

Abstract.
Hyperspectral imaging is a versatile tool that has recently been applied to a variety of biomedical applications, notably live-cell and whole-tissue signaling. Traditional hyperspectral imaging approaches filter the fluorescence emission over a broad wavelength range while exciting at a single band. However, these emission-scanning approaches have shown reduced sensitivity due to light attenuation from spectral filtering. Consequently, emission scanning has limited applicability for time-sensitive studies and photosensitive applications. In this work, we have developed an excitation-scanning hyperspectral imaging microscope that overcomes these limitations by providing high transmission with short acquisition times. This is achieved by filtering the fluorescence excitation rather than the emission. We tested the efficacy of the excitation-scanning microscope in a side-by-side comparison with emission scanning for detection of green fluorescent protein (GFP)-expressing endothelial cells in highly autofluorescent lung tissue. Excitation scanning provided higher signal-to-noise characteristics, as well as shorter acquisition times ( band with excitation scanning versus band with emission scanning). Excitation scanning also provided higher delineation of nuclear and cell borders, and increased identification of GFP regions in highly autofluorescent tissue. These results demonstrate excitation scanning has utility in a wide range of time-dependent and photosensitive applications.
Keywords: hyperspectral imaging, fluorescence spectroscopy, microscopy, fluorescence, thin films, tunable filters
1. Introduction
1.1. Emission-Scanning Hyperspectral Imaging
Traditional fluorescence microscopy approaches utilize several bandpass filters to isolate the excitation and emission wavelengths of fluorescent probes. If multiple probes are used, compensation approaches are often employed to remove cross-talk between the signal from undesired probes and the signal of desired probes. However, many fluorophores share similar peak emission wavelengths, resulting in limited utility of traditional fluorescence microscopy for multilabel studies. Additionally, separation of exogenous fluorescence from tissue autofluorescence is often prohibitively difficult with traditional fluorescence microscopy, as autofluorescence often displays a very broad emission spectrum overlapping that of many fluorescent probes.1,2
Hyperspectral imaging approaches have great promise for overcoming these limitations of traditional fluorescence microscopy. Hyperspectral imaging combines spectroscopy and imaging technologies by sampling image data at many wavelength bands, forming a contiguous spectrum.3–6 Hyperspectral imaging has recently been applied to biomedical applications, notably hyperspectral fluorescence microscopy.4,7–9 In contrast to traditional fluorescence microscopy, hyperspectral microscopy allows isolation of the fluorescence emission from multiple fluorescent probes by utilizing the unique spectral signatures of each probe. Hyperspectral imaging approaches have greatly facilitated multilabel7,8 and multiplexed10,11 studies, including separation of emission from fluorophores sharing similar peak emission wavelengths.9,12 Additionally, hyperspectral imaging approaches have allowed separation of fluorescent signals from autofluorescence, significantly enhancing the specificity of ex vivo13–15 and in vivo16–18 studies.
Hyperspectral imaging is traditionally performed by filtering the fluorescence emission at set increments across a broad wavelength range (emission scanning). Wavelength filtering can be performed using diffraction gratings,6,19 prisms,20,21 tunable filters,9,22–24 or interferometers.25,26 We have previously shown the utility of detecting green fluorescent protein (GFP) in highly autofluorescent lung tissue using both acousto-optic tunable filters (AOTFs)9,15 and thin-film tunable filters (TFTFs).15 Our previous work demonstrated improved specificity for discriminating GFP from tissue autofluorescence using emission-scanning hyperspectral approaches compared with standard epifluorescence approaches. However, because emission light was filtered using narrow-bandwidth tunable filters; the fluorescence signal detected at any wavelength band was low. In these previous studies, acquisition times were increased to accommodate the low signal level. This approach was highly successful for imaging fixed tissues, but has distinct limitations for performing high-speed hyperspectral imaging, needed for live-cell imaging or for minimizing photobleaching artifacts.
1.2. Excitation-Scanning Hyperspectral Imaging
To overcome the limitations of fluorescence emission-scanning hyperspectral imaging, we have developed a new approach to hyperspectral imaging that filters the fluorescence excitation, rather than fluorescence emission. Excitation scanning substantially increases the available signal detected and allows additional spectral information to be acquired. The increase in available signal is due to filtering the excitation light instead of the fluorescence emission, permitting all emitted light (at each excitation wavelength) to be detected. The higher detectable signal reduces the acquisition time required for a hyperspectral image stack and improves signal-to-noise ratio (SNR) characteristics of the acquired images. Reductions in acquisition time greatly facilitate time-dependent assays, and reduce photobleaching artifacts. In addition to increased signal strength, excitation scanning allows detection of excitation spectra that often differ from their corresponding emission spectra, providing additional information that can be used to discriminate amongst fluorescent signals.
To demonstrate the utility of excitation-scanning hyperspectral imaging, we have developed a prototype excitation-scanning hyperspectral imaging microscope system and have compared it with an emission-scanning system. An excitation-scanning spectral imaging system was previously designed by our group using an imaging spectrometer to detect the fluorescence excitation spectrum of fluorescent dyes in nude mice at a fixed emission wavelength.27 However, scanning the fluorescence excitation while detecting emission using a fixed band-pass emission filter did not provide a significant signal increase. Additionally, an AOTF was used to filter fluorescence excitation, which has since been shown to have reduced out-of-band blocking power and light transmission compared with the TFTF technology we use here.15 In this work, we present a procedure and results from implementing and calibrating the excitation-scanning hyperspectral imaging system. In addition, we present a side-by-side comparison with an emission-scanning system: imaging GFP-expressing microvascular endothelial cells in highly-autofluorescent lung tissue. Our results indicate excitation-scanning hyperspectral imaging provides more sensitive detection of GFP, increased delineation of nuclei, and higher structural content of autofluorescence, compared with emission scanning. Additionally, the excitation spectra of autofluorescence provided unique spectral information that differed from the emission spectra, and that were effective in accurately calculating autofluorescence contributions. Because of the significant increase in signal strength, excitation scanning may enable high-speed hyperspectral imaging for time-dependent measurements, such as live-cell Förster resonance energy transfer (FRET) and imaging. Additionally, spectral sampling throughout the fluorescence excitation-emission matrix may provide increase capabilities for fluorophore multiplexing.
2. Methods
2.1. Cell, Animal, and Sample Preparation
All animal work was approved by the University of South Alabama Institutional Animal Care and Use Committee (IACUC) and conforms to standard practices and established guidelines. Cell, animal, and sample preparation for the lung injury model has been described previously.9,15,28 Briefly, highly proliferative pulmonary microvascular endothelial cells (PMVECs) were transduced with a lentivirus encoding GFP. Adult male CD rats were injected intratracheally with Pseudomonas aeruginosa at the in 150-μL saline solution, then injected in the jugular vein with of body weight GFP-positive PMVECs. After 1 week, rats were euthanized and the lungs and heart were excised and perfused with a ventilator at a constant volume of . Samples from the most injured portions of the lung were removed and fixed in formalin for 24 h. Rats not injected with GFP-positive PMVECs were injected with saline and euthanized for use as autofluorescence controls. Lung tissue samples were paraffin-embedded, sectioned at 10 μm, and placed on microscope slides. Microscope slides were deparaffinized using xylene and 70% ethanol treatments. Tissue samples were then stained with 0.225-μM Hoechst-33342 (Life Technologies, Carlsbad, California). Autofluorescence control samples were not stained with Hoechst. Slides were prepared using fluorescence mounting medium (Dako, North America, Inc., Carpinteria, California).
Samples of GFP-positive PMVECs were prepared as confluent monolayers on 25 mm round coverslips and used as controls for the pure GFP spectrum. Confluent monolayers of wild-type PMVECs on 25 mm round coverslips were labeled with 2.4 nM Hoechst-33342 and used as controls for the pure Hoechst spectrum.
2.2. Excitation-Scanning Hyperspectral Microscope Set-up, Calibration, and Spectral Correction
Imaging was performed using an inverted fluorescence microscope (TE2000-U, Nikon Instruments, Melville, New York), equipped with a oil-immersion objective (S Fluor, 40X/1.30 Oil DIC H/N2, Nikon Instruments). A 300-W Xe arc lamp (Titan 300, Sunoptic Technologies, Jacksonville, Florida) provided excitation light. A filter cube comprised of a long-pass fluorescence emission filter (BLP01-495R, Semrock Inc., Rochester, New York) and a long-pass dichroic beamsplitter (FF-495-Di03, Semrock Inc.) was used to separate excitation from emission light. Two arrays of TFTFs (VersaChrome®, Semrock Inc.) were used for spectral filtering—one array for filtering the excitation light (excitation scanning) and another array for filtering the fluorescence emission (emission scanning). Each array consisted of a set of 6 (for excitation) or 5 (for emission) VersaChrome tunable filters, arranged side-by-side. A linear translator motor allowed switching between filters. In addition, the filter assembly could rotate with respect to the incident light, allowing the center wavelength of the pass-band to be tuned. Each filter in the array could be tuned over a given range of center wavelengths, and by switching between adjacent filters a very wide range of wavelengths can be sampled. Both excitation-scanning and emission-scanning light paths are shown in Fig. 1, while the individual filters used in each TFTF array are listed in Table 1.
Fig. 1.

Excitation- and emission-scanning light paths were implemented on an inverted fluorescence microscope. For each configuration, an array of thin-film tunable filters (TFTFs) was used for spectral filtering. Excitation scanning was accomplished by placing a TFTF array following the Xe arc lamp. The excitation center wavelength was scanned in 5-nm increments from 360 to 485 nm, and the fluorescence emission was collected above the cutoff wavelength of the long-pass emission filter. For emission scanning, a 420/17-nm bandpass filter was used to filter excitation light and the emission center wavelength was scanned in 5-nm increments from 475 to 700 nm.
Table 1.
Excitation and emission-scanning tunable filters used in this study (VersaChrome®, Semrock Inc., Rochester, New York). An array of filters was used for each tuning approach. The arrays were placed in custom housings, allowing translation between filters and rotation of each filter.15 Excitation-scanning filters were configured to allow a wavelength-tuning range from 357.9 to 707.6 nm, and emission-scanning filters were configured to allow a wavelength-tuning range from 398.4 to 710.0 nm.
| Part Number | Center Wavelength Range (nm) | Bandwidth (nm) | Refractive Index |
|---|---|---|---|
| Excitation-Scanning Tunable Filters | |||
| TBP01-402-16 | 357.9-403.4 | 21.2 | 1.88 |
| TBP01-451-15 | 397.1-451.5 | 17.4 | 1.82 |
| TBP01-503-15 | 446.3-503.1 | 23.4 | 1.88 |
| TBP01-564-14 | 498.5-564.7 | 19.2 | 1.84 |
| TBP01-632-14 | 556.9-631.7 | 21.5 | 1.83 |
| TBP01-708-13 | 621.7-707.6 | 21.5 | 1.81 |
| Emission-Scanning Tunable Filters | |||
| TBP01-451-15 | 398.4-451.6 | 16.6 | 1.84 |
| TBP01-503-15 | 447.5-504.2 | 16.6 | 1.88 |
| TBP01-564-14 | 497.6-563.0 | 21.2 | 1.85 |
| TBP01-632-14 | 557.5-630.8 | 17.9 | 1.85 |
| TBP01-708-13 | 624.2-710.0 | 21.5 | 1.82 |
| OPEN | — | — | — |
Spectral correction was used to compensate for wavelength-dependent attenuation present in both the excitation- and emission-scanning configurations. To correct the excitation-scanning configuration, a National Institute of Standards and Technology (NIST)-traceable light source (LS-1-CAL-INT, Ocean Optics, Inc., Dunedin, Florida) was used to calibrate a fiber-coupled spectrometer (QE6500, Ocean Optics, Inc.) equipped with an integrating sphere (FOIS-1, Ocean Optics, Inc.). The integrating sphere was then placed on the sample stage, and the excitation spectrum was measured at each center tuning wavelength of the excitation scan (360 to 485 nm, in 5-nm increments). Wavelength scanning for both excitation and emission scanning was achieved by varying the angle of incidence on a tunable filter, allowing variation of the center wavelength of the passband over a broad wavelength range. A 5-nm increment was selected so as to ensure (greater than Nyquist) oversampling of the filter bandwidth for each filter (bandwidths given in Table 1). The integrated area (total optical power) of the excitation spectrum was calculated for each center tuning wavelength providing the excitation intensity profile. A spectral correction factor was then calculated as the inverse of the excitation intensity profile, normalized to a maximum value of unity. All excitation-scanning hyperspectral image stacks were corrected by background (stray-light) subtraction and multiplication by the spectral correction factor. All corrections for background and wavelength-dependent attenuation were made using MATLAB® software (MATLAB® R2012b, Mathworks, Natick, Massachusetts).
2.3. Emission-Scanning Hyperspectral Microscope Set-up and Spectral Correction
The emission-scanning configuration, calibration, and spectral correction has been previously described (lightpath shown in Fig. 1).15 Briefly, a second TFTF array was placed in collimated space prior to the charge-coupled device (CCD) detector. The emission TFTF array operated identically to the excitation array (described above), except that it featured only 5 VersaChrome tunable filters in order to allow one filter position to remain open (to enable the entire fluorescence emission to be detected during the excitation scan). Spectral correction was performed by projecting the NIST-traceable light source onto a blank sample (blank microscope slide). Emission scans were performed by tuning the emission center wavelength from 475 to 700 nm, in 5-nm increments. A fluorescence filter cube with a long-pass dichroic beamsplitter (FF-458-Di02, Semrock, Inc.) and long-pass fluorescence emission filter (BLP01-458R, Semrock, Inc.) was implemented to separate excitation and emission light. A spectral transfer function was calculated from the known (NIST-traceable lamp) spectrum and the detected spectrum. The inverse of the spectral transfer function was used as a spectral correction factor. As in excitation scanning, each emission-scanning hyperspectral image set was corrected through background subtraction and multiplication by the spectral correction factor.
2.4. Hyperspectral Image Acquisition
Hyperspectral image stacks using excitation scanning were acquired at excitation wavelengths from 360 to 485 nm, in 5-nm increments. At each excitation wavelength, a 495-nm long-pass dichroic beamsplitter (BLP01-495R, Semrock, Inc.) and 495-nm long-pass emission filter (FF-495-Di02, Semrock, Inc.) were used to separate excitation light from fluorescence emission. The emission long-pass wavelength was selected to allow an excitation scan that included the peak excitation wavelength of all three fluorescent species in the sample (Hoechst, GFP, and autofluorescence). After each excitation scan, hyperspectral image stacks using emission scanning were acquired at emission wavelengths from 475 to 700 nm, in 5-nm increments. Excitation light was filtered with a 420/17-nm bandpass filter (TBP01-451-15 tuned to 420 nm, Semrock, Inc.). The excitation center wavelength was selected to allow concurrent excitation of GFP, Hoechst, and autofluorescence. The same field-of-view was imaged by both excitation- and emission-scanning systems. Image detection for both systems was performed using an electron-multiplied charge-coupled device (EMCCD) camera (Rolera EM-C2, QImaging, Surrey, British Columbia). An acquisition time of 300 ms and EMCCD gain of 3800 was used for both systems, for all samples, with the exception of acquiring the spectrum of GFP used by the spectral library. For the case of the GFP control, an EMCCD gain of 3600 was used due to the high signal intensity from confluent monolayers of GFP-expressing PMVECs in culture. A background image stack was acquired using both systems from a region of the slide containing no tissue. Background subtraction and spectral correction were performed on all hyperspectral image stacks acquired with both systems.
2.5. Spectral Image Analysis
A spectral library was constructed for both excitation- and emission-scanning systems. Linear unmixing of image stacks was performed using a positively-constrained unmixing algorithm (lsqnonneg, MATLAB 2012a, Mathworks). Spectral data visualization and plotting were performed using Excel (Office 2010, Microsoft, Redmond, Washington). Linear unmixing calculates the abundance of each fluorophore on a pixel-by-pixel basis, with reference to pure (end-member) spectra that are contained in a spectral library. We have previously shown linear unmixing to be accurate for quantifying relative fluorophore concentrations in an image, notably for detecting GFP in highly autofluorescent lung tissue and for quantifying FRET efficiencies.9,15,29
Pure excitation and emission spectra of GFP were measured from image data of a monolayer of GFP-expressing PMVECs. Excitation and emission spectra of Hoechst were measured from image data of a monolayer of Hoechst-labeled PMVECs (not expressing GFP). The excitation and emission spectra of tissue autofluorescence were collected from tissue slides without GFP and Hoechst present. For each control image, all wavelength bands were summed to provide the total fluorescence intensity. Threshold levels were selected to identify regions with high SNR ratio. These regions were then applied to the original hyperspectral image stack and the pixel-averaged spectrum of the region was measured and used as the pure spectrum for the corresponding fluorophore. The final spectral library featured excitation and emission spectra for GFP, Hoechst, and lung autofluorescence.
The spectral library for each hyperspectral scanning method was used to linearly unmix signals from GFP, Hoechst, and autofluorescence in hyperspectral image stacks acquired using either excitation or emission scanning. Linear unmixing was performed on each image stack featuring GFP, Hoechst, and autofluorescence by evaluating the abundance of each known spectra found in the spectral library. Non-negatively constrained linear unmixing was performed using the “lsqnonneg” algorithm in MATLAB (The MathWorks). In brief, this algorithm applies a least-squares approach to fit the measured (unknown) spectrum to a mixture of the known end-member spectra (contained in the spectral library). A constraint is placed so that a negative fit (negative amount of an end-member) is not allowed. An additional custom script was written to calculate the root-mean-square (rms) error—a measure of the residual signal that was not accounted for in the least-squares fitting. In addition, the % rms error was calculated by dividing the rms error by the rms signal. The rms error and % rms error were used to ensure that the linear unmixing algorithm was effectively accounting for the signal present in the spectral image data.
Three separate images were created from the relative abundance of each fluorophore in each pixel. All images were saved as uncompressed tiff files. End-members from each unmixed image, for a given scanning method, were merged and false colored into a composite image using NIS Elements (NIS Elements 3.2, Nikon Instruments, Inc.) software. All images were adjusted equally for inspection.
2.6. SNR Measurements
Differences between excitation and emission-scanning systems were quantified using the SNR. We estimated SNR in each image stack using the method initially described by Amer et al.30 that was further demonstrated for fluorescence microscopy applications by our group and Bernas et al.9,31 Briefly, unmixed images were analyzed to detect the fluorescence intensity in each pixel. Pixels within regions of homogeneous intensity were identified using an 8-way high-pass filter. Surrounding pixels were used to determine the standard deviation associated with each pixel. The mean intensity of each pixel divided by the standard deviation of each pixel provided the SNR. The median SNR from at least 5 regions were used as the SNR for each image.
3. Results
3.1. Spectral Correction
Spectral correction was performed to characterize and correct for wavelength-dependent attenuation in both systems. We have previously demonstrated spectral correction methods using a NIST-traceable lamp to characterize light attenuation in emission-scanning hyperspectral imaging systems.9,15 To ensure consistency, the same NIST-traceable lamp spectrum was used correct both the excitation- and emission-scanning hyperspectral imaging systems used in the current study. For the excitation-scanning system, the NIST-traceable lamp was used to calibrate a high-sensitivity spectrometer that was fiber coupled to an integrating sphere. The spectrometer was then used to measure the spectrally-filtered illumination profile incident upon the sample as a function of the center tuning wavelength of the tunable filter [Fig. 2(a), selected center tuning wavelengths shown]. The integrated intensity (total power) at each center tuning wavelength was calculated and plotted to describe the illumination profile as a function of center tuning wavelength [Fig. 2(b)]. The peak excitation intensity was achieved at 470 nm, indicating that the combination of the Xe arc lamp, tunable filter array, liquid light guide, and intermediate optical components provided higher illumination intensity at these wavelengths. To correct for wavelength-dependent excitation intensity, the inverse of the illumination profile was calculated, normalized to a peak value of unity, and used as a spectral correction factor [Fig. 2(c)]. The highest correction value occurred at 360 nm and the lowest at 470 nm.
Fig. 2.
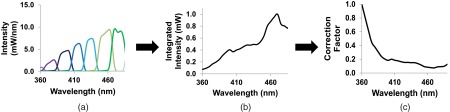
Spectral output and wavelength-dependent attenuation in the excitation-scanning system. (a) Calibrated intensity measurements of the Xe arc lamp, filtered by the excitation tunable filter array (six representative tuning wavelengths shown), as measured at the objective using an integrating sphere and fiber-coupled spectrometer. (b) Integrating the area under each band provided the illumination power for each tuning wavelength (all tuning wavelengths shown). (c) The inverse of the integrated intensity was normalized to a value of unity at the wavelength with the lowest intensity (highest correction factor value) and used to correct for wavelength-dependent illumination intensity in the excitation-scanning system.
Wavelength-dependent attenuation in the emission-scanning system was characterized similarly to the excitation-scanning system. However, instead of performing a calibrated measurement of the excitation spectrum, a NIST-traceable lamp was used as the input to the emission-scanning system [Fig. 3(a)]. The NIST-traceable lamp was spectrally-filtered by the emission tunable filter and was detected using an EMCCD camera [Fig. 3(b)]. There was a high similarity between the input spectrum of the NIST-traceable lamp and the spectrum measured using the emission-scanning system, indicating a high-spectral reproducibility. The ratio of the measured spectrum to the input spectrum was used as the spectral transfer function, whereas the inverse of the spectral transfer function was normalized to a peak value of unity and used as a spectral correction factor [Fig. 3(c)]. The highest correction factor occurred at 470 nm, while the lowest correction factor occurred at 610 nm.
Fig. 3.

Measurements of wavelength-dependent attenuation in the emission-scanning system. (a) The spectrum of a NIST-traceable light source was (b) filtered by an array of TFTFs positioned for emission-scanning hyperspectral imaging. A spectral transfer function was calculated from the measured spectrum and known lamp spectrum. (c) The inverse of the spectral transfer function was normalized to a value of unity at the lowest intensity (highest correction factor value), and used to correct for wavelength-dependent attenuation through the emission-scanning system.
Further spatial correction was used in the emission-scanning system to account wavelength-dependent image shift (typically referred to as “pixel-shift”) from the tunable filters.32 An image stack was acquired without a slide present to measure the illumination profile of a NIST-traceable lamp after filtering by the emission-scanning tunable filter array. The inverse of the illumination profile was used to correct for uneven spatial transmission due to the emission-scanning tunable filter array. Both spectral correction and spectrally-dependent spatial correction were applied to all subsequent spectral image stacks acquired with the emission-scanning system.
3.2. Acquisition of Spectral Libraries
Spectral libraries were constructed from spectra acquired through excitation and emission scans of single-labeled samples: GFP, Hoechst, and unlabeled tissue autofluorescence. Spectra were sampled from regions with high intensity [Figs. 4(a)–4(f)] to obtain a strong SNR ratio for each fluorescent species (end member) in the library. Regions of interest for measuring the average GFP spectrum are shown in green, for either excitation scanning [Fig. 4(a)] or emission scanning [Fig. 4(d)]. Regions of interest for measuring the average Hoechst spectrum are shown in blue, for either excitation scanning [Fig. 4(b)] or emission scanning [Fig. 4(e)]. Regions of interest for measuring the average autofluorescence spectrum are shown in red, for either excitation scanning [Fig. 4(c)] or emission scanning [Fig. 4(f)]. The pixel-averaged spectra for each respective region of interest are shown in Fig. 4, panels (g)–(i). Results are in agreement with previously reported literature values for GFP and Hoechst1,2,4,5 and our previously determined emission spectrum for lung tissue, which was measured using both an AOTF-based system9,15 and a TFTF-based system.15
Fig. 4.

Spectral library data for excitation- and emission-scanning systems. Regions of interest in hyperspectral image sets were selected by intensity thresholding the wavelength-summed image of confluent monolayers of GFP-expressing PMVECs (a and d), confluent monolayers of wild-type PMVECs stained with Hoechst (b and e), and unlabeled tissue autofluorescence (c and f). The average spectrum from each thresholded region for GFP (g), Hoechst, (h), and autofluorescence (i) was used as the respective end-member spectrum in the spectral library.
3.3. Linear Spectral Unmixing
For visualization, a total fluorescence intensity image was calculated by summing the intensity at each wavelength for excitation and emission scanning [Figs. 5(a) and 5(b)]. Image stacks containing GFP, Hoechst, and autofluorescence were then unmixed using non-negatively constrained linear spectral unmixing. Linear unmixing algorithms calculate the amount (abundance) of each fluorescent species (end member) contained in the spectral library for each pixel of a spectral image stack, resulting in an unmixed abundance image for each end member [Figs. 5(c)–5(h)]. GFP and autofluorescence were better discriminated amongst unmixed images acquired with the excitation-scanning system than those acquired with the emission-scanning system [Figs. 5(c) and 5(d)]. Additionally, Hoechst-labeled nuclei were clearly resolved and discriminated from surrounding autofluorescence using the excitation-scanning system [Figs. 5(e) and 5(g)]. By contrast, images from the emission-scanning system displayed degraded quality due to reduced SNR characteristics.
Fig. 5.

Linear unmixing of spectral image stacks allowed identification of GFP, Hoechst, and autofluorescence in both excitation- and emission-scanning hyperspectral image sets. The summed fluorescence intensity (a and b) was used to visualize raw image data and revealed improved clarity of nuclei and GFP in the excitation-scanning image compared with the emission-scanning image. The GFP emission was better localized using excitation scanning compared with emission scanning (c and d). In addition, nuclei were better resolved in the excitation-scanning image compared with the emission-scanning image (e and f). Finally, autofluorescence was more clearly delineated using excitation scanning than emission scanning (g and h). Excitation scanning provided a higher signal-to-noise ratio (SNR) for all three fluorophores measured. In specific, the Hoechst displayed a 20-fold increase in SNR with excitation scanning, compared with emission scanning. Additionally, GFP possessed a 3-fold increase in SNR with excitation scanning, compared with emission scanning.
Linearly unmixed abundance images were merged into a composite, false-colored image for both excitation- and emission-scanning systems [Figs. 6(a) and 6(b)] to allow visualization of end-member features. All images were linearly scaled and displayed identically for both excitation- and emission-scanning spectral image data. GFP and Hoechst regions were better delineated using excitation scanning compared with emission scanning. In addition, linearly unmixed excitation-scanning images displayed higher SNR characteristics than unmixed emission-scanning images.
Fig. 6.

Linearly unmixed images were false-colored and merged for visualization. GFP (green), Hoechst (blue), and autofluorescence (red) were overlayed for image stacks acquired with excitation scanning (a) and emission scanning (b). The unmixed merged image taken with excitation scanning had clearly delineated regions of GFP and Hoechst, whereas the image taken with emission scanning had poorly-delineated GFP and nuclear regions.
4. Discussion
4.1. Excitation- and Emission-Scanning Hyperspectral Imaging
Traditional hyperspectral imaging approaches for biomedical applications filter the fluorescence emission to sample a contiguous emission spectrum (emission scanning).3,9,33,34 These emission-scanning approaches provide a wealth of additional information that allows for highly specific molecular identification. However, emission scanning also provides inherently low-signal intensity due to the spectral filtering technologies employed, which require attenuating the majority of emitted light. This loss in signal intensity can result in long acquisition times, poor SNR characteristics, and photobleaching. Consequently, emission-scanning approaches, while providing significant benefits for spectral discrimination, are not conducive for high-speed, time-dependent measurements, assays with low-signal intensities, or assays that are highly susceptible to photobleaching.29
In this work, we have demonstrated a novel method for hyperspectral imaging using wavelength-filtering of excitation light to sample a contiguous excitation spectrum (excitation scanning). There is inherently higher signal detected using this excitation-scanning approach because the fluorescence emission is not filtered, therefore permitting all emitted light to be detected. This allows a significant increase in signal strength and/or decrease in acquisition time. Consequently, time-dependent spectral measurements are feasible with faster acquisition times than were previously possible. Additionally, excitation scanning provides complimentary data to emission scanning through unique excitation spectra. Excitation spectra are often dissimilar to emission spectra. For example, many fluorophores exhibit multiple peak excitation wavelengths—often a primary excitation peak in the visible range with a secondary peak in the ultraviolet range. These excitation spectral features represent an alternative basis for discriminating amongst fluorophores with similar emission spectra.
It should be noted that the fluorescent labels (GFP and Hoechst) selected for this study are labels that are easily separable using either excitation or emission-scanning procedures due to the large differences in peak excitation and peak emission wavelengths. However, the spectral imaging and linear unmixing approach should also be applicable for fluorophores with overlapping excitation or emission spectra or similar peak intensities. Emission-scanning hyperspectral imaging and linear unmixing approaches have previously been shown to be effective for detecting FRET pairs with overlapping spectra35 and for detecting FRET pairs with additional fluorescence labels.29 As mentioned above, the presence of a secondary excitation peak in many commonly used fluorescent probes may provide additional spectral information that is useful for discriminating amongst fluorophores with similar, or even identical, primary peak excitation wavelengths—lending further utility to the excitation-scanning approach.
4.2. Spectral Calibration for Excitation and Emission Scanning
In previous work, we demonstrated the effectiveness of a TFTF for emission-scanning hyperspectral imaging.15 Here, we have developed a novel microscope configuration utilizing two TFTF modules: one to scan the excitation spectrum and one to scan the emission spectrum. Identical filters were utilized to ensure equivalent spectral filtering characteristics for excitation and emission scanning (Table 1). Each TFTF module allowed a wide wavelength-tuning range (402 to 708 nm for excitation, 451 to 708 nm for emission) and 16 to 20-nm spectral bandwidth. For excitation scanning, a TFTF module was positioned after the arc lamp (Fig. 1). For emission scanning, a second TFTF module was placed in collimated (infinity-focused) space before the CCD detector. A 495-nm dichroic beamsplitter and a 495-nm long-pass emission filter were used to separate excitation and emission light, respectively. During emission-scanning operation, the excitation wavelength was fixed and the emitted light was sampled at 5-nm increments. During excitation-scanning operation, an open slot in the emission TFTF module was used and all emitted light above 495 nm was detected, while the excitation light was tuned at 5 nm increments. As a consequence of collecting all emitted light at each excitation wavelength, a lower acquisition time of 300 ms was required. Emission scans taken with the equivalent acquisition time exhibited much less detectable fluorescence [compare panels (a) and (b), Fig. 6].
Images acquired using each system were corrected for wavelength-dependent attenuation to achieve a flat spectral response across all wavelengths. Significant correction was required from 360 to 400 nm with the excitation-scanning system due to the uneven illumination of the Xe arc lamp and transmission characteristics of the liquid light guide (Fig. 2). In the emission-scanning system, wavelength-dependent attenuation was primarily due to the transmission characteristics of the TFTF module and the quantum efficiency of the CCD detector (Fig. 3). Correction factors for excitation and emission scanning were applied postacquisition to each image in the spectral image stack by multiplying by the spectral correction factor at the corresponding wavelength. However, it could also be possible to achieve spectral correction by adjusting the acquisition time for each image in the image stack or by incorporating a variable attenuator into either the excitation or emission-scanning lightpath. The advantage of achieving a flat spectral response during acquisition would be the ability to acquire consistent signal intensities at all wavelengths. Conversely, characterizing attenuation postacquisition does not require a variable attenuator nor adjusting the acquisition time at each wavelength, hence presents a more streamlined acquisition process.
4.3. Excitation Scanning Provides Improved Signal Intensity and SNR Characteristics
Excitation-scanning hyperspectral image data of samples containing GFP, Hoechst, and lung autofluorescence revealed dramatic improvements in image clarity due to increased signal strength. Notably, there was more sensitive detection of GFP regions using the excitation-scanning system than the emission-scanning system. Additionally, nuclei were more clearly delineated, while autofluorescence regions demonstrated improved contrast with the excitation-scanning system (Fig. 5). The overall improvement in signal strength was easily visualized in the merged image (Fig. 6), where signals from each fluorophore were clearly delineated using excitation scanning. Conversely, nuclei were not easily resolved with emission scanning, while autofluorescence contrast was decreased, with significantly increased noise in the unmixed autofluorescence image. Not all regions of GFP were identified using emission scanning compared with excitation scanning. This is primarily due to the higher signal associated with excitation scanning. Additionally, multiple spectral features comprise the excitation spectrum of the lung autofluorescence, which may contribute additional information content and an increased accuracy of linear unmixing. Based on these improvements in signal intensity and spectral discrimination, we anticipate that excitation scanning will provide increased detection capabilities for high-speed, live-cell imaging or for detecting and discriminating weak fluorescent signatures.
4.4. Excitation Scanning Provides Complementary Spectral Information to Emission Scanning
Implementing excitation scanning and emission scanning on a single-microscope platform allowed acquisition of both the excitation and emission spectra for a given field-of-view. Results from imaging GFP-labeled cells within autofluorescent lung tissues indicate that the excitation and emission spectra for both GFP2,9,15 and Hoechst15,36,37 are consistent with previous values reported in the literature. The emission spectrum from lung autofluorescence is also consistent with our previous studies using AOTF-based and TFTF-based emission-scanning hyperspectral imaging systems.9,15 To our knowledge, the excitation spectrum of lung autofluorescence has not been previously characterized. In the lung, autofluorescence is well characterized with a bulk fluorescence spectrum.9,15,36 This is likely because the lung contains very high concentrations of elastin and collagen, when compared with other tissues. However, autofluorescence is present in many tissue types, and may be attributed to other endogenous fluorophores, such as flavin adenine dinucleotide (FAD), nicotinamide adenine dinucleotide (NADH), and collagen.38–40 For example, in pancreatic islets, metabolic activity has been shown to correspond to dynamic concentrations of FAD or NADH, which can be measured using autofluorescence microscopy.41,42 Consequently, when performing spectral imaging and analysis of tissues with complex or varying autofluorescence properties (such as highly metabolic tissues), it may be necessary to incorporate spectra from multiple endogenous fluorophores into the spectral library. This could be accomplished by obtaining isolated spectra for predominant autofluorescent proteins in the tissue or developing experimental approaches to ensure that the spectrum of each of the endogenous fluorophores could be sampled idependently. With an appropriately constructed spectral library, the non-negatively constrained linear unmixing algorithm, or other spectral analysis algorithms, should be able to account for changes in tissue composition or physiology that have corresponding changes in endogenous fluorophore concentrations.
Based on differences between the spectral library for excitation scanning and that for emission scanning, combining both excitation and emission scanning will allow increased discrimination amongst multiple fluorescent species due to the increase in spectral information content. Notably, the excitation spectrum of autofluorescence differs markedly from the emission spectrum, having peak wavelengths at 360 and 440 nm, compared with a single peak emission wavelength at 510 nm. Because GFP and lung autofluorescence share similar peak emission wavelengths, the presence of more than one autofluorescence peak excitation wavelength increases the ability to discriminate between GFP and autofluorescence using excitation scanning. Additionally, several others have shown that spectroscopic (nonimaging) excitation and emission matrices are effective for discriminating endogenous fluorescence in human tissues.14,39,43 However, implementation of a hyperspectral imaging system capable of measuring both excitation and emission spectra has not been previously described. We anticipate that the excitation- and emission-scanning hyperspectral imaging system described here will allow similar spectroscopic studies to be conducted on an imaging level—offering an unprecedented view of cells and tissues, while providing increased sensitivity and specificity for multilabel studies and characterizing cell and tissue composition.
5. Conclusions
Hyperspectral imaging is an invaluable approach for discriminating amongst and quantifying fluorescent species in biomedical applications. In this work, we have demonstrated a novel excitation-scanning hyperspectral imaging system. We have compared this approach to a previously-reported emission-scanning system based on the efficacy for detecting GFP in highly autofluorescent lung tissue. The excitation-scanning system provided significantly increased signal levels, requiring shorter acquisition times than the emission-scanning system. Consequently, the excitation-scanning system was capable of higher sensitivity in detecting regions of GFP. Additionally, excitation scanning provided increased delineation of nuclei and increased structural information in autofluorescence, when compared with emission scanning. If used in conjunction with emission scanning, the excitation spectra of GFP, Hoechst, and lung autofluorescence can provide complementary spectral information to existing emission spectra. Combined excitation-emission hyperspectral imaging could be especially advantageous for highly multiplexed or multilabel studies. In future work, we anticipate that excitation scanning will allow significant improvements in temporal resolution for live-cell imaging.
Acknowledgments
The authors would like to acknowledge support from NIH grant P01 HL066299, the Abraham Mitchell Cancer Research Fund, the Alabama Space Grant Consortium, and the University Committee on Undergraduate Research (UCUR). VersaChrome filters and tuning hardware for this study were provided by Semrock Inc., a Unit of IDEX.
Biographies
Peter F. Favreau received his BSc in physics from the University of South Alabama in 2010. He is currently pursuing a PhD in basic medical sciences at the University of South Alabama. His research interests include applying spectral imaging technologies to biomedical and clinical applications.
Clarissa Hernandez is currently completing a BSc in chemical and biomolecular engineering from the University of South Alabama. She will attend graduate school at Purdue University in pursuit of a PhD in biomedical engineering.
Tiffany Heaster will receive a BSc in biological engineering from Mississippi State University in May 2014. In August 2014, she will begin pursuing a PhD in biomedical engineering from Vanderbilt University. Her research interests lie in the application of optical techniques for the detection of disease, development of improved therapies, and stimulation of the nervous system.
Diego F. Alvarez received his Medical Degree (1994) from La Universidad Libre de Colombia, Cali, Colombia, and his PhD in basic medical sciences (2005) from The University of South Alabama, Mobile, Alabama. He is currently appointed as an assistant professor of Internal Medicine and Pharmacology. Major scientific interest involves addressing endothelial stress responses. Strength of his research is provided by integrating reductionist approaches using molecular and cellular biology, experimental animal models, and bedside work.
Thomas C. Rich received his BAE and MSc in aerospace engineering from the Georgia Institute of Technology in 1988 and 1990, and his PhD in biomedical engineering from Vanderbilt University in 1996. He is currently an associate professor in the Center for Lung Biology and Department of Pharmacology at the University of South Alabama. His research interests include real time measurement of intracellular signals and understanding the mechanisms underlying signaling specificity.
Prashant Prabhat earned his PhD in EE from UT Dallas and completed graduate research at the UT Southwestern Medical Center at Dallas, where he co-invented multifocal plane microscopy (MUM). Since 2008 Prashant has been with Semrock, where he is a customer facing expert on optical filters, writes and speaks on this topic, and helps develop company’s product roadmap. As a new venture manager, he also has led the development of a tunable filter system at Semrock.
Silas J. Leavesley received a BSc in chemical engineering in 2003 from Florida State University and a PhD in biomedical engineering in 2008 from Purdue University. He is currently an assistant professor of Chemical and Biomolecular Engineering, Pharmacology, and the Center for Lung Biology at the University of South Alabama. His research interests lie in the development of spectral imaging technologies for medical sciences research and clinical applications.
References
- 1.Mansfield J. R., et al. , “Autofluorescence removal, multiplexing, and automated analysis methods for in-vivo fluorescence imaging,” J. Biomed. Opt. 10(4), 041207 (2005). 10.1117/1.2032458 [DOI] [PubMed] [Google Scholar]
- 2.Billinton N., Knight A. W., “Seeing the wood through the trees: a review of techniques for distinguishing green fluorescent protein from endogenous autofluorescence,” Anal. Biochem. 291(2), 175–197 (2001). 10.1006/abio.2000.5006 [DOI] [PubMed] [Google Scholar]
- 3.Garini Y., Young I. T., McNamara G., “Spectral imaging: principles and applications,” Cytometry A 69A(8), 735–747 (2006). 10.1002/(ISSN)1552-4930 [DOI] [PubMed] [Google Scholar]
- 4.Harris A. T., “Spectral mapping tools from the earth sciences applied to spectral microscopy data,” Cytometry A 69A(8), 872–879 (2006). 10.1002/(ISSN)1552-4930 [DOI] [PubMed] [Google Scholar]
- 5.Gat N., “Imaging spectroscopy using tunable filters: a review,” Proc. SPIE 4056, 50–64 (2000). 10.1117/12.381686 [DOI] [Google Scholar]
- 6.Schultz R. A., et al. , “Hyperspectral imaging: a novel approach for microscopic analysis,” Cytometry A 43(4), 239–247 (2001). 10.1002/(ISSN)1097-0320 [DOI] [PubMed] [Google Scholar]
- 7.Nieman L. T., et al. , “Hyperspectral imaging system for quantitative identification and discrimination of fluorescent labels in the presence of autofluorescence,” in 3rd IEEE Int. Symp. Biomed. Imaging Nano Macro, pp. 1288–1291 (2006). [Google Scholar]
- 8.Seekell K., et al. , “Hyperspectral molecular imaging of multiple receptors using immunolabeled plasmonic nanoparticles,” J. Biomed. Opt. 16(11), 116003 (2011). 10.1117/1.3646529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leavesley S. J., et al. , “Hyperspectral imaging microscopy for identification and quantitative analysis of fluorescently‐labeled cells in highly autofluorescent tissue,” J. Biophotonics 5(1), 67–84 (2012). 10.1002/jbio.v5.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pohling C., Buckup T., Motzkus M., “Hyperspectral data processing for chemoselective multiplex coherent anti-Stokes Raman scattering microscopy of unknown samples,” J. Biomed. Opt. 16(2), 021105 (2011). 10.1117/1.3533309 [DOI] [PubMed] [Google Scholar]
- 11.Han M., et al. , “Quantum-dot-tagged microbeads for multiplexed optical coding of biomolecules,” Nat. Biotechnol. 19(7), 631–635 (2001). 10.1038/90228 [DOI] [PubMed] [Google Scholar]
- 12.Davis R. W., et al. , “Hyperspectral image correlation for monitoring membrane protein dynamics in living cells,” Proc. SPIE 7184, 71840J (2009). 10.1117/12.807743 [DOI] [Google Scholar]
- 13.De Beule P. A. A., et al. , “A hyperspectral fluorescence lifetime probe for skin cancer diagnosis,” Rev. Sci. Instrum. 78(12), 123101 (2007). 10.1063/1.2818785 [DOI] [PubMed] [Google Scholar]
- 14.Zuluaga A. F., et al. , “Fluorescence excitation emission matrices of human tissue: a system for in vivo measurement and method of data analysis,” Appl. Spectrosc. 53(3), 302–311 (1999). 10.1366/0003702991946695 [DOI] [Google Scholar]
- 15.Favreau P., et al. , “Thin-film tunable filters for hyperspectral imaging of lung tissue,” J. Biomed. Opt. 19(1), 011017 (2014). 10.1117/1.JBO.19.1.011017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chung A., et al. , “In vivo cytometry: a spectrum of possibilities,” Cytometry A 69A, 142–146 (2006). 10.1002/(ISSN)1552-4930 [DOI] [PubMed] [Google Scholar]
- 17.Radosevich A. J., et al. , “Hyperspectral in vivo two-photon microscopy of intrinsic contrast,” Opt. Lett. 33(18), 2164–2166 (2008). 10.1364/OL.33.002164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vo-Dihn T., et al. , “A hyperspectral imaging system for in vivo optical diagnostics,” IEEE Eng. Med. Biol. Mag. 23(5), 40–49 (2004). 10.1109/MEMB.2004.1360407 [DOI] [PubMed] [Google Scholar]
- 19.Meyer J. N., et al. , “Intracellular uptake and associated toxicity of silver nanoparticles in Caenorhabditis elegans,” Aquat. Toxicol. 100(2), 140–150 (2010). 10.1016/j.aquatox.2010.07.016 [DOI] [PubMed] [Google Scholar]
- 20.Dicker D. T., Lerner J. M., El-Deiry W. S., “Hyperspectral image analysis of live cells in various cell cycle stages,” Cell Cycle 6(20), 2563–2570 (2007). 10.4161/cc [DOI] [PubMed] [Google Scholar]
- 21.Dicker D. T., Lerner J., “Differentiation of normal skin and melanoma using high resolution hyperspectral imaging,” Cancer Biol. Ther. 5(8), 1033–1038 (2006). 10.4161/cbt [DOI] [PubMed] [Google Scholar]
- 22.Gupta N., “Acousto-optic-tunable-filter-based spectropolarimetric imagers for medical diagnostic applications—instrument design point of view,” J. Biomed. Opt. 10(5), 051802 (2005). 10.1117/1.2102507 [DOI] [PubMed] [Google Scholar]
- 23.Stratis D. N., et al. , “Comparison of acousto-optic and liquid crystal tunable filters for laser-induced breakdown spectroscopy,” Appl. Spectrosc. 55(8), 999–1004 (2001). 10.1366/0003702011953144 [DOI] [Google Scholar]
- 24.Wachman E. S., Niu W., Farkas D. L., “AOTF microscope for imaging with increased speed and spectral versatility,” Biophys. J. 73(3), 1215–1222 (1997). 10.1016/S0006-3495(97)78154-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buican T. N., “Real-time Fourier transform spectrometry for fluorescence imaging and flow cytometry,” Proc. SPIE 1205, 126–133 (1990). 10.1117/12.17787 [DOI] [Google Scholar]
- 26.Fox J., et al. , “Computational hyperspectral interferometry for studies of brain function: proof of concept,” Appl. Opt. 45(13), 3009–3021 (2006). 10.1364/AO.45.003009 [DOI] [PubMed] [Google Scholar]
- 27.Leavesley S., et al. , “An excitation wavelength-scanning spectral imaging system for preclinical imaging,” Rev. Sci. Instrum. 79(2), 023707 (2008). 10.1063/1.2885043 [DOI] [PubMed] [Google Scholar]
- 28.King J., et al. , “Structural and functional characteristics of lung macro- and microvascular endothelial cell phenotypes,” Microvasc. Res. 67(2), 139–151 (2004). 10.1016/j.mvr.2003.11.006 [DOI] [PubMed] [Google Scholar]
- 29.Leavesley S. J., et al. , “Assessing FRET using spectral techniques,” Cytom. Part J. Int. Soc. Anal. Cytol. 83(10), 898–912 (2013). 10.1002/cyto.a.22340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Amer A., Mitiche A., Dubois E., “Reliable and fast structure-oriented video noise estimation,” in 2002 Int. Conf. Image Process. Proc., Vol. 1, pp. I–840 (2002). [Google Scholar]
- 31.Bernas T., et al. , “Loss of image quality in photobleaching during microscopic imaging of fluorescent probes bound to chromatin,” J. Biomed. Opt. 10(6), 064015 (2005). 10.1117/1.2136313 [DOI] [PubMed] [Google Scholar]
- 32.Prabhat P., Erdogan T., “Physics of pixel shift in fluorescence microscopy,” Semrock White Pap. Ser., 1–9 (2011).
- 33.Gao L., Hagen N., Tkaczyk T. S., “Quantitative comparison between full-spectrum and filter-based imaging in hyperspectral fluorescence microscopy,” J. Microsc. 246(2), 113–123 (2012). 10.1111/j.1365-2818.2012.03596.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li Q., et al. , “Review of spectral imaging technology in biomedical engineering: achievements and challenges,” J. Biomed. Opt. 18(10), 100901–100901 (2013). 10.1117/1.JBO.18.10.100901 [DOI] [PubMed] [Google Scholar]
- 35.Zimmermann T., et al. , “Spectral imaging and linear un-mixing enables improved FRET efficiency with a novel GFP2–YFP FRET pair,” FEBS Lett. 531(2), 245–249 (2002). 10.1016/S0014-5793(02)03508-1 [DOI] [PubMed] [Google Scholar]
- 36.Annamdevula N. S., et al. , “An approach for characterizing and comparing hyperspectral microscopy systems,” Sensors 13(7), 9267–9293 (2013). 10.3390/s130709267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Żurek-Biesiada D., Kędracka-Krok S., Dobrucki J. W., “UV-activated conversion of Hoechst 33258, DAPI, and Vybrant DyeCycle fluorescent dyes into blue-excited, green-emitting protonated forms,” Cytometry A 83A(5), 441–451 (2013). 10.1002/cyto.a.v83a.5 [DOI] [PubMed] [Google Scholar]
- 38.Wagnieres G. A., Star W. M., Wilson B. C., “In vivo fluorescence spectroscopy and imaging for oncological applications,” Photochem. Photobiol. 68(5), 603–632 (1998). 10.1111/php.1998.68.issue-5 [DOI] [PubMed] [Google Scholar]
- 39.Drezek R., et al. , “Understanding the contributions of NADH and collagen to cervical tissue fluorescence spectra: modeling, measurements, and implications,” J. Biomed. Opt. 6(4), 385–396 (2001). 10.1117/1.1413209 [DOI] [PubMed] [Google Scholar]
- 40.Chance B., et al. , “Intracellular oxidation-reduction states in vivo: the microfluorometry of pyridine nucleotide gives a continuous measurement of the oxidation state,” Science 137(3529), 499–508 (1962). 10.1126/science.137.3529.499 [DOI] [PubMed] [Google Scholar]
- 41.Rocheleau J. V., Head W. S., Piston D. W., “Quantitative NAD(P)H/Flavoprotein autofluorescence imaging reveals metabolic mechanisms of pancreatic islet pyruvate response,” J. Biol. Chem. 279(30), 31780–31787 (2004). 10.1074/jbc.M314005200 [DOI] [PubMed] [Google Scholar]
- 42.Patterson G. H., et al. , “Separation of the glucose-stimulated cytoplasmic and mitochondrial NAD(P)H responses in pancreatic islet β cells,” Proc. Natl. Acad. Sci. U. S. A. 97(10), 5203–5207 (2000). 10.1073/pnas.090098797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.DaCosta R. S., Andersson H., Wilson B. C., “Molecular fluorescence excitation–emission matrices relevant to tissue spectroscopy,” Photochem. Photobiol. 78(4), 384–392 (2003). [DOI] [PubMed] [Google Scholar]