

Summary
CD4+ Forkhead box protein 3 (Foxp3)+ regulatory T cells (Tregs) are the major cell type that mediates dominant tolerance in the periphery. Over the past decade, extensive study of Tregs has revealed that these cells express substantial heterogeneity to maintain tolerance and regulate immune responses. Tregs possess heterogeneity with respect to their origin and processes for development, functional activity, migratory pattern, and activation status. Some of the same environmental cues and molecular pathways utilized to generate specialized T-effector cells are also integrated by Tregs to co-localize and fine-tune suppressive mechanisms to optimally regulate and restrain distinctive self and antigen-specific T-cell responses. Here, we review our current understanding and significance of Treg heterogeneity in maintaining peripheral immune tolerance. We also highlight recent work from our laboratory that has studied the extent phenotypic distinct Treg subsets are related to each other and expand in an ordered fashion to give rise to highly activated short-lived Klrg1+ suppressor cells to optimize immune regulation and maintain homeostasis of the Treg compartment.
Keywords: Tregs, tolerance, subsets, effector/memory, Klrg1
Introduction
CD4+ Forkhead box protein 3 (Foxp3)+ regulatory T cells (Tregs) were initially described as thymus-derived T cells that suppress and normally keep in check peripheral autoreactive T cells that escape thymic negative selection (1). Foxp3 is the signature transcription factor of Tregs that promotes the Treg gene suppressive program while repressing key genes characteristic of T-effector (Teff) cells such as interleukin-2 (IL-2) (2-8) . When Tregs are absent as a consequence of effective deletion or impaired Foxp3 expression, severe rapid lethal multi-organ autoimmune attack occurs (4, 5, 7-10) . Thus, the sum of all other immune regulatory mechanisms is ineffective in controlling autoreactive T cells in the absence of functional Tregs. Intensive study has revealed that Tregs are also important in regulating many other immune responses, including responses to pathogenic agents, environmental allergens and food, gut microbiota, tumors, and transplanted tissues, which has been reviewed elsewhere (11-17). The high T-cell receptor (TCR) diversity of thymic-derived Tregs (tTregs) provides a range of specificities to respond to self and non-self antigens (18-20). In addition, under the proper environmental conditions, conventional peripheral CD4+ T cells can be induced to express Foxp3 and function as suppressor cells (pTregs). These Tregs provide another level of regulation by which various antigen-specific immune responses are regulated and constrained.
Expression of Foxp3 represents a unifying feature of tTreg and pTregs. Nevertheless, it is apparent that Tregs exhibit substantial heterogeneity besides the origin of their development to control immune responses to self and foreign antigens. This review discusses the cellular and molecular basis that contribute to developmental, functional, migratory, and activation-related heterogeneity of Tregs and their importance for proper suppressive function. We also summarize recent studies that begin to assess the inter-relationship between activated and memory Tregs and our work on phenotypic distinct Treg populations, including the role of Klrg1+ Tregs in immune suppression and homeostasis. These studies are generally consistent with the notion that many properties of Tregs are expressed by coopting activities and mechanisms that regulate Teff immune responses. Tregs can be viewed as another subset of activated T cells whose role is to regulate and suppress, rather than to induce and promote, immune responses.
Developmental heterogeneity of Tregs
The large majority of Foxp3+ Tregs that are found in the periphery arise as a consequence of development into the Treg lineage in the thymus (tTregs). After positive selection and development of CD4 single positive thymocytes, tTreg commitment occurs primarily in the medulla through selection events on medullary epithelial and dendritic cells (21, 22). These selection events result in tTregs that express a diverse TCR repertoire with a relatively high affinity for self-antigens that only partially overlaps with that found on conventional CD4+ T cells (18-20, 23-26). The developmental cues that promote tTregs have been recently reviewed (27). In brief, relatively high TCR signaling in conjunction with costimulatory signaling results in activation of nuclear factor of activated T cells (NF-AT), activator protein-1 (AP-1), and CARMA1/Bcl10/Malt1 ()-dependent activation of NF-κB, which directly acts on the Foxp3 gene to induce Foxp3 expression (28-32). Full maturation of tTregs requires further activation from transforming growth factor-β (TGFβ)-dependent xxx (Smad3) and interleukin-2 (IL-2)-dependent signal transducer and activator of transcription 5 (STAT5) that provides survival and growth signals and further acts on the Foxp3 gene to induce heightened levels of Foxp3 required for the expression of the Treg functional program (33-42). Transcriptional regulation of Foxp3 critically depends upon factors that exert their activity on its promoter and conserved non-coding sequence 2 (CNS2) and 3 (CNS3) (32). Mature tTregs seed peripheral immune tissues and are critically required to suppress peripheral autoreactive T cells that were not deleted during thymic negative selection.
Under appropriate environmental cues, conventional CD4+ T cells can develop in the periphery into Foxp3+ Tregs (pTregs). These pTregs regulate antigen-specific immune responses and contribute to tolerance in mucosal tissues and during pregnancy (43, 44). Along with a requirement for TCR and costimulatory signaling, the development of pTregs is highly dependent upon TGFβ (45-48). The mucosa is a preferential site for pTreg induction, in part due to abundant TGFβ and retinoid acid produced locally by epithelial cells, macrophages, and CD103+ DCs (49-54). The development of pTregs is particularly dependent on TGFβ-activation of pSmad3 through its action on CNS1 of Foxp3 (32). Accordingly, after germ-line deletion of Foxp3 CNS1, mice lack pTregs and exhibit Th2-type inflammation in the gut and lung (43). This finding supports the notion that pTregs provide a critical role for mucosal tolerance.
Another factor contributing to the generation of pTregs is commensal bacteria and food antigens in the gut and allergens in lung airways (46, 54-57). These environmental antigens select TCRs with specificities that are unique to the mucosa (58). Some of these specificities are likely derived from conventional T cells because when individual TCR specificities from the gut mucosa were expressed in bone marrow chimeras, they were not selected in the pool of tTregs. In another study, some tTregs, however, have been shown to respond to microflora-derived antigens (59). Thus, mucosal Tregs are comprised of a mixture of tTregs and pTregs, and the activity of both is likely required for effective tolerance. In this regard, the most effective means to prevent autoimmunity associated with Foxp3-deficient mice required the transfer of large number of tTregs and some conventional CD4+ T cells, which developed into pTregs (60).
Although it is commonly accepted that the peripheral conversion of activated T cells to Tregs contributes to peripheral tolerance, the proportion of tTreg and pTregs within mucosal sites have been difficult to precisely determine by TCR repertoire analysis. Another approach is to identify a marker that definitively distinguishes tTregs and pTregs. Two candidates are Helios and neuropilin-1, as these molecules are highly expressed by tTregs but poorly expressed by in vitro-induced Tregs (iTregs) and by pTregs in a number of settings (61-63). These two markers, however, do not perfectly distinguish tTregs from iTregs and pTregs. For example, many pTregs in the central nervous system expressed neuropilin-1 due to inflammation during spontaneous chronic experimental autoimmune encephalomyelitis (62). Likewise, expression of Helios is sometimes found on iTregs and pTregs, depending on the type of antigen-presenting cells and activation signals encountered (64-66).
Recent work supports the notion that tTregs are a stable lineage of dedicated suppressor cells with minimal capacity to de-differentiate and convert into Teff cells. The Foxp3 promoter and the CNS2 are highly demethylated in tTregs, which contribute to their lineage stability (32, 67-70). In contrast, iTregs have Foxp3 CNS2 that is substantially methylated and convert to Th17 cells under conditions of lower TGFβ and increased inflammatory signals (67, 68, 71). In vivo pTregs have also been shown to lose expression of Foxp3 and convert to Th cells (70, 72-74). Considering that tTregs behave as a stable lineage, this plasticity likely reflects the instability of pTregs. Such plasticity might provide a mechanism to help resist pathogenic infections.
Functional heterogeneity of Tregs
Early considerations of Treg suppressive function focused on the hypothesis that there was a single major dominant mediator of suppression. However, extensive study of Tregs has revealed that Tregs express a number of distinct mediators that downregulate immune responses. Some of these mediators include IL-10, TGFβ, IL-35, granzyme, Galectin-1, cytotoxic T-lymphocyte antigen-4 (CTLA4), CD39, CD73, neuropilin-1 (Nrp-1), lymphocyte activation gene-3 (LAG-3), and fibrinogen-like protein-2 (FGL-2). Detailed discussion of their role in Treg-mediated immune suppression has been reviewed recently (75-77). In vivo, these mediators create a localized immune inhibitory environment in part by directly acting on autoreactive T cells and in part by promoting tolerogenic DCs. Suppressive activity of Tregs extends beyond adaptive immunity, because Tregs also suppress innate immune cells, such as mast cells, macrophages, and NK cells (75, 78).
The expression of multiple mediators raises the question concerning the extent an individual mediator is dominant versus multiple mediators cooperating to suppress immune responses. The knockout of several of these mediators only in Tregs provides some answers to this question. Mice engineered to contain Tregs that do not express CTLA4 exhibited severe and lethal autoimmunity, even though Treg development and peripheral numbers were normal (79). Thus, expression of CTLA4 by Tregs is essential for normal immune homeostasis and tolerance and represents a core mechanism of the Treg suppressive program. In contrast, mice that contain Tregs engineered not to express IL-10 spontaneously developed inflammatory bowel disease (IBD) at 3-6 months of age and exhibited lung and skin hypersensitivity (80). These findings point to an important role for Treg production of IL-10 to suppress certain tissues specific responses, but the generally long time frame to obvious pathological changes suggests that other Treg suppressive mechanisms are also active but not sufficient to fully control these unwanted immune responses.
Current studies favor a view that Tregs possess functional heterogeneity and specialization to suppress distinct types of immune responses and to adapt and integrate signals from the local environment present in distinct tissues. One example already discussed is the gut mucosa that is dominated by IL-10-producing highly activated Tregs and that is permissive for development of pTregs to maintain tolerance to commensal microbiota and food antigens. A more recent example is the detection of a specialized population of Tregs in adipose tissue. Genome-wide profiling of Tregs from adipose tissue confirmed their expression of the Treg gene signature but also detected distinct sets of genes associated with a unique trafficking, anti-inflammatory and lipid metabolism profile (81). Depletion of Tregs in fat tissue correlated with increased inflammation and insulin resistance. Reciprocally, expanding Tregs in this tissue increased the anti-flammatory cytokine IL-10 and decreased glucose levels in the blood. The unique property of Tregs in adipose tissue is controlled in part by PPAR-γ, a key regulator of adipocyte differentiation (82). PPAR-γ not only determines the distinctive phenotype of fat Tregs but also regulates local Treg homeostasis. These findings link the failure of specialized Treg activity to a new and alternative mechanism to explain how chronic inflammation contributes to obesity.
There are also substantial additional data that Tregs respond to the local milieu and behave as subtypes that are equipped with distinctive suppressive programs to combat immune responses, in particular, Th1, Th2, and Th17 responses by CD4+ T cells (Table 1). Various cytokines and transcriptional regulators are linked to specific Th activities, e.g. IFNγ activates STAT1 and IL-12 activates STAT4 to drive T-bet expression for Th1 cells, IL-4 activates STAT6 to drive Gata3 and IRF4 expression in Th2 cells, and IL-6 and TGFβ activates STAT3 to drive RORγt expression for Th17 cells (83). Tregs also utilize these same transcriptional regulators to generate suppressive responses to counteract the respective Th responses, which we have designated here as Ts1, Ts2, and Ts17 in Table 1. Correspondingly, Tregs with deletion in T-bet, IRF4, and STAT3 showed selective inability to control Th1, Th2, and Th17 immune responses, respectively (84-86).
Table 1.
Comparison of Th and Treg subtypes
| Cytokine | Transcription factor |
Function | |
|---|---|---|---|
| Th1 | IFN-γ, IL-12 | T-bet | Production of IFN-γ; expression of CXCR3 on T cells |
| Ts1 | IFN-γ, IL-27 | T-bet | Differentiation of CXCR3+ Tregs; high expression of GITR, CTLA4, CD 103, IL-10, TGFβ on Tregs |
| Th2 | IL-4, IL-2 | Gata3, IRF4 | Production of IL-4, IL-5, IL-13, IL-10 cytokines; Ig production |
| Ts2 | ? | IRF4 | Upregulate Icos, Fgl2, IL-10, Gzmb, maf, Ccr8, IL-1R1 on Tregs |
| Th17 | IL-6, TGF-β | RORγt | Production of IL-17A, IL-17F, IL-21, IL-22, GM-CSF |
| Ts17 | IL-10 | STAT3 | Upregulate IL-10, Ebi3, Gzmb, Prf1, Ccr6 expression on Tregs |
| Tfh | IL-6, IL-21 | Bcl6 | Expression of CXCR5 on T cells; germinal center formation; affinity maturation |
| Tfs | ? | Bcl6 | Expression CXCR5 |
The cytokines promoting lineage-specific suppressor activity of Tregs are largely distinct from the pro-inflammatory cytokines that promote Th subsets (87-89) (Table 1). These cytokines, nevertheless, also generate signals that converge on the same transcriptional regulators used by Th cells to activate T-bet, IRF4, and STAT3, leading to expression of CXCR3, CCR8, and CCR6 on Tregs, respectively. This type of control co-localizes the Tregs to appropriate tissues sites to regulate Th1, Th2, and Th17 responses. Along with regulation of homing, these transcriptional regulators also distinctively control the expression of molecules associated with suppressive activity of Tregs. During a Th1 response, optimal expression of CTLA4, IL-10, and TGFβ was dependent upon T-bet (85). Targeted deletion of IRF4 in Tregs resulted in developing severe Th2 autoimmunity (86). Although Treg expression of CTLA4, TGFβ and GITR was unchanged, other Treg functional molecules, including Fgl2, IL-10 and granzyme B, were decreased, linking Treg expression of IRF4 to specific aspects of the Treg gene program and to regulate Th2 responses. Mice with targeted inactivation of STAT3 in Tregs were associated with impaired function to suppress Th17 cells and developed severe intestinal inflammation (84). Gene expression profiling revealed that STAT3-deficient Tregs expressed lower levels of IL-10, granzyme B, Ebi3, and perforin, linking these suppressive mediators to control Th17 responses.
T-follicular helper (Tfh) cells are CD4+ T cells specialized in regulating the germinal center reaction and their function depends on relatively high expression of Bcl6 and CXCR5; the latter is necessary for Tfh cell trafficking into the B-cell follicles (90-93). In Tregs, the transcription factor Bcl6 is also critical for the expression of CXCR5 and is essential for controlling the germinal center reaction (94-96). Tregs lacking either CXCR5 or Bcl6 showed impaired suppressor function and caused enhanced germinal center responses characterized by pronounced antibody production, affinity maturation, and accumulation of plasma cells. Collectively, all these data indicate substantial Treg heterogeneity develops as they respond to antigens in localized environments that also vary with regard to cellular and inflammatory signals.
Phenotypic heterogeneity contributes to Treg migration
Besides the above-mentioned relationship to varied chemokine receptor expression by Th and Treg populations, there is also additional heterogeneity in expression of other chemokine receptors that regulate Treg trafficking. A two-step switch model of chemokine receptor expression has been proposed to depict the migration of Tregs from the thymus to peripheral lymphoid and non-lymphoid tissues (97). The first switch occurs in the thymus where Tregs acquire CCR7 expression for homing to secondary lymphoid tissues. Tregs emerging from the thymus are uniformly CD62Lhi CCR7+ CXCR4low, which promotes migration to secondary lymphoid organs. The second switch occurs in secondary lymphoid tissues where Tregs are educated by local antigenic stimulation to express an array of chemokine receptors and integrins, e.g. CCR2, CCR5, CCR6, CXCR6, CCR4, CCR8, and α4β7, while expression of CD62L and CCR7 is downregulated, which promotes trafficking to various non-lymphoid tissues. Generally, CD62Llo Tregs found in secondary lymphoid tissues express increased levels and diversity of the trafficking molecules, which provide an advantage over conventional T cells to migrate to non-lymphoid tissues to maintain local immune homeostasis.
The types of chemokine receptors expressed by Tregs are influenced by the local environmental cues associated with the regional lymph nodes. For example, expression of CCR4 in peripheral LNs is associated with trafficking to skin and lung, while expression α4β7 and CCR9 in the MLNs is associated with homing to small intestine (98-100). However, homing to the gut mucosa is complicated, because this process involves lymphocyte entry into the lamina propria and across the basement membrane to the intraepithelial lymphocyte compartment. For example, in mice deficient in CCR9 or its ligand CCL25, homing of CD8αα TCRγδ cells to the intraepithelial lymphocyte compartment of small intestine was impaired, but homing of T cells, particularly CD4+ T cells, to the lamina propria was unaffected (101). Other chemokine receptors, such as CXCR3 and CXCR4, likely complement CCR9/CCL25 to facilitate lymphocyte homing to the gut mucosa (102-104). In addition, integrin β7 and αE (CD103), which form the αEβ7 heterodimer, may also contribute to T-cell homing or retention in the gut (105-108). However, the absence of integrin β7 only partially reduced Treg trafficking to the gut mucosa and those Tregs still suppressed immune responses efficiently in a transfer model of colitis, further indicating substantial redundancy in molecules that function in T-cell homing (109). One molecule that may be essential for Treg homing to the large intestine is GPR15 (110). In this regard, Tregs that do not express GPR15 do not efficiently home and prevent colitis. Unlike small intestine tropic homing molecules, which are regulated largely by retinoic acid, GPR15 is TGFβ-dependent and regulated by colonic microbiota, suggesting that the local environment endows Tregs with unique properties that at least in part affect their homing.
Heterogeneity reflecting effector and memory Treg subsets
The discovery early on that CD25 (IL-2Rα) was an excellent, albeit not perfect, marker for Tregs indicated that Tregs express properties of activated T cells (111). When considering Foxp3 expression, which more precisely marks Tregs, typically 70-80% of Tregs are CD25+, firmly establishing the activated phenotype of Tregs. In vivo Tregs express higher levels of CD25 than conventional activated T cells, and this in part reflects the fact the Foxp3 functions to promote CD25 transcription. Most Tregs not only express CD25 but also express CD122 (IL-2Rβ) and CD132 (γc) required for the high affinity IL-2R, which is characteristic of activated T cells. Tregs stringently depend on IL-2 during thymic development and subsequent maintenance in the periphery (112). IL-2R signaling in Tregs results in a classical positive feedback loop with Foxp3, where IL-2-induced STAT5 directly contributes to promote Foxp3 transcription and Foxp3 in turn directly promotes CD25 transcription (4, 37, 39, 40). This positive feedback helps to enforce the Treg suppressive program, which critically depends on Foxp3. In this regard, the functional activity of Foxp3+ CD25neg Tregs is unstable and such cells are prone to lose Foxp3 expression (70, 72, 73).
Constant stimulation with self-antigens, costimulatory molecules, and IL-2 represent important drivers for Tregs to express an activated phenotype. Indeed, Tregs are also characterized by expression of many other activation markers besides the IL-2R, including CD69, CD38, CTLA4, 4-1BB, OX40, CD62Llo, CD45RBlo, GITR, and CD103 (113, 114). Similar to CD25, some of the molecules, e.g. GITR, CTLA4, 4-1BB, CD103, and OX40, are expressed at higher levels on Tregs than conventional T cells, and this property likely endows Tregs with a competitive advantage over autoreactive T cells for accessing their respective ligands, providing a mechanism that would favor immune suppression and tolerance over autoimmunity.
Some markers of T-cell activation on Tregs readily discriminate subpopulation of cells. One that has received considerable attention is CD103, which is expressed on 10-30% of Tregs in secondary lymphoid tissues but is found on 75-85% of Tregs within some tissues sites such as the skin and gut mucosa (115). Gene expression profiling is consistent with CD103+ Tregs favoring migration into tissue sites and expressing enhanced suppressive activity due to high levels of Treg suppressor molecules (116). In line with this finding, CD103+ Tregs showed increased suppressor function when compared to CD103− Tregs in vitro and showed robust suppressive activity when tested in the T-cell-transfer model of colitis (114, 117, 118). In addition, CD103+ but not CD103− Tregs suppressed acute arthritis, consistent with their important role in immune regulation within tissue sites (119). Upon transfer into a lymphopenic environment, many Tregs adopted a CD103+ phenotype and expressed a transcriptional profile that resembled gut Tregs, where most Tregs are CD103+ (116). Thus, the development of CD103+ Tregs in part depends upon high proliferation as reflected as a consequence of lymphopenia or frequent gut antigenic stimulation. All these findings support the notion that CD103 marks a specialized subset of effector Tregs that undergo substantial proliferation. However, besides its role as an adhesion molecule to E-cadherin, it is not known whether CD103 is actively required for suppressive function of this Treg subset.
At the transcriptional level, IRF4 and Blimp-1 are key transcription factors involved in promoting a subpopulation of highly activated effector-phenotypic Tregs (120). Blimp-1+ Tregs are characterized by high expression of CD103, CD44, inducible costimulatory (ICOS), glucocorticoid-induced tumor necrosis factor receptor (GITR), and low expression of CD62L. The expression of Blimp-1 is increased in Tregs stimulated with IL-2 and further upregulated in the presence of inflammatory cytokines, including IL-12, IL-6, and IL-4, consistent with Blimp-1 in regulation of a specialized Treg subpopulation. In the absence of Blimp-1, highly activated Tregs are still readily detected, indicating that Blimp-1 is not required for the development of these effector-phenotypic Tregs, but some of their other activities are impaired, such as production of IL-10. In addition, Blimp-1 regulates Treg homeostasis in mucosal sites. In contrast, these highly activated Tregs are substantially reduced in IRF4-deficient mice, indicating that the development of effector-phenotypic Tregs depends on this transcriptional regulator, which is also important for Th2 immune responses. IRF4 and Blimp-1 have been shown to control a subset of overlapping genes, but IRF4 is required for normal Blimp-1 expression by Tregs. Collectively, these data indicate that the activity of IRF4 is upstream of Blimp-1 in the development of highly activated Tregs.
The pattern of expression of CD44, CD62L, and CD127 (IL-7Rα) by conventional T cells is useful to distinguish naive (CD44lo, CD62Lhi, CD127hi), effector (CD44hi, CD62Llo, CD127lo), effector/memory (Tem) (CD44hi, CD62Llo, CD127hi), and central memory (Tcm) (CD44hi, CD62Lhi, CD127lo) T cells. For Tregs, most are CD44hi and CD127lo, consistent with activated effector cells, although there is heterogeneity in CD62L expression, which is important with respect to the lymphoid versus non-lymphoid localization of Tregs, as discussed above. Notably, there are very few Tregs that are obviously Tem and Tcm based on these markers. However, recent studies are consistent with the presence of a persistent Treg memory pool that is driven by antigen. A sophisticated triple transgenic model was developed where transgenic ovalbumin (OVA)-specific conventional CD4+ T and Treg cells could be followed over time in response to transient expression of OVA in the skin (121). Upon an initial expression of OVA, a pathogenic response toward the skin by the conventional TCR transgenic effector cells was noted that was eventually suppressed by the TCR transgenic Tregs, which showed an activated phenotype. Importantly, in this model system, the transient nature of OVA expression eventually resulted in an OVA-free environment, yet the OVA-specific Tregs persisted in the skin. Upon re-induction of OVA, only a mild form of skin disease was noted and was accompanied by activation, proliferation, and suppression by the OVA-specific persistent Tregs. This study indicates that Tregs exhibit properties of memory cells, i.e. persistence in the absence of antigen and a faster response upon recall. More recently, it has been shown in this system that IL-2 is required to generate effector Tregs, while IL-7 is required for their persistence (122). Evidence for memory Tregs has also been associated with pregnancy (123). Maternal Tregs contribute to maternal-fetal tolerance. Importantly, after delivery, fetus-specific Tregs persisted, and during a second pregnancy they rapidly proliferated in response to the same fetal antigens and promoted tolerance.
The inter-relationship of Treg subsets
Although phenotypic and functional heterogeneity of Tregs are well characterized, the potential relationship between these distinct Treg populations has not been extensively studied. Our laboratory has begun to directly address this issue. One underlying tenet for these studies is that distinct expression of key surface proteins by Tregs marks varied activation states derived from a common precursor as Tregs respond to auto-antigens. This idea implies that Tregs adopt a similar progression upon encountering self-antigen, co-stimulatory, and cytokine signals to move from a relatively quiescent state to a proliferative and more activated state to yield increased number and potency of suppressive Treg cells. Eventually many of the activated Tregs undergo apoptosis to maintain Treg homeostasis, but some Tregs persist as memory cells (Fig. 1). Such a scenario would be analogous to how conventional T cells respond to nominal antigens. The finding that Tregs exhibit properties of effector and memory cells, as discussed above, is in agreement with this view. However, one caveat when considering Treg progression as a result of antigenic stimulation in a normal unmanipulated mouse is that the large majority of Tregs exhibit an activated phenotype whereas most conventional T cells are resting naive T cells.
Fig. 1.

Model of activation and development of Klrg1+ Tregs.
We have initially focused on three markers which discriminate four Tregs subsets, i.e. CD62L, CD69, and Klrg1. When considering conventional T cells, CD62Lhi is represented on naive and central memory T cells, CD62Llo is indicative of effector and effector memory T cells, CD69 represents recently activated T cells, and Klrg1 marks terminally differentiated Teff cells (124-126). With respect to Tregs found in the spleen and lymph nodes, Tregs can be divided into three main subsets, CD62LhiCD69−, CD62loCD69−, and CD62LloCD69+ (127). Although there is mouse to mouse variation, generally each subset individually represents 30% of Tregs, i.e. these subsets comprise 90% of all Tregs in the spleen and lymph nodes. Approximately 10% of the Tregs in these lymphoid tissues are Klrg1+, and most of these are CD69+.
Genome-wide expression profiling was performed for these four Treg subsets as a measure of their relationship between each other. These data showed that CD62LhiCD69− and CD62loCD69− Treg subsets were more related to each other, while CD62LloCD69+ and Klrg1+ subsets were more closely related to each other (127). At one end of the spectrum was CD62Lhi Tregs, which exhibited properties akin to a ‘resting’ lymphoid-tissue residing subset. CD62Lhi Tregs expressed lower levels of proliferative markers such as Ki67, but higher levels of pro-survival molecules such as Bcl-2 (Fig. 1). At the other end of the spectrum was Klrg1+ Tregs, which expressed higher pro-apoptotic molecules, such as caspase-1 and caspase-4, and higher levels of certain Treg suppressive molecules, e.g. granzyme B, Fgl2, IL-10, and IL-35 and chemokine receptors that favor migration into non-lymphoid tissues. We also noted that Blimp-1 expression was primarily expressed in Klrg1+ Tregs. This raises the possibility that Blimp-1 may either be selectively required for the development of Klrg1+ Tregs and/or be required for the regulation of a distinctive effector program associated with these Tregs. Notably, Blimp-1 has been linked to the regulation of IL-10, a Treg suppressive molecule that is highly expressed by this Treg subset (120). The properties of these Klrg1+ Tregs are akin to a highly activated Treg cells that are prone to apoptosis and favor immune regulation within tissue sites. Consistent with this view, CD69+ and Klrg1+ are dominant Treg populations in the lamina propria of the small intestine.
We developed approaches to directly follow the developmental relationship between these Tregs subsets by using adoptive transfer into lymphopenic TCRα−/− or lympho-replete Y3 IL-2Rβ−/− recipients (127). The advantage of TCRα−/− recipients is that donor Tregs are easy to follow through their lymphopenia-driven expansion. This model, however, also requires the co-transfer of conventional Treg-depleted CD4+ T cells as a source of growth and survival factors. An important disadvantage of TCRα−/− recipients is that the rapid lymphopenia-induced Treg proliferation hinders tracking the progression of CFSE-labeled donor cells because CFSE is fully diluted by donor Tregs 3-4 days after transfer. However, Y3 IL-2Rβ−/− recipients do not have this drawback. Y3 is an IL-2Rβ transgene that contains mutations in three key tyrosine residues in the cytoplasmic tail of IL-2Rβ that greatly impairs IL-2-dependent PI3K and STAT5 activation (128). When this transgene was expressed in all T-lineage cells in IL-2Rβ−/− mice, low IL-2-dependent STAT5 signaling associated with the transgene supported Treg development and homeostasis but not IL-2-dependent functions in T-effector and memory cells (128, 129). Y3 IL-2Rβ−/− mice are not lymphopenic, including the Treg compartment, but in a competitive setting, wildtype donor Tregs outcompete recipient-derived Y3 IL-2Rβ−/− Tregs for available IL-2. Thus, the adoptive transfer of wildtype Tregs into Y3 IL-2Rβ−/− recipients results in a greater accumulation of donor Tregs than occurs if wildtype mice are used as recipients. This advantage facilitates identification of the donor Tregs, but their accumulation is not as robust as that detected in TCRα−/− recipients, which allows us to follow Treg subset development as cell division progresses.
Several important observations were made when CFSE-labeled wildtype ‘resting’ CD62Lhi CD69− CD103− Klrg1− Tregs were transferred into Y3 IL-2Rβ−/− mice (127). First, by 3 days post-transfer, virtually all donor Tregs retained the resting phenotype and did not divide, but by 6 days post-transfer, the majority of the donor Tregs fully diluted CFSE, indicative of at least 7-8 cell divisions. This finding suggests that Tregs may undergo a programming step upon encountering self-antigen and environmental cues. Upon integration of these signals, Tregs undergo a rapid proliferative burst. Second, accompanying Treg proliferation, progressive loss of the resting phenotype was noted and activated Tregs accumulated, characterized by downregulation of CD62L and upregulation of CD69, CD103, and Klrg1. Third, the appearance of CD103+ and especially Klrg1+ Tregs was observed only after at least 7-8 cell divisions. Collectively, these findings indicate that the engrafted resting Tregs respond in a lymphoreplete environment and give rise to activated Tregs in a manner analogous to conventional T cells and that expression of Klrg1 represents a late stage of this response (Fig. 1).
We were also interested in the question of what is the capacity of activated Tregs to adopt a resting CD62Lhi cell fate. To begin to evaluate this relationship between Treg subsets, highly purified CD62Lhi CD69− (Fr1), CD62lo CD69− (Fr2), and CD62LloCD69+ (Fr3) Treg subsets, all depleted of Klrg1+ Tregs, from Foxp3/RFP reporter mice were adoptively transferred into TCRα−/− recipients with conventional CD4+ T cells, which provide growth/survival signals, as discussed above. Three to four weeks post-transfer, donor Treg and T-conventional cells were detected in the spleen and MLN. Analysis of the spleen indicated that there was approximately a 12-15 fold expansion of the donor Treg and T-conventional cells (Fig. 2A). The fraction of RFP+ donor Tregs, therefore, still remained approximately 5-10% of the total donor pool of CD4+ T cells (Fig. 2B). Phenotypic analysis of the RFP+ Tregs revealed heterogeneity in expression of CD62L and CD69 for each fraction of donor Tregs, even though the input Tregs were homogenous with respect to the expression of the markers used to select the input cells (Fig. 2C, top). CD62Llo CD69+ Tregs dominated in all recipients, including those recipients that received resting RFP+ CD62Lhi CD69− (Fr1) Tregs, further supporting the notion that resting CD62Lhi Tregs include precursors to activated Tregs. In contrast, most ‘activated’ RFP+ Fr2 and Fr3 Tregs remained as activated cells and did not substantially move towards a resting CD62Lhi phenotype, suggesting that activated Tregs do not have the potential to readily develop into central memory-like cells. Donor RFP+ Tregs readily expressed Klrg1 in all recipients (Fig. 2C, bottom). Thus, these data also show that Klrg1+ Tregs readily develop from Klrg1− Treg cells. A small population of resting CD62Lhi Tregs was detected in all recipients, which fits a scenario that a few central memory-like Tregs may also develop from activated Tregs.
Fig. 2. Inter-relationship between Treg subsets.
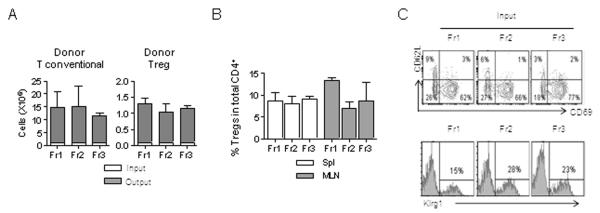
Adult TCRα−/− mice were adoptively transferred with a mixture (1:10) of RFP+ Tregs from purified subsets (Fr1-3, Klrg1−) and Treg-depleted conventional CD4+ T cells. The persistence and phenotype of the donor cells were assessed 3-4 weeks post-transfer. (A) Expansion of the donor T-conventional and Treg cells where the total numbers of injected (input) and recovered (output) cells from the spleen were enumerated. (B) Spleen and MLN were examined for donor Tregs as a fraction of the total CD4+ T-cell compartment. (C) The phenotype of donor Tregs from recipients that received the indicated fractions of Klrg1-depleted Fr1, Fr2, or Fr3 RFP+ donor Treg cells. Shown at the top is the expression of CD62L and CD69 and at the bottom is the expression of Klrg1 by donor Tregs.
Expression of Klrg1 marks short-lived terminally differentiated Tregs
Klrg1 has been primarily studied in NK and CD8+ T cells. Klrg1 is detected on these cells only after they have undergone extensive proliferation and marks cells with replicative senescence (125, 126, 130-132). Klrg1 is associated with mature NK cells with poor proliferative potential that arise from Klrg1− NK cells (133, 134). During a CD8+ T-cell immune response, Klrg1 expression is associated with short-lived highly activated Teffs (125, 126, 132). Analogous to Klrg1+ CD8+ T cells, Klrg1+ Tregs have a gene expression profile that is consistent with cells that exhibit highly suppressive, proliferative, and apoptotic activity. Moreover, we performed direct studies to test whether Klrg1+ Tregs are indeed short-lived terminally differentiated Tregs (127). First, Klrg1+ Tregs turnover faster than Klrg1− Tregs in vivo as assessed by BrdU-labeling experiments and exhibited increased suppressive activity in vitro. Second and more importantly, Klrg1+ Tregs did not expand and poorly survived upon adoptive transfer into TCRα−/− or Y3 IL-2Rβ−/− recipients, indicating that these Tregs are short-lived, even though they show high proliferative levels at the steady state. For the few Klrg1+ Tregs detected in these recipients, they remained Klrg1+ and stably expressed Foxp3. These data indicate that once Klrg1 is expressed on Tregs, most of these cells quickly become exhausted and are in a terminally differentiated state. Collectively, the late developmental progression, the gene expression profile, the preference for non-lymphoid tissue residence, and these direct studies indicate that Klrg1 marks highly functional, short-lived terminally differentiated Tregs that act predominantly to maintain tolerance and homeostasis in non-lymphoid tissues.
Signaling through the IL-2R represents one important requirement for the development of Klrg1+ Tregs. This was first suggested from genome-wide profiling of splenic Tregs with normal and impaired IL-2R signaling, where the level of Klrg1 was one of the most differentially and highly expressed mRNAs by wildtype Tregs (128). Analysis of Tregs with impaired IL-2R signaling showed that Klrg1+ Tregs were highly diminished. This finding was most striking for the lamina propria of the small intestine, where Klrg1+ Tregs comprise approximately 50% of the Tregs in wildtype mice but only about 5% in IL-2R signaling impaired mice (127). Interestingly, direct assessment of the effect of agonist IL-2/anti-IL-2 complex on Klrg1+ and Klrg1− Tregs showed that only the latter cells respond to IL-2. Thus, IL-2 acts on Klrg1− cells to drive the development of Klrg1+ Tregs, but the Klrg1+ Tregs remain senescent and non-proliferative to IL-2. This finding also has important implications concerning the mechanisms in play with respect to IL-2-dependent Treg homeostasis (Fig. 1). Recent studies indicate that expression of pro-survival Mcl-2 by IL-2 contributes to Treg homeostasis (135). However, IL-2-dependent development of Klrg1+ Tregs is also likely an important control point for Treg homeostasis. Klrg1− Tregs respond to IL-2, which contributes to their proliferation. After perhaps >8 cell divisions, Tregs progress and develop into Klrg1+ cells. The Klrg1+ Tregs quickly cease to proliferate, are non-responsive to IL-2, and will die in a short-period of time. Thus, even though Tregs are highly proliferative cells in vivo at the steady state, the death of Klrg1+ Tregs provides a mechanism to link IL-2R signaling to limit and balance Treg numbers, maintaining homeostasis.
Concluding remarks
Current work makes plain that Foxp3+ Tregs in the periphery are mostly activated T cells that exhibit substantial heterogeneity in a manner very analogous to developing and polarized Th subsets. Treg functional and migratory diversity provides a mechanism to optimally match suppressive Tregs to particular T-effector responses. Tregs also exhibit diversity that in part reflects the response to self and nominal antigenic stimulation and other signals to select Tregs with appropriate specificities and expand them to sufficient numbers to regulate T-effector responses. This process requires mechanisms to limit the Treg response, such as development of Klrg1+ Tregs, and produce persistent memory Tregs. Both are important for immune homeostasis, as the former promotes Treg homeostasis and the latter promotes homeostasis of the T-conventional compartment. With this understanding, there remain open a number of key questions. Several that we consider are as follows: what signals promote Treg effector versus memory responses, to what degree are resting and activated Treg subsets related to each other or represent specialized subsets of cells, and to what extent does some of the heterogeneity of Tregs defined using sophisticated mouse models apply to the human immune system?
Acknowledgments
Our work is supported by grants from the National Institutes of Health (R01 AI055815 and R01 DK093866).
Footnotes
The authors declare no competing financial interests
References
- 1.Sakaguchi S. Naturally arising CD4+ regulatory T cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol. 2004;22:531–562. doi: 10.1146/annurev.immunol.21.120601.141122. [DOI] [PubMed] [Google Scholar]
- 2.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 3.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. [PubMed] [Google Scholar]
- 4.Gavin MA, et al. Foxp3-dependent programme of regulatory T-cell differentiation. Nature. 2007;445:771–775. doi: 10.1038/nature05543. [DOI] [PubMed] [Google Scholar]
- 5.Lin W, et al. Regulatory T cell development in the absence of functional Foxp3. Nat Immunol. 2007;8:359–368. doi: 10.1038/ni1445. [DOI] [PubMed] [Google Scholar]
- 6.Marson A, et al. Foxp3 occupancy and regulation of key target genes during T-cell stimulation. Nature. 2007;445:931–935. doi: 10.1038/nature05478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wan YY, Flavell RA. Regulatory T-cell functions are subverted and converted owing to attenuated Foxp3 expression. Nature. 2007;445:766–770. doi: 10.1038/nature05479. [DOI] [PubMed] [Google Scholar]
- 8.Williams LM, Rudensky AY. Maintenance of the Foxp3-dependent developmental program in mature regulatory T cells requires continued expression of Foxp3. Nat Immunol. 2007;8:277–284. doi: 10.1038/ni1437. [DOI] [PubMed] [Google Scholar]
- 9.Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol. 2007;8:191–197. doi: 10.1038/ni1428. [DOI] [PubMed] [Google Scholar]
- 10.Lahl K, et al. Selective depletion of Foxp3+ regulatory T cells induces a scurfy-like disease. J Exp Med. 2007;204:57–63. doi: 10.1084/jem.20061852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Demengeot J, Zelenay S, Moraes-Fontes MF, Caramalho I, Coutinho A. Regulatory T cells in microbial infection. Springer Semin Immunopathol. 2006;28:41–50. doi: 10.1007/s00281-006-0024-5. [DOI] [PubMed] [Google Scholar]
- 12.Wohlfert E, Belkaid Y. Role of endogenous and induced regulatory T cells during infections. J Clin Immunol. 2008;28:707–715. doi: 10.1007/s10875-008-9248-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barnes MJ, Powrie F. Regulatory T cells reinforce intestinal homeostasis. Immunity. 2009;31:401–411. doi: 10.1016/j.immuni.2009.08.011. [DOI] [PubMed] [Google Scholar]
- 14.Weiner HL, da Cunha AP, Quintana F, Wu H. Oral tolerance. Immunol Rev. 2011;241:241–259. doi: 10.1111/j.1600-065X.2011.01017.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adeegbe DO, Nishikawa H. Natural and induced T regulatory cells in cancer. Front Immunol. 2013;4:190. doi: 10.3389/fimmu.2013.00190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beres AJ, Drobyski WR. The role of regulatory T cells in the biology of graft versus host disease. Front Immunol. 2013;4:163. doi: 10.3389/fimmu.2013.00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Veiga-Parga T, Sehrawat S, Rouse BT. Role of regulatory T cells during virus infection. Immunol Rev. 2013;255:182–196. doi: 10.1111/imr.12085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsieh CS, Liang Y, Tyznik AJ, Self SG, Liggitt D, Rudensky AY. Recognition of the peripheral self by naturally arising CD25+ CD4+ T cell receptors. Immunity. 2004;21:267–277. doi: 10.1016/j.immuni.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 19.Pacholczyk R, Ignatowicz H, Kraj P, Ignatowicz L. Origin and T cell receptor diversity of Foxp3+CD4+CD25+ T cells. Immunity. 2006;25:249–259. doi: 10.1016/j.immuni.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 20.Wong J, Obst R, Correia-Neves M, Losyev G, Mathis D, Benoist C. Adaptation of TCR repertoires to self-peptides in regulatory and nonregulatory CD4+ T cells. J Immunol. 2007;178:7032–7041. doi: 10.4049/jimmunol.178.11.7032. [DOI] [PubMed] [Google Scholar]
- 21.Fontenot JD, Dooley JL, Farr AG, Rudensky AY. Developmental regulation of Foxp3 expression during ontogeny. J Exp Med. 2005;202:901–906. doi: 10.1084/jem.20050784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aschenbrenner K, et al. Selection of Foxp3+ regulatory T cells specific for self antigen expressed and presented by Aire+ medullary thymic epithelial cells. Nat Immunol. 2007;8:351–358. doi: 10.1038/ni1444. [DOI] [PubMed] [Google Scholar]
- 23.Jordan MS, et al. Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat Immunol. 2001;2:301–306. doi: 10.1038/86302. [DOI] [PubMed] [Google Scholar]
- 24.Apostolou I, Sarukhan A, Klein L, von Boehmer H. Origin of regulatory T cells with known specificity for antigen. Nat Immunol. 2002;3:756–763. doi: 10.1038/ni816. [DOI] [PubMed] [Google Scholar]
- 25.Kawahata K, et al. Generation of CD4+CD25+ regulatory T cells from autoreactive T cells simultaneously with their negative selection in the thymus and from nonautoreactive T cells by endogenous TCR expression. J Immunol. 2002;168:4399–4405. doi: 10.4049/jimmunol.168.9.4399. [DOI] [PubMed] [Google Scholar]
- 26.Hsieh CS, Zheng Y, Liang Y, Fontenot JD, Rudensky AY. An intersection between the self-reactive regulatory and nonregulatory T cell receptor repertoires. Nat Immunol. 2006;7:401–410. doi: 10.1038/ni1318. [DOI] [PubMed] [Google Scholar]
- 27.Yuan X, Malek TR. Cellular and molecular determinants for the development of natural and induced regulatory T cells. Hum Immunol. 2012;73:773–782. doi: 10.1016/j.humimm.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mantel PY, et al. Molecular mechanisms underlying FOXP3 induction in human T cells. J Immunol. 2006;176:3593–3602. doi: 10.4049/jimmunol.176.6.3593. [DOI] [PubMed] [Google Scholar]
- 29.Long M, Park SG, Strickland I, Hayden MS, Ghosh S. Nuclear factor-κB modulates regulatory T cell development by directly regulating expression of Foxp3 transcription factor. Immunity. 2009;31:921–931. doi: 10.1016/j.immuni.2009.09.022. [DOI] [PubMed] [Google Scholar]
- 30.Molinero LL, Yang J, Gajewski T, Abraham C, Farrar MA, Alegre ML. CARMA1 controls an early checkpoint in the thymic development of FoxP3+ regulatory T cells. J Immunol. 2009;182:6736–6743. doi: 10.4049/jimmunol.0900498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ruan Q, et al. Development of Foxp3+ regulatory t cells is driven by the c-Rel enhanceosome. Immunity. 2009;31:932–940. doi: 10.1016/j.immuni.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K, Rudensky AY. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature. 2010;463:808–812. doi: 10.1038/nature08750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol. 2005;6:1142–1151. doi: 10.1038/ni1263. [DOI] [PubMed] [Google Scholar]
- 34.Marie JC, Letterio JJ, Gavin M, Rudensky AY. TGF-β1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J Exp Med. 2005;201:1061–1067. doi: 10.1084/jem.20042276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li MO, Sanjabi S, Flavell RA. Transforming growth factor-β controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity. 2006;25:455–471. doi: 10.1016/j.immuni.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 36.Bayer AL, Yu A, Malek TR. Function of the IL-2R for thymic and peripheral CD4+CD25+ Foxp3+ T regulatory cells. J Immunol. 2007;178:4062–4071. doi: 10.4049/jimmunol.178.7.4062. [DOI] [PubMed] [Google Scholar]
- 37.Burchill MA, Yang J, Vogtenhuber C, Blazar BR, Farrar MA. IL-2 receptor β-dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J Immunol. 2007;178:280–290. doi: 10.4049/jimmunol.178.1.280. [DOI] [PubMed] [Google Scholar]
- 38.Yao Z, et al. Nonredundant roles for Stat5a/b in directly regulating Foxp3. Blood. 2007;109:4368–4375. doi: 10.1182/blood-2006-11-055756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Burchill MA, et al. Linked T cell receptor and cytokine signaling govern the development of the regulatory T cell repertoire. Immunity. 2008;28:112–121. doi: 10.1016/j.immuni.2007.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lio CW, Hsieh CS. A two-step process for thymic regulatory T cell development. Immunity. 2008;28:100–111. doi: 10.1016/j.immuni.2007.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ouyang W, Beckett O, Ma Q, Li MO. Transforming growth factor-β signaling curbs thymic negative selection promoting regulatory T cell development. Immunity. 2010;32:642–653. doi: 10.1016/j.immuni.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheng G, Yu A, Dee MJ, Malek TR. IL-2R signaling is essential for functional maturation of regulatory T cells during thymic development. J Immunol. 2013;190:1567–1575. doi: 10.4049/jimmunol.1201218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Josefowicz SZ, et al. Extrathymically generated regulatory T cells control mucosal TH2 inflammation. Nature. 2012;482:395–399. doi: 10.1038/nature10772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Samstein RM, Josefowicz SZ, Arvey A, Treuting PM, Rudensky AY. Extrathymic generation of regulatory T cells in placental mammals mitigates maternal-fetal conflict. Cell. 2012;150:29–38. doi: 10.1016/j.cell.2012.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kretschmer K, Apostolou I, Hawiger D, Khazaie K, Nussenzweig MC, von Boehmer H. Inducing and expanding regulatory T cell populations by foreign antigen. Nat Immunol. 2005;6:1219–1227. doi: 10.1038/ni1265. [DOI] [PubMed] [Google Scholar]
- 46.Mucida D, Kutchukhidze N, Erazo A, Russo M, Lafaille JJ, Curotto de Lafaille MA. Oral tolerance in the absence of naturally occurring Tregs. J Clin Invest. 2005;115:1923–1933. doi: 10.1172/JCI24487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tone Y, Furuuchi K, Kojima Y, Tykocinski ML, Greene MI, Tone M. Smad3 and NFAT cooperate to induce Foxp3 expression through its enhancer. Nat Immunol. 2008;9:194–202. doi: 10.1038/ni1549. [DOI] [PubMed] [Google Scholar]
- 48.Josefowicz SZ, Wilson CB, Rudensky AY. Cutting edge: TCR stimulation is sufficient for induction of Foxp3 expression in the absence of DNA methyltransferase 1. J Immunol. 2009;182:6648–6652. doi: 10.4049/jimmunol.0803320. [DOI] [PubMed] [Google Scholar]
- 49.Benson MJ, Pino-Lagos K, Rosemblatt M, Noelle RJ. All-trans retinoic acid mediates enhanced T reg cell growth, differentiation, and gut homing in the face of high levels of co-stimulation. J Exp Med. 2007;204:1765–1774. doi: 10.1084/jem.20070719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Coombes JL, et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-β and retinoic acid-dependent mechanism. J Exp Med. 2007;204:1757–1764. doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Denning TL, Wang YC, Patel SR, Williams IR, Pulendran B. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nat Immunol. 2007;8:1086–1094. doi: 10.1038/ni1511. [DOI] [PubMed] [Google Scholar]
- 52.Mucida D, et al. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science. 2007;317:256–260. doi: 10.1126/science.1145697. [DOI] [PubMed] [Google Scholar]
- 53.Sun CM, et al. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med. 2007;204:1775–1785. doi: 10.1084/jem.20070602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Soroosh P, et al. Lung-resident tissue macrophages generate Foxp3+ regulatory T cells and promote airway tolerance. J Exp Med. 2013;210:775–788. doi: 10.1084/jem.20121849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Round JL, Mazmanian SK. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci USA. 2010;107:12204–12209. doi: 10.1073/pnas.0909122107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Atarashi K, et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science. 2011;331:337–341. doi: 10.1126/science.1198469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Atarashi K, et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature. 2013;500:232–236. doi: 10.1038/nature12331. [DOI] [PubMed] [Google Scholar]
- 58.Lathrop SK, et al. Peripheral education of the immune system by colonic commensal microbiota. Nature. 2011;478:250–254. doi: 10.1038/nature10434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cebula A, et al. Thymus-derived regulatory T cells contribute to tolerance to commensal microbiota. Nature. 2013;497:258–262. doi: 10.1038/nature12079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Haribhai D, et al. A requisite role for induced regulatory T cells in tolerance based on expanding antigen receptor diversity. Immunity. 2011;35:109–122. doi: 10.1016/j.immuni.2011.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Thornton AM, et al. Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J Immunol. 2010;184:3433–3441. doi: 10.4049/jimmunol.0904028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Weiss JM, et al. Neuropilin 1 is expressed on thymus-derived natural regulatory T cells, but not mucosa-generated induced Foxp3+ T reg cells. J Exp Med. 2012;209:1723–1742. S1721. doi: 10.1084/jem.20120914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yadav M, et al. Neuropilin-1 distinguishes natural and inducible regulatory T cells among regulatory T cell subsets in vivo. J Exp Med. 2012;209:1713–1722. S1711–1719. doi: 10.1084/jem.20120822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Akimova T, Beier UH, Wang L, Levine MH, Hancock WW. Helios expression is a marker of T cell activation and proliferation. PloS One. 2011;6:e24226. doi: 10.1371/journal.pone.0024226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gottschalk RA, Corse E, Allison JP. Expression of Helios in peripherally induced Foxp3+ regulatory T cells. J Immunol. 2012;188:976–980. doi: 10.4049/jimmunol.1102964. [DOI] [PubMed] [Google Scholar]
- 66.Yadav M, Stephan S, Bluestone JA. Peripherally induced Tregs - role in immune homeostasis and autoimmunity. Front Immunol. 2013;4:232. doi: 10.3389/fimmu.2013.00232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Floess S, et al. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol. 2007;5:e38. doi: 10.1371/journal.pbio.0050038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Polansky JK, et al. DNA methylation controls Foxp3 gene expression. Eur J Immunol. 2008;38:1654–1663. doi: 10.1002/eji.200838105. [DOI] [PubMed] [Google Scholar]
- 69.Huehn J, Polansky JK, Hamann A. Epigenetic control of FOXP3 expression: the key to a stable regulatory T-cell lineage? Nat Rev Immunol. 2009;9:83–89. doi: 10.1038/nri2474. [DOI] [PubMed] [Google Scholar]
- 70.Zhou X, et al. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat Immunol. 2009;10:1000–1007. doi: 10.1038/ni.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yang XO, et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity. 2008;29:44–56. doi: 10.1016/j.immuni.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Duarte JH, Zelenay S, Bergman ML, Martins AC, Demengeot J. Natural Treg cells spontaneously differentiate into pathogenic helper cells in lymphopenic conditions. Eur J Immunol. 2009;39:948–955. doi: 10.1002/eji.200839196. [DOI] [PubMed] [Google Scholar]
- 73.Komatsu N, Mariotti-Ferrandiz ME, Wang Y, Malissen B, Waldmann H, Hori S. Heterogeneity of natural Foxp3+ T cells: a committed regulatory T-cell lineage and an uncommitted minor population retaining plasticity. Proc Natl Acad Sci USA. 2009;106:1903–1908. doi: 10.1073/pnas.0811556106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tsuji M, et al. Preferential generation of follicular B helper T cells from Foxp3+ T cells in gut Peyer’s patches. Science. 2009;323:1488–1492. doi: 10.1126/science.1169152. [DOI] [PubMed] [Google Scholar]
- 75.Shevach EM. Mechanisms of Foxp3+ T regulatory cell-mediated suppression. Immunity. 2009;30:636–645. doi: 10.1016/j.immuni.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 76.Yamaguchi T, Wing JB, Sakaguchi S. Two modes of immune suppression by Foxp3+ regulatory T cells under inflammatory or non-inflammatory conditions. Semin Immunol. 2011;23:424–430. doi: 10.1016/j.smim.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 77.Schmidt A, Oberle N, Krammer PH. Molecular mechanisms of Treg-mediated T cell suppression. Front Immunol. 2012;3:51. doi: 10.3389/fimmu.2012.00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lund JM, Hsing L, Pham TT, Rudensky AY. Coordination of early protective immunity to viral infection by regulatory T cells. Science. 2008;320:1220–1224. doi: 10.1126/science.1155209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wing K, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322:271–275. doi: 10.1126/science.1160062. [DOI] [PubMed] [Google Scholar]
- 80.Rubtsov YP, et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity. 2008;28:546–558. doi: 10.1016/j.immuni.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 81.Feuerer M, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med. 2009;15:930–939. doi: 10.1038/nm.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cipolletta D, et al. PPAR-γ is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature. 2012;486:549–553. doi: 10.1038/nature11132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*) Annu Rev Immunol. 2010;28:445–489. doi: 10.1146/annurev-immunol-030409-101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chaudhry A, et al. CD4+ regulatory T cells control TH17 responses in a Stat3-dependent manner. Science. 2009;326:986–991. doi: 10.1126/science.1172702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol. 2009;10:595–602. doi: 10.1038/ni.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zheng Y, et al. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature. 2009;458:351–356. doi: 10.1038/nature07674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chaudhry A, et al. Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity. 2011;34:566–578. doi: 10.1016/j.immuni.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hall AO, et al. The cytokines interleukin 27 and interferon-γ promote distinct Treg cell populations required to limit infection-induced pathology. Immunity. 2012;37:511–523. doi: 10.1016/j.immuni.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Koch MA, Thomas KR, Perdue NR, Smigiel KS, Srivastava S, Campbell DJ. T-bet+ Treg cells undergo abortive Th1 cell differentiation due to impaired expression of IL-12 receptor β2. Immunity. 2012;37:501–510. doi: 10.1016/j.immuni.2012.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Breitfeld D, et al. Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J Exp Med. 2000;192:1545–1552. doi: 10.1084/jem.192.11.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Johnston RJ, et al. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science. 2009;325:1006–1010. doi: 10.1126/science.1175870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nurieva RI, et al. Bcl6 mediates the development of T follicular helper cells. Science. 2009;325:1001–1005. doi: 10.1126/science.1176676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yu D, et al. The transcriptional repressor Bcl-6 directs T follicular helper cell lineage commitment. Immunity. 2009;31:457–468. doi: 10.1016/j.immuni.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 94.Chung Y, et al. Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat Med. 2011;17:983–988. doi: 10.1038/nm.2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Linterman MA, et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat Med. 2011;17:975–982. doi: 10.1038/nm.2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wollenberg I, et al. Regulation of the germinal center reaction by Foxp3+ follicular regulatory T cells. J Immunol. 2011;187:4553–4560. doi: 10.4049/jimmunol.1101328. [DOI] [PubMed] [Google Scholar]
- 97.Lee JH, Kang SG, Kim CH. FoxP3+ T cells undergo conventional first switch to lymphoid tissue homing receptors in thymus but accelerated second switch to nonlymphoid tissue homing receptors in secondary lymphoid tissues. J Immunol. 2007;178:301–311. doi: 10.4049/jimmunol.178.1.301. [DOI] [PubMed] [Google Scholar]
- 98.Hamann A, Andrew DP, Jablonski-Westrich D, Holzmann B, Butcher EC. Role of α4-integrins in lymphocyte homing to mucosal tissues in vivo. J Immunol. 1994;152:3282–3293. [PubMed] [Google Scholar]
- 99.Svensson M, et al. CCL25 mediates the localization of recently activated CD8αβ+ lymphocytes to the small-intestinal mucosa. J Clin Invest. 2002;110:1113–1121. doi: 10.1172/JCI15988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sather BD, et al. Altering the distribution of Foxp3+ regulatory T cells results in tissue-specific inflammatory disease. J Exp Med. 2007;204:1335–1347. doi: 10.1084/jem.20070081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wurbel MA, Malissen M, Guy-Grand D, Malissen B, Campbell JJ. Impaired accumulation of antigen-specific CD8 lymphocytes in chemokine CCL25-deficient intestinal epithelium and lamina propria. J Immunol. 2007;178:7598–7606. doi: 10.4049/jimmunol.178.12.7598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wright N, et al. The chemokine stromal cell-derived factor-1α modulates α4β7 integrin-mediated lymphocyte adhesion to mucosal addressin cell adhesion molecule-1 and fibronectin. J Immunol. 2002;168:5268–5277. doi: 10.4049/jimmunol.168.10.5268. [DOI] [PubMed] [Google Scholar]
- 103.Annunziato F, et al. CXCR3 and αEβ7 integrin identify a subset of CD8+ mature thymocytes that share phenotypic and functional properties with CD8+ gut intraepithelial lymphocytes. Gut. 2006;55:961–968. doi: 10.1136/gut.2005.077560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Oyama T, et al. CXCL12 and CCL20 play a significant role in mucosal T-lymphocyte adherence to intestinal microvessels in mice. Microcirculation. 2007;14:753–766. doi: 10.1080/10739680701409993. [DOI] [PubMed] [Google Scholar]
- 105.Wagner N, et al. Critical role for β7 integrins in formation of the gut-associated lymphoid tissue. Nature. 1996;382:366–370. doi: 10.1038/382366a0. [DOI] [PubMed] [Google Scholar]
- 106.Lefrancois L, et al. The role of β7 integrins in CD8 T cell trafficking during an antiviral immune response. J Exp Med. 1999;189:1631–1638. doi: 10.1084/jem.189.10.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Schon MP, et al. Mucosal T lymphocyte numbers are selectively reduced in integrin αE (CD103)-deficient mice. J Immunol. 1999;162:6641–6649. [PubMed] [Google Scholar]
- 108.Casey KA, et al. Antigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. J Immunol. 2012;188:4866–4875. doi: 10.4049/jimmunol.1200402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Denning TL, Kim G, Kronenberg M. Cutting edge: CD4+CD25+ regulatory T cells impaired for intestinal homing can prevent colitis. J Immunol. 2005;174:7487–7491. doi: 10.4049/jimmunol.174.12.7487. [DOI] [PubMed] [Google Scholar]
- 110.Kim SV, et al. GPR15-mediated homing controls immune homeostasis in the large intestine mucosa. Science. 2013;340:1456–1459. doi: 10.1126/science.1237013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor α-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151–1164. [PubMed] [Google Scholar]
- 112.Malek TR. The biology of interleukin-2. Annu Rev Immunol. 2008;26:453–479. doi: 10.1146/annurev.immunol.26.021607.090357. [DOI] [PubMed] [Google Scholar]
- 113.Thornton AM, Shevach EM. Suppressor effector function of CD4+CD25+ immunoregulatory T cells is antigen nonspecific. J Immunol. 2000;164:183–190. doi: 10.4049/jimmunol.164.1.183. [DOI] [PubMed] [Google Scholar]
- 114.McHugh RS, et al. CD4+CD25+ immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity. 2002;16:311–323. doi: 10.1016/s1074-7613(02)00280-7. [DOI] [PubMed] [Google Scholar]
- 115.Suffia I, Reckling SK, Salay G, Belkaid Y. A role for CD103 in the retention of CD4+CD25+ Treg and control of Leishmania major infection. J Immunol. 2005;174:5444–5455. doi: 10.4049/jimmunol.174.9.5444. [DOI] [PubMed] [Google Scholar]
- 116.Feuerer M, Hill JA, Kretschmer K, von Boehmer H, Mathis D, Benoist C. Genomic definition of multiple ex vivo regulatory T cell subphenotypes. Proc Natl Acad Sci USA. 2010;107:5919–5924. doi: 10.1073/pnas.1002006107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lehmann J, et al. Expression of the integrin αEβ7 identifies unique subsets of CD25+ as well as CD25− regulatory T cells. Proc Natl Acad Sci USA. 2002;99:13031–13036. doi: 10.1073/pnas.192162899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Banz A, Peixoto A, Pontoux C, Cordier C, Rocha B, Papiernik M. A unique subpopulation of CD4+ regulatory T cells controls wasting disease, IL-10 secretion and T cell homeostasis. Eur J Immunol. 2003;33:2419–2428. doi: 10.1002/eji.200324205. [DOI] [PubMed] [Google Scholar]
- 119.Huehn J, et al. Developmental stage, phenotype, and migration distinguish naive- and effector/memory-like CD4+ regulatory T cells. J Exp Med. 2004;199:303–313. doi: 10.1084/jem.20031562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cretney E, et al. The transcription factors Blimp-1 and IRF4 jointly control the differentiation and function of effector regulatory T cells. Nat Immunol. 2011;12:304–311. doi: 10.1038/ni.2006. [DOI] [PubMed] [Google Scholar]
- 121.Rosenblum MD, Gratz IK, Paw JS, Lee K, Marshak-Rothstein A, Abbas AK. Response to self antigen imprints regulatory memory in tissues. Nature. 2011;480:538–542. doi: 10.1038/nature10664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gratz IK, et al. Cutting Edge: memory regulatory t cells require IL-7 and not IL-2 for their maintenance in peripheral tissues. J Immunol. 2013;190:4483–4487. doi: 10.4049/jimmunol.1300212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rowe JH, Ertelt JM, Xin L, Way SS. Pregnancy imprints regulatory memory that sustains anergy to fetal antigen. Nature. 2012;490:102–106. doi: 10.1038/nature11462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Beyersdorf NB, Ding X, Karp K, Hanke T. Expression of inhibitory “killer cell lectin-like receptor G1” identifies unique subpopulations of effector and memory CD8 T cells. Eur J Immunol. 2001;31:3443–3452. doi: 10.1002/1521-4141(200112)31:12<3443::aid-immu3443>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 125.Voehringer D, Blaser C, Brawand P, Raulet DH, Hanke T, Pircher H. Viral infections induce abundant numbers of senescent CD8 T cells. J Immunol. 2001;167:4838–4843. doi: 10.4049/jimmunol.167.9.4838. [DOI] [PubMed] [Google Scholar]
- 126.Joshi NS, et al. Inflammation directs memory precursor and short-lived effector CD8+ T cell fates via the graded expression of T-bet transcription factor. Immunity. 2007;27:281–295. doi: 10.1016/j.immuni.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Cheng G, Yuan X, Tsai MS, Podack ER, Yu A, Malek TR. IL-2 receptor signaling is essential for the development of Klrg1+ terminally differentiated T regulatory cells. J Immunol. 2012;189:1780–1791. doi: 10.4049/jimmunol.1103768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yu A, Zhu L, Altman NH, Malek TR. A low interleukin-2 receptor signaling threshold supports the development and homeostasis of T regulatory cells. Immunity. 2009;30:204–217. doi: 10.1016/j.immuni.2008.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Castro I, Yu A, Dee MJ, Malek TR. The basis of distinctive IL-2- and IL-15-dependent signaling: weak CD122-dependent signaling favors CD8+ T central-memory cell survival but not T effector-memory cell development. J Immunol. 2011;187:5170–5182. doi: 10.4049/jimmunol.1003961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Badovinac VP, Harty JT. Manipulating the rate of memory CD8+ T cell generation after acute infection. J Immunol. 2007;179:53–63. doi: 10.4049/jimmunol.179.1.53. [DOI] [PubMed] [Google Scholar]
- 131.Rubinstein MP, et al. IL-7 and IL-15 differentially regulate CD8+ T-cell subsets during contraction of the immune response. Blood. 2008;112:3704–3712. doi: 10.1182/blood-2008-06-160945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wilson DC, Matthews S, Yap GS. IL-12 signaling drives CD8+ T cell IFN-γ production and differentiation of KLRG1+ effector subpopulations during Toxoplasma gondii Infection. J Immunol. 2008;180:5935–5945. doi: 10.4049/jimmunol.180.9.5935. [DOI] [PubMed] [Google Scholar]
- 133.Robbins SH, Nguyen KB, Takahashi N, Mikayama T, Biron CA, Brossay L. Cutting edge: inhibitory functions of the killer cell lectin-like receptor G1 molecule during the activation of mouse NK cells. J Immunol. 2002;168:2585–2589. doi: 10.4049/jimmunol.168.6.2585. [DOI] [PubMed] [Google Scholar]
- 134.Robbins SH, Tessmer MS, Mikayama T, Brossay L. Expansion and contraction of the NK cell compartment in response to murine cytomegalovirus infection. J Immunol. 2004;173:259–266. doi: 10.4049/jimmunol.173.1.259. [DOI] [PubMed] [Google Scholar]
- 135.Pierson W, et al. Antiapoptotic Mcl-1 is critical for the survival and niche-filling capacity of Foxp3+ regulatory T cells. Nat Immunol. 2013;14:959–965. doi: 10.1038/ni.2649. [DOI] [PMC free article] [PubMed] [Google Scholar]