

Summary
This study advances mechanistic understanding for the anticancer effects of a natural plant flavone apigenin that can inhibit prostate tumor growth and block metastasis by targeting PI3K/Akt/FoxO-mediated survival pathway.
Abstract
Forkhead box O (FoxO) transcription factors play an important role as tumor suppressor in several human malignancies. Disruption of FoxO activity due to loss of phosphatase and tensin homolog and activation of phosphatidylinositol-3 kinase (PI3K)/Akt are frequently observed in prostate cancer. Apigenin, a naturally occurring plant flavone, exhibits antiproliferative and anticarcinogenic activities through mechanisms, which are not fully defined. In the present study, we show that apigenin suppressed prostate tumorigenesis in transgenic adenocarcinoma of the mouse prostate (TRAMP) mice through the PI3K/Akt/FoxO-signaling pathway. Apigenin-treated TRAMP mice (20 and 50 μg/mouse/day, 6 days/week for 20 weeks) exhibited significant decrease in tumor volumes of the prostate as well as completely abolished distant organ metastasis. Apigenin treatment resulted in significant decrease in the weight of genitourinary apparatus (P < 0.0001), dorsolateral (P < 0.0001) and ventral prostate (P < 0.028), compared with the control group. Apigenin-treated mice showed reduced phosphorylation of Akt (Ser473) and FoxO3a (Ser253), which correlated with its increased nuclear retention and decreased binding of FoxO3a with 14-3-3. These events lead to reduced proliferation as assessed by Ki-67 and cyclin D1, along with upregulation of FoxO-responsive proteins BIM and p27/Kip1. Complementing in vivo results, similar observations were noted in human prostate cancer LNCaP and PC-3 cells after apigenin treatment. Furthermore, binding of FoxO3a with p27/Kip1 was markedly increased after 10 and 20 μM apigenin treatment resulting in G0/G1-phase cell cycle arrest, which was consistent with the effects elicited by PI3K/Akt inhibitor, LY294002. These results provide convincing evidence that apigenin effectively suppressed prostate cancer progression, at least in part, by targeting the PI3K/Akt/FoxO-signaling pathway.
Introduction
Prostate cancer remains the most common non-cutaneous malignancy and the second leading cause of cancer-related death in American males (1,2). According to an estimate by the American Cancer Society, ~238 590 new cases of prostate cancer will be diagnosed in the USA and 29 720 men will die of this fatal disease in 2013 (2,3). Although prostate cancer is curable at an early stage by surgery or radiation therapy, many patients present with locally advanced or metastatic disease for which there are currently no curative treatment options available. Therefore, effective targets and therapies, which could inhibit proliferation of localized tumors and prevent disease progression, are needed.
Loss of phosphatase and tensin homolog (PTEN) and activation of phosphatidylinositol-3 kinase (PI3K)/Akt cascades are common events in human prostate cancer (4,5). Defective PTEN activates Akt signaling by preventing conversion of phosphatidylinositol-3,4,5-triphosphate back to phosphatidylinositol 4,5-bisphosphate (5,6). PI3K/Akt is an oncogenic kinase which suppresses the apoptotic response, undermines cell cycle control and offers resistance to chemotherapeutic drugs and radiation therapy through downstream signaling and target molecules (7). The mammalian forkhead box subgroup ‘O’ (FoxO) of forkhead transcription factors consists of FoxO1, FoxO3a, FoxO4 and FoxO6, which function downstream of PI3K/Akt-signaling pathways and act as important regulators of cell death, promoting cell survival (8,9). Specifically, FoxO3a activity is negatively regulated by Akt, which phosphorylates FoxO3a at multiple sites, facilitating its association with 14-3-3 protein, thereby leading to its transport out of the nucleus and retention in the cytoplasm (10,11). In fact, anticancer drugs such as docetaxel and paclitaxel induce apoptosis in tumor cells by enhancing FoxO3a activity and stimulating its nuclear translocation (12). These events lead to overexpression of FoxO-responsive genes such as BIM, p27/Kip1 and p21/waf1.
FoxO family proteins, including FoxO1a and FoxO3a, are often deregulated in human prostate cancer (13,14). Most studies on FoxO3a in prostate cancer have been conducted on prostate cancer cell lines and clinical specimens. Loss of FoxO3a and reduced p27/Kip1 expression promote prostate cancer progression from androgen dependence to androgen independence (15). Our studies on FoxO3a in prostate cancer patient samples have demonstrated increased cytoplasmic expression of phosphorylated FoxO3a (Ser253), which correlates with disease progression (14). Overexpression of FoxO3a in prostate cancer cells causes apoptosis and induction of genes that affect cell proliferation (16,17). In recent years, regulation of FoxO protein by natural dietary agents has been the focus of much cancer research (18–20).
Apigenin (4′,5,7-trihydroxyflavone) is a biflavonoid present in common fruits, vegetables, herbs and spices. It is abundantly present in plants such as celery, parsley and dried chamomile flowers (ref. 21, and references therein). Apigenin is a potent inhibitor of PI3K-Akt, mitogen-activated protein kinases (ERK1/2, c-jun-N-terminal kinase and p38), casein kinase-2, focal adhesion kinase and other upstream kinases involved in the development and progression of cancer (22–24). Our previous studies have shown that apigenin inhibits the growth of human prostate cancer cells by causing cell cycle arrest and induction of apoptosis (25). In this study, our results show that oral intake of apigenin at doses equivalent to human consumption of a healthy diet of flavonoids (6–64mg/day of flavones and flavonols) significantly suppresses prostate cancer progression in transgenic adenocarcinoma of the mouse prostate (TRAMP) mice. The tumor growth suppression by apigenin was associated with increased nuclear retention and transcriptional activation of FoxO3a in the prostate of TRAMP mice. Further, apigenin-mediated inhibition of Akt/PKB phosphorylation activates FoxO3a binding to p27/Kip1, thereby enhancing its antiproliferative and proapoptotic effects in human prostate cancer LNCaP and PC-3 cells.
Materials and methods
Cell lines and treatments
Androgen-responsive human prostate cancer LNCaP cells and androgen-refractory PC-3 cells obtained from American Type Culture Collection (Manassas, VA) were used in the study. These cell lines possess high constitutive levels of Akt/PKB activity due to defect in PTEN gene (7,11). The cells were maintained in RPMI 1640 containing 2.05mM l-glutamine (Lonza Walkersville, Walkersville, MD) with 10% fetal bovine serum, respectively, supplemented with 1% penicillin and streptomycin in a humidified incubator at 37°C with an atmosphere of 5% CO2. For experimental studies, these cells were grown to 70% confluence in monolayer and treated with 10 or 20 μM apigenin (cat. no. A3145; >97% purity) and 10 μM LY294002 (both from Sigma, St Louis, MO) for 16h in dimethyl sulfoxide as vehicle. The final concentration of the vehicle dimethyl sulfoxide did not exceed 0.1% in all the treatments.
Animals
Male and female heterozygous C57BL/TGN TRAMP mice, Line PB Tag 8247NG, were purchased as breeding pairs from The Jackson Laboratory (Ann Arbor, MI). The animals were bred and maintained at the Association for Assessment and Accreditation of Laboratory Animals-accredited Animal Resource Facility of Case Western Reserve University. Housing and care of the animals were in accordance with the guidelines established by the University’s Animal Research Committee and with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. Transgenic males for the studies were routinely obtained as (TRAMP × C57BL/6) F1 or as (TRAMP × C57BL/6) F2 offspring. Identity of transgenic mice was established by PCR-based DNA screening as described previously (26).
Animal experimentation
Approximately 8-week-old male TRAMP mice were used in the studies. The animals received autoclaved Teklad 8760 high-protein diet and tap water ad libitum throughout the study. Apigenin (10mg) was suspended in 1 ml vehicle material (0.5% methyl cellulose and 0.025% Tween 20) by sonication for 30 s at 4°C and further diluted for appropriate concentration. Apigenin at 20 and 50 μg/mouse/day (wt/vol) was administered by gavage in 0.2 ml of a vehicle consisting of 0.5% methyl cellulose and 0.025% Tween 20 to TRAMP mice beginning at 8 weeks of age and was continued until the animals were 28 weeks old, at which time the experiment was terminated. These doses are comparable with the daily consumption of flavonoid in humans as reported in previously published studies (27).
Tissue preparation and histology
The genitourinary (GU) apparatus was isolated followed by excision of the dorsolateral and ventral prostate and weighed separately. A small portion of the dorsolateral and ventral prostates was fixed overnight in 10% zinc-buffered formalin and then transferred to 70% ethanol. Sections (4 μm) were cut from paraffin-embedded tissue and mounted on slides. The sections were stained with hematoxylin and eosin as described previously (26) and were evaluated for the presence or absence of the following lesions: well-differentiated adenocarcinoma, moderately differentiated adenocarcinoma and poorly differentiated adenocarcinoma. The histological characteristics of these lesions have been well established and described in a previous publication (28).
Metastasis examination
Microscopic examinations of lymph nodes, liver and lungs were performed to evaluate for the presence of metastases. The India ink method was used to examine the lungs for metastasis as described previously (29).
Immunohistochemistry
Immunohistochemistry (IHC) for Ki-67 and FoxO3a was performed on formalin-fixed, paraffin-embedded prostate tissue sections using a standard protocol as described previously using 3,3′-diaminobenzidine and counterstaining with Mayer’s hematoxylin (14). Sections were examined with an inverted Olympus BX51 microscope and images were acquired with Olympus MicroSuite™ Five Software (Soft Imaging System, Lakewood, CO).
Western blot analysis
Dorsolateral prostate tissue and cells from treated and control groups were subjected to preparation of total lysate and isolation of cytosolic and nuclear fractions as described previously (26,28). For western blotting, 25 μg of protein was resolved over 4–20% Tris-glycine polyacrylamide gel and then transferred onto the nitrocellulose membrane. The blots were blocked using 5% non-fat dry milk and probed using appropriate primary antibodies overnight at 4°C. The membrane was then incubated with appropriate secondary antibody horseradish peroxidase conjugate (Santa Cruz Biotechnology, Santa Cruz, CA) followed by detection using chemiluminescence ECL kit (GE Healthcare Biosciences). For equal loading of proteins, the membrane was probed with appropriate loading controls. The antibodies used were anti-p-Akt-Ser473 (SC-7985), anti-Akt (SC8312), anti-BIM (SC-11425) and anti-14-3-3 (SC-629) from Santa Cruz Biotechnology. Anti-p-FoxO3a-Ser253 was purchased from Upstate Biotechnology, Danvers, MA. Anti-FoxO3a (9467) was procured from Cell Signaling Technology, Danvers, MA. Anti-p27/Kip1 (clone DCS-72.F6) and anti-cyclin D1 (MS-210.PL) were obtained from Neomarkers, Fremont, CA and anti-Ki-67 was procured from Bethyl Laboratories, Montgomery, TX. Densitometric measurement of the bands in western blot analysis was performed using digitalized scientific software program using Kodak 2000R imaging system.
Immunoprecipitation
Total tissue lysate from the prostates of treated and untreated mice and nuclear lysate from human prostate cancer cells (200 μg) were immunoprecipitated with 2 μg appropriate primary antibody and were incubated at 4°C for 3h. Protein A/G beads (20 μl) were added and incubated overnight at 4°C. Immunoprecipitated proteins were washed four times with lysis buffer, electrophoresed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and analyzed by western blotting.
FoxO3A DNA-binding activity
Prostate tissue and human prostate cancer cells were processed for isolation of nuclear fraction and evaluation for FoxO3A DNA binding using FoxO3a EZ-TFA Transcription Factor Assay Chemiluminescent Kit (70–653; Upstate Biotechnology) according to the manufacturer’s protocol.
Cell cycle analysis
The cells (70% confluent) were starved for 36h to arrest them in G1 phase of the cell cycle, after which they were treated with 10 and 20 μM apigenin and 10 μM LY294002 in RPMI-1640 complete media for 16h. After treatment cells were collected, washed twice with chilled phosphate-buffered saline (PBS) and spun in a cold centrifuge at 600g for 10min. The pellet was fixed and resuspended in 50 μl PBS and 450 μl chilled methanol for 1h at 4°C. The cells were washed twice with PBS at 600g for 5min and again suspended in 500 μl PBS and incubated with 5 ml RNase (20 μg/ml final concentration) for 30min at 37°C. The cells were chilled over ice for 10min and stained with propidium iodide (50 μg/ml final concentration) for 1h and analyzed by flow cytometry and evaluated using Cell Quest & ModFit cell cycle analysis software.
Statistical analysis
Data were summarized as mean ± SD and visualized using box and bar plots. The difference of the data including GU weight, ventral lobe prostate, dorsolateral lobe prostate among three treatment groups (control, 20 and 50 μg apigenin) was examined using analysis of variance followed by Turkey multiple comparison procedure. All tests are two tailed and P value <0.05 were considered to be statistically significant.
Results
Apigenin treatment inhibits development of prostate carcinogenesis in TRAMP mice
The autochthonous TRAMP mice exhibit both histological and morphological features that mimic human prostate carcinogenesis (26,28). TRAMP mice exhibit low-grade prostatic intraepithelial neoplasia at 6 weeks that progresses to high-grade PIN by 12 weeks. Focal adenocarcinoma develops between 12 and 18 weeks and progresses to poorly differentiated carcinoma by 28 weeks. After 28 weeks, these mice develop occasional metastasis to lungs, lymph nodes, liver and bone. TRAMP mice have been widely used for the investigation of molecular targets and therapeutic agents (26,29). In our experiments, we used TRAMP/C57BL6 mice. Apigenin treatment at the doses of 20 and 50 μg/day for 6 days a week was initiated when the mice were of 8 weeks of age. Control group of mice received vehicle only. No apparent toxicity or loss of weight was observed with apigenin administration during the entire period of the experiment. All the mice were examined at 28 weeks of age for the growth of prostate cancer (Figure 1A and B). As shown in Figure 1C, apigenin intake for 20 weeks elicited a significant decrease in GU weight (P < 0.0001) and in the dorsolateral (P < 0.0001) and ventral prostate (P < 0.028), compared with the control group.
Fig. 1.
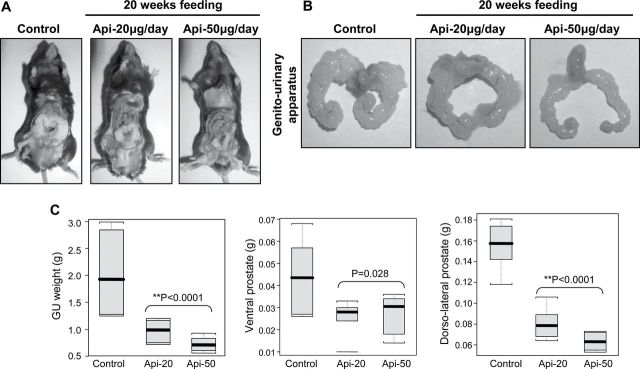
Effect of apigenin administration on prostate cancer progression in TRAMP mice. Apigenin was administered at 20 and 50 μg/mouse/day (wt/vol) by gavage in 0.2 ml of a vehicle consisting of 0.5% methyl cellulose and 0.025% Tween 20 to TRAMP mice beginning at 8 weeks of age for 20 weeks till experiment was terminated. (A) Representative pictures of dissected control and apigenin-treated TRAMP mice at 28 weeks of age. (B) Representative pictures of excised GU apparatus from control and apigenin-treated TRAMP mice at 28 weeks of age. (C) Boxplot of GU apparatus, ventral and dorsolateral lobes weight of control and apigenin-treated TRAMP mice at 28 weeks of age. Black bar = median, gray box = 25th–75th percentiles, bars = entire range of six mice. P < 0.0001, P = 0.028 and P < 0.0001, respectively, TRAMP control versus TRAMP apigenin (analysis of variance). Details are described in Materials and methods.
Apigenin intake inhibits the progression and invasiveness of adenocarcinoma in TRAMP mice
Histopathological analysis of the excised prostate tumor tissues of both control and apigenin treatment at 20 and 50 μg doses for 20 weeks is shown in Figure 2A. Composite analysis of the prostates of control group mice at 28 weeks of age exhibited predominantly well-differentiated adenocarcinoma (63.9%), along with moderately differentiated cancer (17.95%) and, infrequently, poorly differentiated adenocarcinoma (4.1%) in the prostate. Approximately 15.18% of the prostate tissue did not exhibit detectable cancer in control mice. Four of six mice (66.6%) in the control group exhibited metastasis to lymph nodes, whereas one mouse showed liver (16.6%) and lung metastases (16.6%), respectively. At 28 weeks of age, the histological findings in the prostates of TRAMP mice given 20 μg of apigenin per day were notably different from findings in the control group, showing a decrease in the percentage of well-differentiated carcinoma (40.8%), moderately differentiated cancer (10.04%) and poorly differentiated cancer (1.52%). Approximately 48.5% of the prostate tissue of TRAMP mice given 20 μg of apigenin per day appeared to be non-cancerous. The prostates of TRAMP mice receiving a higher dose of 50 μg apigenin exhibited 25.1% of well-differentiated cancer, 4.52% of moderately differentiated cancer and 0.478% of poorly differentiated cancer, and ~70.2% of the prostate tissue appeared to be non-cancerous (Figure 2B). No metastases were recorded in any of the mice treated with apigenin. These data suggest the inhibitory potential of apigenin against prostate cancer progression.
Fig. 2.

Effect of apigenin administration on the progression of prostate cancer in TRAMP mice. Apigenin was administered at 20 and 50 μg/mouse/day (wt/vol) by gavage in 0.2 ml of a vehicle consisting of 0.5% methyl cellulose and 0.025% Tween 20 to TRAMP mice beginning at 8 weeks of age for 20 weeks till experiment was terminated. (A) TRAMP mice (control) exhibited adenocarcinoma with extensive epithelial stratification, crowded cribriform structures accompanied with marked thickening, remodeling and hypercellularity of the fibromuscular stroma. Apigenin administration to TRAMP mice resulted in a marked reduction in epithelial stratification and cribriform structures. Magnification, ×20 and ×40. (B) Distribution of pathologic findings after apigenin intake in the dorsolateral lobes of TRAMP mice. Hematoxylin and eosin-stained slides were evaluated by three independent scientists. Prostatic lobe was scored for percentage of each pathologic finding present in the dorsolateral lobe in TRAMP mice at 28 weeks of age. The scores of the evaluators were averaged as shown in the table. Pathologic findings: WD, well-differentiated cancer; MD, moderately differentiated cancer; PD, poorly differentiated cancer. Data in the table represent mean ± SD. Details are described in Materials and methods.
Apigenin intake inhibits phosphorylated Akt and FoxO3a expression in TRAMP mice
Expression levels of p-Akt (Ser473) and p-FoxO3a (Ser253) correlate with prostate cancer progression (13,14). We have previously reported that loss of PTEN caused increased Akt phosphorylation, which correlates with disease progression in clinical tumor specimens and in vivo prostate cancer models (26,30). A more recent study from our group demonstrates that an increase in p-Akt (Ser473) caused increased phosphorylation of FoxO3a at Ser253 and its deregulation (31). Results from previous studies involving subcellular distribution, immunoprecipitation, pharmacological and genetic knockdown of Akt using human prostate cancer cells indicate that FoxO3a phosphorylation is critical for its DNA-binding and transcriptional activity (14). To determine whether apigenin targets the expression of FoxO3a and its phosphorylation during prostate cancer progression in TRAMP mice, we determined the expression of FoxO3a and its phosphorylation by IHC and western blot analysis in excised prostate tissue from control and apigenin-treated TRAMP mice. Apigenin intake of TRAMP mice resulted in an increase in FoxO3a expression, especially in the nucleus in excised prostate tissue, compared with the control group (Figure 3A). These results were further confirmed by western blot analysis. As shown in Figure 3B, apigenin treatment resulted in increase in FoxO3a expression along with concomitant decrease in p-FoxO3a (Ser253). In the same experiment, apigenin intake inhibited phosphorylation of Akt at Ser473 at 20 and 50 μg apigenin intake levels in prostate tumor tissue compared with control group. No significant effect was observed on the protein levels of total Akt and 14-3-3.
Fig. 3.

Effect of apigenin administration on the expression of FoxO3a, p-FoxO3a (Ser253), Akt, p-Akt (Ser473) and 14-3-3 in the dorsolateral prostate of TRAMP mice. Apigenin was administered at 20 and 50 μg/mouse/day (wt/vol) by gavage in 0.2 ml of a vehicle consisting of 0.5% methyl cellulose and 0.025% Tween 20 to TRAMP mice beginning at 8 weeks of age for 20 weeks till experiment was terminated. (A) IHC of FoxO3a in the dorsolateral prostate from control and apigenin-treated TRAMP mice. An increase in the nuclear presence of FoxO3a is observed after apigenin treatment. (B) Protein expression of FoxO3a, Akt and their phosphorylated forms, 14-3-3 as determined by western blot analysis. A significant decrease in Akt and FoxO3a phosphorylation is observed after apigenin treatment. Bands were quantitated by densitometric analysis. Numeric values represent the protein level normalized to the loading control (actin). (C) Coimmunoprecipitation analysis of FoxO3a and p-FoxO3a (Ser253) with Akt and 14-3-3 chaperone. A significant decrease in FoxO3a binding with Akt and 14-3-3 is observed after apigenin treatment. (D) Nuclear FoxO3A DNA binding in the dorsolateral prostate of TRAMP mice after apigenin feeding. A significant increase in the nuclear FoxO3a binding was observed after apigenin treatment. **P < 0.001 TRAMP control versus TRAMP apigenin. Mean ± SD of four mice. Details are described in Materials and methods.
Apigenin regulates nuclear retention of FoxO3a protein in TRAMP mice
Once phosphorylated by Akt, FoxO3a binds to 14-3-3 chaperone protein, resulting in its nuclear exclusion and inability to bind DNA (10,11). Hence, we determined the interaction of FoxO3a with Akt and 14-3-3. Immunoprecipitation studies using tissue lysate was performed with Akt and 14-3-3 and the levels of FoxO3a and its phosphorylation were analyzed. As shown in Figure 3C, apigenin treatment at 20 and 50 μg doses resulted in significant decrease in FoxO3a binding and its phosphorylation at Ser253 with Akt and 14-3-3, compared with the control group. To confirm the nuclear localization of FoxO3a, next DNA-binding assay was performed on the nuclear protein obtained from the prostate of control and apigenin-treated mice. An increase in DNA-binding activity was noted in the prostate of apigenin-treated mice, compared with control group (Figure 3D).
Apigenin intake causes alteration in FoxO3a downstream target proteins in TRAMP mice
Next, we determined the expression of Ki-67 protein, which is strictly associated with cell proliferation. Ki-67 protein is present during all active phases of the cell cycle including G1, S, G2/M and mitosis, but is absent in the resting cells at G0 phase, designating it an excellent proliferation marker. Here, we analyzed the effect of apigenin on the expression of Ki-67. Immunohistochemical analysis indicated that apigenin treatment inhibited Ki-67 expression in the prostates of TRAMP mice, compared with the control group (Figure 4A). These results were further confirmed with western blotting. Since nuclear translocation and increased FoxO3a DNA binding are expected to upregulate the transcription of FoxO-responsive genes such as Bim and p27/Kip1, we determined the expression of these proteins after apigenin intake in TRAMP mice. As expected, our results clearly demonstrate that apigenin intake substantially enhanced the expression of Bim and p27/Kip1 in the prostate, and also decreased the levels of cyclin D1, which is overexpressed during prostate cancer progression (Figure 4B).
Fig. 4.

Effect of apigenin administration on the expression of Ki-67, cyclin D1, p27/Kip1 and BIM in the dorsolateral prostate of TRAMP mice. Apigenin was administered at 20 and 50 μg/mouse/day (wt/vol) by gavage in 0.2 ml of a vehicle consisting of 0.5% methyl cellulose and 0.025% Tween 20 to TRAMP mice beginning at 8 weeks of age for 20 weeks till experiment was terminated. (A) IHC of Ki-67 in the dorsolateral prostate from control and apigenin-treated TRAMP mice. A marked decrease in the expression of Ki-67 is observed after apigenin treatment. (B) Protein expression of Ki-67, cyclin D1, p27/Kip1 and BIM as determined by western blot analysis. A marked decrease in Ki-67 and cyclin D1 was noted after apigenin treatment along with increase in the protein expression of BIM and p27/Kip1. Numeric values represent the protein level normalized to the loading control (actin). Details are described in Materials and methods.
Apigenin downregulates Akt-FoxO3a-signaling pathway in human prostate cancer LNCaP and PC-3 cells
To model and elucidate the molecular observations made in vivo, we treated LNCaP and PC-3 cells with varying concentrations of apigenin or LY294002, a pharmacological inhibitor of PI3K/Akt, for 16h. As shown in Figure 5A, apigenin treatment significantly suppressed phosphorylation of FoxO3a at Ser253, Akt at Ser473 and chaperone protein 14-3-3 in the nuclear fraction, compared with the control group in both cell lines. These events resulted in marked increase in FoxO3a expression in the nucleus. Treatment of LNCaP and PC-3 cells with LY294002 resulted in decreased p-Akt expression, as expected, and increased the levels of FoxO3a in the nucleus. No significant effect was observed on the protein levels of total Akt and 14-3-3. Next, we determined whether the increased FoxO3a in the nucleus is transcriptionally active. For this, DNA-binding assay was performed. As shown in Figure 5B, exposure of LNCaP and PC-3 cells to apigenin caused an increase in FoxO3a DNA-binding activity, compared with untreated cells.
Fig. 5.

Effect of apigenin treatment on the protein expression of FoxO3a, p-FoxO3a (Ser253), Akt, p-Akt (Ser473) and 14-3-3 in human prostate cancer LNCaP and PC-3 cells. The cells were treated with 10 or 20 μM apigenin and 10 μM LY294002 for 16h and subjected to preparation of cytosolic and nuclear lysates. (A) A significant decrease in Akt and FoxO3a phosphorylation is observed along with increase in nuclear retention of FoxO3a after apigenin and LY294002 treatment. (B) FoxO3A DNA binding in the nuclear fractions. A significant increase in the nuclear FoxO3a binding was observed after apigenin treatment. **P < 0.001 control versus apigenin treatment. Mean ± SD of four samples. Details are described in Materials and methods.
Apigenin caused increased binding of FoxO3a to p27/Kip in human prostate cancer LNCaP and PC-3 cells
Next, we sought to determine whether retention of FoxO3a in the nucleus causes increased DNA binding resulting in the transcription of responsive gene, p27/Kip1. Indeed, immunoprecipitation results exhibit that apigenin treatment of both LNCaP and PC-3 cells markedly increased binding of FoxO3a to p27/Kip1. Similar results where increased association of FoxO3a with p27/Kip1 was noted with LY294002 exposure to prostate cancer cell lines (Figure 6A).
Fig. 6.

Effect of apigenin on the binding of FoxO3a to p27/Kip1 and reduced proliferation of human prostate cancer LNCaP and PC-3 cells. The cells were treated with 20 μM apigenin and 10 μM LY294002 for 16h and subjected to preparation of nuclear lysate. (A) A marked increase in the binding of FoxO3a with p27/Kip1 was observed after apigenin treatment. Similar results were observed with LY294002. (B) Cell cycle analysis. The cells were serum starved for 36h followed by treatment with 10 and 20 μM apigenin and 10 μM LY294002 in RPMI-1640 complete media for 16h and subjected for cell cycle analysis. A significant increase in G0/G1 phase of the cell cycle was observed after apigenin and LY294002 treatment. Details are described in Materials and methods.
Apigenin causes cell cycle arrest in human prostate cancer LNCaP and PC-3 cells
Lastly, we determined whether increased FoxO3a binding to p27/Kip1 caused decrease in proliferation of human prostate cancer cells. We ascertained the effect of apigenin on the cell cycle. The cells were synchronized by serum deprivation for 36h and later incubated with 10% fetal bovine serum with varying concentrations of apigenin for 16h. Compared with the vehicle-treated controls, apigenin treatment resulted in an appreciable arrest of LNCaP cells in G0-G1 phase of cell cycle after 24h of the treatment. The treatment caused an arrest of 84.6% cells in G0-G1 phase of the cell cycle at 10 μM concentration that further increased to 89.1% at 20 μM in these cells, compared with vehicle-treated control (77.8%). This increase in G0-G1 cell population was accompanied with a concomitant decrease of cell number in S phase and G2-M phase of the cell cycle (Figure 6B, left panel). Similarly, apigenin treatment to PC-3 cells caused an arrest of 72.4% cells in G0-G1 phase of the cell cycle at 10 μM concentration that further increased to 80.6% at 20 μM in these cells, compared with vehicle-treated control (68.2%). LY294002 treatment at 10 μM concentration to LNCaP and PC-3 cells caused 86.3 and 77.7% arrest in G0-G1 phase of the cell cycle (Figure 6B, right panel).
Discussion
Apigenin has been shown to exert anticancer and antiproliferative activities in cell culture and in various animal models of cancer (ref. 21 and references therein). We have shown previously that apigenin suppresses the in vivo growth of prostate cancer by targeting β-catenin and insulin-like growth factor-I-signaling pathways (29,32). In the present study, we demonstrate tumor growth suppression by apigenin intake was associated with reduced proliferation of tumor cells, which in turn was linked with the inhibition of Akt/PKB and FoxO3a activation. Proapoptotic protein, BIM, which was negatively regulated by Akt through FoxO3a, was upregulated in the prostates of apigenin-supplemented mice. Inhibition of activated levels of Akt and FoxO3a were confirmed in LNCaP and PC-3 prostate cancer cells by western blotting, immunoprecipitation, IHC and DNA-binding assay. To the best of our knowledge, our results for the first time demonstrate the involvement of the Akt/FoxO3a-signaling axis in apigenin-mediated prostate cancer suppression.
The PI3K/Akt-signaling pathway is constitutively activated in various human cancers, including prostate cancer (33,34). Hyperactive Akt as a result of reduced PTEN expression or loss of heterozygosity is commonly observed in human prostate cancer (35). Targeted deletion of PTEN in prostate cancer increases the oncogenic activity of PI3K/Akt, leading to the development of PIN and rapidly progressing to invasive carcinoma (4,5,36). We have previously demonstrated constitutive activation of PI3K/Akt in human prostate cancer and in TRAMP mice, suggesting it as an attractive molecular target (26). Furthermore, many cancers acquire drug resistance due to activation of the PI3K/Akt pathway. Acquired drug resistance due to hyperactivation of PI3K/Akt survival pathway has been observed during administration of paclitaxel and doxorubicin in breast cancer (12), docetaxel in prostate cancer (37), cisplatin in ovarian cancer (38) and gemcitabine in pancreatic cancer (39). Our results showed that apigenin treatment substantially suppressed Akt phosphorylation at Ser473 in LNCaP and PC-3 cells. In agreement with these results, prostates from apigenin-supplemented TRAMP mice showed marked decrease in Akt phosphorylation at Ser473. Our results further indicate that apigenin suppresses prostate cancer by targeting constitutively activated Akt without affecting the protein levels.
FoxO proteins are important targets of PI3K/Akt-signaling pathway (10,11). The Akt-mediated phosphorylation of FoxO3a on different residues is known to facilitate its association with 14-3-3 protein, thereby leading to the transport of FoxO3a out of the nucleus and its retention in the cytoplasm (11). The cytosolic retention of FoxO3a prevents the transactivation of downstream target genes such as BIM and p27/Kip1. We hypothesized that inhibition of Akt phosphorylation by apigenin would lead to nuclear sequestration of FoxO3a and increased transcription of responsive genes. Studies have shown that FoxO3a is dephosphorylated and activated by LY294002, which correlates with upregulation of p27/Kip1 (40). In agreement with the hypothesis, our results demonstrate that phosphorylated levels of FoxO3a were decreased after apigenin intake resulting in the nuclear retention of these proteins. These observations were supported by the results exhibiting reduced binding of FoxO3a with 14-3-3 in the prostate of apigenin-supplemented group. Furthermore, increase in the levels of BIM and p27/Kip1 and increased binding of FoxO3a to p27/Kip1 provide evidence that apigenin intake causes localization of FoxO3a in the nucleus, reducing proliferation of tumor cells.
Posttranslational modification including phosphorylation of FoxO proteins by various upstream kinases are important mechanisms regulating cellular processes through a wide range of target genes implicated in survival, apoptosis, cell cycle inhibition and resistance to oxidative stress (8–11). Increased level of p-FoxO3a (Ser253) and its association with 14-3-3 looses the ability of FoxO3a to bind to the DNA, a phenomenon frequently observed in human prostate tumors and cancer cells (11). The enhanced DNA-binding activity may also serve to limit the availability of FoxO3a for its phosphorylation by Akt. It is possible, that rather than enhancing Akt-mediated dephosphorylation of FoxO3a, apigenin might inhibit the rephosphorylation of FoxO3a. The results of the present study demonstrate that apigenin treatment inhibits accumulation of p-FoxO3a, either by inhibiting the phosphorylation of FoxO3a in the nucleus or by promoting the dephosphorylation of phosphorylated FoxO3a in the cytosol. Further detailed studies are warranted in determining the effect of apigenin on various kinases such as IκB kinase and mitogen-activated protein kinase, which could phosphorylate FoxO3a at various residues and to elucidate the mechanism of action.
Akt promotes cell survival by indirectly regulating oxidative stress response through regulation of antioxidant enzymes such as manganese superoxide dismutase through phosphorylation of FoxO transcription factors (41). In fact, cysteine residues in FoxO can also act as sensors of the local redox state. The cysteine-dependent redox switch can regulate reactive oxygen species-signaling pathway upstream of FoxO (42). Our previous studies have shown that apigenin causes oxidative stress in prostate cancer cells by reactive oxygen species generation (43). In fact, it would be worthwhile to study the effect of apigenin in reactive oxygen species-mediated regulation of FoxO3a in prostate cancer cells. Furthermore, the role of FoxO3a in cellular stress response to apigenin leads to induction of BIM and p27/Kip1 (10). Activated FoxO3a has been shown to induce cell cycle arrest at G1 and G2/M phase in response to oxidative stress. Our studies demonstrate that apigenin treatment to LNCaP and PC-3 cells resulted in G1-phase arrest further suggests that FoxO3a may function as a modulator of stress response.
Epidemiologic and case–control studies have reported that higher intake of plant flavonoids reduces the risk of certain chronic diseases, including cancer (44). Reports have shown a strong inverse association between flavone intake and risk of breast, colorectal, prostate and epithelial ovarian cancer (45–48). There is still lack of pharmacokinetic study of apigenin in humans. Although apigenin in natural forms have been given to human subjects and its systemic distribution is quantified, however, at this point, no reported clinical trial on the possible beneficial effects of apigenin in human cancer patients has been conducted. A study on the bioavailability of apigenin from apiin-rich parsley in healthy human subjects has been demonstrated (49). Consumption of single oral bolus of 2g blanched parsley corresponding to 65.8±15.5 μM apigenin per kilogram body weight resulted in 127±81nM/l apigenin plasma concentration after 7.2 h. Our studies on TRAMP mice at 20 and 50 μg/day corresponds to ~50 and 120mg/day of flavone intake by an adult human with the plasma apigenin concentration ranging from 0.63 to 1.15 μM/l. These results indicate that the bioavailable concentration of apigenin has a therapeutic effect. Nonetheless, detailed pharmacokinetic studies on apigenin especially its accumulation in the prostate gland is required.
In summary, the data presented demonstrate that apigenin treatment in prostate cancer can activate FoxO3a and its DNA binding is able to induce the expression of downstream target proteins BIM and p27/Kip1 resulting in cell cycle arrest and reduced proliferation in prostate tumors. This finding may indicate a prognostic role of both p27/Kip1 and FoxO3a in predicting apigenin responsiveness in prostate cancer. Interestingly, the expression of two FoxO targets, namely p27/Kip1 and cyclin D1, are already regarded as prognostic indicators of prostate cancer (50). We conclude that apigenin could be used as a potential preventive/therapeutic agent in the management of prostate cancer in humans.
Funding
United States Public Health Services (RO1CA108512, RO1AT002709 to S.G., RO3CA1376676 to S.S.). Ruth L. Kirschstein Metabolism Training Program for Pre-Doctoral Fellowship (5T32DK007319 to M.A.B.).
Conflict of Interest Statement: The authors have no competing interest.
Glossary
Abbreviations:
- FoxO
forkhead box O
- GU
genitourinary
- IHC
immunohistochemistry
- PBS
phosphate-buffered saline
- PI3K
phosphatidylinositol-3 kinase
- PTEN
phosphatase and tensin homolog
- TRAMP
transgenic adenocarcinoma of the mouse prostate.
References
- 1. Siegel R, et al. (2013). Cancer statistics, 2013. Cancer J. Clin., 63, 11–30 [DOI] [PubMed] [Google Scholar]
- 2. Siegel R, et al. (2012). Cancer treatment and survivorship statistics, 2012. Cancer J. Clin., 62, 220–241 [DOI] [PubMed] [Google Scholar]
- 3.Prostate cancer facts and statistics on the American Cancer Society website. http://www.cancer.org/cancer/prostatecancer/index http://www.cancer.org/cancer/prostatecancer/index
- 4. Sircar K, et al. (2009). PTEN genomic deletion is associated with p-Akt and AR signalling in poorer outcome, hormone refractory prostate cancer. J. Pathol., 218, 505–513 [DOI] [PubMed] [Google Scholar]
- 5. Blando J, et al. (2009). PTEN deficiency is fully penetrant for prostate adenocarcinoma in C57BL/6 mice via mTOR-dependent growth. Am. J. Pathol., 174, 1869–1879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nardella C, et al. (2010). Faithfull modeling of PTEN loss driven diseases in the mouse. Curr. Top. Microbiol. Immunol., 347, 135–168 [DOI] [PubMed] [Google Scholar]
- 7. West K.A, et al. (2002). Activation of the PI3K/Akt pathway and chemotherapeutic resistance. Drug Resist. Updat., 5, 234–248 [DOI] [PubMed] [Google Scholar]
- 8. Katoh M, et al. (2013). Cancer genetics and genomics of human FOX family genes. Cancer Lett., 328, 198–206 [DOI] [PubMed] [Google Scholar]
- 9. Zhang Y, et al. (2011). FoxO family members in cancer. Cancer Biol. Ther., 12, 253–259 [DOI] [PubMed] [Google Scholar]
- 10. Zhao Y, et al. (2011). Applications of post-translational modifications of FoxO family proteins in biological functions. J. Mol. Cell Biol., 3, 276–282 [DOI] [PubMed] [Google Scholar]
- 11. Tzivion G, et al. (2011). FoxO transcription factors; Regulation by AKT and 14-3-3 proteins. Biochim. Biophys. Acta, 1813, 1938–1945 [DOI] [PubMed] [Google Scholar]
- 12. Sunters A, et al. (2003). FoxO3a transcriptional regulation of Bim controls apoptosis in paclitaxel-treated breast cancer cell lines. J. Biol. Chem., 278, 49795–49805 [DOI] [PubMed] [Google Scholar]
- 13. Li R, et al. (2007). Forkhead protein FKHR and its phosphorylated form p-FKHR in human prostate cancer. Hum. Pathol., 38, 1501–1507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shukla S, et al. (2009). Deregulation of FOXO3A during prostate cancer progression. Int. J. Oncol., 34, 1613–1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lynch R.L, et al. (2005). The progression of LNCaP human prostate cancer cells to androgen independence involves decreased FOXO3a expression and reduced p27KIP1 promoter transactivation. Mol. Cancer Res., 3, 163–169 [DOI] [PubMed] [Google Scholar]
- 16. Liu J.W, et al. (2005). Induction of prosurvival molecules by apoptotic stimuli: involvement of FOXO3a and ROS. Oncogene, 24, 2020–2031 [DOI] [PubMed] [Google Scholar]
- 17. Yang L, et al. (2005). Induction of androgen receptor expression by phosphatidylinositol 3-kinase/Akt downstream substrate, FOXO3a, and their roles in apoptosis of LNCaP prostate cancer cells. J. Biol. Chem., 280, 33558–33565 [DOI] [PubMed] [Google Scholar]
- 18. Chen Q, et al. (2010). Resveratrol induces growth arrest and apoptosis through activation of FOXO transcription factors in prostate cancer cells. PLoS One, 5, e15288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shankar S, et al. (2008). Sulforaphane enhances the therapeutic potential of TRAIL in prostate cancer orthotopic model through regulation of apoptosis, metastasis, and angiogenesis. Clin. Cancer Res., 14, 6855–6866 [DOI] [PubMed] [Google Scholar]
- 20. Li Y, et al. (2007). Regulation of FOXO3a/beta-catenin/GSK-3beta signaling by 3,3’-diindolylmethane contributes to inhibition of cell proliferation and induction of apoptosis in prostate cancer cells. J. Biol. Chem., 282, 21542–21550 [DOI] [PubMed] [Google Scholar]
- 21. Shukla S, et al. (2010). Apigenin: a promising molecule for cancer prevention. Pharm. Res., 27, 962–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shukla S, et al. (2007). Apigenin-induced cell cycle arrest is mediated by modulation of MAPK, PI3K-Akt, and loss of cyclin D1 associated retinoblastoma dephosphorylation in human prostate cancer cells. Cell Cycle, 6, 1102–1114 [DOI] [PubMed] [Google Scholar]
- 23. Franzen C.A, et al. (2009). The chemopreventive bioflavonoid apigenin inhibits prostate cancer cell motility through the focal adhesion kinase/Src signaling mechanism. Cancer Prev. Res. (Phila)., 2, 830–841 [DOI] [PubMed] [Google Scholar]
- 24. Wang G, et al. (2008). Impact of protein kinase CK2 on inhibitor of apoptosis proteins in prostate cancer cells. Mol. Cell. Biochem., 316, 91–97 [DOI] [PubMed] [Google Scholar]
- 25. Shukla S, et al. (2004). Molecular mechanisms for apigenin-induced cell-cycle arrest and apoptosis of hormone refractory human prostate carcinoma DU145 cells. Mol. Carcinog., 39, 114–126 [DOI] [PubMed] [Google Scholar]
- 26. Shukla S, et al. (2005). Constitutive activation of P I3 K-Akt and NF-kappaB during prostate cancer progression in autochthonous transgenic mouse model. Prostate, 64, 224–239 [DOI] [PubMed] [Google Scholar]
- 27. Hollman P.C, et al. (1999). Dietary flavonoids: intake, health effects and bioavailability. Food Chem. Toxicol., 37, 937–942 [DOI] [PubMed] [Google Scholar]
- 28. Kaplan-Lefko P.J, et al. (2003). Pathobiology of autochthonous prostate cancer in a pre-clinical transgenic mouse model. Prostate, 55, 219–237 [DOI] [PubMed] [Google Scholar]
- 29. Shukla S, et al. (2007). Blockade of beta-catenin signaling by plant flavonoid apigenin suppresses prostate carcinogenesis in TRAMP mice. Cancer Res., 67, 6925–6935 [DOI] [PubMed] [Google Scholar]
- 30. Shukla S, et al. (2007). Activation of PI3K-Akt signaling pathway promotes prostate cancer cell invasion. Int. J. Cancer, 121, 1424–1432 [DOI] [PubMed] [Google Scholar]
- 31. Shukla S, et al. (2013). Deregulation of FoxO3a accelerates prostate cancer progression in TRAMP mice. Prostate, 73, 1507–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shukla S, et al. (2012). Apigenin attenuates insulin-like growth factor-I signaling in an autochthonous mouse prostate cancer model. Pharm. Res., 29, 1506–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Carnero A. (2010). The PKB/AKT pathway in cancer. Curr. Pharm. Des., 16, 34–44 [DOI] [PubMed] [Google Scholar]
- 34. Majumder P.K, et al. (2005). Akt-regulated pathways in prostate cancer. Oncogene, 24, 7465–7474 [DOI] [PubMed] [Google Scholar]
- 35. Trotman L.C, et al. (2006). Identification of a tumour suppressor network opposing nuclear Akt function. Nature, 441, 523–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang S, et al. (2003). Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell, 4, 209–221 [DOI] [PubMed] [Google Scholar]
- 37. Zhong B, et al. (2010). Induction of clusterin by AKT–role in cytoprotection against docetaxel in prostate tumor cells. Mol. Cancer Ther., 9, 1831–1841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang H, et al. (2013). Molecular imaging reveals a role for AKT in resistance to cisplatin for ovarian endometrioid adenocarcinoma. Clin. Cancer Res., 19, 158–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kagawa S, et al. (2012). Akt/mTOR signaling pathway is crucial for gemcitabine resistance induced by Annexin II in pancreatic cancer cells. J. Surg. Res., 178, 758–767 [DOI] [PubMed] [Google Scholar]
- 40. Chandramohan V, et al. (2004). Reciprocal control of Forkhead box O 3a and c-Myc via the phosphatidylinositol 3-kinase pathway coordinately regulates p27Kip1 levels. J. Immunol., 172, 5522–5527 [DOI] [PubMed] [Google Scholar]
- 41. van Gorp A.G, et al. (2006). Chronic protein kinase B (PKB/c-akt) activation leads to apoptosis induced by oxidative stress-mediated Foxo3a transcriptional up-regulation. Cancer Res., 66, 10760–10769 [DOI] [PubMed] [Google Scholar]
- 42. Kops G.J, et al. (2002). Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature, 419, 316–321 [DOI] [PubMed] [Google Scholar]
- 43. Shukla S, et al. (2008). Apigenin-induced prostate cancer cell death is initiated by reactive oxygen species and p53 activation. Free Radic. Biol. Med., 44, 1833–1845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Romagnolo D.F, et al. (2012). Flavonoids and cancer prevention: a review of the evidence. J. Nutr. Gerontol. Geriatr., 31, 206–238 [DOI] [PubMed] [Google Scholar]
- 45. Hui C, et al. (2013). Flavonoids, flavonoid subclasses and breast cancer risk: a meta-analysis of epidemiologic studies. PLoS One, 8, e54318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kyle J.A, et al. (2010). Dietary flavonoid intake and colorectal cancer: a case-control study. Br. J. Nutr., 103, 429–436 [DOI] [PubMed] [Google Scholar]
- 47. Bosetti C, et al. (2006). Flavonoids and prostate cancer risk: a study in Italy. Nutr. Cancer, 56, 123–127 [DOI] [PubMed] [Google Scholar]
- 48. Gates M.A, et al. (2009). Flavonoid intake and ovarian cancer risk in a population-based case-control study. Int. J. Cancer, 124, 1918–1925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Meyer H, et al. (2006). Bioavailability of apigenin from apiin-rich parsley in humans. Ann. Nutr. Metab., 50, 167–172 [DOI] [PubMed] [Google Scholar]
- 50. Abdulkader I, et al. (2005). Cell-cycle-associated markers and clinical outcome in human epithelial cancers: a tissue microarray study. Oncol. Rep., 14, 1527–1531 [DOI] [PubMed] [Google Scholar]