

Abstract
Background
Autosomal dominant polycystic kidney disease (ADPKD) is one of the most common human inherited diseases. Modifier genes seem to modulate the disease progression and might therefore be promising drug targets. Although a number of modifier loci have been already identified, no modifier gene has been proven to be a real modifier yet.
Methods
Gene expression profiling of two substrains of the Han:SPRD rat, namely PKD/Mhm and PKD/US, both harboring the same mutation, was conducted in 36-day-old animals. Catechol-O-methyltransferase (Comt) was identified as a potential modifier gene. A 3-month treatment with tolcapone, a selective inhibitor of Comt, was carried out in PKD/Mhm and PKD/US (cy/+) animals.
Results
Comt is localized within a known modifier locus of PKD (MOP2). The enzyme encoding gene was found upregulated in the more severely affected PKD/Mhm substrain and was hence presumed to be a putative modifier gene of PKD. The treatment with tolcapone markedly attenuated the loss of renal function, inhibited renal enlargement, shifted the size distribution of renal cysts and retarded cell proliferation, apoptosis, inflammation and fibrosis development in affected (cy/+) male and female PKD/Mhm and PKD/US rats.
Conclusions
Comt has been confirmed to be the first reported modifier gene for PKD and tolcapone offers a promising drug for treating PKD.
Keywords: catechol-O-methyltransferase, gene expression profiling, modifier, polycystic kidney disease, tolcapone
INTRODUCTION
Autosomal dominant polycystic kidney disease (ADPKD) is the most commonly inherited renal disorder in humans (prevalence between 1:400 and 1:1000) [1]. The majority of affected patients develop renal failure and insufficiency by the fifth or sixth decade. At present, ∼8–10% of ADPKD patients with end-stage renal disease require renal replacement therapy including haemodialysis, peritoneal dialysis or renal transplantation.
ADPKD should be considered as a systemic disease with clinical presentation in multiple organ systems [2]. However, the development of bilateral large fluid-filled cysts throughout the renal nephron accompanied by interstitial inflammation and fibrosis are considered as the main features of ADPKD [3]. The formation and growth of cysts are attended by the progressive destruction of healthy renal tissue with deteriorating renal function. The pathways involved in the pathogenesis of polycystic kidney disease (PKD) are numerous and to date only a fraction of them have been conceived. Increased epithelial cell proliferation and apoptosis, immoderate tubular secretion of fluid due to an aberration of planar cell polarity and extracellular matrix remodelling has been specified as the fundamental mechanisms of cystogenesis [4].
ADPKD is a genetically heterogeneous disease and can arise from mutations in at least two different genes, PKD1 (∼85% of cases) and PKD2 (∼10% of cases). Additionally, rare mutations in a third not yet identified gene (PKD3) are suspected. The gene products, polycystin-1 (PKD1) and polycystin-2 (PKD2), associate in a complex, localized at the primary cilium of kidney epithelial cells [5]. The complex plays a role in calcium homeostasis, mechano- and chemosensation, and in a number of signalling pathways (JAK/STAT, AP1 and Wnt) [6, 7]. While polycystin-2 functions as a Ca2+-permeable selective channel, polycystin-1 acts as a mechanosensor that controls Ca2+ influx [8, 9]. One of the remarkable features of ADPKD is the great variance in the severity and progression of the disease not only interfamilial but also intrafamilial [10]. This phenomenon is well known for both humans [11] and even inbred animal models [12–15]. Predisposing factors such as affected gene [16], position of mutation [17, 18], mosaicism [19], hypomorphic or incomplete penetration of alleles and dosage of functional protein [20], gender and hormonal effects [14, 21–23], environmental factors as well as genetic modifiers may control disease progression and/or severity. A variety of modifier loci in different murine models of PKD and in humans affected with ADPKD have been described [12, 13, 15, 24–29]. However, no proposed modifier gene has been proven to be a real modifier yet. The identification of such modifier genes and the search for pharmacological agonists/antagonists of the respective gene products represent a promising therapy approach for PKD.
The Hannover Sprague-Dawley (Han:SPRD) rat is a well-documented animal model of PKD [30], which is widely used to study the mechanism of cystogenesis and the effect of therapeutic interventions. A spontaneous missense mutation in Anks6 leads to aberrant expression and mislocalization of Anks6(p.R823W) [31, 32]. The cellular function of Anks6 and the role of Anks6(p.R823W) in cyst formation are still not completely understood and are, therefore, the subject of various studies [33–36]. Two inbred substrains, the PKD/Mhm and the PKD/US rat, originating from this Han:SPRD rat might have developed over the last 20 years due to separate breeding in two different facilities. Although both substrains harbour the same genetic mutation, they differ in disease progression and severity [32]. This finding gives reason to the search for possible modifier genes through the evaluation of differential gene expression between these two substrains. Gene expression profiling revealed that catechol-O-methyltransferase (Comt) was highly upregulated in the more severely affected PKD/Mhm substrain. The Comt mRNA codes for two different isoforms, MB-Comt (membrane-bound) and S-Comt (soluble) [37]. Both proteins are expressed in various tissues; however, S-Comt is usually dominant by a factor of ≥3 [37]. The general function of Comt is the elimination of biologically active or toxic catechols and some other hydroxylated metabolites [37]. Several studies indicate a role for sex hormones in the regulation of Comt activity [38]; however, oestrogen exposure has opposed effects in different tissues [39, 40]. Comt activity as a risk factor for different types of cancer (e.g. breast and renal cancer) is controversially discussed in the literature [41, 42]. Until now, there is no evidence for an involvement of catecholamines and Comt in the pathogenesis of PKD [43]. However, due to upregulation of Comt in the more severely affected PKD/Mhm substrain, the selective Comt inhibitor Tolcapone was, therefore, investigated for its potential to treat PKD.
MATERIALS AND METHODS
Animals
The Han:SPRD rat model was first discovered in 1986 as a spontaneous mutation in a Sprague-Dawley colony in Hannover [44]. A colony of the Han:SPRD rat is maintained in the animal care facility of the Medical Research Center of the University of Heidelberg in Mannheim, Germany, since 1990. After ∼40 generations of inbreeding, an inbred substrain formed and was registered as PKD/Mhm (cy/+) [31]. A second subgroup of the Han:SPRD rats was sent to the animal care facility at the University of Kansas Medical Center, USA, ∼25 years ago [3, 45], and has since matured into an ADPKD model distinct from the PKD/Mhm colony. For this study, animals from the US substrain were obtained from Ph.D. M.D. Vicente E. Torres (Mayo Clinic College of Medicine, Rochester, MN) and will be termed as PKD/US in the following experiments.
All experiments were performed in accordance with federal and local laws, as well as institutional regulations. The animals were kept under standard laboratory conditions (12 h light cycle, 55 ± 5% humidity, 20 ± 2°C room temperature) in an animal care facility at the Medical Research Center, Medical Faculty Mannheim, University of Heidelberg, Germany. Animals had free access to tap water and to standard 19% protein rodent pellet feed (ssniff R/M-H, ssniff Spezialdiäten GmbH, Soest, Germany), if not stated otherwise.
Expression profiling using microarrays
Expression profiling in kidneys of 36-day-old rats was performed using Affymetrix GeneChip® rat expression array 230A (Affymetrix, Santa Clara, CA). Nine rats were investigated for each combination of rat subgroup (PKD/Mhm or PKD/US), genotype (affected cy/+ or wild-type +/+) and gender (male or female). Total RNA was prepared using TRIzol® (Invitrogen Corp., Carlsbad, CA). RNA was further processed as described in the Affymetrix manual. Statistical analysis was performed with the software package Microarray Solution version 1.0 (SAS Institute, Cary, NC) using standard settings, except the following specifications: log-linear mixed models [46] were fitted for values of perfect matches with gender, substrain and genotype considered to be constant and the array-id random. Customer annotation [47] Version 8 was applied to annotate the probes on the microarray to Unigene. A false discovery rate of 0.05 with Bonferroni correction was used to determine the significance of differential gene expression.
Real-time PCR
A commercial real-time PCR assay (Applied Biosystems, Forster City, CA) was performed to analyse Comt gene expression on an ABI PRISM® 7000 sequence detection system (Applied Biosystems). Ppia (peptidyl-isomerase A) served as an endogenous control.
Immunoblot
In a kidney homogenate, the soluble (S-) and the membrane bound (MB-) isoforms of Comt (Chemicon International, Temecula, CA) and Raf-B (Santa Cruz Biotechnology, Heidelberg, Germany) were investigated. Albumin (Cappel, MP Biomedicals, Inc., Aurora, OH) served as a control.
Tolcapone treatment
PKD/Mhm (cy/+) and PKD/US (cy/+) animals of both sexes were assigned to tolcapone treatment and vehicle control groups with 10–12 animals per group, respectively. Animals were provided with a phytoestrogen low 19% protein rat pellet diet (ssniff R/M-H phytoestrogen low, ssniff Spezialdiäten GmbH, Soest, Germany) at all times of the experiments. The minimal dose of 3.0 mg tolcapone per 100 g body weight per 24 h was administered orally in three doses a day starting at the 10th day of life. The drug was prepared in standard solvent vehicle (0.5% carboxymethylcellulose, 0.9% NaCl, 0.4% Tween-80, 0.5% benzyl alcohol) at a concentration of 0.1 mg/mL tolcapone. For better acceptance, sugar syrup was added. Suckling animals were individually fed the drug containing 60% (v/v) sugar syrup into the rodents' cheek pockets using a 1 mL syringe. The body weight was determined daily in suckling animals and administered doses adjusted daily accordingly. From the 36th day of life, the drug was supplied via drinking water [5% (v/v) sugar syrup]. Due to the added sugar, the administration was accepted freely. Animals in control groups obtained the drug vehicle alone. After 3 months of tolcapone treatment, the experiment was terminated.
Biochemical analysis
In the 15th week of life, blood samples were taken and a 24-h urine output was collected. Biochemical parameters of plasma and urine samples were determined by standard laboratory methods (Hitachi 911 Autoanalyzer, Roche Diagnostics, Mannheim, Germany). Renal cyclic adenosine monophosphate (cAMP) levels were measured using the cAMP Biotrak enzyme immunoassay (EIA, GE Healthcare, Freiburg, Germany) after termination of the study.
Morphological and immunohistochemical assessments
All morphological and immunohistochemical assessments of the kidneys were performed in paraffin sections under light microscopy. Cyst scoring was done on haematoxylin–eosin stained kidney sections using a previously described scoring system [13, 14]. Additionally, cystic areas were measured and fibrosis was assessed on Heidenhain's azan stained sections using the software Leica QWin (Leica Mikrosysteme Vertrieb GmbH, Bensheim, Germany). Cell proliferation was assessed by counting Ki-67-positive cells (Dako Deutschland GmbH, Hamburg, Germany), apoptosis was assessed using the DeadEnd™ colorimetric TUNEL system (Promega GmbH, Mannheim, Germany), and inflammation was assessed by counting CD43-positive cells (Acris Antibodies GmbH, Hiddenhausen, Germany).
Statistics
Data were analysed with the SAS System Version 9.1 (SAS Institute Inc.). The following procedures were applied: PROC TTEST (t-test), PROC FREQ (Fisher's exact test) and PROC MEANS (mean and SD). A P value of <0.05 was used to determine the significance of substrain or treatment effects.
RESULTS
Two phenotypic different substrains—PKD/Mhm rats exhibited a more severely progression of PKD compared with PKD/US rats
Considering that both PKD/Mhm and PKD/US originated from the same colony of Han:SPRD and harbour the same mutation for PKD, possible functional and structural differences were investigated in 36-day-old animals. Both wild-type (+/+) PKD/Mhm males and females were 15% heavier (P = 0.001 and P = 0.011, respectively) than their US counterparts. Heterozygously affected PKD/Mhm (cy/+) males were 8% heavier (P = 0.004) and females were 7% heavier (P = 0.064) than their US counterparts. The relative kidney weight (total kidney weight per 100 g body weight) of heterozygously affected PKD/Mhm (cy/+) rats was 62% higher (P < 0.001) in males and was 21% higher (P = 0.023) in female animals than those of PKD/US rats (Table 1). Plasma creatinine levels of PKD/Mhm (cy/+) animals were 15% higher (P = 0.008) in males and 14% higher (P = 0.043) in females compared with the levels of the corresponding PKD/US animals. The plasma urea levels of PKD/Mhm (cy/+) male animals were 21% higher (P = 0.014), whereas the plasma urea levels did not significantly differ (P = 0.855) between female animals of the subgroups (Table 1). No difference was observed in the relative kidney weight, plasma creatinine as well as plasma urea comparing wild-type (+/+) PKD/Mhm and PKD/US animals. Furthermore, cyst formation was more pronounced in (cy/+) PKD/Mhm than in PKD/US rats as measured by higher cyst scores (Figure 1). These results confirmed the formation of two different substrains of the Han:SPRD rat, PKD/Mhm and PKD/US, during the years of separated inbreeding.
Table 1:
Higher relative kidney weights, higher plasma creatinine levels and higher plasma urea levels in 36-day-old (cy/+) PKD/Mhm compared with PKD/US rats
| Parameter | Sex | Rats | n | Mean | SD | P value |
|---|---|---|---|---|---|---|
| Relative kidney weight (g/100 g body weight) | Male | PKD/Mhm | 41 | 1.46 | 0.33 | <0.001 |
| PKD/US | 15 | 1.11 | 0.07 | |||
| Female | PKD/Mhm | 29 | 1.22 | 0.27 | 0.023 | |
| PKD/US | 10 | 1.01 | 0.1 | |||
| Plasma creatinine (mg/dL) | Male | PKD/Mhm | 41 | 0.15 | 0.03 | 0.008 |
| PKD/US | 15 | 0.13 | 0.02 | |||
| Female | PKD/Mhm | 29 | 0.16 | 0.04 | 0.043 | |
| PKD/US | 10 | 0.14 | 0.03 | |||
| Plasma urea (mg/dL) | Male | PKD/Mhm | 41 | 46 | 11 | 0.014 |
| PKD/US | 15 | 38 | 6 | |||
| Female | PKD/Mhm | 29 | 44 | 9 | 0.855 | |
| PKD/US | 10 | 43 | 6 |
n, number of investigated animals; SD, standard deviation; P value, P value of t-test comparing PKD/Mhm versus PKDUS rats.
FIGURE 1:

More pronounced cyst formation in PKD/Mhm compared with PKD/US. Shown are the relative frequencies of cyst scores of 36-day-old heterozygously affected (cy/+) rats. Cyst scores compromise a grading between 1 (mildly affected), 2, 3 or 4 (severely affected).
Comt was strongly upregulated in PKD/Mhm compared with PKD/US
In order to identify possible modifier genes for PKD, renal gene expression profiles of 36-day-old rats of both substrains were generated. Differential gene expression between PKD/Mhm and PKD/US was detected for at least 104 genes. However, the expression changes in most of these genes were marginal. Only three genes exhibited expression changes of more than 2-fold, namely Comt (catechol-O-methyltransferase), RT1-Aw2 (RT1 class Ib, locus Aw2) and an expressed sequence tag (Figure 2). Comt exhibited a more than 5-fold upregulation in PKD/Mhm compared with their PKD/US counterparts. This conspicuous upregulation of Comt in PKD/Mhm (cy/+) animals was confirmed on both mRNA and protein levels (Figures 2 and 3).
FIGURE 2:
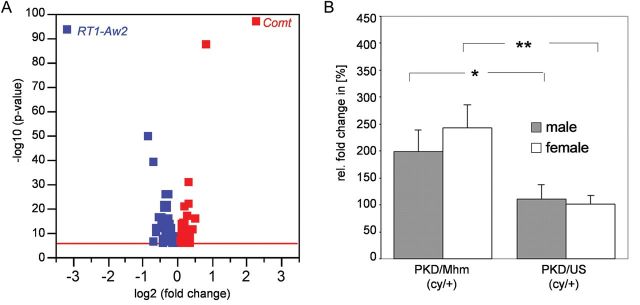
Gene expression profiling revealed Comt as the most upregulated gene in PKD/Mhm compared with PKD/US. (A) Shown is the volcano plot of the significantly regulated genes found with the microarray experiment. Renal gene expression was analysed in 36-day-old PKD/Mhm and PKD/US rats. Red squares indicate overexpressed and blue squares underexpressed genes in PKD/Mhm compared with PKD/US. Red line: cut-off for P = 10−6, corresponding to a false discovery rate of 0.05 with Bonferroni correction for multiple testing. (B) Real-time PCR revealed a 2-fold upregulation of Comt mRNA in PKD/Mhm rats compared with PKD/US animals. Ppia was taken as endogenous control. P values between 0.001 < P < 0.01 are indicated by **, and P values between 0.01 < P < 0.05 indicated by *.
FIGURE 3:

Upregulation of Comt in PKD/Mhm compared with PKD/US confirmed by western blot. (A) Conspicuous higher protein amounts of S-Comt in the PKD/Mhm substrain for both gender and genotypes were found using western blot when compared with the PKD/US substrain. (B+C) MB-Comt was equally expressed in all groups. Albumin was taken as an endogenous control. Analyses were performed with kidneys of 36-day-old rats. Lysates of six animals were pooled per genotype, sex and substrain.
Comt inhibition with tolcapone attenuated PKD progression
With the objective of proving Comt as a putative modifier gene, the influence of tolcapone on PKD severity and progression was investigated. Both substrains were treated with tolcapone (3.0 mg per 100 g body weight per 24 h) for 3 months starting at the 10th day of life. Upon treatment, the relative kidney weight, plasma creatinine, plasma urea and the albumin excretion were reduced in all treated compared with control rats (Table 2 and Figure 4). Reduced cell proliferation, apoptosis, inflammation and fibrosis were detected in the kidney sections after the treatment (Figures 5 and 6). The percentage of cyst area of the total kidney area was reduced by 19% (P = 0.065), 11% (P = 0.319), 14% (P = 0.075) and 18% (P = 0.073) upon treatment in PKD/Mhm males and females and PKD/US males and females, respectively. Although the number of renal cysts was not significantly changed by tolcapone treatment, the cyst size distribution was significantly shifted after 3 months of therapy (Table 3). Proportions of small cysts (<0.3 mm2) were larger (P < 0.001) in treated animals compared with their control littermates. All these results provide evidence of attenuating PKD progression by tolcapone treatment.
Table 2:
Changes in relative kidney weight, plasma creatinine and plasma urea after 3 months of tolcapone treatment in male and female PKD/Mhm and PKD/US animals
| Parameter | Sex | Rats | Group | n | Mean | SD | P value | Change by treatment (%) |
|---|---|---|---|---|---|---|---|---|
| Relative kidney weight (g/100 g body weight) | Male | PKD/Mhm | T | 12 | 0.98 | 0.10 | <0.001 | −21 |
| C | 10 | 1.24 | 0.13 | |||||
| PKD/US | T | 11 | 1.28 | 0.16 | <0.001 | −22 | ||
| C | 12 | 1.64 | 0.19 | |||||
| Female | PKD/Mhm | T | 12 | 1.24 | 0.19 | 0.152 | −8 | |
| C | 12 | 1.35 | 0.18 | |||||
| PKD/US | T | 10 | 1.26 | 0.11 | 0.005 | −12 | ||
| C | 11 | 1.44 | 0.15 | |||||
| Plasma creatinine (mg/dL) | Male | PKD/Mhm | T | 12 | 1.04 | 0.13 | 0.006 | −21 |
| C | 10 | 1.31 | 0.28 | |||||
| PKD/US | T | 11 | 1.08 | 0.21 | 0.094 | −14 | ||
| C | 12 | 1.26 | 0.27 | |||||
| Female | PKD/Mhm | T | 12 | 0.71 | 0.17 | 0.265 | +11 | |
| C | 12 | 0.64 | 0.15 | |||||
| PKD/US | T | 10 | 0.54 | 0.14 | 0.801 | −2 | ||
| C | 11 | 0.55 | 0.07 | |||||
| Plasma urea (mg/dL) | Male | PKD/Mhm | T | 12 | 106 | 23 | 0.003 | −31 |
| C | 10 | 154 | 43 | |||||
| PKD/US | T | 11 | 103 | 23 | 0.039 | −17 | ||
| C | 12 | 123 | 19 | |||||
| Female | PKD/Mhm | T | 12 | 72 | 13 | 0.377 | −8 | |
| C | 12 | 78 | 16 | |||||
| PKD/US | T | 10 | 56 | 15 | 0.505 | −7 | ||
| C | 11 | 60 | 8 | |||||
| Albumin excretion (µg/24 h) | Male | PKD/Mhm | T | 12 | 38 | 32 | 0.042 | −46 |
| C | 10 | 70 | 37 | |||||
| PKD/US | T | 11 | 57 | 66 | 0.358 | −42 | ||
| C | 12 | 98 | 60 | |||||
| Female | PKD/Mhm | T | 12 | 21 | 20 | 0.176 | −36 | |
| C | 12 | 33 | 20 | |||||
| PKD/US | T | 10 | 34 | 24 | 0.358 | −26 | ||
| C | 11 | 46 | 31 |
FIGURE 4:

Tolcapone reduced the relative kidney weight, plasma creatinine, plasma urea and albumin excretion. Shown are the effects of 3 months tolcapone treatment in PKD/Mhm and PKD/US males and females on (A) relative kidney weight (B) plasma creatinine, (C) plasma urea and (D) albumin excretion (mean ± SD). P values <0.001 are denoted by ***, between 0.001 < P < 0.01 by **, and P values between 0.01 < P < 0.05 by *.
FIGURE 5:
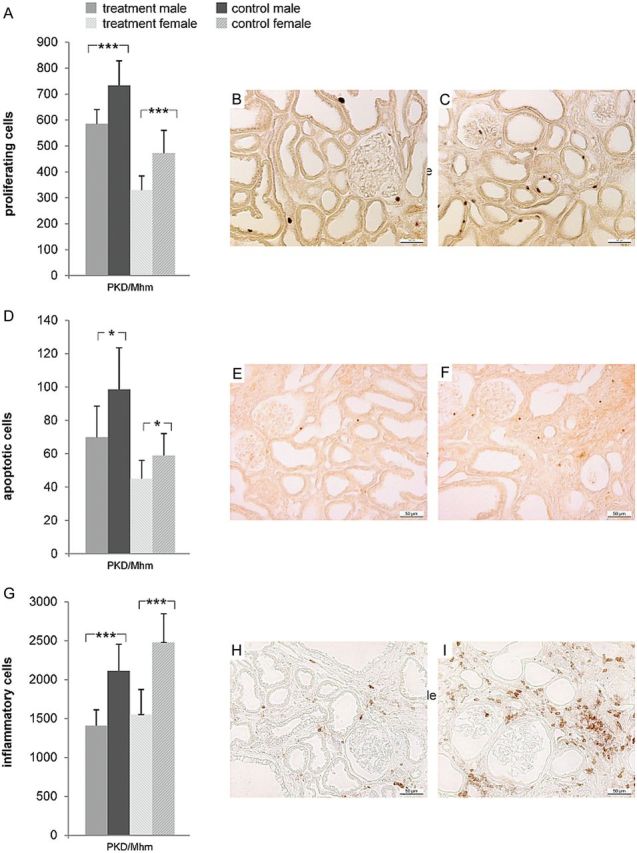
Tolcapone reduced cell proliferation, apoptosis and inflammation in PKD/Mhm substrain. Shown are the effects of 3 months of tolcapone treatment in PKD/Mhm males and females on (A–C) cell proliferation, (D–F) apoptosis and (G–I) inflammation. Shown are the mean counts ± SD of (A) Ki-67 positive cells, (D) TUNEL-positive cells and (G) CD43-positive cells (P = 0.006, P < 0.001 is denoted by * and ***, respectively). (B, E, H) and (C, F, I) are representative pictures of the (immuno-) histochemical staining with (B, E, H) showing kidney sections of tolcapone-treated animals, and (C, F, I) showing kidney sections of vehicle controls, respectively. Pictures were taken at a ×20 magnification.
FIGURE 6:
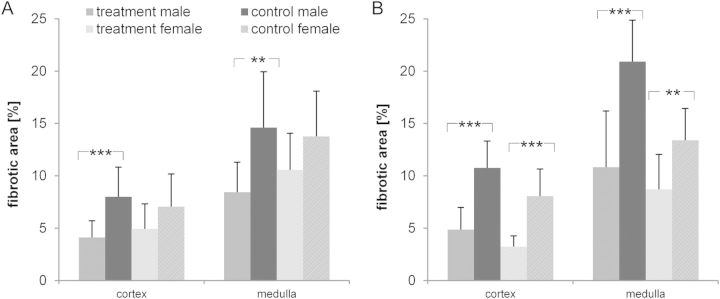
Tolcapone reduced fibrotic alterations in cortex and medulla. Shown are the effects of 3 months of tolcapone in male and female(A) PKD/Mhm and (B) PKD/US animals on the development of fibrosis after 3 months of tolcapone treatment (mean ± SD, P values <0.001 are denoted by ***, and P values between 0.001 < P < 0.01 by **).
Table 3:
Cyst size distribution was shifted towards higher proportions of small cysts after 3 months of tolcapone treatment
| Relative group size in (%) |
Differences between the relative group sizes |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Substrain | PKD/Mhm |
PKD/US |
PKD/Mhm |
PKD/US |
||||||||
| Sex | Male |
Female |
Male |
Female |
Male | Female | Male | Female | ||||
| Cyst size in (mm2) | T | C | T | C | T | C | T | C | Difference (treated-control) |
|||
| <0.010 | 18.3 | 16.6 | 6.9 | 29.4 | 11.6 | 5.9 | 6.0 | 9.1 | 1.6 | −22.5 | 5.8 | −3.1 |
| 0.010–0.019 | 22.5 | 16.8 | 17.8 | 15.1 | 17.4 | 10.2 | 11.6 | 17.2 | 5.7 | 2.7 | 7.1 | −5.6 |
| 0.020–0.029 | 16.9 | 16.2 | 18.4 | 14.2 | 19.5 | 17.1 | 22.4 | 21.6 | 0.7 | 4.2 | 2.3 | 0.8 |
| 0.030–0.049 | 16.4 | 17.8 | 22.3 | 16.3 | 19.8 | 22.5 | 25.7 | 22.0 | −1.4 | 6.1 | −2.7 | 3.6 |
| 0.050–0.100 | 12.9 | 17.5 | 20.8 | 14.9 | 17.1 | 23.2 | 23.5 | 19.8 | −4.6 | 5.9 | −6.2 | 3.7 |
| >0.100 | 13.0 | 15.1 | 13.7 | 10.1 | 14.7 | 21.1 | 10.8 | 10.2 | −2.1 | 3.7 | −6.4 | 0.6 |
| total | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | ||||
Cyst sizes were divided to obtain six groups with approximately equal group sizes in the control animals.
Tolcapone treatment downregulated cAMP signalling
In order to analyse the effects of Comt inhibition on cellular pathways, protein expression levels of cAMP and Raf-B were determined after 3 months of tolcapone treatment. Renal cAMP levels were 48% lower (P = 0.048) in PKD/Mhm males and protein levels of Raf-B were also markedly reduced in treated compared with control rats of both substrains and gender (Figure 7). These findings confirm the supposition that Tolcapone exerts influence on the progression of PKD by affecting the cAMP signalling pathway.
FIGURE 7:
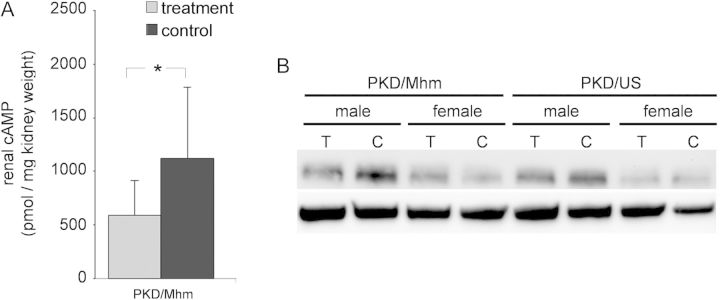
Three months of tolcapone treatment downregulated cAMP signalling. (A) Renal cAMP levels of PKD/Mhm males (mean ± SD, P values between 0.01 < P < 0.05 by *). (B) Western blot analysis. Shown are the protein expression levels of the Raf-B (a downstream molecule of cAMP signalling) of all groups. Protein lysates of all animals in the study were pooled according to gender, genotype and treatment (see Table 2 for n per group). Albumin was used as endogenous control. (C) Vehicle control rats, T: tolcapone-treated rats.
DISCUSSION
Inbred strains of rodent animal models are commonly used in biomedical research and substrain divergence is a widespread phenomenon. The genetic variability between substrains exhibiting diversified phenotypes can be used as a unique possibility to identify modifier genes. Although originating from the same colony of Han:SPRD rats [44] and harbouring the same mutation [32], two substrains with obvious differences in PKD progression have formed due to the separated inbreeding over the last 25 years. PKD/Mhm rats compared with PKD/US rats exhibited the more severe PKD progression, indicated by more aggressive kidney growth, faster deteriorating renal function and more pronounced cyst formation (Figure 1).
Differential gene expression between the two substrains was analysed to study the underlying mechanism for their phenotypic divergence. Gene expression profiling using microarrays revealed three genes, which were highly differentially expressed (with fold changes higher than two) (Figure 2). Comt was strongly upregulated in the more severely affected PKD/Mhm (cy/+) animals (Figures 2 and 3). Furthermore, the Comt gene is localized on chromosome 16 within a known modifier locus of PKD (MOP2), which was demonstrated to strongly modulate PKD progression in pcy mice [15]. These findings hence suggest Comt as a modifier for PKD and the more pronounced disease severity in PKD/Mhm animals is due to the strong overexpression of Comt in PKD/Mhm animals.
Although ADPKD is a significant cause of chronic renal failure in adults, an effective therapy does not yet exist and thus to date treatment of PKD has been limited to renal replacement therapy by dialysis or by renal transplantation coupled with aggressive management of hypertension. Concerning this aspect, the selective Comt inhibitor tolcapone was tested for its potential to attenuate PKD progression. Starting at the 10th day of life, male and female affected PKD/Mhm (cy/+) and PKD/US (cy/+) animals were treated with tolcapone for 3 months.
Judging by physical appearance, the treatment with tolcapone was well tolerated without any side effects or mortalities. Body weight, liver weight and the liver mass index were not significantly different between the treatment and control groups. All investigated liver toxicity parameters (alkaline phosphatase, glutamic-oxaloacetic transaminase and glutamic-pyruvic transaminase) did not reach pathological levels (data not shown).
During the tolcapone treatment, a phytoestrogen low diet was fed to the animals. As oestrogens have been described to be renoprotective by attenuating glomerulosclerosis and tubulointerstitial fibrosis [48, 49] and downregulating inflammatory responses [50], this diet was chosen to eliminate diet-related contributions to phenotypic changes. Several studies have demonstrated that PKD is ameliorated with dietary soy protein substitution [51] and soy proteins are the major source of phytoestrogens [52]. The two major dietary phytoestrogens are daidzein and genistein [53] and the latter one was shown to inhibit Comt activity [54]. Therefore, favourable effects (anti-inflammatory and anti-fibrotic) of estrogens and the decrease of Comt activity by the existence of genistein by dietary phytoestrogens could have influenced the treatment study.
Tolcapone treatment attenuated progression of PKD by modulating cell proliferation and apoptosis, ameliorating renal injury and improving renal function (Figures 4 and 5). A precisely controlled balance between cellular proliferation and programmed cell death (apoptosis) is essential for normal growth and differentiation of the kidney. These fundamental processes are disturbed in PKDs [55, 56]. Reduced cell proliferation and apoptosis were recorded in treated rats compared with control animals. Renal injury including interstitial inflammation and fibrosis accompany the course of chronic renal failure. Significantly less CD43-positive inflammatory cells were present in the kidneys of treated rats compared with the untreated control group. Also, the areas affected by fibrosis in the renal cortex and the medulla were significantly smaller after treatment. The total kidney volume of polycystic kidneys may be a useful surrogate marker for disease progression. Indeed, currently the existence of a significant correlation between the rate of growth of renal volume and the rate of decline of renal function in patients with PKD is still controversially discussed [57, 58]. Upon 3 months of tolcapone treatment, the relative kidney weight was significantly reduced in treated animals compared with untreated controls. Accordingly, the size distribution of renal cysts has been shifted towards smaller cysts after tolcapone treatment. The improvement of renal function by tolcapone treatment was indicated by the lower levels of plasma creatinine, plasma urea and albumin excretion in treated animals.
An increased level of cAMP is common in animal models of PKD [59, 60] and was shown to be associated with renal cyst formation in ADPKD [61, 62]. The attenuation of PKD by tolcapone treatment most probably involves the downregulation of cellular cAMP signalling. Alterations in the polycystin pathway in PKD result in decreased intracellular calcium levels. Adenylyl cyclase VI is, in turn, stimulated and the intracellular cAMP level elevated. An elevated cAMP level further stimulates the ERK pathway via Raf-B and prompts cyst formation by increasing cell proliferation and Cl-dependent fluid secretion [63, 64]. Tolcapone treatment downregulated both cAMP and the protein level of the ERK pathway component Raf-B (Figure 7). Further putatively involved pathways through which Comt inhibitor tolcapone markedly attenuated PKD progression are summarized in Figure 8.
FIGURE 8:

Pathways regulated in PKD are influenced by tolcapone. The polycystin pathway is disrupted in PKD resulting in decreased intracellular calcium levels. Adenylyl cyclase VI is in turn simulated and intracellular cAMP is increased. cAMP further stimulates the ERK pathway via Raf-B increasing cell proliferation. Mechanisms and components upregulated in PKD are shown in blue and downregulation is shown in yellow, effects observed in male PKD/Mhm rats upon 3 months of Comt inhibition with tolcapone in orange. PC 1: polycystin 1, PC 2: polycystin 2, R: G protein-coupled receptor, AC VI: adenylyl cyclase VI, PKA: protein kinase A, CFTR: cystic fibrosis transmembrane conductance regulator, V2R: vasopressin 2 receptor, ERK: extracellular signal-regulated kinase, Gi and Gs: G-proteins, cAMP: cyclic adenosine monophosphate.
Also antioxidative mechanisms are most likely involved in the improvement of PKD through Comt inhibition by tolcapone. Upregulated Comt was shown to be involved in Ang II-induced oxidative stress and inflammation. Comt inhibitors have been described to scavenge peroxyl radicals, nitric oxide and superoxide, inhibit lipid peroxidation, as well as NF-κB activation [65–67]. Comt inhibition by Entacapone improved Ang II-induced vascular inflammation and glomerular damage in a rat model transgenic for human renin and human angiotensinogen genes [68, 69]. The Comt inhibitor Nitecapone mediates renoprotective effects in diabetic rats. Hyperfiltration, focal glomerulosclerosis and albuminuria were reversed by Nitecapone. The protective mechanisms are ascribed to antioxidative and Comt inhibitory properties of the drug [70]. Tolcapone is chemically similar to entacapone and nitecapone and it is hence likely that the anti-inflammatory and anti-fibrotic effects observed are ascribable to antioxidant properties of tolcapone.
Comparing the two substrains, Comt is upregulated in the PKD/Mhm strain compared with the PKD/US in both affected and unaffected animals. However, both strains harbour the same missense mutation responsible for the polycystic phenotype. Different starting levels of Comt in the both strains, however, result in a different ability of the strains to respond to the treatment, e.g. Comt inhibition. The effect of tolcapone is thus more pronounced in PKD/Mhm animals compared with PKD/US animals.
Most promising therapeutic interventions for PKD target cAMP signalling and cell cycle control. Some of them such as Rapamycin and Tolvaptan have already entered phases of clinical trials. The mTOR inhibitor and potent antiproliferative substance rapamycin was found to reduce kidney volume and cyst volume density, decrease proliferation in cystic and non-cystic tubules and prevent the loss of renal function [71]. Vasopressin V2 receptor antagonists (e.g. tolvaptan or OPC-31260) are effective agents to reduce cyst and kidney volumes by reducing cAMP levels [59, 72]. Inhibitors of the cyclin-dependent kinase, roscovitine and fisetin, attenuated cystogenesis through cell-cycle arrest and transcriptional inhibition [73, 74]. Also, the renoprotective function of tolcapone seems to be mediated by cAMP signalling and cell-cycle control. Reduced cAMP levels and less proliferating cells were also documented in our current study in tolcapone-treated rats compared with control animals. The improvements in renal function and morphology achieved after 3 months of tolcapone treatment were quite comparable with that reported for roscovitine [73], rapamycin and sirolimus [71, 75]. These data suggest tolcapone as a promising alternative therapeutic strategy to other common effective therapy approaches for PKD.
In summary, in PKD/Mhm and PKD/US, two inbred substrains both originating from Han:SPRD rats but differing in PKD progression, Comt was identified as the first modifier gene of PKD. Furthermore a selective Comt inhibitor was documented to markedly downregulate cAMP signalling, inhibit inflammation, cell proliferation, apoptosis and fibrosis, shift the cyst size distribution and prevent renal failure in PKD/Mhm rats. Therefore, tolcapone offers a promising drug for treating PKD. Further studies are in progress to confirm the usefulness of tolcapone also in genetically different animal models of PKD.
SUPPLEMENTARY DATA
Supplementary data are available online at http://ndt.oxfordjournals.org.
ACKNOWLEDGEMENTS
We thank Elisabeth Wühl, Viktoria Skude, Mareike Gill, and other colleagues for excellent technical support. This work was supported by the DFG Research Training Group 886 ‘Molecular Imaging methods for the analysis of gene and protein expression’.
CONFLICT OF INTEREST STATEMENT
None declared. The presented results in this paper have not been published previously in whole or part, except in abstract format.
REFERENCES
- 1.Torres VE, Harris PC. Autosomal dominant polycystic kidney disease: the last 3 years. Kidney Int. 2009;76:149–168. doi: 10.1038/ki.2009.128. doi:10.1038/ki.2009.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gabow PA. Autosomal dominant polycystic kidney disease—more than a renal disease. Am J Kidney Dis. 1990;16:403–413. doi: 10.1016/s0272-6386(12)80051-5. [DOI] [PubMed] [Google Scholar]
- 3.Cowley BD, Jr., Gudapaty S, Kraybill AL, et al. Autosomal-dominant polycystic kidney disease in the rat. Kidney Int. 1993;43:522–534. doi: 10.1038/ki.1993.79. doi:10.1038/ki.1993.79. [DOI] [PubMed] [Google Scholar]
- 4.Igarashi P, Somlo S. Genetics and pathogenesis of polycystic kidney disease. J Am Soc Nephrol. 2002;13:2384–2398. doi: 10.1097/01.asn.0000028643.17901.42. doi:10.1097/01.ASN.0000028643.17901.42. [DOI] [PubMed] [Google Scholar]
- 5.Yoder BK, Hou X, Guay-Woodford LM. The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J Am Soc Nephrol. 2002;13:2508–2516. doi: 10.1097/01.asn.0000029587.47950.25. doi:10.1097/01.ASN.0000029587.47950.25. [DOI] [PubMed] [Google Scholar]
- 6.Veland IR, Awan A, Pedersen LB, et al. Primary cilia and signaling pathways in mammalian development, health and disease. Nephron Physiol. 2009;111:39–53. doi: 10.1159/000208212. doi:10.1159/000208212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Distefano G, Boca M, Rowe I, et al. Polycystin-1 regulates extracellular signal-regulated kinase-dependent phosphorylation of tuberin to control cell size through mTOR and its downstream effectors S6K and 4EBP1. Mol Cell Biol. 2009;29:2359–2371. doi: 10.1128/MCB.01259-08. doi:10.1128/MCB.01259-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xia S, Li X, Johnson T, et al. Polycystin-dependent fluid flow sensing targets histone deacetylase 5 to prevent the development of renal cysts. Development. 2010;137:1075–1084. doi: 10.1242/dev.049437. doi:10.1242/dev.049437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chapin HC, Rajendran V, Caplan MJ. Polycystin-1 surface localization is stimulated by polycystin-2 and cleavage at the G protein-coupled receptor proteolytic site. Mol Biol Cell. 2010;21:4338–4348. doi: 10.1091/mbc.E10-05-0407. doi:10.1091/mbc.E10-05-0407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peters DJ, Sandkuijl LA. Genetic heterogeneity of polycystic kidney disease in Europe. Contrib Nephrol. 1992;97:128–139. doi: 10.1159/000421651. [DOI] [PubMed] [Google Scholar]
- 11.Peters DJ, Breuning MH. Autosomal dominant polycystic kidney disease: modification of disease progression. Lancet. 2001;358:1439–1444. doi: 10.1016/S0140-6736(01)06531-X. doi:10.1016/S0140-6736(01)06531-X. [DOI] [PubMed] [Google Scholar]
- 12.Guay-Woodford LM, Wright CJ, Walz G, et al. Quantitative trait loci modulate renal cystic disease severity in the mouse bpk model. J Am Soc Nephrol. 2000;11:1253–1260. doi: 10.1681/ASN.V1171253. [DOI] [PubMed] [Google Scholar]
- 13.Bihoreau MT, Megel N, Brown JH, et al. Characterization of a major modifier locus for polycystic kidney disease (Modpkdr1) in the Han:SPRD(cy/+) rat in a region conserved with a mouse modifier locus for Alport syndrome. Hum Mol Genet. 2002;11:2165–2173. doi: 10.1093/hmg/11.18.2165. doi:10.1093/hmg/11.18.2165. [DOI] [PubMed] [Google Scholar]
- 14.Gretz N, Ceccherini I, Kranzlin B, et al. Gender-dependent disease severity in autosomal polycystic kidney disease of rats. Kidney Int. 1995;48:496–500. doi: 10.1038/ki.1995.319. doi:10.1038/ki.1995.319. [DOI] [PubMed] [Google Scholar]
- 15.Woo DD, Nguyen DK, Khatibi N, et al. Genetic identification of two major modifier loci of polycystic kidney disease progression in pcy mice. J Clin Invest. 1997;100:1934–1940. doi: 10.1172/JCI119724. doi:10.1172/JCI119724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harris PC. Molecular basis of polycystic kidney disease: PKD1, PKD2 and PKHD1. Curr Opin Nephrol Hypertens. 2002;11:309–314. doi: 10.1097/00041552-200205000-00007. doi:10.1097/00041552-200205000-00007. [DOI] [PubMed] [Google Scholar]
- 17.Hateboer N, Veldhuisen B, Peters B, et al. Location of mutations within the PKD2 gene influences clinical outcome. Kidney Int. 2000;57:1444–1451. doi: 10.1046/j.1523-1755.2000.00989.x. doi:10.1046/j.1523-1755.2000.00989.x. [DOI] [PubMed] [Google Scholar]
- 18.Rossetti S, Burton S, Strmecki L, et al. The position of the polycystic kidney disease 1 (PKD1) gene mutation correlates with the severity of renal disease. J Am Soc Nephrol. 2002;13:1230–1237. doi: 10.1097/01.asn.0000013300.11876.37. doi:10.1097/01.ASN.0000013300.11876.37. [DOI] [PubMed] [Google Scholar]
- 19.Connor A, Lunt PW, Dolling C, et al. Mosaicism in autosomal dominant polycystic kidney disease revealed by genetic testing to enable living related renal transplantation. Am J Transplant. 2008;8:232–237. doi: 10.1111/j.1600-6143.2007.02030.x. [DOI] [PubMed] [Google Scholar]
- 20.Rossetti S, Kubly VJ, Consugar MB, et al. Incompletely penetrant PKD1 alleles suggest a role for gene dosage in cyst initiation in polycystic kidney disease. Kidney Int. 2009;75:848–855. doi: 10.1038/ki.2008.686. doi:10.1038/ki.2008.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gretz N. Progression of chronic renal failure in a rat strain with autosomal dominant polycystic kidney disease. Nephron Physiol. 1994;68:462–467. doi: 10.1159/000188308. [DOI] [PubMed] [Google Scholar]
- 22.Nagao S, Kusaka M, Nishii K, et al. Androgen receptor pathway in rats with autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2005;16:2052–2062. doi: 10.1681/ASN.2004070595. doi:10.1681/ASN.2004070595. [DOI] [PubMed] [Google Scholar]
- 23.Anderson S, Oyama TT, Lindsley JN, et al. 2-Hydroxyestradiol slows progression of experimental polycystic kidney disease. Am J Physiol Renal Physiol. 2011 doi: 10.1152/ajprenal.00265.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iakoubova OA, Dushkin H, Beier DR. Localization of a murine recessive polycystic kidney disease mutation and modifying loci that affect disease severity. Genomics. 1995;26:107–114. doi: 10.1016/0888-7543(95)80088-4. doi:10.1016/0888-7543(95)80088-4. [DOI] [PubMed] [Google Scholar]
- 25.Kuida S, Beier DR. Genetic localization of interacting modifiers affecting severity in a murine model of polycystic kidney disease. Genome Res. 2000;10:49–54. [PMC free article] [PubMed] [Google Scholar]
- 26.Iakoubova OA, Dushkin H, Beier DR. Genetic analysis of a quantitative trait in a mouse model of polycystic kidney disease. Am J Respir Crit Care Med. 1997;156:S72–S77. doi: 10.1164/ajrccm.156.4.12-tac-0. [DOI] [PubMed] [Google Scholar]
- 27.Upadhya P, Churchill G, Birkenmeier EH, et al. Genetic modifiers of polycystic kidney disease in intersubspecific KAT2J mutants. Genomics. 1999;58:129–137. doi: 10.1006/geno.1999.5830. doi:10.1006/geno.1999.5830. [DOI] [PubMed] [Google Scholar]
- 28.Sommardahl C, Cottrell M, Wilkinson JE, et al. Phenotypic variations of orpk mutation and chromosomal localization of modifiers influencing kidney phenotype. Physiol Genomics. 2001;7:127–134. doi: 10.1152/physiolgenomics.00089.2001. [DOI] [PubMed] [Google Scholar]
- 29.Mrug M, Li R, Cui X, et al. Kinesin family member 12 is a candidate polycystic kidney disease modifier in the cpk mouse. J Am Soc Nephrol. 2005;16:905–916. doi: 10.1681/ASN.2004121083. doi:10.1681/ASN.2004121083. [DOI] [PubMed] [Google Scholar]
- 30.Schafer K, Gretz N, Bader M, et al. Characterization of the Han:SPRD rat model for hereditary polycystic kidney disease. Kidney Int. 1994;46:134–152. doi: 10.1038/ki.1994.253. doi:10.1038/ki.1994.253. [DOI] [PubMed] [Google Scholar]
- 31.Bihoreau MT, Ceccherini I, Browne J, et al. Location of the first genetic locus, PKDr1, controlling autosomal dominant polycystic kidney disease in Han:SPRD cy/+ rat. Hum Mol Genet. 1997;6:609–613. doi: 10.1093/hmg/6.4.609. doi:10.1093/hmg/6.4.609. [DOI] [PubMed] [Google Scholar]
- 32.Brown JH, Bihoreau MT, Hoffmann S, et al. Missense mutation in sterile alpha motif of novel protein SamCystin is associated with polycystic kidney disease in (cy/+) rat. J Am Soc Nephrol. 2005;16:3517–3526. doi: 10.1681/ASN.2005060601. doi:10.1681/ASN.2005060601. [DOI] [PubMed] [Google Scholar]
- 33.Stagner EE, Bouvrette DJ, Cheng J, et al. The polycystic kidney disease-related proteins Bicc1 and SamCystin interact. Biochem Biophys Res Commun. 2009;383:16–21. doi: 10.1016/j.bbrc.2009.03.113. doi:10.1016/j.bbrc.2009.03.113. [DOI] [PubMed] [Google Scholar]
- 34.Kugita M, Nishii K, Morita M, et al. Global gene expression profiling in early-stage polycystic kidney disease in the Han:SPRD Cy rat identifies a role for RXR signaling. Am J Physiol Renal Physiol. 2011;300:F177–F188. doi: 10.1152/ajprenal.00470.2010. doi:10.1152/ajprenal.00470.2010. [DOI] [PubMed] [Google Scholar]
- 35.Nagao S, Morita M, Kugita M, et al. Polycystic kidney disease in Han:SPRD Cy rats is associated with elevated expression and mislocalization of SamCystin. Am J Physiol Renal Physiol. 2010;299:F1078–F1086. doi: 10.1152/ajprenal.00504.2009. doi:10.1152/ajprenal.00504.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Neudecker S, Walz R, Menon K, et al. Transgenic overexpression of Anks6(p.R823W) causes polycystic kidney disease in rats. Am J Pathol. 2010;177:3000–3009. doi: 10.2353/ajpath.2010.100569. doi:10.2353/ajpath.2010.100569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Männistö PT, Kaakkola S. Catechol-O-methyltransferase (COMT): biochemistry, molecular biology, pharmacology, and clinical efficacy of the new selective COMT inhibitors. Pharmacol Rev. 1999;51:593–628. [PubMed] [Google Scholar]
- 38.Schendzielorz N, Rysa A, Reenila I, et al. Complex estrogenic regulation of catechol-O-methyltransferase (COMT) in rats. J Physiol Pharmacol. 2011;62:483–490. [PubMed] [Google Scholar]
- 39.Weisz J, Fritz-Wolz G, Clawson GA, et al. Induction of nuclear catechol-O-methyltransferase by estrogens in hamster kidney: implications for estrogen-induced renal cancer. Carcinogenesis. 1998;19:1307–1312. doi: 10.1093/carcin/19.7.1307. doi:10.1093/carcin/19.7.1307. [DOI] [PubMed] [Google Scholar]
- 40.Cohn CK, Axelrod J. The effect of estradiol on catechol-O-methyltransferase activity in rat liver. Life Sci I. 1971;10:1351–1354. doi: 10.1016/0024-3205(71)90335-3. doi:10.1016/0024-3205(71)90335-3. [DOI] [PubMed] [Google Scholar]
- 41.Lajin B, Hamzeh AR, Ghabreau L, et al. Catechol-O-methyltransferase Val 108/158 Met polymorphism and breast cancer risk: a case–control study in Syria. Breast Cancer. 2011 doi: 10.1007/s12282-011-0309-y. [DOI] [PubMed] [Google Scholar]
- 42.Chang I, Liu J, Majid S, et al. Catechol-O-methyltransferase-mediated metabolism of 4-hydroxyestradiol inhibits the growth of human renal cancer cells through the apoptotic pathway. Carcinogenesis. 2012;33:420–426. doi: 10.1093/carcin/bgr294. doi:10.1093/carcin/bgr294. [DOI] [PubMed] [Google Scholar]
- 43.Belibi FA, Edelstein CL. Novel targets for the treatment of autosomal dominant polycystic kidney disease. Expert Opin Investig Drugs. 2010;19:315–328. doi: 10.1517/13543781003588491. doi:10.1517/13543781003588491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kaspareit-Rittinghausen J, Deerberg F, Rapp KG, et al. A new rat model for polycystic kidney disease of humans. Transplant Proc. 1990;22:2582–2583. [PubMed] [Google Scholar]
- 45.Torres VE, Bengal RJ, Litwiller RD, et al. Aggravation of polycystic kidney disease in Han:SPRD rats by buthionine sulfoximine. J Am Soc Nephrol. 1997;8:1283–1291. doi: 10.1681/ASN.V881283. [DOI] [PubMed] [Google Scholar]
- 46.Chu TM, Weir B, Wolfinger R. A systematic statistical linear modeling approach to oligonucleotide array experiments. Math Biosci. 2002;176:35–51. doi: 10.1016/s0025-5564(01)00107-9. doi:10.1016/S0025-5564(01)00107-9. [DOI] [PubMed] [Google Scholar]
- 47.Dai M, Wang P, Boyd AD, et al. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005;33:e175. doi: 10.1093/nar/gni179. doi:10.1093/nar/gni179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maric C, Sandberg K, Hinojosa-Laborde C. Glomerulosclerosis and tubulointerstitial fibrosis are attenuated with 17beta-estradiol in the aging Dahl salt sensitive rat. J Am Soc Nephrol. 2004;15:1546–1556. doi: 10.1097/01.asn.0000128219.65330.ea. doi:10.1097/01.ASN.0000128219.65330.EA. [DOI] [PubMed] [Google Scholar]
- 49.Mankhey RW, et al. 17beta-estradiol supplementation reduces tubulointerstitial fibrosis by increasing MMP activity in the diabetic kidney. Am J Physiol Regul Integr Comp Physiol. 2007;292:R769–R777. doi: 10.1152/ajpregu.00375.2006. doi:10.1152/ajpregu.00375.2006. [DOI] [PubMed] [Google Scholar]
- 50.Josefsson E, Tarkowski A, Carlsten H. Anti-inflammatory properties of estrogen. I. In vivo suppression of leukocyte production in bone marrow and redistribution of peripheral blood neutrophils. Cell Immunol. 1992;142:67–78. doi: 10.1016/0008-8749(92)90269-u. doi:10.1016/0008-8749(92)90269-U. [DOI] [PubMed] [Google Scholar]
- 51.Ogborn MR, Bankovic-Calic N, Shoesmith C, et al. Soy protein modification of rat polycystic kidney disease. Am J Physiol. 1998;274:F541–F549. doi: 10.1152/ajprenal.1998.274.3.F541. [DOI] [PubMed] [Google Scholar]
- 52.Thigpen JE, Setchell KD, Goelz MF, et al. The phytoestrogen content of rodent diets. Environ Health Perspect. 1999;107:A182–A183. doi: 10.1289/ehp.107-1566530. doi:10.2307/3434577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thigpen JE, Setchell KD, Ahlmark KB, et al. Phytoestrogen content of purified, open- and closed-formula laboratory animal diets. Lab Anim Sci. 1999;49:530–536. [PubMed] [Google Scholar]
- 54.van Duursen MB, Sanderson JT, de Jong PC, et al. Phytochemicals inhibit catechol-O-methyltransferase activity in cytosolic fractions from healthy human mammary tissues: implications for catechol estrogen-induced DNA damage. Toxicol Sci. 2004;81:316–324. doi: 10.1093/toxsci/kfh216. doi:10.1093/toxsci/kfh216. [DOI] [PubMed] [Google Scholar]
- 55.Calvet JP, Grantham JJ. The genetics and physiology of polycystic kidney disease. Semin Nephrol. 2001;21:107–123. doi: 10.1053/snep.2001.20929. doi:10.1053/snep.2001.20929. [DOI] [PubMed] [Google Scholar]
- 56.Moser M, Pscherer A, Roth C, et al. Enhanced apoptotic cell death of renal epithelial cells in mice lacking transcription factor AP-2beta. Genes Dev. 1997;11:1938–1948. doi: 10.1101/gad.11.15.1938. doi:10.1101/gad.11.15.1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fick-Brosnahan GM, Belz MM, McFann KK, et al. Relationship between renal volume growth and renal function in autosomal dominant polycystic kidney disease: a longitudinal study. Am J Kidney Dis. 2002;39:1127–1134. doi: 10.1053/ajkd.2002.33379. doi:10.1053/ajkd.2002.33379. [DOI] [PubMed] [Google Scholar]
- 58.Grantham JJ, Torres VE, Chapman AB, et al. Volume progression in polycystic kidney disease. N Engl J Med. 2006;354:2122–2130. doi: 10.1056/NEJMoa054341. doi:10.1056/NEJMoa054341. [DOI] [PubMed] [Google Scholar]
- 59.Gattone VH, II, Wang X, Harris PC, et al. Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med. 2003;9:1323–1326. doi: 10.1038/nm935. doi:10.1038/nm935. [DOI] [PubMed] [Google Scholar]
- 60.Torres VE. Cyclic AMP, at the hub of the cystic cycle. Kidney Int. 2004;66:1283–1285. doi: 10.1111/j.1523-1755.2004.00945.x. doi:10.1111/j.1523-1755.2004.00945.x. [DOI] [PubMed] [Google Scholar]
- 61.Hanaoka K, Guggino WB. cAMP regulates cell proliferation and cyst formation in autosomal polycystic kidney disease cells. J Am Soc Nephrol. 2000;11:1179–1187. doi: 10.1681/ASN.V1171179. [DOI] [PubMed] [Google Scholar]
- 62.Li H, Findlay IA, Sheppard DN. The relationship between cell proliferation, Cl-secretion, and renal cyst growth: a study using CFTR inhibitors. Kidney Int. 2004;66:1926–1938. doi: 10.1111/j.1523-1755.2004.00967.x. doi:10.1111/j.1523-1755.2004.00967.x. [DOI] [PubMed] [Google Scholar]
- 63.Nagao S, Yamaguchi T, Kusaka M, et al. Renal activation of extracellular signal-regulated kinase in rats with autosomal-dominant polycystic kidney disease. Kidney Int. 2003;63:427–437. doi: 10.1046/j.1523-1755.2003.00755.x. doi:10.1046/j.1523-1755.2003.00755.x. [DOI] [PubMed] [Google Scholar]
- 64.Wallace DP. Cyclic AMP-mediated cyst expansion. Biochim Biophys Acta. 2011;1812:1291–1300. doi: 10.1016/j.bbadis.2010.11.005. doi:10.1016/j.bbadis.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Marcocci L, Maguire JJ, Packer L. Nitecapone: a nitric oxide radical scavenger. Biochem Mol Biol Int. 1994;34:531–541. [PubMed] [Google Scholar]
- 66.Suzuki YJ, Packer L. Inhibition of NF-kappa B transcription factor by catechol derivatives. Biochem Mol Biol Int. 1994;32:299–305. [PubMed] [Google Scholar]
- 67.Suzuki YJ, et al. Antioxidant properties of nitecapone (OR-462) Free Radic Biol Med. 1992;13:517–525. doi: 10.1016/0891-5849(92)90146-8. doi:10.1016/0891-5849(92)90146-8. [DOI] [PubMed] [Google Scholar]
- 68.Mervaala E, Muller DN, Schmidt F, et al. Blood pressure-independent effects in rats with human renin and angiotensinogen genes. Hypertension. 2000;35:587–594. doi: 10.1161/01.hyp.35.2.587. doi:10.1161/01.HYP.35.2.587. [DOI] [PubMed] [Google Scholar]
- 69.Helkamaa T, Finckenberg P, Louhelainen M, et al. Entacapone protects from angiotensin II-induced inflammation and renal injury. J Hypertens. 2003;21:2353–2363. doi: 10.1097/00004872-200312000-00025. doi:10.1097/00004872-200312000-00025. [DOI] [PubMed] [Google Scholar]
- 70.Lal MA, Korner A, Matsuo Y, et al. Combined antioxidant and COMT inhibitor treatment reverses renal abnormalities in diabetic rats. Diabetes. 2000;49:1381–1389. doi: 10.2337/diabetes.49.8.1381. doi:10.2337/diabetes.49.8.1381. [DOI] [PubMed] [Google Scholar]
- 71.Tao Y, Kim J, Schrier RW, et al. Rapamycin markedly slows disease progression in a rat model of polycystic kidney disease. J Am Soc Nephrol. 2005;16:46–51. doi: 10.1681/ASN.2004080660. doi:10.1681/ASN.2004080660. [DOI] [PubMed] [Google Scholar]
- 72.Wang X, Gattone V, 2nd, Harris PC, et al. Effectiveness of vasopressin V2 receptor antagonists OPC-31260 and OPC-41061 on polycystic kidney disease development in the PCK rat. J Am Soc Nephrol. 2005;16:846–851. doi: 10.1681/ASN.2004121090. doi:10.1681/ASN.2004121090. [DOI] [PubMed] [Google Scholar]
- 73.Bukanov NO, Smith LA, Klinger KW, et al. Long-lasting arrest of murine polycystic kidney disease with CDK inhibitor roscovitine. Nature. 2006;444:949–952. doi: 10.1038/nature05348. doi:10.1038/nature05348. [DOI] [PubMed] [Google Scholar]
- 74.Sung B, Pandey MK, Aggarwal BB. Fisetin, an inhibitor of cyclin-dependent kinase 6, down-regulates nuclear factor-kappaB-regulated cell proliferation, antiapoptotic and metastatic gene products through the suppression of TAK-1 and receptor-interacting protein-regulated IkappaBalpha kinase activation. Mol Pharmacol. 2007;71:1703–1714. doi: 10.1124/mol.107.034512. doi:10.1124/mol.107.034512. [DOI] [PubMed] [Google Scholar]
- 75.Wahl PR, Serra AL, Le Hir M, et al. Inhibition of mTOR with sirolimus slows disease progression in Han:SPRD rats with autosomal dominant polycystic kidney disease (ADPKD) Nephrol Dial Transplant. 2006;21:598–604. doi: 10.1093/ndt/gfi181. doi:10.1093/ndt/gfi181. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.