0.02% sodium azide) and resuspended in 200 μl FACS buffer for analysis by flow cytometry (FACSCalibur with CellQuest Pro BD Bioscience). Data analysis was performed using FlowJo 7.2.5 (Tree Star). To examine the effects of HTT knock-down on cytokine production, macrophages were treated with either scrambled or anti-HTT small interfering RNA containing GeRPs at a 1:10 cell: particle ratio on Day 3 of the differentiation protocol; stimulation of the cells took place 3 days later. To examine the effects of HTT knock-down on transcriptional dysregulation, monocytes were treated with either scrambled or anti-HTT small interfering RNA containing GeRPs at a 1:10 cell: particle ratio, before quantitative PCR analysis 3 days later.
Cytokine profiling
All cells were seeded at 5 × 105 cells per well in 24-well plates and isolated, differentiated and transduced as described above. For stimulation, medium was changed to fresh cell culture medium containing 10 ng/ml IFNγ (R & D Systems) and 2 μg/ml lipopolysaccharide (Sigma-Aldrich, E.coli 055:B5, strain 1644-70. Cat. number L6529). After 24 h, supernatants were harvested and analysed using MSD multiplex assays, according to manufacturer’s instructions (MesoScale Discovery). For monocytes the pro-inflammatory (7-plex) assay was used, however, IFNγ measures were not analysed as we used IFNγ as stimulus. For all other cell types, the pro-inflammatory II (4-plex) assay was used and all data are shown. Monocyte data were adjusted to basal cytokine levels, whereas all other cell types were normalized to total protein concentration in each well. Cells were lysed in 50 mM Tris pH 8, 150 mM NaCl, 0.5% sodium deoxycholate, 0.5% Triton™ X-100 and assayed for total protein concentration using a Bradford-based protein assay (Bio-Rad).
Time resolved fluorescence resonance energy transfer quantification of HTT
TR-FRET immunoassay quantification of total HTT and soluble mutant HTT was performed as previously described (Baldo et al., 2012) and is detailed in the Supplementary material.
Polymerase chain reaction arrays
SABioscience Human NFκB Signaling Pathway RT2Profiler™ PCR Arrays were used in combination with the QIAGEN RNeasy® Mini Kit for RNA isolation from 2 × 106 cells. RNA integrity was evaluated using 2100 RNA Bioanalyser chips (Agilent). RNA was reverse transcribed using the RT2 First Strand kit for complementary DNA transcription, before the RT2 SYBR® Green qPCR Mastermix and pre-primer coated PCR plates were used for quantitative PCR (SABioscience). All kits were used according to the manufacturer’s instructions.
For standard SYBR® Green protocols and bioinformatics used for Supplementary Fig. 6, see Supplementary material.
Proximity ligation assays
Proximity ligation assays were conducted on monocyte-derived macrophages seeded on 13 mm coverslips. Cells were fixed in 4% paraformaldehyde for 10 min and permeabilized with 100% ice cold methanol at −20°C for 15 min. Coverslips were blocked with 10% bovine serum albumin for 30 min at 37°C before staining with primary antibodies was performed for 1 h at 37°C (mouse anti-HTT 4C9, 1:300, kind gift from Novartis; rabbit anti-IKKγ, 1:100, Santa Cruz; rabbit anti-IKKα/β, 1:25, Santa Cruz). Instead of using fluorescently labelled secondary antibodies, a proximity ligation approach was applied following manufacturer’s instructions (Sigma). Briefly, samples were incubated with secondary antibodies conjugated with DNA probes (minus anti-mouse and plus anti-rabbit DNA probes). Probes were hybridized and ligated before amplification of the DNA template in a rolling circle amplification reaction. Detection solution was added to identify amplified DNA. Signals were detected using a Zeiss LSM 710 confocal microscope (objective plan-apachromat ×40/1.4 oil DIC M27, 1024 × 1024). Spots were quantified using Volocity (PerkinElmer) on at least seven fields of view per subject, taken of random sides of each coverslips.
Immunoblot analysis of IκB degradation
Monocytes were seeded at 1 × 106 cells/well into 24-well plates and rested for 16 h. Cells were stimulated with 2 µg/ml lipopolysaccharide over a 2 h or 24 h time course before extraction of lysates for western blotting (see Supplementary material for detail).
NFκB RelA translocation
CD14+ monocytes were seeded at 2 × 106 into 6-well plates and left to rest for 16 h. The cells were stimulated with 2 µg/ml lipopolysaccharide before being scraped off the plates. Pelleted cells were fixed for 15 min and permeabilized for 10 min using the eBioscience Fix/Perm solutions, before NFκB p65/RelA XP antibody (1:200; Cell Signaling) diluted in permeabilization buffer was added. After 30 min incubation shaking at 4°C, cells were washed twice with FACS buffer and spun for 5 min at 300g. Secondary anti-rabbit IgG phycoerythrin (eBioscience) was added at 1:100 in FACS buffer and incubated for 30 min before washing the cells twice with FACS buffer. Cells were resuspended in 80 µl FACS buffer and stained with 1 µg/ml DAPI just before analysis. Samples were run on the ImageStreamX (Amnis) and analysed using the IDEAS software. Briefly, gating on single cells, the similarity feature {Similarity_Erode [Object (M04,BF,Tight)2]_Dapi _RelA} was used to establish the rate of RelA translocation by measuring the overlap of DAPI and RelA staining. Translocation rate was normalized to baseline levels for each subject.
Statistical analysis
For cytokine profiling data, inter-group differences were identified by one-way ANOVA with post hoc Tukey Honestly Significant Difference testing to allow for multiple comparisons. Data were corrected for age and gender before analysis. Linear regression with log10 transformed data was used to establish whether cytokine production by primary human monocytes and macrophages correlates with CAG repeat length. Cytokine profiling data from U937 cells and knock-down cells were analysed by two-way ANOVA with Bonferroni post-tests. Gene expression changes measured by quantitative PCR were analysed using unpaired two-tailed student t-tests. Paired two-tailed student t-tests were used to analyse the effects of anti-HTT small interfering RNA compared to scrambled small interfering RNA in cells from the same individual. All error bars represent standard error of the mean.
Results
Huntington’s disease patient monocytes and macrophages are hyper-reactive after lipopolysaccharide stimulation
Previously we have shown that Huntington’s disease patient monocytes produce increased levels of IL-6 upon stimulation with lipopolysaccharide (Björkqvist et al., 2008). To extend these findings to other cytokines, we collected blood samples from a large cohort (n = 53) of HTT gene carriers ranging from pre-manifest to moderate disease stages and control subjects (n = 27) (Supplementary Table 1). CD14+ monocytes were isolated, primed with IFNγ and stimulated with lipopolysaccharide ex vivo. Monocytes from Huntington’s disease gene carriers at each disease stage were found to produce more IL-6 and TNFα than control cells (Fig. 1A). Furthermore, IL-1β production by pre-manifest monocytes was significantly increased. IL-8, IL-10 and IL-12 levels did not differ between Huntington’s disease and control cells (Fig. 1A).
Figure 1.
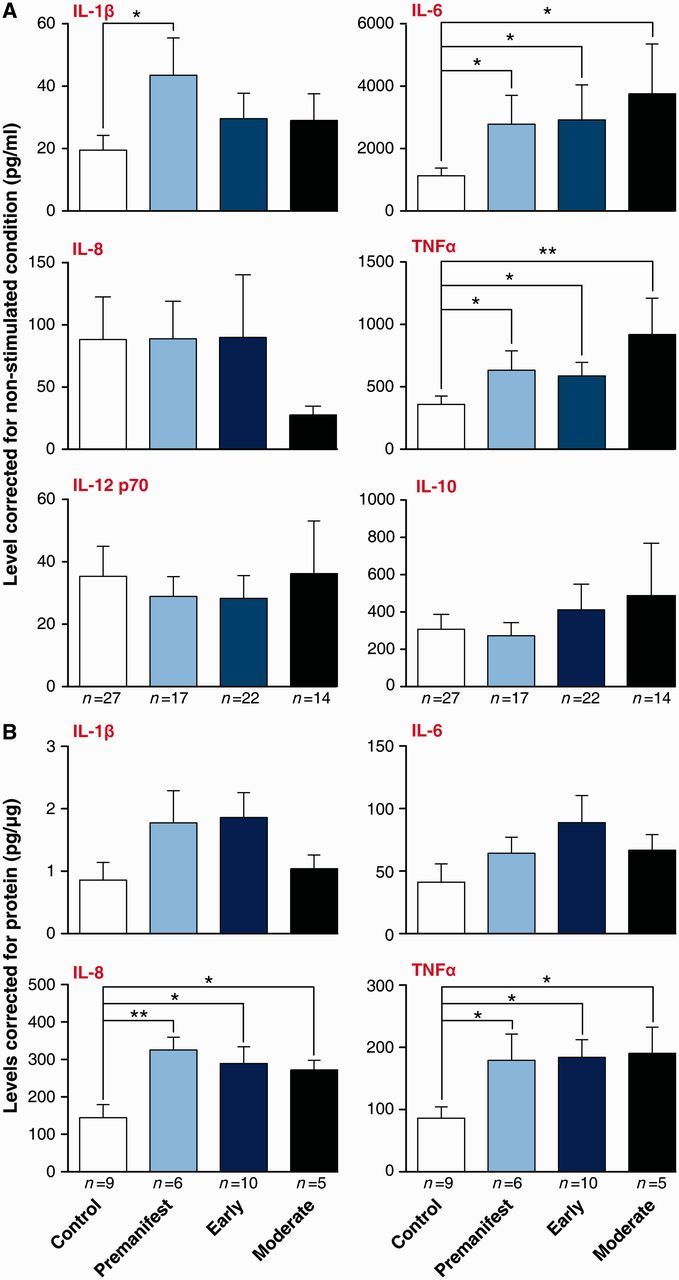
Pro-inflammatory cytokine production by monocytes and macrophages is elevated in patients with Huntington’s disease. Innate immune regulators such as IL-6, IL-8 and TNFα were elevated in Huntington’s disease patients (A) blood monocytes and (B) macrophages collected from two independent patient cohorts, stimulated in vitro with 10 ng/ml IFNγ and 2 µg/ml lipopolysaccharide for 24 h. Data show mean concentrations ± SEM, n = individual biological repeats, ANOVA with post hoc Tukey HSD test. *P < 0.05; **P < 0.01.
When monocytes migrate into tissues, they differentiate into macrophages capable of eliciting effective immune responses to localized inflammatory signals (Gordon and Taylor, 2005). To test whether Huntington’s disease macrophages are abnormal too, blood monocytes were differentiated into macrophages using granulocyte macrophage-colony stimulating factor and stimulated with IFNγ and lipopolysaccharide to assess their cytokine profile. In keeping with our previous findings, macrophages from all Huntington’s disease stages produced significantly higher levels of TNFα than control cells (Fig. 1B). Furthermore, IL-8 levels were also significantly increased in Huntington’s disease macrophages (Fig. 1B). This marks a shift in the pro-inflammatory cytokines elevated, when compared to the pattern seen in monocytes (Fig. 1A), where IL-8 was not changed. This is likely because of the distinct functions of the two cell types.
These data show that myeloid cells isolated from patients with Huntington’s disease are hyper-reactive, producing elevated levels of several key pro-inflammatory cytokines following stimulation.
Correlating production of individual cytokines to CAG repeat length showed a significant association (P = 0.048) of CAG repeat length with TNFα produced by Huntington’s disease monocytes (Supplementary Fig. 1). There was no correlation between CAG repeat length and levels of any other cytokine in either Huntington’s disease monocytes or macrophages.
Lowering HTT levels reverses Huntington’s disease myeloid cell hyper-reactivity
Lowering HTT expression using small interfering RNA is a promising therapeutic approach for Huntington’s disease (Sah and Aronin, 2011). Therefore, we investigated whether lowering total HTT levels can reverse the hyper-reactive phenotype in primary Huntington’s disease monocytes and macrophages. Using a novel approach that takes advantage of these cells’ ability to phagocytose (Aouadi et al., 2009), GeRPs were packaged with previously validated (DiFiglia et al., 2007) anti-HTT small interfering RNA for delivery into human ex vivo monocytes or monocyte-derived macrophages. Monocytes cultured with the GeRPs readily ingested them through phagocytosis (Fig. 2A), without effect on cell viability (Supplementary Fig. 2). Testing different macrophage:GeRP ratios, up to 90% of macrophages phagocytosed the green fluorescent GeRPs when they were added at a 10-fold particle to cell ratio, demonstrating high transfection efficiency at this concentration (Fig. 2B).
Figure 2.
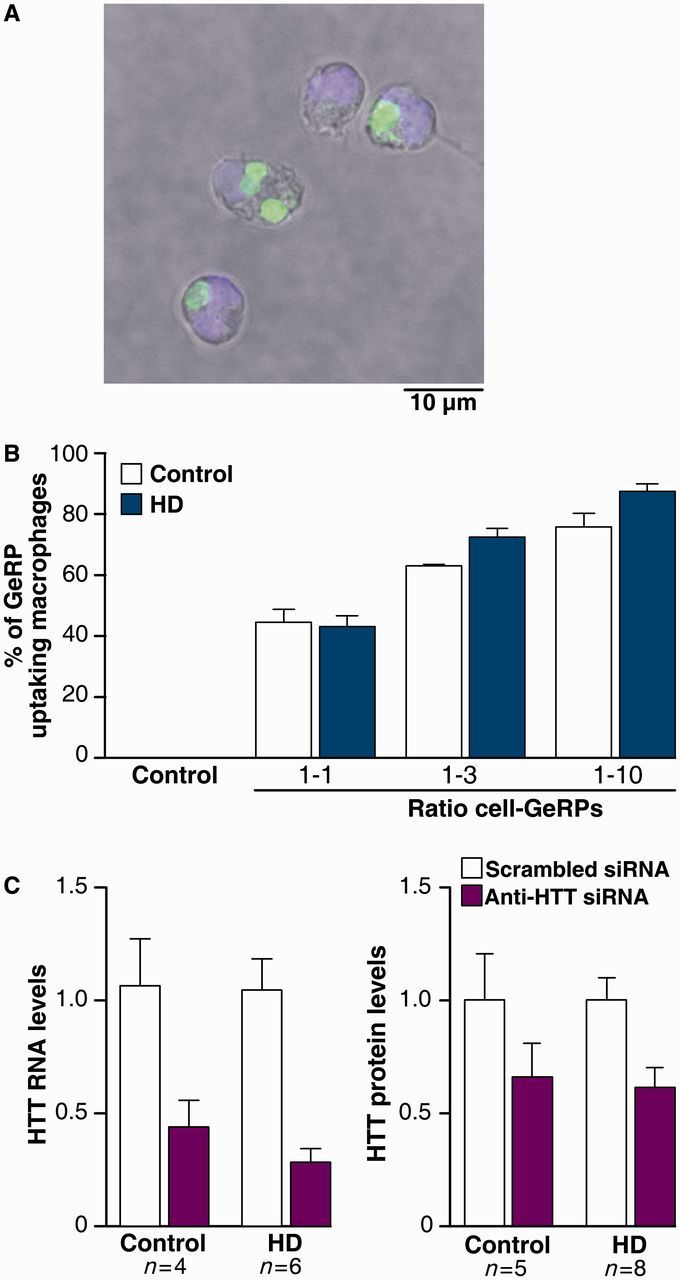
Glucan encapsulated small interfering RNA particles (GeRPs) can effectively knock-down total HTT in primary human immune cells. (A) GeRPs deliver small interfering RNA (siRNA) efficiently when phagocytosed by myeloid cells, as shown in primary human monocytes after 12 h incubation in culture (GeRPs = green; DAPI = blue). (B) Ninety per cent of macrophages take up GeRPs when incubated at 1:10 cell: particle ratio for 12 h as quantified by flow cytometry. Data shown as mean [n = 2 for controls and n = 3 for Huntington’s disease (HD)] ± SEM. (C) Total HTT RNA measured by quantitative PCR and protein levels measured by TR-FRET were reduced by 70% and 50%, respectively, in macrophages treated for 3 days with GeRPs containing anti-HTT small interfering RNA. Data shown as mean HTT levels (each combining two independent experiments, n = individual biological repeats) ± SEM. Data are normalized to the scrambled small interfering RNA treated condition for each genotype.
The efficacy of the anti-HTT small interfering RNA GeRPs was tested 3 days after small interfering RNA delivery in macrophages, using both quantitative PCR for HTT RNA levels and TR-FRET immunoassay for HTT protein levels. Macrophages treated with anti-HTT small interfering RNA GeRPs had 60–70% less HTT messenger RNA and 50% less HTT protein, compared with macrophages treated with scrambled small interfering RNA-containing GeRPs (Fig. 2C). As expected, the decrease in HTT levels was the same in both control and disease macrophages (Fig. 2C).
Next, we examined the effect of lowering total HTT levels on cytokine production. After treating primary human monocyte-derived macrophages with anti-HTT or scrambled small interfering RNA GeRPs for 3 days, IFNγ-primed macrophages were stimulated with lipopolysaccharide and cytokine production was measured. Validating our previous findings, IL-8 and TNFα levels were significantly higher in Huntington’s disease than in control cells, when both had been treated with scrambled small interfering RNA (Fig. 3). However, lowering HTT levels in Huntington’s disease macrophages using anti-HTT GeRPs rescued this increase by significantly decreasing the production of IL-6, IL-8 and TNFα (Fig. 3). IL-1β production showed a similar trend that did not reach significance. Interestingly, lowering HTT levels also significantly reduced IL-6, IL-8 and TNFα levels in control cells, suggesting a role of wild-type HTT in cytokine production.
Figure 3.
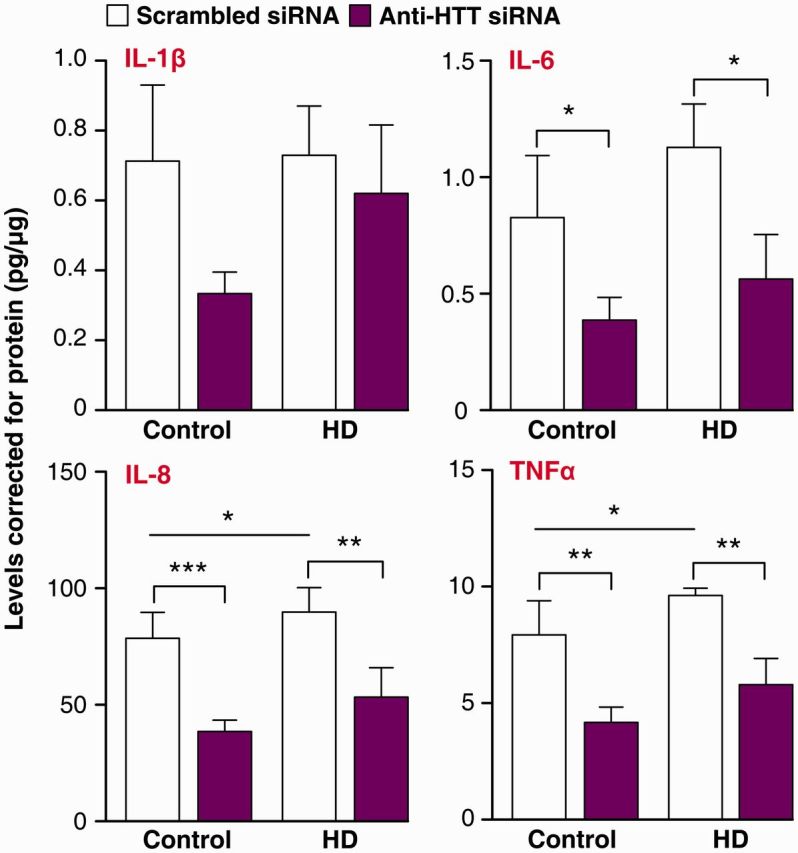
Knock-down of total HTT reverses the hyper-reactive cytokine production by Huntington’s disease macrophages. Huntington’s disease (HD) and control macrophages were treated with either anti-HTT or scrambled small interfering RNA (siRNA) for 3 days, before the cells were stimulated with 10 ng/ml IFNγ and 2 µg/ml lipopolysaccharide for 24 h. Measuring cytokine production with multiplex ELISA assays showed that lowering HTT levels reduces IL-6, IL-8 and TNFα levels after stimulation. Data shown as mean concentrations (n = 9 for controls and n = 8 for Huntington’s disease, combined from three independent experiments, n = individual biological repeats) ± SEM, two-way ANOVA with Bonferroni post-tests. *P < 0.05; **P < 0.01, ***P < 0.001.
Thus, lowering HTT levels by 50% using this novel method of small interfering RNA delivery can reverse the hyper-reactivity of Huntington’s disease patient macrophages. The use of GeRPs to achieve cell-targeted gene knock-down has to date shown significant promise in mice, but this is the first report showing efficient small interfering RNA delivery, pathogenic gene knock-down and rescue of a deleterious phenotype using this method in primary human immune cells.
The use of HTT-lowering in Huntington’s disease patient myeloid cells demonstrates that their production of cytokines in response to stimuli is regulated by HTT. This suggests that immune cell dysfunction is caused by the cell-intrinsic expression of mutant HTT, rather than being secondary to extracellular disease-associated factors. To confirm this, we transduced histiocytic lymphoma U937 cells, a commonly used model of monocytes (Alciato et al., 2010), with lentiviral vectors expressing human HTT exon 1. The constructs contained either wild-type HTT exon 1 with 29 CAG repeats, or mutant HTT exon 1 with 71 or 129 CAG repeats. Sorting the transduced cells using the co-expressed GFP resulted in 99% pure cultures (Supplementary Fig. 3) and HTT expression in the transduced cells was confirmed using TR-FRET (Fig. 4A). The HTT exon 1-expressing U937 cell lines were differentiated with PMA for 3 days to induce a mature monocyte phenotype before stimulating the cells with IFNγ and lipopolysaccharide, and analysing their cytokine profile. Stimulated U937 cells expressing either 71 or 129Q mutant HTT exon 1 produced significantly increased levels of IL-6 and TNFα compared with those expressing the 29Q wild-type HTT exon 1 construct (Fig. 4B). Cells expressing 129Q produced significantly higher IL-1β levels compared with control cells, whereas IL-8 levels did not differ.
Figure 4.
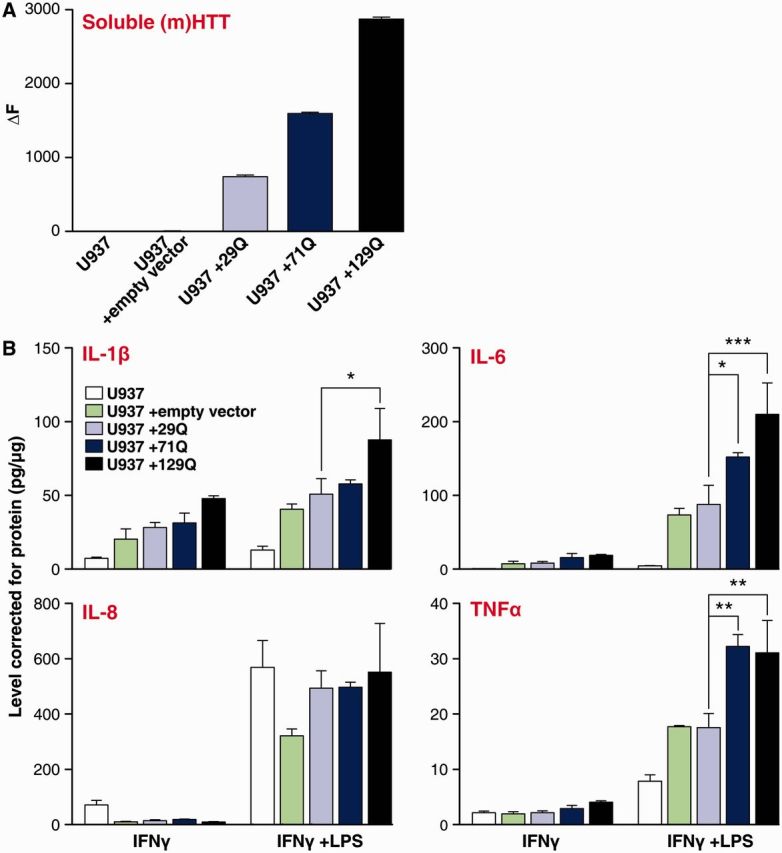
Expression of mutant HTT induces elevated cytokine production. U937 cells were lentivirally-transduced with mutant (m)HTT exon 1 containing either 29, 71 or 129 glutamine (Q) repeats or an empty vector. (A) Expression of mutant HTT protein post-transduction was confirmed, with increased levels of soluble HTT in all three cell lines expressing exogenous HTT. (B) Innate immune regulators were elevated in PMA-differentiated mutant HTT expressing U937 cells stimulated for 24 h with 10 ng/ml IFNγ and 2 µg/ml lipopolysaccharide (LPS). Data shown as mean concentrations (n = 3 technical repeats for all conditions) ± SEM, two-way ANOVA with Bonferroni post-tests, *P < 0.05; **P < 0.01, ***P < 0.001. The experiment was repeated three times independently and showed similar results.
Taken together, modulating HTT levels via overexpression of mutant HTT exon 1 in a myeloid cell line and by knock-down of HTT in primary human peripheral immune cells demonstrates that cell-intrinsic expression of mutant HTT causes the hyper-reactive immune phenotype observed in monocytes and macrophages from patients with Huntington’s disease.
Mutant HTT interacts with the NFκB pathway in human Huntington’s disease myeloid cells
That Huntington’s disease patient monocytes and macrophages resemble normal cells when unstimulated, but are hyper-reactive in response to lipopolysaccharide, suggests that mutant HTT affects the signalling cascade induced by lipopolysaccharide. Expression of the main lipopolysaccharide receptor, TLR4, was unaltered (Supplementary Fig. 4), suggesting downstream effects. The NFκB pathway, a key signalling cascade downstream of TLR4, has previously been shown to interact with mutant HTT exon 1 in mice (Khoshnan et al., 2004).
To test whether this interaction occurs in human primary immune cells, peripheral blood mononuclear cells from patients with early-stage Huntington’s disease and control subjects were isolated for co-immunoprecipitation experiments. Full-length HTT was detectable in both the control and Huntington’s disease samples with two anti-HTT antibodies (2B7 and MAB2166), whereas co-precipitation of IKKγ was observed only in the Huntington’s disease sample (Supplementary Fig. 5A). Given the high background signal in these experiments because of poor antibody performance in the immunoprecipitation, we performed more sensitive proximity ligation assays to detect native IKKγ-HTT interactions in the cells. As shown in Fig. 5A, specific IKKγ-HTT protein interactions, represented by red spots can be detected in both control and disease macrophages. Quantification of the number of spots per cell demonstrated more interaction between IKKγ and HTT in Huntington’s disease patient cells compared with controls (Fig. 5B). Further evidence for a CAG repeat dependent interaction between HTT and the IKK complex was given by an increased number of interactions between HTT and the IKKα/β subunits in Huntington’s disease samples (Fig. 5B). The fact that classical immunoprecipitation did not pick up an interaction of the proteins in control individual’s cells is likely to be because the method being less sensitive. These data demonstrate for the first time a direct interaction between the IKK complex and full-length HTT expressed at normal allelic expression levels in primary human cells.
Figure 5.
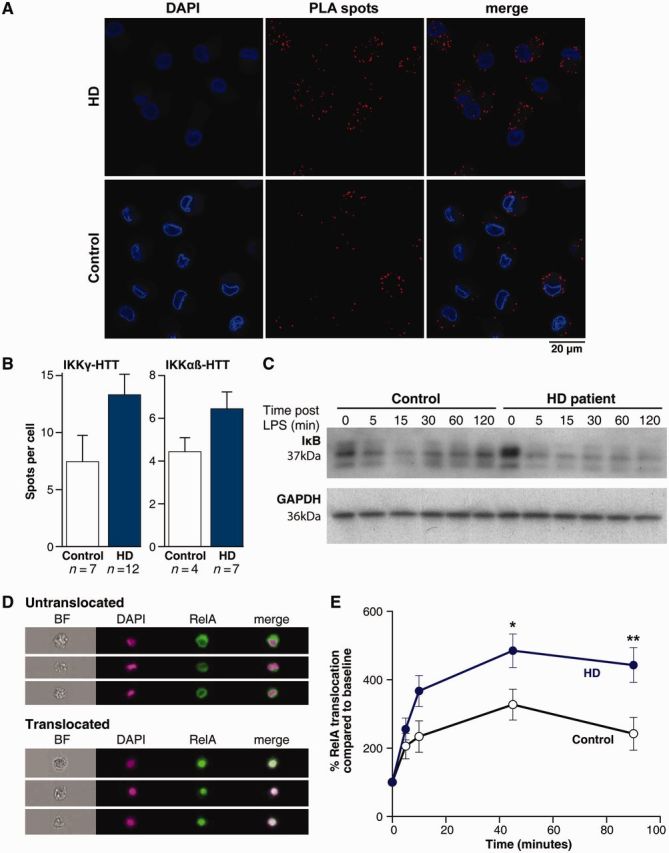
HTT interacts directly with the NFκB pathway, which is dysregulated in Huntington’s disease (HD). (A) HTT interacts directly with IKK, as shown by proximity ligation assays. Monocyte-derived macrophages were differentiated on glass cover-slips and stained for HTT and IKKγ or IKKα/β before antibodies binding in close proximity were visualized using proximity ligation assay (PLA) probes as red spots, shown here. Cells stained with a single primary antibody did not result in red spots. (B) Quantification of the number of spots per cell using the Volocity software shows increased binding between IKKγ and HTT in Huntington’s disease compared with control cells (P = 0.06). Binding of HTT to the α and β subunit of IKK showed a similar, but smaller trend (P = 0.1). Two-tailed unpaired t-test used for statistical analysis. (C) In control cells lipopolysaccharide (LPS)-induced degradation of IκB occurred within 15 min of stimulation and recovered within 2 h, whereas Huntington’s disease monocytes demonstrated a more rapid loss of IκB and no recovery of the protein. Shown is an example blot of samples from one control subject and one patient with Huntington’s disease. (D) Translocation of the NFκB transcription factor RelA to the nucleus after lipopolysaccharide stimulation was measured using imaging flow cytometry; example images are shown here. In untranslocated cells the green RelA staining surrounds the nuclear DAPI staining, whereas in cells demonstrating translocation of RelA the colours merge. (E) Increased RelA translocation into the nucleus following lipopolysaccharide stimulation was observed in Huntington’s disease monocytes (n = 7) compared to controls (n = 8). n = individual biological repeats. Data shown as mean concentrations ± SEM, two-way ANOVA with Bonferroni post-tests, *P < 0.05; **P < 0.01. All experiments were repeated at least twice with the same results.
Activation of the IKK complex leads to the phosphorylation and degradation of IκB, the endogenous inhibitor of NFκB (Hayden and Ghosh, 2012). To evaluate whether the increased interaction of mutant HTT with IKKγ leads to increased IKK complex activation and subsequent changes in IκB degradation, we stimulated Huntington’s disease and control monocytes with lipopolysaccharide over a time course of 2 h to analyse IκB levels by western blot. Control monocytes demonstrated a drop in IκB levels over the first 15 min, before a recovery of IκB levels over the next 2 h, representing a normal pattern of NFκB activation on stimulation (Fig. 5C) (Gross and Piwnica-Worms, 2005). After stimulation of Huntington’s disease monocytes, we observed a different pattern: IκB levels dropped within 5 mins and did not recover to baseline levels within the 2 h time course (Fig. 5C). This demonstrates that IκB is degraded more rapidly and over a prolonged period of time in primary human Huntington’s disease monocytes as a result of IKK activation. Similarly, levels of phosphorylated IκB were increased over the 2 h period in monocytes isolated from patients with Huntington’s disease compared with control subjects (Supplementary Fig. 5B). To investigate by which time IκB levels return to baseline in patients with Huntington’s disease, we performed a prolonged time course over 24 h and found that IκB levels returned to baseline levels or above (because of high level re-synthesis of the protein) by 4 h post-stimulation (Supplementary Fig. 5C). These findings demonstrate a transient effect of mutant HTT expression on IκB levels after stimulation.
Under steady-state conditions, IκB binds NFκB and blocks its translocation to the nucleus. Degradation of IκB allows the NFκB transcription factors to enter the nucleus and influence transcription (Beinke and Ley, 2004). To test whether increased IκB degradation in Huntington’s disease monocytes leads to more rapid nuclear translocation of NFκB, we analysed translocation of RelA, one of five DNA-binding NFκB subunits, in Huntington’s disease and control monocytes using imaging flow cytometry. ImageStream technology, combining the high image content information of microscopy with the high throughput analysis of flow cytometry, is used to overcome the limitations of conventional assays to produce highly reproducible and statistically robust data (Maguire et al., 2011). Cells were stained with DAPI to mark the nucleus and with anti-RelA antibodies (Fig. 5D). Analysis of the levels of RelA and DAPI co-localization showed significantly higher levels of RelA translocation in Huntington’s disease than in control monocytes at 45 and 90 min post lipopolysaccharide stimulation (Fig. 5E).
Thus, we demonstrate in primary Huntington’s disease patient cells that mutant HTT binds IKKγ and causes increased NFκB activity by increased IκB degradation and subsequent NFκB translocation. We hypothesize that this causes altered transcription of NFκB target genes, leading to increased cytokine production by immune cells.
Transcriptional changes affect signalling pathways in Huntington’s disease myeloid cells
Transcriptional dysregulation is a central pathogenic mechanism in Huntington’s disease (Hodges et al., 2006). Therefore, we tested whether basal differences in transcription play a role in mutant HTT induced immune hyper-reactivity by analysing differences in the expression of genes related to the NFκB pathway. The messenger RNA expression of 84 genes was tested in untreated human monocytes using the SABioscience NFκB signalling pathway PCR array. We identified seven genes that were significantly upregulated (TLR2, LTBR, CD40, TMED4, AKT1, IL10, FR2) and one gene that was significantly downregulated (CHUK) in Huntington’s disease compared with control monocytes (Table 1). Four of the upregulated genes showed a ≥1.5-fold change: CD40 (1.5); AKT1 (1.5); IL10 (1.85) and F2R (2.23). Furthermore, the adaptor molecules IRAK1, TICAM2, MYD88 and TRADD, were also upregulated (Table 1 and Supplementary Fig. 6). Interestingly, CHUK, which encodes for IKKα, was found to be downregulated, whereas all other parts of the IKK complex, IκB and the NFκB transcription factors were unchanged.
Table 1.
The top 20 gene changes within the NFκB pathway observed in Huntington’s disease monocytes
| Gene name | Fold change | P-value |
|---|---|---|
| TLR2 | 1.483 | 0.010 |
| LTBR | 1.355 | 0.014 |
| CHUK | 0.771 | 0.015 |
| CD40 | 1.513 | 0.016 |
| TMED4 | 1.227 | 0.021 |
| AKT1 | 1.535 | 0.031 |
| IL10 | 1.850 | 0.037 |
| F2R | 2.232 | 0.041 |
| IRAK1 | 2.004 | 0.051 |
| JUN | 2.215 | 0.051 |
| TICAM2 | 1.319 | 0.051 |
| MYD88 | 1.262 | 0.053 |
| FOS | 1.867 | 0.059 |
| TRADD | 1.251 | 0.062 |
| RAF1 | 1.121 | 0.065 |
| SLC44A2 | 1.292 | 0.066 |
| ATF1 | 1.156 | 0.071 |
| IL6 | 2.486 | 0.075 |
| GJA1 | 0.612 | 0.080 |
| CCL2 | 1.574 | 0.160 |
Data presented as fold change calculated from ΔΔ-CT values, unpaired two-tailed t-test used as statistical method.
The array also screened intracellular signalling pathways closely linked to the NFκB pathway, such as MAPK and PI3K/AKT pathways. Increased AKT protein levels have been found in Huntington’s disease patient lymphoblasts (Colin et al., 2005) and AKT1 is one of the genes upregulated in Huntington’s disease monocytes (fold change = 1.5, P = 0.031). Moreover, the genes composing the transcription factor AP-1, JUN and FOS, are also upregulated in Huntington’s disease monocytes (Table 1 and Supplementary Fig. 6). Therefore, both of these pathways may also contribute to the increased immune response observed after stimulation of Huntington’s disease monocytes.
To validate our findings, six candidate genes chosen on the basis of array fold changes and their importance within the NFκB signalling cascade (CD40, AKT1, IRAK1, JUN, IL6 and IL10) were quantified by quantitative PCR using different primer sets and cells from a different patient cohort. The relative changes in gene expression when comparing Huntington’s disease and control human monocytes matched our previous findings for all six genes (Supplementary Fig. 7). Expression levels for CD40, IRAK1 and IL10 were significantly increased in Huntington’s disease compared with control monocytes, whereas expression changes in AKT1, JUN and IL6 demonstrated an upward trend, not reaching statistical significance because of large interindividual differences.
Lowering HTT levels reverses transcriptional changes in Huntington’s disease myeloid cells
Finally, we investigated whether lowering HTT using anti-HTT small interfering RNA GeRPs could reverse the transcriptional changes observed in Huntington’s disease monocytes. IRAK1 (encoding the main adapter molecule between TLR4 and NFκB), CD40 (immunomodulatory molecule giving co-stimulatory signals to both innate and adaptive immune cells) and JUN (part of the AP-1 transcription factor) expression were all increased in Huntington’s disease monocytes (Table 1). After 3 days of treatment with GeRPs containing either scrambled or anti-HTT small interfering RNA, we analysed the expression of these candidate genes by quantitative PCR. HTT messenger RNA levels were assessed to validate the knock-down. Treatment with anti-HTT small interfering RNA resulted in a 50% reduction in HTT levels in both Huntington’s disease and control monocytes (Fig. 6A and B). However, only the Huntington’s disease monocytes demonstrated a significant 20–30% reduction in IRAK1, CD40 and JUN expression when treated with anti-HTT small interfering RNA (Fig. 6A). Lowering HTT in control cells did not affect levels of IRAK1, CD40 and JUN transcript expression (Fig. 6B), suggesting that the transcriptional dysregulation of these genes in Huntington’s disease myeloid cells is caused specifically by a gain of mutant HTT function, rather than loss of wild-type protein function.
Figure 6.

Lowering total HTT levels reverses transcriptional changes found in Huntington’s disease monocytes. Huntington’s disease and control monocytes were incubated with either scrambled or anti-HTT small interfering RNA (siRNA) containing GeRPs for 3 days before RNA isolation. Using quantitative PCR, efficient HTT knock-down was demonstrated as well as lowering of key NFκB pathway molecules IRAK1, CD40 and JUN in (A) Huntington’s disease patient cells but not (B) controls. Data shown as relative gene expression (n = 10 individual biological repeats for controls and Huntington’s disease) ± SEM, paired t-test. *P < 0.05; **P < 0.01.
Discussion
Plasma pro-inflammatory cytokine levels are elevated in patients with Huntington’s disease, even in the pre-manifest stages of the disease (Björkqvist et al., 2008). Here we demonstrate that Huntington’s disease peripheral blood mononuclear cells are the likely source of the increased pro-inflammatory cytokines, as both monocytes and macrophages isolated from patients with Huntington’s disease and stimulated with lipopolysaccharide produce significantly more IL-6, IL-8 and TNFα compared with control subjects. Supporting our previous finding that plasma cytokine levels are already elevated in pre-manifest subjects with a mean of 16 years to clinical onset (Björkqvist et al., 2008), myeloid cells isolated from patients with pre-manifest Huntington’s disease were hyper-reactive to the same degree as cells isolated from late-stage disease patients. Cytokine production seems CAG repeat length independent and suggests an early deficit that is already present many years before disease onset, which may be a marker of when to intervene with potential modulatory therapies. Modulating HTT expression by overexpression of mutant HTT exon 1 in a monocyte-like cell line and lowering HTT levels in primary human monocytes/macrophages demonstrated that this hyper-reactive phenotype is because of a cell-intrinsic effect of mutant HTT expression and not non-cell autonomous secondary factors.
Importantly, we have been able to show that lowering total HTT levels partially rescues this hyper-reactive phenotype, with a reversal of both elevated cytokine production and transcriptional changes observed in human Huntington’s disease myeloid cells ex vivo. This is the first report showing that lowering HTT in cells freshly isolated from patients with Huntington’s disease can reverse cellular dysfunction caused by mutant HTT expression—an important first demonstration of the reversibility of cellular dysfunction after HTT-lowering in human tissue. HTT-lowering was achieved using a novel phagocytosis dependent approach, in which small interfering RNAs are packaged into glucan particles isolated from yeast (Aouadi et al., 2009). This study is the first to use this technique in primary human macrophages and demonstrates that a 90% transfection rate can be achieved, much higher than the 10–20% transfection rate achieved by traditional methods such as lentiviral transduction.
Our findings, that lowering total HTT levels by only 50% in primary human Huntington’s disease macrophages and monocytes can reverse the increased cytokine production and transcriptional changes, respectively, validate the potential of HTT-lowering therapy as well as the possibility of using peripheral cells to test small interfering RNA efficiency, safety and efficacy. Interestingly, cytokine release was also decreased in control macrophages treated with anti-HTT small interfering RNA, indicating either that HTT regulates cytokine production in a CAG dependent manner or that wild-type HTT influences cytokine production in parallel with mutant HTT. Wild-type HTT has been shown to play a role in both actin remodelling (Munsie et al., 2011; Kwan et al., 2012b) and microtubule-mediated transport (Gauthier et al., 2004). As both processes are needed for the trafficking of cytokines to the cell surface membrane for release (Lacy and Stow, 2011), a reduction of wild-type HTT levels might exert a loss of function by hindering normal actin and microtubule remodelling causing changes in cytokine release. A future study using allele-specific silencing of mutant but not wild-type HTT will help determine the exact contributions that loss of wild-type HTT and gain of mutant HTT function have on the myeloid cell dysfunction in Huntington’s disease.
The NFκB pathway has been previously implicated in Huntington’s disease in murine studies (Khoshnan et al., 2004; Thompson et al., 2009; Steffan, 2010; Hsiao et al., 2013). Investigating this pathway, we found that HTT binds the IKK complex in a CAG repeat length dependent manner. Testing HTT binding to both IKKγ and IKKα/β subunits, we detected a stronger interaction between HTT and IKKγ, suggesting this subunit as the direct interaction partner. IKKγ is the regulatory subunit of the IKK trimer, consisting of one regulatory (γ) and two kinase subunits (α and β), and is a critical component without which cells are unresponsive to all upstream stimuli (Israël, 2000). During signal transduction, polyubiquitin chains form the scaffold on which TAK1/TAB2/3 and IKKα/β/γ complexes are formed to induce TAK1 dependent activation of IKKβ (Miyamoto, 2011). In agreement with previously described findings using non-primary human cell model systems (Khoshnan and Patterson, 2011), we have shown in primary human cells that HTT can function as an alternative scaffold for the NFκB pathway. The CAG repeat dependent binding of HTT to IKKγ is associated with increased IKK complex formation and downstream signal transduction following lipopolysaccharide stimulation in Huntington’s disease myeloid cells (Fig. 7). Previously, this interaction has only been observed in cultured tumour cells (Khoshnan et al., 2004) or mouse models expressing exon 1 mutant HTT (Khoshnan et al., 2004; Hsiao et al., 2013). Here we demonstrate that the interaction also takes place in primary human ex vivo cells expressing the full-length protein at normal allelic expression levels. That mutant HTT exon 1 fragments have previously been shown to bind IKKγ is consistent, however, with our finding that an N-terminal human exon 1 mutant HTT fragment can induce elevated cytokine production in a histiocytic cell line and our recent report demonstrating increasing N-terminal fragmentation of mutant HTT in human myeloid cells as the disease progresses (Weiss et al., 2012).
Figure 7.
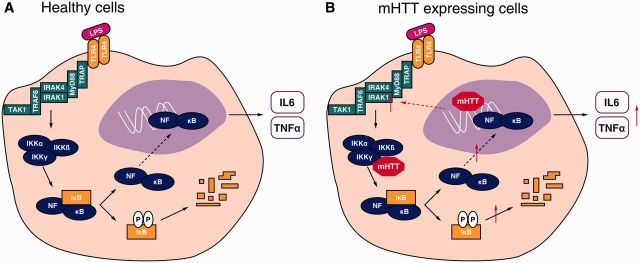
Mechanism of immune dysfunction in Huntington’s disease. (A) In normal wild-type HTT expressing myeloid cells, lipopolysaccharide binds the TLR4 receptor activating the NFκB pathway triggering production of pro-inflammatory cytokines such as IL-6 and TNFα. (B) Mutant (m)HTT interferes with the NFκB pathway by two distinct mechanisms. The mutant protein binds IKKγ to directly cause increased IκB degradation and NFκB transcription factor translocation, allowing increased transcription of target genes such as IL-6 and TNFα. Moreover, mutant HTT causes transcriptional changes leading to increased expression of key molecules within the signalling cascade likely to increase signalling transduction rate.
Interestingly, a recent study showed that activating the immune modulator cannabinoid receptor 2, which is thought to dampen NFκB signalling (Rajesh et al., 2007), reduces increased serum IL-6 levels while extending life span and reducing motor deficits in Huntington’s disease mouse models (Bouchard et al., 2012).
Our finding that mutant HTT alters the NFκB pathway in human Huntington’s disease monocytes is likely to be relevant to other cell types and tissues, including those of the CNS. We previously showed that hyper-reactivity of Huntington’s disease peripheral myeloid cells is mirrored in microglia (Björkqvist et al., 2008). The NFκB pathway is present and active in both neurons and glial cells (O'Neill and Kaltschmidt, 1997). Pharmacological inhibition of NFκB impairs memory and learning (Mattson and Meffert, 2006) and NFκB pathway activation is critical for neuronal survival and neurite outgrowth (Teng and Tang, 2010). Increased levels of NFκB activity have been shown in both Alzheimer’s disease (Kaltschmidt et al., 1997) and Parkinson’s disease (Hunot et al., 1997). Blocking NFκB function in mutant HTT exon 1 expressing PC12 cells leads to reduced mutant HTT toxicity, implying that the NFκB pathway contributes to neurotoxicity in Huntington’s disease (Khoshnan et al., 2004). Indeed, a recent study showing that mutant HTT enhances NFκB-mediated inflammation in astrocytes to cause toxicity in the brain of Huntington’s disease mice underlines the potential importance of NFκB in non-neuronal cells during neurodegeneration (Hsiao et al., 2013).
Given the manifold roles NFκB signalling plays in the different cell types, inhibiting the pathway to lower hyper-reactive immune function in Huntington’s disease may also affect other cell types. For example, compounds that target NFκB activity need to be closely evaluated as to whether they cross the blood–brain barrier and with regard to negative effects on synaptic activity and plasticity. However, drugs that target this pathway will not necessarily have negative effects. Laqinimod for example, an immunomodulatory compound inhibiting NFκB activity (Brück et al., 2012), was well tolerated and showed decreased progression rates in patients with multiple sclerosis in clinical trials (Comi et al., 2012). Furthermore, targeting the NFκB pathway further downstream, for example at the level of cytokine secretion is also a possible therapeutic target. In a Huntington’s disease mouse model, peripherally administrated anti-IL-6 antibody treatment has shown improvement of disease progression (Bouchard et al., 2012), while perispinal administration of a TNFα inhibitor improves disease in patients with Alzheimer’s disease (Tobinick et al., 2006), clearly demonstrating the positive effect of immunomodulatory therapy for neurodegeneration.
We have identified gene expression changes in key molecules involved in immune signalling in Huntington’s disease patients’ monocytes. Several adapter proteins downstream of TLR4, such as IRAK1, TICAM2 and MyD88 were found to be slightly elevated in native Huntington’s disease patients’ monocytes. A cumulative baseline increase in expression of several of these adapter proteins may lead to increased signal transduction from TLR4 to NFκB, further increasing NFκB pathway dysregulation. Another gene found to be upregulated in Huntington’s disease monocytes was CD40 which, together with its ligand CD154, mainly expressed on T cells, regulates the immune response on several levels. Monocytes are activated leading to upregulated cytokine production and antigen presentation, and priming of the adaptive immune system (Grewal and Flavell, 1998). This points to further functional abnormalities in the immune system of patients with Huntington’s disease, suggesting a possible deficit in the communication between antigen presenting cells and the adaptive immune system. Furthermore, CD40 mediates cell adhesion needed for leucocyte trafficking (Alderson et al., 1993). Given recent studies showing defective migration in Huntington’s disease because of defective actin remodelling (Kwan et al., 2012b), the increase in CD40 expression could be a compensatory response of immune cells to counteract their decreased migrative ability. FOS and JUN, subunits of the AP-1 transcription factor, were also upregulated in primary human myeloid Huntington’s disease cells. Interestingly, FOS and JUN levels have been found to be increased in brain of patients with Alzheimer’s disease (Anderson et al., 1994), and the MAP kinase needed for JUN activation, JNK, is elevated and involved in neurotoxicity in Huntington’s disease mouse (Fan et al., 2012) and rat models (Perrin et al., 2009). Thus, we cannot exclude that dysregulation in these signalling pathways may also contribute to the Huntington’s disease immune phenotype.
In addition to the pathways identified in this study, we cannot exclude other previously described mechanisms, which may contribute to the transcriptional dysregulation we found in primary human Huntington’s disease myeloid cells. For example, mutant HTT is known to bind and thereby deplete transcription factors such as CBP and p53 from their normal location causing changes in the genes they control (Steffan et al., 2000; Nucifora et al., 2001). Furthermore, HTT may alter DNA conformation upon direct binding, affecting transcription factors binding to their promoter regions (Benn et al., 2008).
This study demonstrates that the cellular dysregulation observed in hyper-reactive immune cells in Huntington’s disease can be reversed by HTT-lowering and represents the first demonstration of phenotypic reversibility on HTT-lowering in primary human cells in Huntington’s disease. It also identifies the underlying intracellular mechanisms of immune dysfunction in human cells in Huntington’s disease. This is important as the immune system has been shown to be a powerful modifier of Huntington’s disease pathogenesis in various mouse models (Zwilling et al., 2011; Bouchard et al., 2012; Kwan et al., 2012a). There is currently a search for genetic and environmental modifiers of Huntington’s disease as the CAG repeat expansion only explains 50–70% of variance in age of onset, and its role in modulating disease progression is variable (Andrew et al., 1993; Brinkman et al., 1997). The remainder of the variance is likely due to environmental and other genetic factors (Wexler et al., 2004). The immune system may be a powerful modifier of Huntington’s disease age of onset and progression, with an interaction of both genetic and environmental factors. This has already been shown to be the case in large genome-wide association studies in Alzheimer’s disease where several key genes involved in the innate immune system were shown to increase susceptibility to developing Alzheimer’s disease (Harold et al., 2009; Lambert et al., 2009; Guerreiro et al., 2013).
Our novel method of small interfering RNA delivery has potential therapeutic relevance to Huntington’s disease and other diseases where immune dysregulation is a feature. Glucan particles are a versatile phagocytic cell targeted delivery system and have been administered by oral, subcutaneous, intraperitoneal and intravenous routes in mice and rats. In our future studies, we plan to administer GeRPs loaded with anti-HTT small interfering RNA to reverse the inflammatory phenotype through intrathecal administration to directly target phagocytic microglial cells and infiltrating monocyte/macrophages, and through intravenous administration to target circulating monocytes and peripheral mononuclear cells, a precursor pool for inflammatory cells trafficking into inflamed brain sites.
Finally, our work also suggests a potential new therapeutic target for Huntington’s disease through modulating NFκB activation and downstream targets. The muscle wasting, weight loss and depression that occurs in Huntington’s disease (van der Burg et al., 2009) may be related to increased peripheral cytokine levels. Therefore, modulating the immune system may have beneficial effects in both the CNS and the periphery. Indeed, a peripherally administered anti-inflammatory, anti-IL-6 antibody treatment in R6/2 mice has already been show to improve both weight loss and disease progression (Bouchard et al., 2012). This work therefore has implications for both understanding the role of the innate immune system as a modifier of neurodegeneration and modulation of the immune system as a possible therapeutic in Huntington’s disease.
Supplementary Material
Acknowledgements
We thank the patients and control subjects who donated samples, and the staff of the multidisciplinary Huntington’s disease clinic in London; Dr Peter Klöhn for his help with fluorescence-activated cell sorting; P.J. Chana for his assistance with the imaging flow cytometry; Dr Christian Landles for his advice on HTT immunoprecipitations; Dr Edward Wild for his help with editing the manuscript and Ray Young for his help with graphics.
Glossary
Abbreviations
- GeRP
β1,3-d-glucan-encapsulated small interfering RNA particle
- TR-FRET
time resolved fluorescence resonance energy transfer
Funding
This study was supported financially by UCL/UCLH Biomedical Research Centre (PhD studentship to UT), Medical Research Council, CHDI Foundation, EU FP7 grant (Paddington consortium), the UK Dementia and Neurodegenerative Diseases Network (DeNDRoN) and supported by the National Institute for Health Research University College London Hospitals Biomedical Research Centre. N.A. was supported by NHI: NS 38194 and the CHDI foundation. The authors acknowledge financial support from the Department of Health via the National Institute for Health Research (NIHR) comprehensive Biomedical Research Centre award to Guy’s & St Thomas’ NHS Foundation Trust in partnership with King’s College London and King’s College Hospital NHS Foundation Trust.
Supplementary material
Supplementary material is available at Brain online.
References
- Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci. 2007;10:1538–43. doi: 10.1038/nn2014. [DOI] [PubMed] [Google Scholar]
- Alciato F, Sainaghi PP, Sola D, Castello L, Avanzi GC. TNF-alpha, IL-6, and IL-1 expression is inhibited by GAS6 in monocytes/macrophages. J Leukoc Biol. 2010;87:869–75. doi: 10.1189/jlb.0909610. [DOI] [PubMed] [Google Scholar]
- Alderson MR, Armitage RJ, Tough TW, Strockbine L, Fanslow WC, Spriggs MK. CD40 expression by human monocytes: regulation by cytokines and activation of monocytes by the ligand for CD40. J Exp Med. 1993;178:669–74. doi: 10.1084/jem.178.2.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson AJ, Cummings BJ, Cotman CW. Increased immunoreactivity for Jun- and Fos-related proteins in Alzheimer's disease: association with pathology. Exp Neurol. 1994;125:286–95. doi: 10.1006/exnr.1994.1031. [DOI] [PubMed] [Google Scholar]
- Andrew SE, Goldberg YP, Kremer B, Telenius H, Theilmann J, Adam S, et al. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington's disease. Nat Genet. 1993;4:398–403. doi: 10.1038/ng0893-398. [DOI] [PubMed] [Google Scholar]
- Aouadi M, Tesz GJ, Nicoloro SM, Wang M, Chouinard M, Soto E, et al. Orally delivered siRNA targeting macrophage Map4k4 suppresses systemic inflammation. Nature. 2009;458:1180–4. doi: 10.1038/nature07774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldo B, Paganetti P, Grueninger S, Marcellin D, Kaltenbach LS, Lo DC, et al. TR-FRET-based duplex immunoassay reveals an inverse correlation of soluble and aggregated mutant huntingtin in huntington's disease. Chem Biol. 2012;19:264–75. doi: 10.1016/j.chembiol.2011.12.020. [DOI] [PubMed] [Google Scholar]
- Beinke S, Ley SC. Functions of NF-kappaB1 and NF-kappaB2 in immune cell biology. Biochem J. 2004;382 (Pt 2):393–409. doi: 10.1042/BJ20040544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benn CL, Sun T, Sadri-Vakili G, McFarland KN, DiRocco DP, Yohrling GJ, et al. Huntingtin modulates transcription, occupies gene promoters in vivo, and binds directly to DNA in a polyglutamine-dependent manner. J Neurosci. 2008;28:10720–33. doi: 10.1523/JNEUROSCI.2126-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björkqvist M, Wild EJ, Thiele J, Silvestroni A, Andre R, Lahiri N, et al. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington's disease. J Exp Med. 2008;205:1869–77. doi: 10.1084/jem.20080178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard J, Truong J, Bouchard K, Dunkelberger D, Desrayaud S, Moussaoui S, et al. Cannabinoid receptor 2 signaling in peripheral immune cells modulates disease onset and severity in mouse models of Huntington’s disease. J Neurosci. 2012;32:18259–68. doi: 10.1523/JNEUROSCI.4008-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman RR, Mezei MM, Theilmann J, Almqvist E, Hayden MR. The likelihood of being affected with Huntington disease by a particular age, for a specific CAG size. Am J Hum Genet. 1997;60:1202–10. [PMC free article] [PubMed] [Google Scholar]
- Brück W, Pförtner R, Pham T, Zhang J, Hayardeny L, Piryatinsky V, et al. Reduced astrocytic NF-κB activation by laquinimod protects from cuprizone-induced demyelination. Acta Neuropathol. 2012;124:411–24. doi: 10.1007/s00401-012-1009-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colin E, Régulier E, Perrin V, Dürr A, Brice A, Aebischer P, et al. Akt is altered in an animal model of Huntington's disease and in patients. Eur J Neurosci. 2005;21:1478–88. doi: 10.1111/j.1460-9568.2005.03985.x. [DOI] [PubMed] [Google Scholar]
- Comi G, Jeffery D, Kappos L, Montalban X, Boyko A, Rocca MA, et al. Placebo-controlled trial of oral laquinimod for multiple sclerosis. N Engl J Med. 2012;366:1000–9. doi: 10.1056/NEJMoa1104318. [DOI] [PubMed] [Google Scholar]
- Dalrymple A, Wild EJ, Joubert R, Sathasivam K, Björkqvist M, Petersén A, et al. Proteomic profiling of plasma in Huntington's disease reveals neuroinflammatory activation and biomarker candidates. J Proteome Res. 2007;6:2833–40. doi: 10.1021/pr0700753. [DOI] [PubMed] [Google Scholar]
- DiFiglia M, Sena-Esteves M, Chase K, Sapp E, Pfister E, Sass M, et al. Therapeutic silencing of mutant huntingtin with siRNA attenuates striatal and cortical neuropathology and behavioral deficits. Proc Natl Acad Sci USA. 2007;104:17204–9. doi: 10.1073/pnas.0708285104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Gladding CM, Wang L, Zhang LY, Kaufman AM, Milnerwood AJ, et al. P38 MAPK is involved in enhanced NMDA receptor-dependent excitotoxicity in YAC transgenic mouse model of Huntington disease. Neurobiol Dis. 2012;45:999–1009. doi: 10.1016/j.nbd.2011.12.019. [DOI] [PubMed] [Google Scholar]
- Gauthier LR, Charrin BC, Borrell-Pagès M, Dompierre JP, Rangone H, Cordelières FP, et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–38. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–64. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- Grewal IS, Flavell RA. CD40 and CD154 in cell-mediated immunity. Annu Rev Immunol. 1998;16:111–35. doi: 10.1146/annurev.immunol.16.1.111. [DOI] [PubMed] [Google Scholar]
- Gross S, Piwnica-Worms D. Real-time imaging of ligand-induced IKK activation in intact cells and in living mice. Nat Methods. 2005;2:607–14. doi: 10.1038/nmeth779. [DOI] [PubMed] [Google Scholar]
- Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, et al. TREM2 variants in Alzheimer's disease. N Engl J Med. 2013;368:117–27. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet. 2009;41:1088–93. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 2012;26:203–34. doi: 10.1101/gad.183434.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges A, Strand AD, Aragaki AK, Kuhn A, Sengstag T, Hughes G, et al. Regional and cellular gene expression changes in human Huntington's disease brain. Hum Mol Genet. 2006;15:965–77. doi: 10.1093/hmg/ddl013. [DOI] [PubMed] [Google Scholar]
- Hsiao HY, Chen YC, Chen HM, Tu PH, Chern Y. A critical role of astrocyte-mediated nuclear factor-κB-dependent inflammation in Huntington's disease. Hum Mol Genet. 2013;22:1826–42. doi: 10.1093/hmg/ddt036. [DOI] [PubMed] [Google Scholar]
- Hunot S, Brugg B, Ricard D, Michel PP, Muriel MP, Ruberg M, et al. Nuclear translocation of NF-kappaB is increased in dopaminergic neurons of patients with parkinson disease. Proc Natl Acad Sci USA. 1997;94:7531–6. doi: 10.1073/pnas.94.14.7531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israël A. The IKK complex: an integrator of all signals that activate NF-kappaB? Trends Cell Biol. 2000;10:129–33. doi: 10.1016/s0962-8924(00)01729-3. [DOI] [PubMed] [Google Scholar]
- Kaltschmidt B, Uherek M, Volk B, Baeuerle PA, Kaltschmidt C. Transcription factor NF-kappaB is activated in primary neurons by amyloid beta peptides and in neurons surrounding early plaques from patients with Alzheimer disease. Proc Natl Acad Sci USA. 1997;94:2642–7. doi: 10.1073/pnas.94.6.2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoshnan A, Ko J, Watkin EE, Paige LA, Reinhart PH, Patterson PH. Activation of the IkappaB kinase complex and nuclear factor-kappaB contributes to mutant huntingtin neurotoxicity. J Neurosci. 2004;24:7999–8008. doi: 10.1523/JNEUROSCI.2675-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoshnan A, Patterson PH. The role of IκB kinase complex in the neurobiology of Huntington's disease. Neurobiol Dis. 2011;43:305–11. doi: 10.1016/j.nbd.2011.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan W, Magnusson A, Chou A, Adame A, Carson MJ, Kohsaka S, et al. Bone marrow transplantation confers modest benefits in mouse models of Huntington's disease. J Neurosci. 2012a;32:133–42. doi: 10.1523/JNEUROSCI.4846-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan W, Träger U, Davalos D, Chou A, Bouchard J, Andre R, et al. Mutant huntingtin impairs immune cell migration in Huntington disease. J Clin Invest. 2012b;122:4737–47. doi: 10.1172/JCI64484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacy P, Stow JL. Cytokine release from innate immune cells: association with diverse membrane trafficking pathways. Blood. 2011;118:9–18. doi: 10.1182/blood-2010-08-265892. [DOI] [PubMed] [Google Scholar]
- Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet. 2009;41:1094–9. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- Li SH, Schilling G, Young WS, Li XJ, Margolis RL, Stine OC, et al. Huntington's disease gene (IT15) is widely expressed in human and rat tissues. Neuron. 1993;11:985–93. doi: 10.1016/0896-6273(93)90127-d. [DOI] [PubMed] [Google Scholar]
- Maguire O, Collins C, O'Loughlin K, Miecznikowski J, Minderman H. Quantifying nuclear p65 as a parameter for NF-κB activation: correlation between ImageStream cytometry, microscopy, and Western blot. Cytometry A. 2011;79:461–9. doi: 10.1002/cyto.a.21068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Meffert MK. Roles for NF-kappaB in nerve cell survival, plasticity, and disease. Cell Death Differ. 2006;13:852–60. doi: 10.1038/sj.cdd.4401837. [DOI] [PubMed] [Google Scholar]
- Miyamoto S. Nuclear initiated NF-κB signaling: NEMO and ATM take center stage. Cell Res. 2011;21:116–30. doi: 10.1038/cr.2010.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munsie L, Caron N, Atwal RS, Marsden I, Wild EJ, Bamburg JR, et al. Mutant huntingtin causes defective actin remodeling during stress: defining a new role for transglutaminase 2 in neurodegenerative disease. Hum Mol Genet. 2011;20:1937–51. doi: 10.1093/hmg/ddr075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nucifora FC, Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, et al. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science. 2001;291:2423–8. doi: 10.1126/science.1056784. [DOI] [PubMed] [Google Scholar]
- O'Neill LA, Kaltschmidt C. NF-kappa B: a crucial transcription factor for glial and neuronal cell function. Trends Neurosci. 1997;20:252–8. doi: 10.1016/s0166-2236(96)01035-1. [DOI] [PubMed] [Google Scholar]
- Perrin V, Dufour N, Raoul C, Hassig R, Brouillet E, Aebischer P, et al. Implication of the JNK pathway in a rat model of Huntington's disease. Exp Neurol. 2009;215:191–200. doi: 10.1016/j.expneurol.2008.10.008. [DOI] [PubMed] [Google Scholar]
- Rajesh M, Mukhopadhyay P, Bátkai S, Haskó G, Liaudet L, Huffman JW, et al. CB2-receptor stimulation attenuates TNF-alpha-induced human endothelial cell activation, transendothelial migration of monocytes, and monocyte-endothelial adhesion. Am J Physiol Heart Circ Physiol. 2007;293:H2210–8. doi: 10.1152/ajpheart.00688.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol. 2009;27:119–45. doi: 10.1146/annurev.immunol.021908.132528. [DOI] [PubMed] [Google Scholar]
- Sah DW, Aronin N. Oligonucleotide therapeutic approaches for Huntington disease. J Clin Invest. 2011;121:500–7. doi: 10.1172/JCI45130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapp E, Kegel KB, Aronin N, Hashikawa T, Uchiyama Y, Tohyama K, et al. Early and progressive accumulation of reactive microglia in the Huntington disease brain. J Neuropathol Exp Neurol. 2001;60:161–72. doi: 10.1093/jnen/60.2.161. [DOI] [PubMed] [Google Scholar]
- Simard AR, Rivest S. Bone marrow stem cells have the ability to populate the entire central nervous system into fully differentiated parenchymal microglia. FASEB J. 2004;18:998–1000. doi: 10.1096/fj.04-1517fje. [DOI] [PubMed] [Google Scholar]
- Soulet D, Cicchetti F. The role of immunity in Huntington's disease. Mol Psychiatry. 2011;16:889–902. doi: 10.1038/mp.2011.28. [DOI] [PubMed] [Google Scholar]
- Steffan JS. Does Huntingtin play a role in selective macroautophagy? Cell Cycle. 2010;9:3401–13. doi: 10.4161/cc.9.17.12718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffan JS, Kazantsev A, Spasic-Boskovic O, Greenwald M, Zhu YZ, Gohler H, et al. The Huntington's disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci USA. 2000;97:6763–8. doi: 10.1073/pnas.100110097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundström C, Nilsson K. Establishment and characterization of a human histiocytic lymphoma cell line (U-937) Int J Cancer. 1976;17:565–77. doi: 10.1002/ijc.2910170504. [DOI] [PubMed] [Google Scholar]
- Tai YF, Pavese N, Gerhard A, Tabrizi SJ, Barker RA, Brooks DJ, et al. Microglial activation in presymptomatic Huntington's disease gene carriers. Brain. 2007;130(Pt 7):1759–66. doi: 10.1093/brain/awm044. [DOI] [PubMed] [Google Scholar]
- Teng FY, Tang BL. NF-kappaB signaling in neurite growth and neuronal survival. Rev Neurosci. 2010;21:299–313. doi: 10.1515/revneuro.2010.21.4.299. [DOI] [PubMed] [Google Scholar]
- The Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell. 1993;72:971–83. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Thompson LM, Aiken CT, Kaltenbach LS, Agrawal N, Illes K, Khoshnan A, et al. IKK phosphorylates Huntingtin and targets it for degradation by the proteasome and lysosome. J Cell Biol. 2009;187:1083–99. doi: 10.1083/jcb.200909067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobinick E, Gross H, Weinberger A, Cohen H. TNF-alpha modulation for treatment of Alzheimer's disease: a 6-month pilot study. MedGenMed. 2006;8:25. [PMC free article] [PubMed] [Google Scholar]
- van der Burg JM, Björkqvist M, Brundin P. Beyond the brain: widespread pathology in Huntington's disease. Lancet Neurol. 2009;8:765–74. doi: 10.1016/S1474-4422(09)70178-4. [DOI] [PubMed] [Google Scholar]
- Weiss A, Träger U, Wild EJ, Grueninger S, Farmer R, Landles C, et al. Mutant huntingtin fragmentation in immune cells tracks Huntington's disease progression. J Clin Invest. 2012;122:3731–6. doi: 10.1172/JCI64565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wexler NS, Lorimer J, Porter J, Gomez F, Moskowitz C, Shackell E, et al. Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington's disease age of onset. Proc Natl Acad Sci USA. 2004;101:3498–503. doi: 10.1073/pnas.0308679101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild E, Magnusson A, Lahiri N, Krus U, Orth M, Tabrizi SJ, et al. Abnormal peripheral chemokine profile in Huntington's disease. PLoS Curr. 2011;3:RRN1231. doi: 10.1371/currents.RRN1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwilling D, Huang SY, Sathyasaikumar KV, Notarangelo FM, Guidetti P, Wu HQ, et al. Kynurenine 3-monooxygenase inhibition in blood ameliorates neurodegeneration. Cell. 2011;145:863–74. doi: 10.1016/j.cell.2011.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.