

Abstract
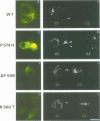
The effects of the mild cystic fibrosis (CF) mutation P574H were analysed and compared with those of three severe ones (delta I507, delta F508 and R560T). Immunochemical and functional analyses indicate that the rank order of CFTR expression at the cell surface is: wild type CFTR > P574H >> delta F508 >> R560T approximately 0. Patch-clamp analysis indicates that the open probability of P574H Cl- channels is almost twice as high as that of the wild type CFTR-Cl- channel. This increased intrinsic activity of individual P574H CFTR-Cl- channels compensates for the lower number of P574H CFTR-Cl- channels reaching the cell surface, and probably explains the milder form of CF associated with the P574H mutation. NS004, a recently described activator, restores near normal CFTR activity in cells expressing the P574H-CFTR channel. The P574H mutation modifies the gating mode of the channel with a large increase (approximately x 7) in the mean channel open time. Proline 574 might play an important role in the process connecting ATP hydrolysis at the nucleotide binding domain and opening and closing events of the CFTR-Cl- channel.
Full text
PDF

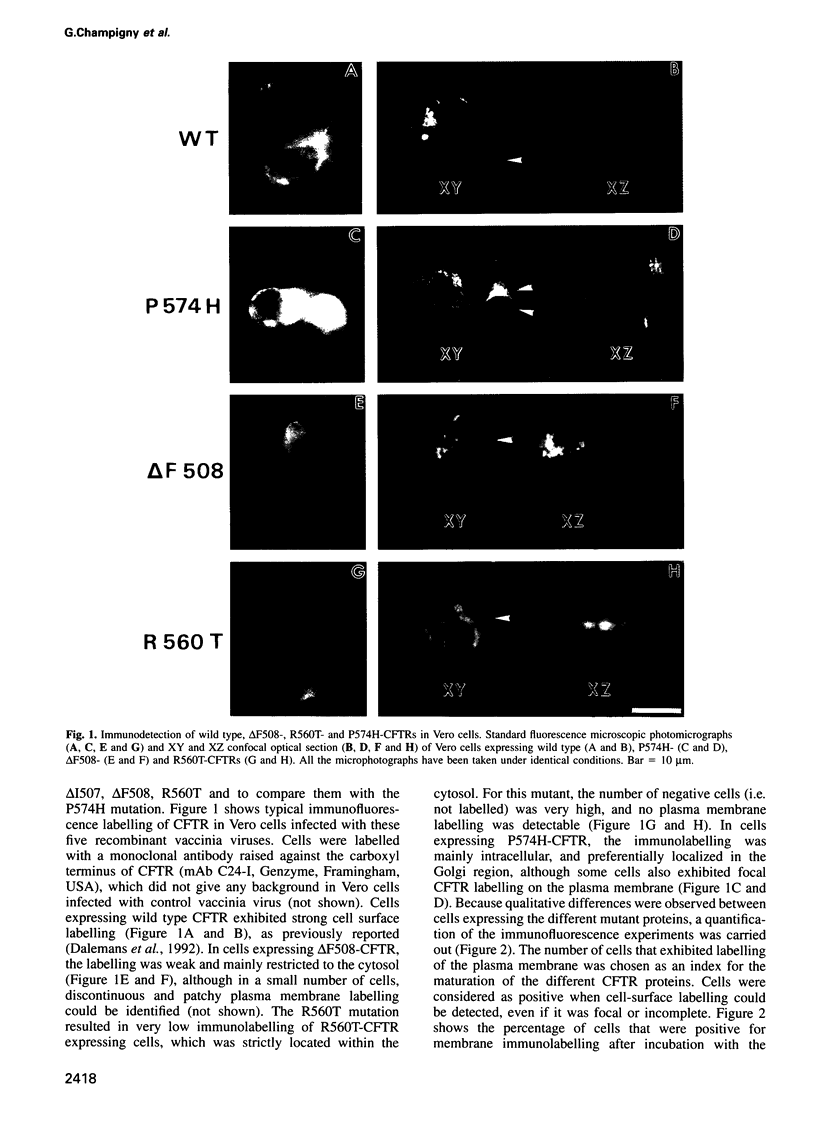
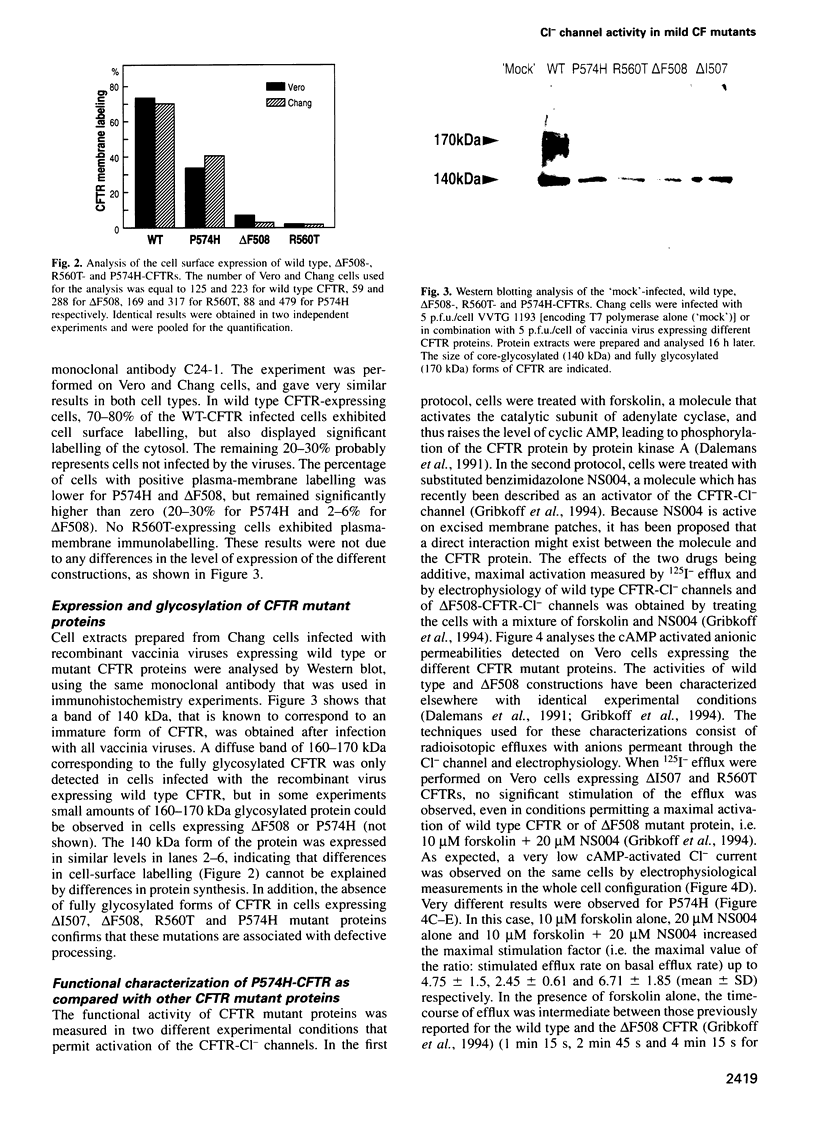
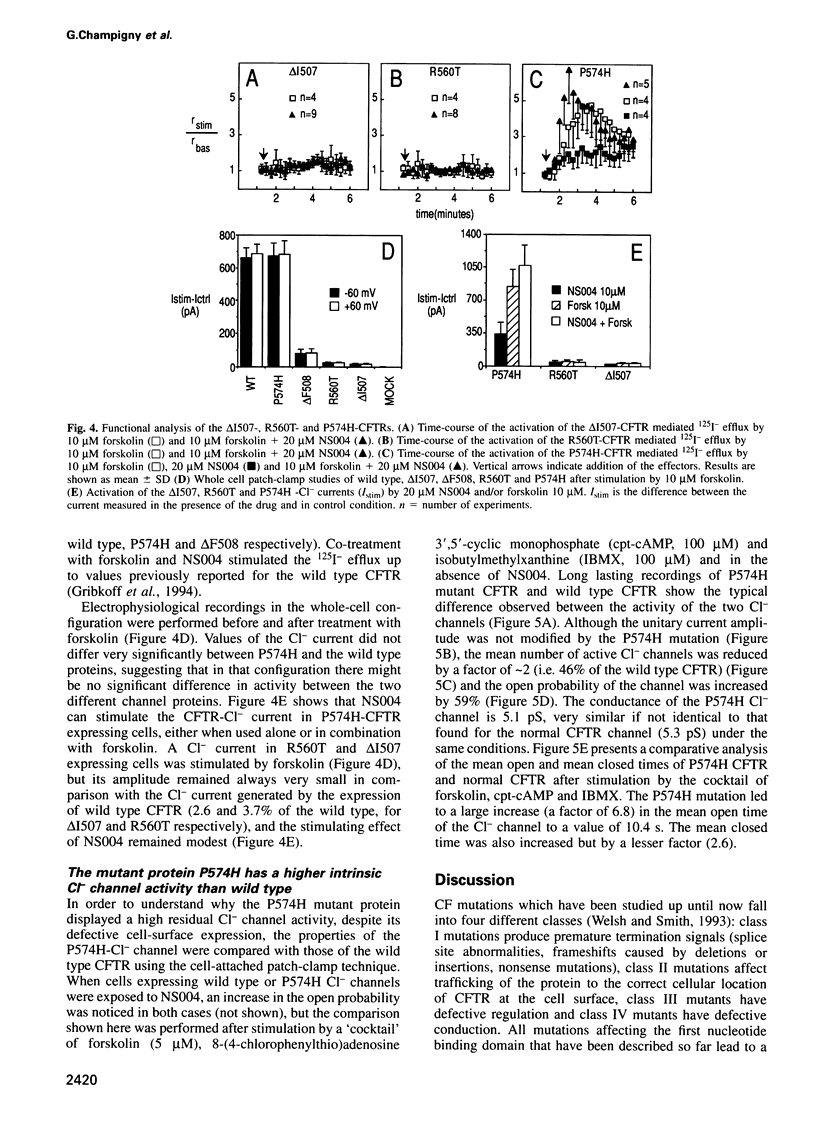
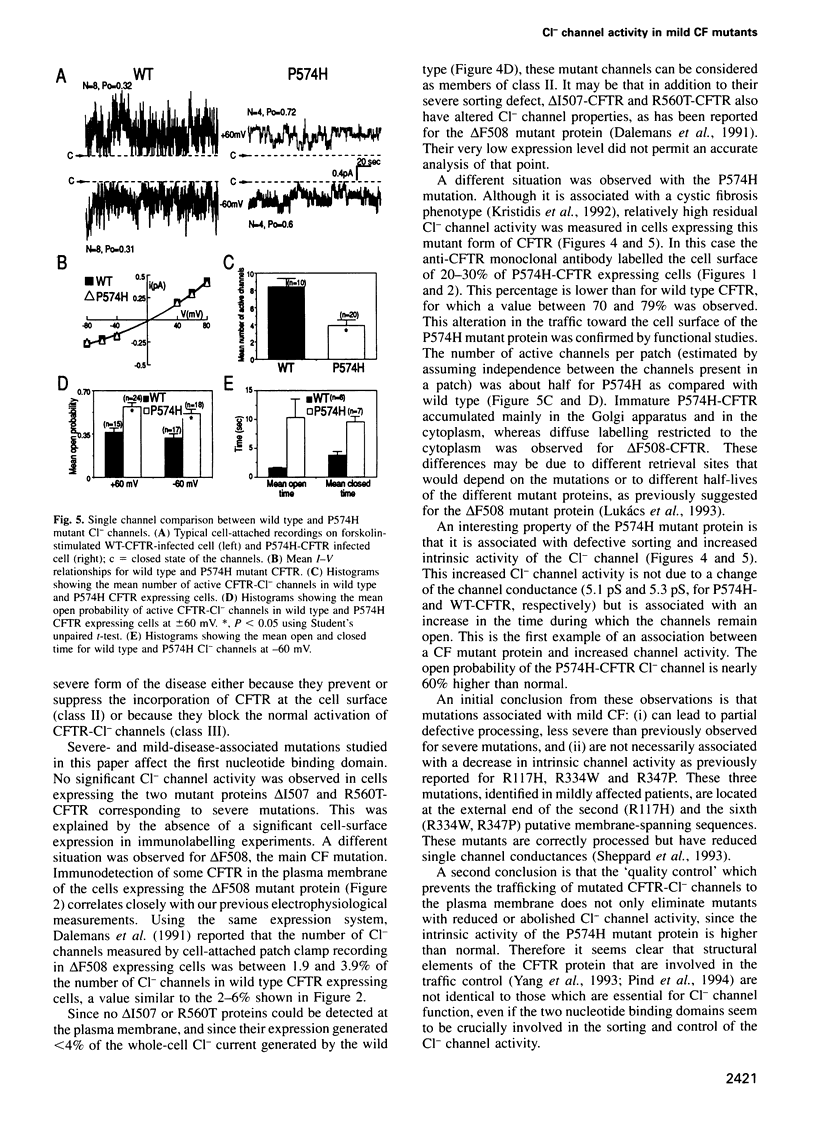


Images in this article

Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Anderson M. P., Berger H. A., Rich D. P., Gregory R. J., Smith A. E., Welsh M. J. Nucleoside triphosphates are required to open the CFTR chloride channel. Cell. 1991 Nov 15;67(4):775–784. doi: 10.1016/0092-8674(91)90072-7. [DOI] [PubMed] [Google Scholar]
- Baukrowitz T., Hwang T. C., Nairn A. C., Gadsby D. C. Coupling of CFTR Cl- channel gating to an ATP hydrolysis cycle. Neuron. 1994 Mar;12(3):473–482. doi: 10.1016/0896-6273(94)90206-2. [DOI] [PubMed] [Google Scholar]
- Berger H. A., Anderson M. P., Gregory R. J., Thompson S., Howard P. W., Maurer R. A., Mulligan R., Smith A. E., Welsh M. J. Identification and regulation of the cystic fibrosis transmembrane conductance regulator-generated chloride channel. J Clin Invest. 1991 Oct;88(4):1422–1431. doi: 10.1172/JCI115450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S. H., Gregory R. J., Marshall J., Paul S., Souza D. W., White G. A., O'Riordan C. R., Smith A. E. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell. 1990 Nov 16;63(4):827–834. doi: 10.1016/0092-8674(90)90148-8. [DOI] [PubMed] [Google Scholar]
- Dalemans W., Barbry P., Champigny G., Jallat S., Dott K., Dreyer D., Crystal R. G., Pavirani A., Lecocq J. P., Lazdunski M. Altered chloride ion channel kinetics associated with the delta F508 cystic fibrosis mutation. Nature. 1991 Dec 19;354(6354):526–528. doi: 10.1038/354526a0. [DOI] [PubMed] [Google Scholar]
- Dalemans W., Hinnrasky J., Slos P., Dreyer D., Fuchey C., Pavirani A., Puchelle E. Immunocytochemical analysis reveals differences between the subcellular localization of normal and delta Phe508 recombinant cystic fibrosis transmembrane conductance regulator. Exp Cell Res. 1992 Jul;201(1):235–240. doi: 10.1016/0014-4827(92)90368-i. [DOI] [PubMed] [Google Scholar]
- Denning G. M., Ostedgaard L. S., Welsh M. J. Abnormal localization of cystic fibrosis transmembrane conductance regulator in primary cultures of cystic fibrosis airway epithelia. J Cell Biol. 1992 Aug;118(3):551–559. doi: 10.1083/jcb.118.3.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer H., Machen T. E. CFTR displays voltage dependence and two gating modes during stimulation. J Gen Physiol. 1994 Sep;104(3):541–566. doi: 10.1085/jgp.104.3.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribkoff V. K., Champigny G., Barbry P., Dworetzky S. I., Meanwell N. A., Lazdunski M. The substituted benzimidazolone NS004 is an opener of the cystic fibrosis chloride channel. J Biol Chem. 1994 Apr 15;269(15):10983–10986. [PubMed] [Google Scholar]
- Hamill O. P., Marty A., Neher E., Sakmann B., Sigworth F. J. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981 Aug;391(2):85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hwang T. C., Nagel G., Nairn A. C., Gadsby D. C. Regulation of the gating of cystic fibrosis transmembrane conductance regulator C1 channels by phosphorylation and ATP hydrolysis. Proc Natl Acad Sci U S A. 1994 May 24;91(11):4698–4702. doi: 10.1073/pnas.91.11.4698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kartner N., Augustinas O., Jensen T. J., Naismith A. L., Riordan J. R. Mislocalization of delta F508 CFTR in cystic fibrosis sweat gland. Nat Genet. 1992 Aug;1(5):321–327. doi: 10.1038/ng0892-321. [DOI] [PubMed] [Google Scholar]
- Kerem B., Rommens J. M., Buchanan J. A., Markiewicz D., Cox T. K., Chakravarti A., Buchwald M., Tsui L. C. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989 Sep 8;245(4922):1073–1080. doi: 10.1126/science.2570460. [DOI] [PubMed] [Google Scholar]
- Kristidis P., Bozon D., Corey M., Markiewicz D., Rommens J., Tsui L. C., Durie P. Genetic determination of exocrine pancreatic function in cystic fibrosis. Am J Hum Genet. 1992 Jun;50(6):1178–1184. [PMC free article] [PubMed] [Google Scholar]
- Kunkel T. A., Roberts J. D., Zakour R. A. Rapid and efficient site-specific mutagenesis without phenotypic selection. Methods Enzymol. 1987;154:367–382. doi: 10.1016/0076-6879(87)54085-x. [DOI] [PubMed] [Google Scholar]
- Lukacs G. L., Chang X. B., Bear C., Kartner N., Mohamed A., Riordan J. R., Grinstein S. The delta F508 mutation decreases the stability of cystic fibrosis transmembrane conductance regulator in the plasma membrane. Determination of functional half-lives on transfected cells. J Biol Chem. 1993 Oct 15;268(29):21592–21598. [PubMed] [Google Scholar]
- Moss B. Vaccinia virus: a tool for research and vaccine development. Science. 1991 Jun 21;252(5013):1662–1667. doi: 10.1126/science.2047875. [DOI] [PubMed] [Google Scholar]
- Pind S., Riordan J. R., Williams D. B. Participation of the endoplasmic reticulum chaperone calnexin (p88, IP90) in the biogenesis of the cystic fibrosis transmembrane conductance regulator. J Biol Chem. 1994 Apr 29;269(17):12784–12788. [PubMed] [Google Scholar]
- Puchelle E., Gaillard D., Ploton D., Hinnrasky J., Fuchey C., Boutterin M. C., Jacquot J., Dreyer D., Pavirani A., Dalemans W. Differential localization of the cystic fibrosis transmembrane conductance regulator in normal and cystic fibrosis airway epithelium. Am J Respir Cell Mol Biol. 1992 Nov;7(5):485–491. doi: 10.1165/ajrcmb/7.5.485. [DOI] [PubMed] [Google Scholar]
- Quinton P. M. Cystic fibrosis: a disease in electrolyte transport. FASEB J. 1990 Jul;4(10):2709–2717. doi: 10.1096/fasebj.4.10.2197151. [DOI] [PubMed] [Google Scholar]
- Rigot J. M., Lafitte J. J., Dumur V., Gervais R., Manouvrier S., Biserte J., Mazeman E., Roussel P. Cystic fibrosis and congenital absence of the vas deferens. N Engl J Med. 1991 Jul 4;325(1):64–65. doi: 10.1056/NEJM199107043250116. [DOI] [PubMed] [Google Scholar]
- Riordan J. R., Rommens J. M., Kerem B., Alon N., Rozmahel R., Grzelczak Z., Zielenski J., Lok S., Plavsic N., Chou J. L. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989 Sep 8;245(4922):1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- Riordan J. R. The cystic fibrosis transmembrane conductance regulator. Annu Rev Physiol. 1993;55:609–630. doi: 10.1146/annurev.ph.55.030193.003141. [DOI] [PubMed] [Google Scholar]
- Sheppard D. N., Rich D. P., Ostedgaard L. S., Gregory R. J., Smith A. E., Welsh M. J. Mutations in CFTR associated with mild-disease-form Cl- channels with altered pore properties. Nature. 1993 Mar 11;362(6416):160–164. doi: 10.1038/362160a0. [DOI] [PubMed] [Google Scholar]
- Tabcharani J. A., Chang X. B., Riordan J. R., Hanrahan J. W. Phosphorylation-regulated Cl- channel in CHO cells stably expressing the cystic fibrosis gene. Nature. 1991 Aug 15;352(6336):628–631. doi: 10.1038/352628a0. [DOI] [PubMed] [Google Scholar]
- Tsui L. C. The spectrum of cystic fibrosis mutations. Trends Genet. 1992 Nov;8(11):392–398. doi: 10.1016/0168-9525(92)90301-j. [DOI] [PubMed] [Google Scholar]
- Venglarik C. J., Bridges R. J., Frizzell R. A. A simple assay for agonist-regulated Cl and K conductances in salt-secreting epithelial cells. Am J Physiol. 1990 Aug;259(2 Pt 1):C358–C364. doi: 10.1152/ajpcell.1990.259.2.C358. [DOI] [PubMed] [Google Scholar]
- Welsh M. J., Smith A. E. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell. 1993 Jul 2;73(7):1251–1254. doi: 10.1016/0092-8674(93)90353-r. [DOI] [PubMed] [Google Scholar]
- Yang Y., Janich S., Cohn J. A., Wilson J. M. The common variant of cystic fibrosis transmembrane conductance regulator is recognized by hsp70 and degraded in a pre-Golgi nonlysosomal compartment. Proc Natl Acad Sci U S A. 1993 Oct 15;90(20):9480–9484. doi: 10.1073/pnas.90.20.9480. [DOI] [PMC free article] [PubMed] [Google Scholar]