

Key Points
Targeting of CD123 via CAR-engineered T cells results in rejection of human AML and myeloablation in mouse models.
Abstract
Many patients with acute myeloid leukemia (AML) are incurable with chemotherapy and may benefit from novel approaches. One such approach involves the transfer of T cells engineered to express chimeric antigen receptors (CARs) for a specific cell-surface antigen. This strategy depends upon preferential expression of the target on tumor cells. To date, the lack of AML-specific surface markers has impeded development of such CAR-based approaches. CD123, the transmembrane α chain of the interleukin-3 receptor, is expressed in the majority of AML cells but is also expressed in many normal hematopoietic cells. Here, we show that CD123 is a good target for AML-directed CAR therapy, because its expression increases over time in vivo even in initially CD123dim populations, and that human CD123-redirected T cells (CART123) eradicate primary AML in immunodeficient mice. CART123 also eradicated normal human myelopoiesis, a surprising finding because anti-CD123 antibody-based strategies have been reportedly well tolerated. Because AML is likely preceded by clonal evolution in “preleukemic” hematopoietic stem cells, our observations support CART123 as a viable AML therapy, suggest that CART123-based myeloablation may be used as a novel conditioning regimen for hematopoietic cell transplantation, and raise concerns for the use of CART123 without such a rescue strategy.
Introduction
The standard treatment of acute myeloid leukemia (AML) has changed little in the past 30 years. In contrast with other hematologic malignancies, few novel agents have been successfully developed for AML. Despite an initially high complete response rate, many patients relapse and die of their disease. Relapsing patients or those with a priori poor prognostic features can potentially achieve long-term disease-free survival with an allogeneic hematopoietic cell transplant, but at the cost of substantial transplant-related mortality often related to infections or graft-versus-host disease.1,2 Increasing transplant conditioning regimen dose intensity has been shown in retrospective studies to be associated with lower rates of relapse posttransplant, and these observations have generally been attributed to the cytotoxic effect of chemotherapy or radiotherapy upon residual leukemia blasts.2-4 However, recent data showing that AML is in some cases preceded by clonal evolution in “preleukemic” hematopoietic stem cells may offer an intriguing new interpretation of the data on the importance of dose intensity in AML by suggesting that eradication of the surrounding morphologically normal bone marrow could play a role in reducing the risk of relapse.5-8
In the last 15 years, specific targeting of cells bearing a particular surface receptor has been shown to be feasible using monoclonal antibody therapy. However, even where supplemented by a cytotoxic payload, single-agent monoclonal antibody therapy rarely leads to durable remissions.9,10 A more recently realized treatment modality combines the specificity of antibody target recognition with the potent effector mechanisms of a T cell, leading to an entity known as a chimeric antigen receptor (CAR)-transduced T cell (CART).11-15
CARs are synthetic transmembrane constructs composed of an extracellular single-chain variable fragment (scFv) linked to intracellular T-cell signaling domains, usually the CD3ζ chain, and with one or more costimulation domains such as 4-1BB (CD137), CD28, or ICOS (CD278).16 Recent clinical data demonstrate that T cells engineered with anti-CD19 CARs engender potent and durable antitumor activity in B-cell malignancies.12,13,17
Anti-CD19 CART therapy as proof-of-concept has been successful in part due to the tissue restriction of CD19 to B cells and by the clinical tolerability of prolonged B-cell depletion. However, in other settings, CART-based targeting of antigens expressed at low levels by normal tissues has led to significant toxicities.18,19 The paucity of well-characterized, truly tumor-specific surface antigens in AML has necessitated consideration of CART tumor-targeting strategies that may also affect normal tissues, such as bone marrow. CD123, the transmembrane α chain of the interleukin-3 receptor, is expressed on the majority of AML blasts,20-22 but it is also expressed on many normal hematopoietic cells, where it is involved in hematopoietic differentiation.23 Although antibody-based targeting of CD123 has been reportedly well tolerated24,25 and a recently published preclinical model study using CART targeting of CD123 did not report significant hematopoietic toxicity,26 we show in this work that more potent targeting of CD123 with a lentiviral anti-CD123 vector costimulated via 4-1BB (1) leads to rejection of primary human AML in vivo regardless of baseline CD123 expression level, (2) markedly impairs human hematopoiesis in a xenograft model, and (3) could potentially be used as a novel myeloablative conditioning regimen for hematopoietic cell transplant.
Methods
T-cell transduction
Generation of the pELNS anti-CD19-41BB-CD3ζ (CAR19) plasmid was previously reported.27 In brief, the plasmid backbone is based on a third-generation self-inactivating lentiviral vector plasmid, pRRL-SIN-CMV-eGFP-WPRE, where the EF-1α promoter replaces the cytomegalovirus promoter. The pELNS anti-CD123-41BB-CD3ζ (CAR123) plasmid DNA was similarly generated by cloning light-to-heavy or heavy-to-light chain orientations of the mouse anti-human CD123 scFv (clone 32716 or clone 2629228), custom synthesized by Geneart, into the CAR19 construct.
Normal donor T cells were positively selected from leukapheresis packs using anti-CD4 and CD8 microbeads (Miltenyi), expanded in vitro with anti-CD3/CD28 beads (Invitrogen) and interleukin-2 100 U/mL (Chiron) for up to 15 days, and transduced with lentiviral supernatant from 293T cells transfected with pELNS anti-CD123-41BB-CD3ζ plasmid DNA or with pELNS anti-CD19-41BB-CD3ζ plasmid DNA as previously reported27 beginning on day +1 of expansion at a multiplicity of infection of 5.
Cells
The MOLM14 cell line was obtained from the ATCC and maintained in RPMI media supplemented with 10% fetal calf serum, penicillin, and streptomycin (R10). In bioluminescent xenograft models, MOLM14 cells were transduced with a luc2-gfp-luciferase lentiviral construct and sorted twice by positive-selection flow cytometry to >99% purity. Deidentified primary human AML specimens were obtained from the University of Pennsylvania Stem Cell and Xenograft Core facility. Some AML specimens were obtained directly from patients via an institutional review board–approved protocol for research studies. These studies were conducted in accordance with the Declaration of Helsinki. Cells were viably cryopreserved in 90% fetal calf serum and 10% dimethylsulfoxide until required for use. For all functional studies, AML cells were thawed at least 12 hours before analysis and rested overnight at 1 × 106 per mL in R10. In all xenograft studies, primary cells were thawed, washed once in phosphate-buffered saline (PBS), and injected via the lateral tail vein.
Flow cytometry
Anti-human antibodies were purchased from BioLegend, eBioscience, or Becton Dickinson. Cells were isolated from in vitro culture or from animals, washed once in PBS supplemented with 2% fetal calf serum, and stained on ice after blockade of Fc receptors. For quantitation, Countbright beads were used according to the manufacturer’s instructions (Invitrogen). In all analyses, the population of interest was gated based on forward vs side scatter characteristics followed by singlet gating, and live cells were gated using Live Dead Aqua (Invitrogen). CD107a degranulation assays and intracellular cytokine production assays were performed as previously described.29 Anti-CAR19 idiotype antibody was a gift of Dr L. Cooper (MD Anderson Cancer Center). Surface expression of anti-CD123 CAR was detected by secondary staining with an Alexa Fluor 647–conjugated goat anti-mouse F(ab′)2 antibody (Jackson ImmunoResearch). Flow cytometry was performed on a 4-laser Fortessa or 2-laser Accuri C6 cell analyzer (both from Becton-Dickinson).
Proliferation
T cells were washed and resuspended at up to 1 × 107 per mL in 100 μL of PBS and stained with 100 μL of carboxyfluorescein diacetate succinimidyl ester (CFSE) 2.5 μM (Life Technologies) for a final concentration of 1.25 μM for 5 minutes at 37°C. The reaction was quenched with cold R10, and the cells were washed thrice. T cells were incubated at a 1:1 ratio with irradiated target cells for 96 hours.
Killing assay
Killing assays were performed as previously described.30 In brief, CFSE-labeled targets were incubated at the indicated ratios with effector T cells for 4 or 16 hours. Cells were then harvested, and Countbright beads and 7-AAD were added prior to flow cytometric analysis. Residual live target cells were CFSE+ 7-AAD−.
Cytokine secretion
Effector and target cells were incubated at a 1:1 ratio in X-Vivo media with 10% human serum for 24 or 48 hours as indicated. Supernatant was analyzed by 30-plex Luminex array according to the manufacturer’s instructions (Invitrogen).
Animals
Nonobese diabetic severe combined immunodeficiency γchain−/− (NSG) and NSG mice transgenic for human interleukin-3 (IL-3), stem cell factor, and granulocyte macrophage colony-stimulating factor (NSGS) originally obtained from Jackson Laboratories were provided by Dr G. Danet-Desnoyers (University of Pennsylvania) and Dr S. Grupp (Children’s Hospital of Philadelphia). All experiments were performed on protocols approved by the institutional animal care and use committees of the University of Pennsylvania or the Children’s Hospital of Philadelphia.
In vivo models
Schemas of the used xenograft models are delineated in the relevant figures in “Results.” MOLM14 cells were injected in 200 μL of PBS at a concentration of 5 × 106 per mL into tail veins of mice, followed by BLI on a Xenogen IVIS-200 Spectrum camera as previously described.31 Primary AML cells were injected in 200 μL of PBS at a concentration of 25 to 50 × 106 per mL. CD123-redirected T cells (CART123), CART19, or untransduced (UTD) human T cells were injected in 200 μL of PBS at a concentration of 0.5 to 20 × 106 per mL into the tail vein. Primary leukemia engraftment was defined as >1% human CD45+ cells in the peripheral blood by flow cytometry. Mice were sacrificed according to protocol when moribund or upon the development of hind-limb paralysis.
Humanized immune system (HIS) mice were created by the laboratory of Dr G. Danet-Desnoyers by injection of fetal liver CD34+ cells into newborn NSG mice.23 Engraftment of human hematopoiesis, defined as >1% human CD45+ cells in the peripheral blood by flow cytometry, was confirmed at 6 to 8 weeks after injection and prior to release of the mice for downstream experiments.
Methylcellulose colonies
Sorted CD34+ adult human bone marrow or cord blood was resuspended in MethoCult Optimum (STEMCELL Technologies) according to the manufacturer’s instructions and plated in 6-well plates at 1 × 103 cells per well for 14 days. In some experiments, CD34+ cells were first cultured for 4 hours with CART123 or control T cells. After 14 days, colonies were scored on an inverted microscope (Zeiss; 4X), followed by solubilisation of the colonies in R10 media overnight and harvest of single-cell suspensions for flow cytometry.
Results
CD123 is a suitable cell-surface target in AML
In order to select an appropriate AML cell-surface target for CART therapy, we first compared the expression of CD123 to other immature myeloid markers in a panel of primary human AML specimens. Consistent with previous reports, CD123 was expressed in most AML specimens at high levels, and we found it to be expressed more frequently than CD33 or CD3420,22,32 (Figure 1A; supplemental Table 1 available at the Blood Web site). Although some of the analyzed AML samples appeared to be CD123dim/− by strictly drawn gates, expression in these was slightly higher than the residual normal lymphocytes within each sample (Figure 1B). AML samples were flow-sorted into CD123dim and CD123bright populations, and both populations were observed to form colonies in semisolid media, suggesting that virtually all AML blasts are functionally CD123+ (Figure 1C). In contrast to previously published studies,20 expression of CD123 was no higher on phenotypically defined leukemic stem cells than on bulk leukemia cells (supplemental Figure 1).
Figure 1.

CD123 is frequently expressed in primary AML. (A) Primary patient AML samples express CD123. AML blasts were gated using standard side scatterlow CD45dim characteristics (n = 35-46, from a diverse range of AML subtypes; see supplemental Table 1). (B) CD123 expression levels vary among leukemia samples, as revealed by gating on blasts and using residual normal lymphocytes or isotype-matched controls (not shown) to establish negative and positive gating for CD123. Two examples of blasts stained with CD123 and CD34 are shown (top), and a panel of 9 representative leukemias is shown (bottom). The median fluorescence intensity (MFI) for CD123 was adjusted for cell size by dividing MFI by the forward scatter (FSC). (C) Sorted CD123dim and CD123bright leukemia cells form methylcellulose colonies with an identical CD123 phenotype. MFI of CD123 is shown adjacent to each peak. PE, phycoerythrin.
Anti-CART123 exhibit multiple antigen-specific effector functions
We designed 8 different CAR123 constructs utilizing scFv sequences from 2 different antibodies linked to the 4-1BB and CD3ζ chain signaling domains in a lentiviral vector, as in our anti-CD19 CART studies (supplemental Figure 2).12,27,28 Each construct was evaluated for the ability to target the human CD123+ AML cell line MOLM14, and the construct that combined the highest degranulation and cytokine production (construct #72) was chosen for further preclinical development (supplemental Figure 3). T cells lentivirally transduced with the anti-CD123-41BB-CD3ζ construct (CART123) demonstrated robust antigen-specific degranulation, cytokine production, proliferation, and cytotoxicity when incubated with MOLM14 cells or with primary AML cells (Figure 2A-D; supplemental Figure 4). Furthermore, when compared with control CART19 cells, CART123s produced a variety of effector and homeostatic cytokines and chemokines. These included interferon-γ, macrophage inflammatory protein (MIP) 1α, MIP1β, interleukin-2, and granulocyte macrophage colony-stimulating factor, among others, underscoring the ability of CART123 to orchestrate a potent immune response17 (Figure 2E).
Figure 2.

CART123 cells manifest multiple effector functions upon in vitro exposure to CD123-expressing targets. (A) Activation of CART123 cells by AML targets. CART123 cells were coincubated with normal bone marrow (NBM) cells, primary AML cells, or with CD123+ MOLM14 cells, and CD107a degranulation was measured via flow cytometry. CAR-expressing cells were identified by staining with goat anti-mouse F(ab′)2 reagent. CART123 cells alone and the CD123− Jurkat cell line were used as negative controls; P < .0001 (Student t test). (B) Antigen-specific cytokine production in response to CD123+ AML. CARs were detected using a goat anti-mouse F(ab′)2 (for CART123) or an anti-CAR19 idiotypic antibody (for CART19). Intracellular cytokines were interferon-γ, MIP1β, and tumor necrosis factor α. (C) Proliferation of CART123 in response to CD123+ primary AML. CART123 or CART19 cells were labeled with CFSE and exposed to primary AML cells for 96 hours. Proliferation was assessed by CFSE dilution; P < .001 (Student unpaired t test). (D) Cytotoxic targeting of primary AML blasts by CART123 after incubation for 16 hours at the indicated effector-to-target (E:T) ratios; CART19 cells were used as controls. Two representative examples are shown. (E) Cytokine profiling of CART123 (black) or CART19 (white) cells in response to 24-hour coincubation with MOLM14 cells. All results are representative of at least 2 experiments with similar results. IFN-γ, interferon-γ; IL-2, interleukin-2; GM-CSF, granulocyte macrophage colony-stimulating factor; TNF-α, tumor necrosis factor α.
CART123 cells mediate a potent in vivo antileukemic effect
The in vivo efficacy of CART123 was next evaluated in therapeutically relevant xenograft models of human AML. NSG mice were engrafted with luciferase-expressing MOLM14 cells and then treated with 1 dose of CART123 or control CART19 cells, because CD19 is not generally expressed on AML or on normal myeloid cells (Figure 3A). Bioluminescent imaging (BLI) was used to quantify AML burden over time (Figure 3B-C). Whereas CART19 treatment had no effect on leukemia progression, leukemia burden in CART123-treated mice began to diminish within 1 week and was eliminated in most mice within 2 weeks of T-cell infusion. A single administration of CART123 led to the long-term survival of the majority of animals (Figure 3D). Late disease recurrence in a minority of CART123-treated animals occurred mostly at facial sites, as has been previously reported in a similar leukemia model and is hypothesized to be a possible leukemia sanctuary site or a location poorly targeted by CART cells in xenografted mice.33 In our studies, a very small population of residual CD123+ leukemia was detected in the marrow of animals with recurrent disease despite a larger population of persistent CART123s (supplemental Figure 5).
Figure 3.

CART123 cells eliminate human AML in xenograft models. (A) Schematic of the MOLM14 xenograft model. NSG mice were sublethally irradiated (200 cGy) on day −1 and then injected via tail vein with 1 × 106 green fluorescent protein/luciferase+ MOLM14 on day 0. BLI was performed on day 6 to quantify engraftment and for randomization of treatment groups. Saline vehicle, CART19 cells (1 × 106), or CART123 (1 × 106) cells were injected IV on day 7, and mice were followed with serial BLI. Quantification of BLI radiance was used as a surrogate measurement of AML burden. (B) Elimination of MOLM14 occurred only in xenografted mice treated with CART123 cells, as measured by BLI radiance and displayed colorimetrically. (C) Summary BLI data from 3 MOLM14 xenograft experiments demonstrated rapid leukemic progression in vehicle-treated (black) and CART19-treated (blue) mice, whereas AML rejection was observed in CART123-treated mice (red). Mean radiance (symbols) with standard error of the mean (whiskers) are depicted at each time point. (D) Survival analysis of MOLM14 xenograft mice revealed significant survival for CART123-treated mice in comparison with vehicle- and CART19-treated mice. Attrition of CART123 T-cell–treated mice was primarily due to BLI-detectable AML progression in facial bones and subsequent anorexia and weight loss. Data were summarized from 4 independent experiments.
Primary patient-derived AML samples are susceptible to CART123 in vivo
Because AML is clonally heterogeneous, it is critical to evaluate the ability of CART123 to eradicate human AML in vivo.8 For our primary AML xenograft studies, we used NSGS mice, because this strain has been reported to facilitate more rapid and higher levels of human AML engraftment.34 Cohorts of NSGS mice were injected with 1 of 3 primary AML specimens that had different levels of surface CD123 expression (high, intermediate, or low; representative data in supplemental Figure 6). Following leukemia engraftment at 2 to 3 weeks, mice were treated with CART123 or control CART19 cells (Figure 4A). CART123 treatment resulted in elimination of AML in blood and marrow and in enhanced survival (Figure 4B-C; supplemental Figure 7). Surprisingly, we observed considerable early attrition of some CART123-treated mice in the primary AML xenograft studies. Animals appeared ill at 7 to 14 days after CART123 treatment, and we discovered that this attrition was due to rapid leukemia progression and/or to systemic toxicity from the CART123s, such as tumor lysis or cytokine release syndrome similar to that reported in patients with B-cell leukemias treated with CART19 cells.12,17 We observed eradication of primary AML by CART123 regardless of baseline CD123 expression, such as in a leukemia that was clearly CD123dim at baseline (UPN034; supplemental Figure 6B). This phenomenon was possibly explained by CD123 upregulation on CD123dim blasts during in vivo (Figure 4D) and in vitro propagation (Figure 1C).
Figure 4.

CD123 is an excellent target in primary AML in vivo. (A) Primary AML xenograft model. NSGS mice were sublethally irradiated on day −1 and injected with 5 to 10 × 106 primary AML blasts via the tail vein on day 0. Engraftment was confirmed by flow cytometric measurement of live mouse CD45neg human CD45dim CD123+ cells in the peripheral blood, usually occurring around day 14. Mice were then injected with CART123 cells or control T cells (CART19 or UTD T cells) and bled weekly to quantify AML burden. (B) Analysis of peripheral blood from mice 8 to 15 days after receiving T cells demonstrated marked reduction of circulating UPN 024 blasts. Note that residual CD45bright T cells are poorly detectable in some CART123 mice at this time point, correlating with the rapid rise and fall of peripheral CART123 cells in response to clearance of AML; P < .0001 (Student t test). (C) Composite survival plot of mice from 3 independent experiments. (D) In vivo upregulation of CD123 on AML blasts. Upon injection into NSGS mice treated with control UTD T cells, the UPN034 sample upregulated CD123 expression in comparison with blood measurements obtained prior to (day 13 [D13], left) and after (day 27 [D27], right) T-cell injection. The CD45bright CD123− population represents adoptively transferred human T cells. MFI of CD123 is shown at top right. All mice receiving control T cells subsequently died of disease, and all CART123-treated mice were long-term survivors (these are a subset of the mice in Figure 4C). Results are representative of 3 independent experiments.
CART123 cells establish immunologic memory and a recall response
Establishment of a population of antigen-specific memory cells is considered to be a prerequisite for successful cancer immunotherapy. To evaluate whether CART123 were capable of persisting in vivo and establishing memory, NSG mice were injected with MOLM14 cells (group A) or with saline (group B) and then injected 10 days later with CART123 (Figure 5A) as described above. All primarily MOLM14-engrafted mice in group A eradicated leukemia with CART123 treatment as assessed by BLI. Mice were then challenged with a second (group A) or first (group B) infusion of MOLM14 cells. Essentially no leukemia engraftment was detectable by BLI in either the CART123-treated, leukemia-rechallenged mice (group A) or in the previously CART123-treated, MOLM14-naïve control mice (group B) (Figure 5B). However, CART123 cells expanded significantly more quickly upon MOLM14 rechallenge in leukemia-experienced compared with leukemia-naïve animals, consistent with the establishment of a CART123 memory T-cell subset (Figure 5C), with a trend toward more central memory T cells and fewer effector memory T cells in group A (supplemental Figure 8). Although a rare event, it is instructive that eventual rejection of leukemia in mice with less robust initial antitumor responses (as exemplified by the data points with increased radiance; Figure 5B, *) correlated with the emergence of CART123 (Figure 5D).
Figure 5.

CART123 cells exhibit characteristics of immunologic memory. (A) In a challenge/rechallenge model, NSG mice were sublethally irradiated and then injected via tail vein with saline or with 1 × 106 green fluorescent protein/luciferase+ MOLM14 cells on day 0. After BLI quantification of AML burden and randomization into treatment groups, CART123 cells (1 × 106) were injected IV on day 10, and mice were followed with serial BLI until AML eradication. Approximately 2 weeks after AML clearance, all mice were challenged (right) or rechallenged (left) with 1 × 106 MOLM14 cells and followed with weekly retro-orbital venous bleeding for T-cell quantification and by BLI for AML burden. (B) Summary of BLI radiance in CART123-treated mice following rechallenge or primary challenge with MOLM14. Each symbol represents an individual animal. The dashed line depicts the mean radiance measurement of untreated NSG mice. (C) CART123-treated, MOLM14-treated mice that are rechallenged with MOLM14 demonstrate increased numbers of peripheral CART123 cells in comparison with CART123-treated mice administered MOLM14 cells for the first time. Mice were bled 7 days prior to (day 28) and 7 days following (day 42) injection of MOLM14 cells. CART123 cells were identified as live, singlet, human CD45+ CD3+ CAR+ cells per the gating strategy in supplemental Figure 7 and quantified using Countbright beads. (D) Representative mouse (the highest blue data point from Figure 5B) showing that initial failure to reject MOLM14 is associated with low CART123 cell numbers in peripheral blood and that late rejection is accompanied by emergence of CART123 cells. Results are representative of 2 to 5 independent experiments. NS, nonsignificant.
CART123 treatment ablates normal human hematopoiesis
CD123 is expressed on some normal hematopoietic cells, including myeloid progenitors, dendritic cells, some B cells, and megakaryocytes, and IL-3 signaling plays an important role in myeloid differentiation.35,36 We thus investigated the impact of CART123 treatment on normal hematopoiesis. In agreement with published reports, CD123 was expressed on the majority of lineage-negative hematopoietic progenitors in bone marrow specimens from healthy donors (supplemental Figure 9 and Figure 6A).37 Both CD123− and CD123+ populations sorted from normal marrow precursors produced hematopoietic colonies with equivalent CD123 surface expression, suggesting that CD123 may be expressed over time during hematopoietic development (Figure 6B). Analogously, short-term exposure of cord-blood–derived human CD34+ cells to CART123 led to marked reduction in myeloid colony formation quantified by colony number (data not shown) as well as total cell number, demonstrating a potent effect of CART123 on myeloid progenitor function; in contrast, following short-term exposure of cord-blood–derived human CD34+ cells to control T cells, myeloid colony formation (20 ± 1) was well within expected values for the assay (54 GM-CFU/1000 CD34+ cells; range, 7-101; www.stemcell.com/∼/media/Technical%20Resources/8/9/A/A/C/28404MAN_4_0_0.pdf?la=en; Figure 6C).
Figure 6.
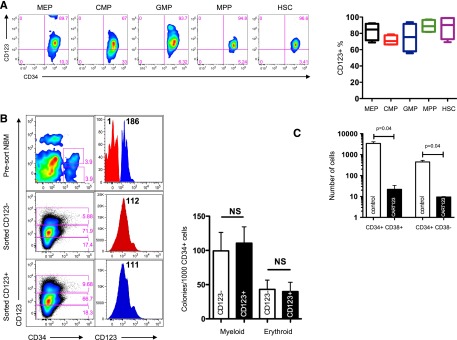

Eradication of normal hematopoiesis by CART123 in a xenograft model. (A) Healthy bone marrow progenitor populations exhibit moderate to bright expression of CD123. Bone marrow from 4 normal donors was stained for CD123 after gating on live singlet lineage-negative CD45dim cells, and the indicated progenitor subpopulations were identified using CD34, CD38, CD45RA, and CD90 (gating strategy is shown in supplemental Figure 8). CD123 gating was based on normal lymphocytes and confirmed with fluorescence-minus-one controls. MEP, megakaryocyte-erythroid progenitors; CMP, common myeloid progenitors; GMP, granulocyte-monocyte progenitors; MPP, multipotent progenitors; HSC, hematopoietic stem cells. (B) CD123dim/− bone marrow progenitors differentiate to CD123+ in semisolid culture. CD123dim (top panel, red histogram) or CD123intermediate/+ (top panel, blue histogram) CD34+ cells were sorted from normal bone marrow (NBM) and cultured in MethoCult Optimum medium for 14 days. The middle and lower panels show the phenotype of colonies that developed from sorted CD123dim/− and CD123intermediate/+ populations, respectively. The sorted cultured populations exhibited similar CD123 expression and an indistinguishable ability to form myeloid or erythroid colonies. MFI of CD123 is shown at top right. (C) CART123 cells markedly impair hematopoietic function. CD34+ cells selected from normal human cord blood were incubated at a 1:10 target-to-effector ratio with CART123 or control UTD T cells for 4 hours, followed by a 14-day culture in Methocult Optimum. Coculture with UTD T cells was used to control for the allogeneic effect. Hematopoietic function was assessed by manual colony counts (not shown) or quantified by flow cytometry for the indicated cell populations using Countbright beads. (D) Cycling bone marrow cells upregulate CD123. Mice previously engrafted with human CD34+ cells were treated with 5-fluorouracil (5-FU) or vehicle. Fourteen days later, bone marrow was harvested from these mice and analyzed for the intracellular proliferation marker Ki67 and for CD123 after gating on live lineage-negative human cells; P < .01 (Student t test). (E) Schematic of xenograft model to evaluate potential CART123-mediated myeloablation. NSG mice were engrafted with human fetal liver CD34+ cells (HIS mice) and bled for confirmation of engraftment after 6 to 8 weeks. On day 0, mice received CART123, control UTD T cells, or saline vehicle. Flow cytometric quantification of human hematopoietic cells in peripheral blood (days 7, 14, 21) and in bone marrow (day 28) was performed. (F) Specific decline in circulating human B cells, myeloid cells, monocytes, and platelets is seen after treatment with CART123. Representative plots are shown after control (top) or CART123 (bottom) infusion and quantified in the lower panel (control T cells, open column; CART123, solid column). (G) Specific myeloablation of human bone marrow in CART123 mice. On day 28 after T-cell injection, bone marrow was harvested and analyzed for human progenitor cell populations after gating on live singlet human lineage-negative cells. (H) Sections of femur taken from HIS mice 1 month after treatment with control (top) or CART123 (bottom). Hematoxylin and eosin staining; Zeiss microscope original magnification ×1 and ×10 shown. Results are representative of at least 2 independent experiments. NS, nonsignificant.
The most likely clinical implementation of CART123 therapy would be in the setting of recent chemotherapy. To investigate the effect of postchemotherapy marrow recovery upon CD123 expression on marrow progenitor cells, human-marrow–engrafted mice were treated with 5-fluorouracil, a chemotherapeutic agent with an established role in inducing hematopoietic cycling in preclinical studies.38 As predicted, postchemotherapy recovery was associated with an increase in proliferating cells, which had higher frequency of CD123 expression than did nonproliferating cells (Figure 6D).
To investigate the effect of CART123 therapy on hematopoiesis in vivo, NSG mice engrafted with normal human CD34+ cells were treated with CART123 or UTD human T cells (Figure 6E). As predicted from the expression of CD123 on normal circulating B cells and myeloid cells (supplemental Figure 10) and on megakaryocytes, decreased numbers of human B cells, monocytes, myeloid cells, and platelets were observed in the peripheral blood of these mice following CART123 treatment (Figure 6F). Phenotypically defined human stem/progenitor cells were essentially absent in CART123-treated animals at 1 month posttreatment in comparison with UTD controls, correlating with the demonstrated expression of CD123 on progenitor cells (Figure 6A,G; supplemental Figure 11). In addition, marrow cellularity was significantly reduced in the CART123-treated mice (Figure 6H). Although we note that we cannot exclude a minor contributing role of high T-cell engraftment in the observed impact on HSC niches and hematopoiesis, these results highlight the profound myeloablative potential of CART123.
Discussion
Recent data suggest that most bone marrow cells in patients with AML and myelodysplasia are clonal, regardless of the myeloblast count.7,8 These observations could explain why many AML patients are incurable with standard therapy and highlight the importance of myeloablation in AML therapy. Here, we report the preclinical evaluation and development of a myeloablative CAR-based therapy for AML. We show that T cells genetically engineered with a CAR that recognizes CD123 and is activated via CD3ζ and 4-1BB signals (CART123) mediate potent effector activity against cell-line and primary AML, evincing antigen-specific proliferation, degranulation, cytotoxicity, and elaboration of multiple effector cytokines. We demonstrate that CART123 eliminate leukemia and lead to long-term survival of mice engrafted with an AML cell line or with primary AML. Given the clonal heterogeneity of cancer in general and of AML in particular8 and the proven potential of AML to develop antigen-escape mutations after targeted therapy,39 we considered it essential to test CART cell therapy against primary AML in vivo. We found that CART123 could eliminate even a CD123dim leukemia (UPN 034), and we speculate that CD123 is a particularly important antigen in AML given its role as a cytokine receptor. Our finding that CD123 expression increases over time in a CD123dim leukemia supports this possibility. Furthermore, we show that infusion of CART123 can result in the establishment of a T-cell memory pool capable of rejecting disease with enhanced kinetics of expansion in the recall compared with the primary response.
Although others have reported the feasibility of CART cell approaches for AML,40,41 we demonstrate in these studies that CART123 manifests remarkable potency, persistence, and bioactivity, which are surely essential components of successful CART therapy. Our results are particularly striking when compared with the results described in the recent publication by Mardiros et al, who showed an incremental improvement in survival analyses, but no long-term animal survival, after the injection of 5 × 106 CART123 cells into mice previously injected with 0.5 × 106 KG1a AML cells. In contrast, we injected 1 × 106 CART123 into mice previously injected with 1 × 106 MOLM14 AML cells and obtained a majority of long-term survivors. Although a direct comparison across groups and across different tumor cell lines is impossible, it is noteworthy that both of these studies used CAR generated using the same CD123-specific scFv.26 Our differing findings may result from disparities in vector design, incorporation of different secondary signaling domains, and distinct T-cell transduction and expansion strategies.16 Mardiros and the City of Hope group used a retroviral vector incorporating CD28 costimulation in T cells expanded with OKT3, whereas we used a lentiviral vector incorporating 4-1BB costimulation in T cells expanded with anti-CD3/CD28 beads. Although there has been no direct comparison among the constructs used by the different groups currently treating patients with CART19, data from our clinical studies show that our construct and CART19 cells have impressive expansion, persistence, and in vivo activity. The current anti-CD123 construct used in these studies is based on our anti-CD19 construct and may thus induce similar potent clinical activity. Differences in clinical outcomes related to potency and persistence of infused CART products have also been noted in clinical trials of CART19 therapies. Hence, systematic investigation of the optimal T-cell product(s) will likely be an important goal of the CAR-redirected T-cell immunotherapy field in the near future.12,14-17,42
Appreciation of the myeloablative potential of CART123 cells is particularly critical at the current preclinical stage of AML CART immunotherapy. Previous trials targeting the IL-3 receptor with antibodies or cytokine-based modalities did not report bone marrow suppression, and we speculate that this phenomenon is due to the ability of CART123 to eliminate CD123dim targets more effectively than antibody-based approaches tested to date.24,43 Experimental design and biological issues thus likely explain the discrepancy between our findings and those of Mardiros et al and Tettamanti et al.26,44 Because our CAR appears particularly potent, it is likely that short-term incubation with cord blood progenitors was sufficient to induce robust killing, leading to a statistically significant impairment in colony growth (as in Figure 6C). Furthermore, our more clinically relevant in vivo xenograft model of human hematopoiesis permits the integration of anti-CD123 pressure over time as likely occurs in patients, rather than a brief in vitro coincubation prior to culture in semisolid culture that may not evoke significantly detectable effects upon hematopoiesis. We noted a significant reduction in B cells and platelets and a trend of reduced myeloid cell numbers, the latter likely reflecting the low circulating numbers of human myeloid cells in the particular xenograft model used. Although this an imperfect system to model human hematopoiesis in vivo, it is currently among the best preclinical standards for this particular application.23
We currently select an optimal CAR construct for future study based solely on the demonstration of superior in vitro potency against antigen-bearing targets. However, such an approach may result in the design of overly potent CART cells. Optimization of the construct selection process is likely an important goal for the next few years of preclinical and clinical studies in the CART cell therapy arena.
Our on-target/off-tumor toxicity findings and those reported recently by Casucci et al in their preclinical anti-CD44 CART AML studies highlight the importance of careful study of potential off-tumor toxicities of this powerful therapeutic modality.41 Our observations that a substantial proportion of NSGS mice bearing primary AML develop systemic toxicity after treatment with CART123 cells is analogous to our recent clinical reports with CART19 and merit careful future investigation.12,17
These observations lead us to highlight a potential novel application for CART123 as a chemotherapy-free myeloablative conditioning regimen for hematopoietic stem cell transplantation. This would likely be of particular importance in conditions where hematopoiesis is itself diseased, such as in AML or myelodysplasia. CD123 is a particularly attractive target due to the importance of IL-3 signaling in the developing marrow.36 Our observation that CD123 is upregulated over time in both normal and malignant hematopoiesis likely reflects its important biological role as a hematopoietic cytokine receptor. Furthermore, our observation that essentially all AML blasts are CD123+ may obviate the need to target selectively the putative AML stem cell.21 Nonetheless, the feasibility of CART123 therapy for AML will depend on our ability to provide transient rather than permanent, or perhaps less potent, anti-CD123 targeting. Such approaches would include the use of “biodegradable” CART-engineered cells such as T cells electroporated with CAR messenger RNA developed by our group and others, which transiently express the CAR transgenes and thus have a finite and short lifespan; the use of “biodegradable” CAR-engineered T cells modified with messenger RNA; the use of inducible suicide systems to ablate engineered cells when desired; the use of costimulatory molecules less potent than CD137; or the use of T-cell–depleting antibodies such as antithymocyte globulin or alemtuzuzmab.45-47 A subsequent infusion of healthy hematopoietic stem cells may be then needed to reconstitute normal hematopoiesis.
Collectively, our results suggest that CD123 is a viable target in AML and that caution and careful planning will be required for future clinical trials of CART123.
Supplementary Material
Acknowledgments
The authors thank Jamie Song for assistance with figure preparation.
This work was funded by a Leukemia and Lymphoma Society Specialized Centers of Research grant to M.K., M.C., and C.H.J., an American Society of Hematology Scholar Award to S.G., a National Institutes of Health Career Development K12 award to S.T. (K12CA076931), and a research agreement with Novartis Pharmaceuticals.
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: S.G. designed and performed research, analyzed data, and wrote the manuscript; S.K.T. designed and performed research, analyzed data, and wrote the manuscript; M.R. designed and performed research; O.S. performed research; Y.L. performed research; D.L.P. designed research and wrote the manuscript; M.C. designed research and contributed reagents; G.D.-D. designed research and contributed reagents; J.S. contributed reagents and performed research; S.A.G. designed research, contributed reagents, and wrote the manuscript; C.H.J. designed research, contributed reagents, analyzed data, and wrote the manuscript; and M.K. designed research, analyzed data, and wrote the manuscript.
Conflict-of-interest disclosure: C.H.J. D.L.P., and M.K. have filed patent applications related to CAR technology and could potentially receive licensing royalties from Novartis corporation; S.G., J.S., C.H.J., and M.K. have filed patent applications on CART123 technology. The remaining authors declare no competing financial interests.
The current affiliation for M.K. is Lilly Research Laboratories, Eli Lilly and Company, 450 East 29th St, New York, NY.
Correspondence: Michael Kalos, Lilly Research Laboratories, Eli Lilly and Company, 450 East 29t Street, New York, NY 10016; e-mail: kalos_michael_d@lilly.com; or Saar Gill, Division of Hematology-Oncology, Room 9-188, Smilow Translational Research Center, University of Pennsylvania, 3400 Civic Center Blvd, Philadelphia, PA 19104; e-mail: saar.gill@uphs.upenn.edu.
References
- 1.Forman SJ, Rowe JM. The myth of the second remission of acute leukemia in the adult. Blood. 2013;121(7):1077–1082. doi: 10.1182/blood-2012-08-234492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gill S, Porter DL. Reduced-intensity hematopoietic stem cell transplants for malignancies: harnessing the graft-versus-tumor effect. Annu Rev Med. 2013;64:101–117. doi: 10.1146/annurev-med-121411-103452. [DOI] [PubMed] [Google Scholar]
- 3.Shimoni A, Hardan I, Shem-Tov N, et al. Allogeneic hematopoietic stem-cell transplantation in AML and MDS using myeloablative versus reduced-intensity conditioning: the role of dose intensity. Leukemia. 2006;20(2):322–328. doi: 10.1038/sj.leu.2404037. [DOI] [PubMed] [Google Scholar]
- 4.Luger SM, Ringdén O, Zhang M-J, et al. Similar outcomes using myeloablative vs reduced-intensity allogeneic transplant preparative regimens for AML or MDS. Bone Marrow Transplant. 2012;47(2):203–211. doi: 10.1038/bmt.2011.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weissman IL. Stem cell research: paths to cancer therapies and regenerative medicine. JAMA. 2005;294(11):1359–1366. doi: 10.1001/jama.294.11.1359. [DOI] [PubMed] [Google Scholar]
- 6.Jan M, Snyder TM, Corces-Zimmerman MR, et al. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci Transl Med. 2012;4(149):149ra118. [DOI] [PMC free article] [PubMed]
- 7.Welch JS, Ley TJ, Link DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150(2):264–278. doi: 10.1016/j.cell.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walter MJ, Shen D, Ding L, et al. Clonal architecture of secondary acute myeloid leukemia. N Engl J Med. 2012;366(12):1090–1098. doi: 10.1056/NEJMoa1106968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burnett AK, Hills RK, Milligan D, et al. Identification of patients with acute myeloblastic leukemia who benefit from the addition of gemtuzumab ozogamicin: results of the MRC AML15 trial. J Clin Oncol. 2011;29(4):369–377. doi: 10.1200/JCO.2010.31.4310. [DOI] [PubMed] [Google Scholar]
- 10.Schrama D, Reisfeld RA, Becker JC. Antibody targeted drugs as cancer therapeutics. Nat Rev Drug Discov. 2006;5(2):147–159. doi: 10.1038/nrd1957. [DOI] [PubMed] [Google Scholar]
- 11.Jensen MC, Popplewell L, Cooper LJ, et al. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transplant. 2010;16(9):1245-1256. [DOI] [PMC free article] [PubMed]
- 12.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365(8):725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kalos M, Levine BL, Porter DL, et al. T Cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3(95):95ra73-95ra73. [DOI] [PMC free article] [PubMed]
- 14.Kochenderfer JN, Dudley ME, Feldman SA, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119(12):2709–2720. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brentjens RJ, Rivière I, Park JH, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118(18):4817–4828. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kalos M, June CH. Adoptive T cell transfer for cancer immunotherapy in the era of synthetic biology. Immunity. 2013;39(1):49–60. doi: 10.1016/j.immuni.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grupp SA, Kalos M, Barrett D, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368(16):1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lamers CH, Sleijfer S, van Steenbergen S, et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther. 2013;21(4):904–912. doi: 10.1038/mt.2013.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18(4):843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jordan CT. Targeting myeloid leukemia stem cells. Sci Transl Med. 2010;2(31):31ps21. doi: 10.1126/scitranslmed.3000914. [DOI] [PubMed] [Google Scholar]
- 21.Jin L, Lee EM, Ramshaw HS, et al. Monoclonal antibody-mediated targeting of CD123, IL-3 receptor alpha chain, eliminates human acute myeloid leukemic stem cells. Cell Stem Cell. 2009;5(1):31–42. doi: 10.1016/j.stem.2009.04.018. [DOI] [PubMed] [Google Scholar]
- 22.Muñoz L, Nomdedéu JF, López O, et al. Interleukin-3 receptor alpha chain (CD123) is widely expressed in hematologic malignancies. Haematologica. 2001;86(12):1261–1269. [PubMed] [Google Scholar]
- 23.Rongvaux A, Takizawa H, Strowig T, et al. Human hemato-lymphoid system mice: current use and future potential for medicine. Annu Rev Immunol. 2013;31:635-674. [DOI] [PMC free article] [PubMed]
- 24.Roberts AW, He S, Bradstock KF, et al. A phase 1 and correlative biological study of CSL360 (anti-CD123 mAb) in AML. Paper presented at the Annual Meeting of the American Society of Hematology. December 6-8, 2008. San Francisco, CA. [Google Scholar]
- 25.Frankel A, Liu J-S, Rizzieri D, Hogge D. Phase I clinical study of diphtheria toxin-interleukin 3 fusion protein in patients with acute myeloid leukemia and myelodysplasia. Leuk Lymphoma. 2008;49(3):543–553. doi: 10.1080/10428190701799035. [DOI] [PubMed] [Google Scholar]
- 26.Mardiros A, Dos Santos C, McDonald T, et al. T cells expressing CD123-specific chimeric antigen receptors exhibit specific cytolytic effector functions and antitumor effects against human acute myeloid leukemia. Blood. 2013;122(18):3138–3148. doi: 10.1182/blood-2012-12-474056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Milone MC, Fish JD, Carpenito C, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. 2009;17(8):1453–1464. doi: 10.1038/mt.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Du X, Ho M, Pastan I. New immunotoxins targeting CD123, a stem cell antigen on acute myeloid leukemia cells. J Immunother. 2007;30(6):607–613. doi: 10.1097/CJI.0b013e318053ed8e. [DOI] [PubMed] [Google Scholar]
- 29.Betts MR, Koup RA. Detection of T-cell degranulation: CD107a and b. Methods Cell Biol. 2004;75:497–512. doi: 10.1016/s0091-679x(04)75020-7. [DOI] [PubMed] [Google Scholar]
- 30.Cao L-F, Krymskaya L, Tran V, et al. Development and application of a multiplexable flow cytometry-based assay to quantify cell-mediated cytolysis. Cytometry A. 2010;77(6):534–545. doi: 10.1002/cyto.a.20887. [DOI] [PubMed] [Google Scholar]
- 31.Edinger M, Cao YA, Verneris MR, Bachmann MH, Contag CH, Negrin RS. Revealing lymphoma growth and the efficacy of immune cell therapies using in vivo bioluminescence imaging. Blood. 2003;101(2):640–648. doi: 10.1182/blood-2002-06-1751. [DOI] [PubMed] [Google Scholar]
- 32.Testa U, Riccioni R, Militi S, et al. Elevated expression of IL-3Ralpha in acute myelogenous leukemia is associated with enhanced blast proliferation, increased cellularity, and poor prognosis. Blood. 2002;100(8):2980–2988. doi: 10.1182/blood-2002-03-0852. [DOI] [PubMed] [Google Scholar]
- 33.Brentjens RJ, Santos E, Nikhamin Y, et al. Genetically targeted T cells eradicate systemic acute lymphoblastic leukemia xenografts. Clin Cancer Res. 2007;13(18 Pt 1):5426–5435. doi: 10.1158/1078-0432.CCR-07-0674. [DOI] [PubMed] [Google Scholar]
- 34.Wunderlich M, Chou F-S, Link KA, et al. AML xenograft efficiency is significantly improved in NOD/SCID-IL2RG mice constitutively expressing human SCF, GM-CSF and IL-3. Leukemia. 2010;24(10):1785–1788. doi: 10.1038/leu.2010.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Emerson SG, Yang YC, Clark SC, Long MW. Human recombinant granulocyte-macrophage colony stimulating factor and interleukin 3 have overlapping but distinct hematopoietic activities. J Clin Invest. 1988;82(4):1282–1287. doi: 10.1172/JCI113727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Metcalf D. Hematopoietic cytokines. Blood. 2008;111(2):485–491. doi: 10.1182/blood-2007-03-079681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taussig DC, Pearce DJ, Simpson C, et al. Hematopoietic stem cells express multiple myeloid markers: implications for the origin and targeted therapy of acute myeloid leukemia. Blood. 2005;106(13):4086–4092. doi: 10.1182/blood-2005-03-1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kunisaki Y, Bruns I, Scheiermann C, et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature. 2013;502(7473):637–643. doi: 10.1038/nature12612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith CC, Wang Q, Chin CS, et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature. 2012;485(7397):260–263. doi: 10.1038/nature11016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ritchie DS, Neeson PJ, Khot A, et al. Persistence and efficacy of second generation CAR T cell against the LeY antigen in acute myeloid leukemia. Mol Ther. 2013;21(11):2122–2129. doi: 10.1038/mt.2013.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Casucci M, Nicolis di Robilant B, Falcone L, et al. CD44v6-targeted T cells mediate potent antitumor effects against acute myeloid leukemia and multiple myeloma. Blood. 2013;122(20):3461–3472. doi: 10.1182/blood-2013-04-493361. [DOI] [PubMed] [Google Scholar]
- 42.Brentjens RJ, Davila ML, Riviere I, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5(177):177ra38. [DOI] [PMC free article] [PubMed]
- 43.Cohen KA, Liu TF, Cline JM, Wagner JD, Hall PD, Frankel AE. Safety evaluation of DT388IL3, a diphtheria toxin/interleukin 3 fusion protein, in the cynomolgus monkey. Cancer Immunol Immunother. 2005;54(8):799–806. doi: 10.1007/s00262-004-0643-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tettamanti S, Marin V, Pizzitola I, et al. Targeting of acute myeloid leukaemia by cytokine-induced killer cells redirected with a novel CD123-specific chimeric antigen receptor. Br J Haematol. 2013;161(3):389–401. doi: 10.1111/bjh.12282. [DOI] [PubMed] [Google Scholar]
- 45.Barrett DM, Zhao Y, Liu X, et al. Treatment of advanced leukemia in mice with mRNA engineered T cells. Hum Gene Ther. 2011;22(12):1575–1586. doi: 10.1089/hum.2011.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Di Stasi A, Tey S-K, Dotti G, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365(18):1673–1683. doi: 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao Y, Moon E, Carpenito C, et al. Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res. 2010;70(22):9053–9061. doi: 10.1158/0008-5472.CAN-10-2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.