

Abstract
Trisomy for chromosome 7 is frequently observed as an initiating event in sporadic colorectal cancer. Although unstable chromosome numbers and recurrent aneuploidies drive a large fraction of human cancers, targeted therapies selective to pre-neoplastic trisomic cells are non-existent. We have previously characterized a trisomy 7 cell line (1CT + 7) spontaneously derived from normal diploid human colonic epithelial cells that aberrantly expresses the epidermal growth factor receptor (EGFR, chromosome 7p11). Recent studies identified AICAR (5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside) as a pharmacological inhibitor of aneuploid murine fibroblast proliferation. Here, we report that AICAR induces profound cytostatic and metabolic effects on 1CT + 7 cells, but not on their isogenic diploid counterpart. Dose–response experiments indicate that 1CT + 7 cells are fourfold preferentially sensitive to AICAR compared to diploid cells. Unexpectedly, treatment of 1CT + 7 cells with AICAR led to a reversible 3.5-fold reduction (P = 0.0025) in EGFR overexpression. AICAR-induced depletion of EGFR protein can be abrogated through inhibition of the proteasome with MG132. AICAR also heavily promoted EGFR ubiquitination in cell-based immunoprecipitation assays, suggesting enhanced degradation of EGFR protein mediated by the proteasome. Moreover, treatment with AICAR reduced EGFR protein levels in a panel of human colorectal cancer cells in vitro and in xenograft tumors in vivo. Our data collectively support the pharmacological compound AICAR as a novel inhibitor of EGFR protein abundance and as a potential anticancer agent for aneuploidy-driven colorectal cancer.
Keywords: aneuploidy, EGFR, AICAR, chromosomal instability, trisomy
Introduction
Chromosomal instability (CIN) leading to aneuploidy, an abnormal number of chromosomes, has long been observed in solid human tumors throughout various tumorigenic stages. Aneuploidy arises when chromosomes fail to segregate properly during mitotic cell division.1 Although there is debate as to whether aneuploidy drives or merely serves as a consequence of cancer progression,2 targeting cells with an inappropriate number of chromosomes may be a practical chemopreventive or therapeutic approach.3,4 Tumors typically acquire non-random gains, losses or translocations of chromosomes that may permit selective benefits that are currently not well understood.5 One of the most common and recurrent cytogenetic alterations in sporadic colorectal cancer (CRC) is the appearance of trisomy for chromosome 7, detected in approximately 40% of early-staged adenomas and increases with disease progression.6,7 Although aneuploidy has been extensively studied in yeast and some mammalian cells, selective therapies targeting cells with such chromosomal abnormalities have only recently been explored.8
We have previously described the spontaneous generation of a trisomy 7 cell line (1CT + 7) derived from originally diploid (46,XY karyotype) human colonic epithelial cells (HCEC 1CT).9 The HCEC 1CT line, which is stably diploid when propagated in 2% serum, originated from non-malignant tissue of a previous CRC patient undergoing routine colonoscopy and immortalized with ectopic expression of CDK4 and hTERT.10 Under defined, serum-free culture conditions, HCECs with the acquisition of a third copy of chromosome 7 emerged from the diploid 1CT line, thus representing an isogenic model to examine the effects of trisomy 7 in vitro. Such isogenic cell lines can serve as useful cellular reagents to identify antigrowth or apoptosis-inducing compounds specific to aneuploid human cells. Interestingly, 1CT + 7 HCECs (along with trisomy 7 cells derived from breast,11 brain12 and esophageal13 tissue) aberrantly overexpresses the epidermal growth factor receptor (EGFR),9 conveniently located on chromosome 7p11.
EGFR, a membrane-localized receptor tyrosine kinase, becomes activated in the presence of EGF ligands14 and promotes cell proliferation through a cascade of signal-transduction events. Upon ligand binding to the extracellular domain of the receptor, autophosphorylation of C-terminal tyrosine residues (Y1173) initiates EGFR activation and internalization through endocytosis. These events lead to the recycling of receptors back to the cell surface for re-use or receptor degradation by the ubiquitin– proteasome and/or ubiquitin–lysosomal pathways.15–18 As many types of human cancers display overexpression19 or mutations in the EGFR gene, a number of antibody-based therapies targeting EGFR are currently utilized for therapeutic purposes.20 For example, the EGFR inhibitor cetuximab is used in the treatment of metastatic colorectal and head/neck cancers. We have previously shown that cetuximab (trade name Erbitux) is more effective at inhibiting HCEC 1CT+7 proliferation compared to karyotypically normal cells, suggesting that cells with trisomy 7 also acquire an increased dependency on EGFR signaling.9
The adenoside analog compound AICAR (5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside) has recently been found to elicit a selective apoptotic response in trisomic mouse embryonic fibroblasts and CIN-driven CRC lines,21 as well as anti-growth responses in EGFR-vIII-mutated glioblastoma cells.22 Immunocompromised mice treated daily with AICAR also displayed reduced xenograft tumor volume using a variety of karyotypically abnormal human cancer cell lines.21 The canonical effects of AICAR include a cellular energy stress response that induces AMP-activated protein kinase (AMPK) phosphorylation (T172) by the kinase LKB1 and subsequent inactivation of acetyl-CoA carboxylase (ACC). AMPK and ACC phosphorylation halts cell proliferation through inhibition of the mammalian target of rapamycin (mTOR) pathway.23 For these reasons, AICAR has been extensively studied as a potential anticancer drug.
In this study, we tested the effects of AICAR on the growth of diploid versus trisomic HCECs. We found that AICAR is selectively and potently cytostatic towards 1CT + 7 cells, but largely ineffective against diploid 1CT cells. Surprisingly, treatment of 1CT + 7 cells dramatically reduced EGFR overexpression and inhibited proliferation in an EGF-dependent manner. We propose that AICAR accelerates ubiquitination and subsequent degradation of EGFR in 1CT + 7 cells through an AMPK phosphorylation-independent mechanism. AICAR-mediated depletion of EGFR proteins is further confirmed in vitro with a panel of human CRC cells as well as in xenograft tumor models in vivo. Taken together, the anticancer properties of AICAR may be potentially beneficial as a chemopreventive agent to patients predisposed to sporadic CRC through the proliferative inhibition of trisomy 7 cells.
Results
AICAR induces selective cytostatic and metabolic effects on HCEC 1CT + 7
Previous studies indicate that mouse embryonic fibroblasts harboring one additional chromosome are more sensitive to apoptosis induced by AICAR exposure compared to diploid mouse embryonic fibroblasts.21 To determine whether 1CT (diploid) or 1CT + 7 (trisomy 7) HCECs are preferentially sensitive to treatment with AICAR, cells were cultured in increasing concentrations of AICAR for 5 days. This dose–response experiment revealed a fourfold lower EC50 value for 1CT + 7 cells compared to 1CT (0.225 and 1.026 mm, respectively) (Figure 1a). The growth inhibitory potency of 0.25 mm AICAR was further evaluated over a 6-day period by collecting sequential cell counts every 3 days after treatment. We found that AICAR completely prevented the proliferation of 1CT + 7, but not 1CT cells, compared to vehicle control (Figure 1b). This block in cell cycling could be attributed to an increase in the S-phase arrest in asynchronous cells (Supplementary Figure S1). Treatment with AICAR did not induce apoptosis, as all cells remain attached to the culture plate. When cells were plated at clonal density, AICAR completely and potently abolished the clonogenic potential of 1CT + 7 cells compared to 1CT cells (Figures 1c and d).
Figure 1.
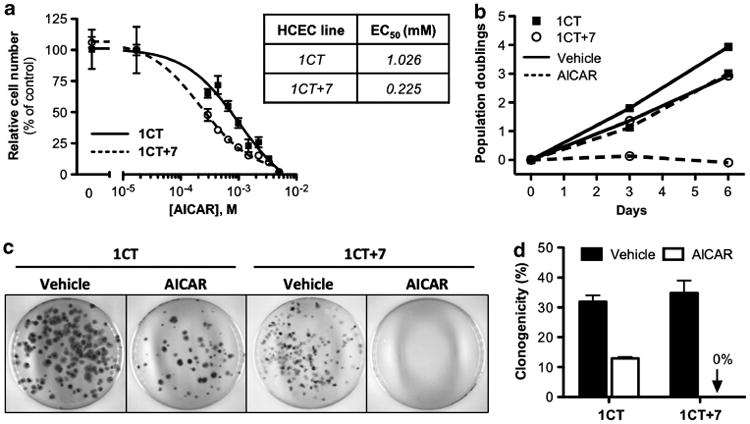
AICAR selectively and potently inhibits growth of 1CT + 7 HCECs. (a) HCEC 1CT and 1CT + 7 were treated with the indicated concentrations of AICAR and cell numbers were assessed 5 days post-treatment by CTG assay. Cell viability is normalized to vehicle control. (b) 1CT and 1CT + 7 cells were cultured in 0.25 mm AICAR or vehicle. Doublings were calculated from total cell counts obtained after 3 and 6 days of treatment. (c) HCECs were seeded at clonal density (200–400 cells per 10 cm2) and treated with 0.25 mm AICAR 24 h later. Colonies were stained after 2 weeks with crystal violet. Representative images of plates are shown. (d) Quantification of clonogenicity from (c). Columns represent mean ± s.e.m.
As the metabolic effects of AICAR on mammalian cells have previously been investigated,24 we next inquired whether AICAR can selectively perturb critical metabolic processes in either of the HCEC lines. 1CT and 1CT + 7 cells were treated with vehicle or AICAR-containing medium for 48 h and cell culture supernatants were collected for analysis. We found that treatment with AICAR selectively decreased both the consumption of glucose (Figure 2a) and the production of lactate (Figure 2b) in 1CT + 7 cells, whereas no significant effects were observed in diploid cells. Furthermore, 1CT + 7 cells have a noticeably higher basal rate of both metabolic processes compared to diploid cells, similar to observations published elsewhere.25
Figure 2.
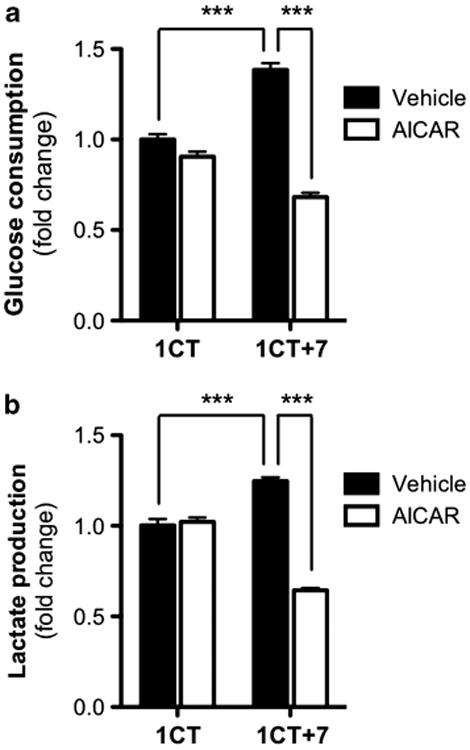
AICAR impairs the metabolism of 1CT + 7 HCECs. AICAR treatment (0.25 mm) selectively decreases (a) glucose consumption and (b) lactate production in 1CT + 7 HCECs. Glucose/lactate levels were measured in cell culture supernatants collected after 48-h treatment and normalized to both cell-free medium and cell counts. Columns represent mean ± s.e.m. (***P<0.0001, by two-tailed, unpaired Student's t-test).
AICAR negatively regulates EGFR protein levels in 1CT + 7 HCECs As 1CT + 7 cells express high levels of EGFR compared to 1CT cells, we tested whether the mechanism by which AICAR inhibits 1CT + 7 proliferation could be mediated through EGFR. Both 1CT and 1CT + 7 cells were treated for 24 h with 0.25 mm AICAR and whole cell lysates were probed for EGFR by western blot analyses. Surprisingly, we show that AICAR treatment led to a 3.5-fold decrease in 1CT + 7 EGFR levels, but had only marginal effects on diploid 1CT cells (Figures 3a and b). Phosphorylation of well-known AICAR targets, such as AMPK (Supplementary Figure S2), ACC and mTOR (Figure 3a), were negligible between control and compound-treated cells (with slight increases in ACC phosphorylation in the presence of AICAR). To determine whether LKB1, the kinase responsible for AMPK activation during energy stress, is required for EGFR depletion, 1CT + 7 cells were transfected with control or LKB1 small interfering RNA (siRNA) for 3 days to deplete endogenous LKB1 levels. siRNA-transfected 1CT + 7 cells were then treated with vehicle or AICAR for 24 h and whole-cell lysates were resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE). We show that AICAR-induced EGFR depletion is independent of LKB1, as knockdown of LKB1 has no effect on EGFR levels regulated by AICAR treatment (Supplementary Figure S3). In addition, treatment with AICAR for a short-term period (30 min) also did not alter EGFR levels in HCECs (Supplementary Figure S4), suggesting that AICAR does not facilitate rapid EGFR turnover through ligand-independent receptor activation. The depletion of EGFR proteins is further verified by immunofluourescent staining on AICAR-treated 1CT + 7 cells (Figure 3c).
Figure 3.
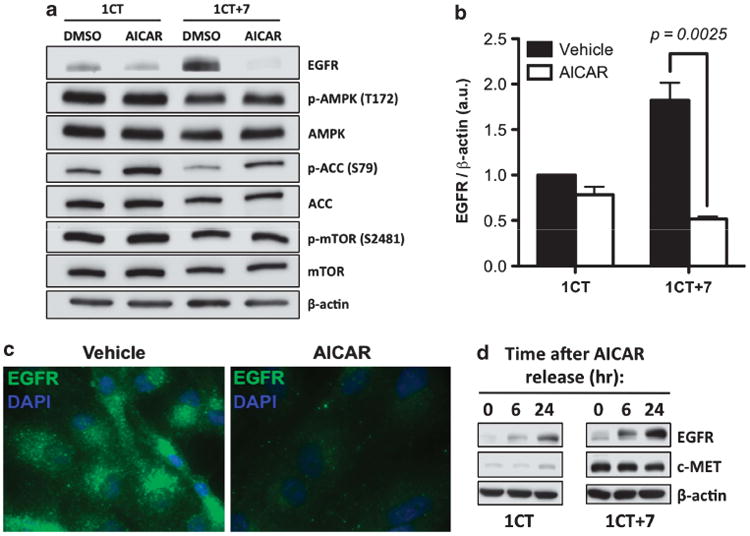
AICAR negatively regulates EGFR protein levels. (a) HCECs were treated for 24 h with 0.25 mm AICAR. Whole cell lysates were resolved by SDS-PAGE and probed for the indicated proteins. No significant changes in the phosphorylation status of known AICAR targets were observed, with the exception of slight increases in p-ACC levels. (b) Quantification of (a) from three independent experiments and normalized to loading control. AICAR treatment induced a 3.5-fold reduction in EGFR protein levels. Columns represent mean ± s.e.m. (c) Confirmation of EGFR repression by immunostaining (× 100 magnification) of 24-h 0.25 mm AICAR-treated 1CT + 7 cells. (d) EGFR levels are restored following 24-h release from AICAR. Cells were treated overnight with AICAR, thoroughly washed and replenished with fresh medium for the indicated time points.
To determine whether depletion of EGFR by AICAR is a permanent or transient effect, HCECs were treated with AICAR for 24 h, washed and replaced with serum- and AICAR-free culture medium for the indicated time points. The 24 h release of cells from AICAR resulted in the restoration of EGFR expression (Figure 3d) by western blot analysis. AICAR also did not affect c-MET protein levels; another chromosome 7-located receptor tyrosine kinase overexpressed in 1CT + 7 cells. These results support EGFR specificity rather than a global repression of receptor tyrosine kinases.
To establish whether AICAR reduces EGFR at the transcriptional level, we examined steady-state EGFR mRNA transcripts by semiquantitative reverse transcription–polymerase chain reaction. Total RNA isolated from cells treated with AICAR was reverse transcribed and the EGFR tyrosine kinase domain was detected by polymerase chain reaction. EGFR transcripts were unaltered in both cell types following treatment with AICAR (Supplementary Figure S5), suggesting a regulation at the protein level.
AICAR treatment induces ligand-dependent EGFR degradation
We subsequently asked whether the availability of EGF ligands is required for AICAR-induced EGFR depletion in 1CT + 7 cells. To determine if the presence of EGF is necessary for this mechanism of action, we measured EGFR protein levels in 1CT + 7 cells treated with AICAR with and without 20 ng/ml EGF. Culturing cells in the absence of EGF abolished EGFR depletion induced by AICAR, indicating a requirement for EGF ligand and most likely EGFR activation/turnover (Figure 4a). Ligand stimulation is known to induce EGFR internalization, followed by either receptor recycling or degradation.26 To establish whether AICAR can induce EGFR degradation due to ligand stimulation, we conducted a time-point experiment in which EGF-starved 1CT + 7 cells were stimulated with EGF ligand to follow receptor degradation kinetics. In Figure 4b, we show that 48 h EGF-starved 1CT + 7 cells treated with vehicle induced only a slight degree of EGFR degradation due to EGF stimulation for the indicated time points. However, EGF-starved 1CT + 7 cells treated with AICAR for 24 h before ligand stimulation provoked rapid and potent EGFR degradation (Figure 4b). These results suggest that AICAR treatment leads to accelerated EGFR degradation in a ligand-dependent manner in contrast to receptor recycling back to the cell surface.
Figure 4.
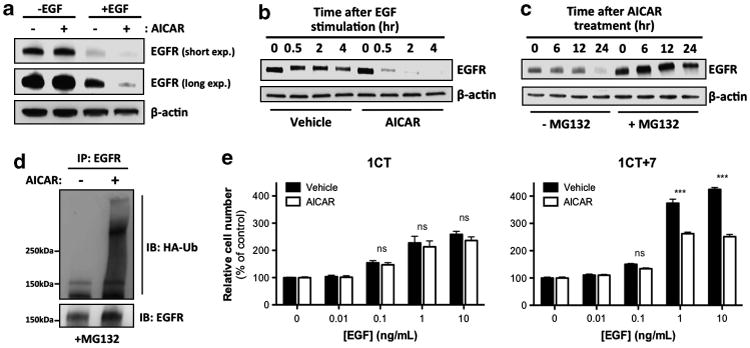
AICAR accelerates ubiquitination and ligand-dependent degradation of EGFR. (a) 1CT + 7 cells were treated with AICAR in the presence or absence of 20 ng/ml EGF for 24 h and lysates were analyzed by SDS–PAGE. In the absence of EGF ligand, AICAR has no effect on EGFR protein levels. (b) EGF-starved 1CT + 7 cells were treated with AICAR for 24 h, followed by 20 ng/ml EGF stimulation for the indicated time points. AICAR accelerates ligand-induced receptor degradation compared to vehicle-treated cells. (c) 1CT + 7 cells were incubated with 10 μm of the proteasome inhibitor MG132 for 1 h before AICAR treatment for the following time points. Whole cell lysates were collected and analyzed by SDS–PAGE and probed for EGFR. (d) 1CT + 7 cells were transfected with HA-tagged ubiquitin and treated with 10μm MG132 and 0.25 mm AICAR for 24 h. Lysates were subjected to immunoprecipitation with an EGFR agarose-conjugated antibody and analyzed by SDS– PAGE. (e) HCECs were treated with AICAR in increasing EGF concentrations for 5 days and cell numbers were assessed by CTG assay. AICAR selectively prevents EGF-induced proliferation in 1CT + 7 cells. Columns represent mean ± s.e.m. (***P<0.0001, by two-tailed, unpaired Student's t-test).
We next examined whether AICAR treatment led to an increase in proteasomal-mediated degradation of EGFR protein. 1CT + 7 cells were pre-treated with the proteasome inhibitor MG132 (10 μm) for 1 h before 24-h AICAR treatment and collection of lysates at the indicated time points. MG132 pre-treatment resulted in a rescue of EGFR levels (Figure 4c), suggesting that AICAR-induced EGFR depletion may be, at least partially, attributed to an increase in protein degradation by the proteasome (at a time frame between 12 and 24 h post-treatment). Although the majority of EGFR proteins is stabilized because of proteasome inhibition in the presence of AICAR, the slight decrease in EGFR levels following 24-h AICAR exposure in MG132-treated 1CT + 7 cells may be attributed to the contributions by the lysosomal degradative pathway. To determine if AICAR enhances ubiquitination of EGFR, we performed an ubiquitination assay in 24-h AICAR-treated 1CT + 7 cells transiently expressing hemagglutinin (HA) epitope-tagged ubiquitin. MG132 was used to prevent EGFR degradation and lysates were immunoprecipitated with an EGFR agarose-conjugated antibody. We observed a considerable increase in EGFR ubiquitination (most likely polyubiquitination as indicated by an HA-positive smear) following AICAR treatment (Figure 4d).
Next, HCECs were cultured with increasing concentrations of EGF to determine if AICAR can prevent growth stimulated by EGF ligands. Our data indicate that AICAR selectively inhibits EGF-induced proliferation in 1CT + 7 cells (Figure 4e), but not in diploid 1CT cells. These results demonstrate that EGF ligands contribute to the specific antiproliferative effects of AICAR on aneuploid human cells. Treatment of 1CT cells with 10 ng/ml EGF induced an approximate 2.6-fold growth response compared to a 4.3-fold increase in 1CT + 7 cells. However, 1CT + 7 cells treated with AICAR resulted in only a 2.5-fold increase in proliferation, reducing EGF-induced growth rates closer to those of diploid cells. Comparably, knockdown of EGFR in 1CT + 7 cells with siRNAs causes a growth inhibitory effect similar to AICAR treatment (data not shown).
Downregulation of EGFR by AICAR in human CRC cells in vitro and in vivo
To determine if AICAR-induced depletion of EGFR is specific towards HCECs, we treated a panel of CIN and microsatellite unstable (MIN) human CRC cell lines with increasing concentrations of AICAR for 36 h. A dose-dependent downregulation of EGFR upon treatment with AICAR was observed (Figure 5a) in five out of six cancer cell lines tested. This effect also appears to be independent of CIN (SW480, HT29) versus MIN (HCT116, LoVo, DLD-1) status as EGFR downregulation was observed in cancer cells of both genetic backgrounds, although similar results were not detected in the pseudodiploid cell line HCT15. As shown previously,21 AICAR effectively reduces tumor volume in mouse xenograft models using CRC cell lines compared to mice treated with phosphate-buffered saline (PBS) control. Daily intraperitoneal injection of AICAR for 18 days reduces xenograft tumor volume by roughly 33 and 45% compared to vehicle-treated mice using the CRC cell lines LoVo and HT29, respectively.21 These tumors were then harvested, fixed and histologically sectioned for EGFR analysis by immunohistochemistry. In agreement with our in vitro data, AICAR treatment reduced the intensity of EGFR staining in xenograft tumors in vivo compared to control-injected animals (Figure 5b).
Figure 5.
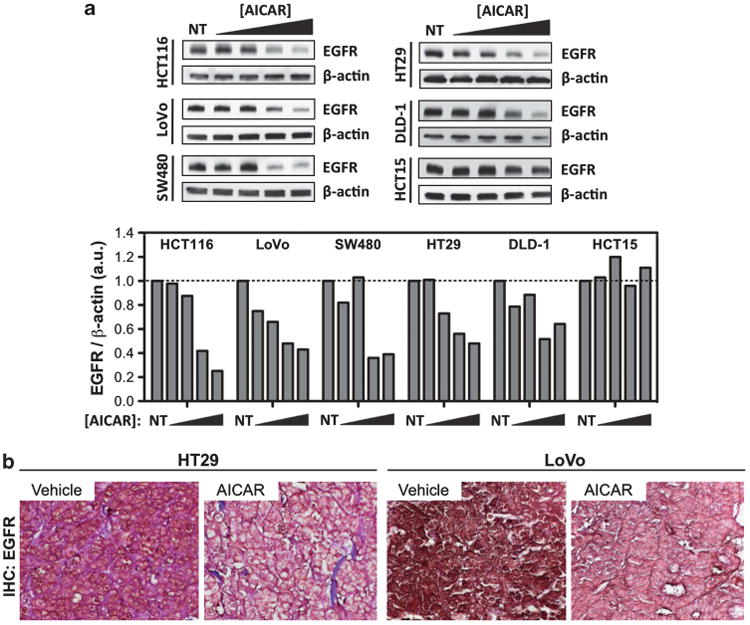
AICAR represses EGFR levels in a panel of human colorectal cancer cell lines in a dose-dependent manner in vitro and in xenograft tumor models in vivo. (a) Six colon cancer cell lines were treated with increasing doses of AICAR (0, 0.1, 0.2, 0.4 and 0.8 mm) for 36 h and lysates were probed for EGFR. Bands were normalized to loading control and quantified relative to non-treated (NT) cells. (b) Xenograft tumors were established from HT29 and LoVo cells and mice were treated daily with PBS vehicle or AICAR for 18 days after injection. Representative images of harvested tumor sections stained for EGFR by immunohistochemistry are shown (× 63 magnification).
It is important to note that most MIN cell lines also display some degree of aneuploidy, amplifications and/or chromosomal rearrangements.27 As the sensitivity of cell lines to AICAR does not appear to strongly correlate with EGFR expression levels or modal number of chromosomes per cell (Supplementary Figure S6), further studies are required to more carefully evaluate the mechanisms involved in AICAR sensitivity and EGFR regulation. As reported previously21 and in this study, the increased sensitivity of non-transformed mouse and human cells to AICAR appears to correlate with an additional gain of a single chromosome (trisomic, 2n + 1), whereas cancer cells represent a plethora of chromosomal abnormalities and other genetic alterations. Nonetheless, these data serve as initial observations in the use of AICAR as a chemopreventive or therapeutic agent against trisomy 7 cells through EGFR downregulation and perhaps in combination with AMPK activation.
Discussion
The discovery of aneuploid-specific compounds for the treatment of CIN-driven cancers is reliant on useful models to recapitulate the aneuploid state. We have employed an isogenic HCEC line that is distinctive at the karyotypic level: normal diploid versus trisomy 7 derived from identical cell populations. In this study, we provide data supporting AICAR as a prospective chemopreventive compound and further reveal for the first time that EGFR is a major target of AICAR in a trisomic, yet otherwise normal, non-malignant human epithelial cell line. Whether or not AICAR may be an effective therapeutic agent against EGFR-driven cancers (for example, lung cancers) remains unknown.
Under optimal growth conditions, aneuploidy has been shown to be detrimental to cellular fitness in terms of proliferation rates compared to diploid cells.25,28 Evidence suggests that aneuploid cells develop an intrinsic stress response, independent of the alteration to a specific chromosome, which may assist them in thriving under selective pressures.29 One hypothesis to explain impaired proliferation is that additional chromosomes generate excess proteins30 that in turn induce proteotoxic stress.28 These proteomic changes could therefore lead to compensatory genetic alterations in protein degradation pathways (for example, the ubiquitin–proteasome pathway) to counteract the effects of imbalanced protein levels. In budding yeast, aneuploid strains develop mutations in the deubiquitinating enzyme UBP6 to compensate for the increased intracellular protein composition and this alteration is sufficient to improve their proliferation.31 In this work, it is plausible that AICAR treatment may also be attenuating similar protein degradation pathways to revert overexpressed proteins generated by extra chromosomes to either normal or low levels that would otherwise provide proliferative cues (for example, EGFR).
Figure 6 illustrates our current working model on the mechanism behind AICAR-induced growth inhibition. We have previously reported an enhanced dependency of 1CT + 7 cells on EGFR signaling compared to diploid cells.9 In the proposed schematic, EGF-ligand-induced receptor activation and internalization is required for the intracellular activity of AICAR. Upon ligand binding and receptor sorting by endosomes, we hypothesize that AICAR indirectly accelerates EGFR ubiquitination and subsequent proteolysis. This reduces the recycling of receptors back to the cell surface and therefore diminishes sensitivity to EGF ligand. Further biochemical analysis is required to elucidate the exact mechanism of action and to uncover any unidentified pathways that may be involved in these events. It is also currently unclear the extent of which the lysosomal degradative pathway contributes to AICAR-mediated downregulation of EGFR.
Figure 6.
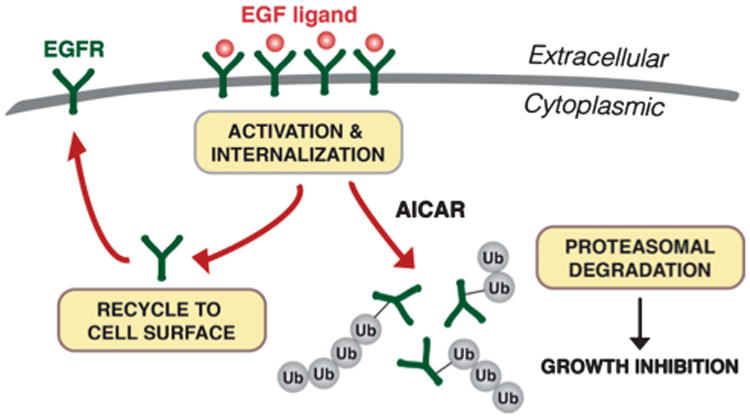
Proposed model for AICAR-induced EGFR depletion and growth inhibition. In the presence of EGF ligand, EGFR becomes autophosphorylated and internalized for sorting (endocytic pathway not shown for purpose of clarity). Rather than recycling receptors to the cell surface, we propose that AICAR promotes the ubiquitination and degradation of EGFR via the proteasome, leading to subsequent growth inhibition. The increased dependency of trisomy 7 cells on EGFR signaling may result in a differential toxicity to AICAR.
As trisomy 7 is found in a large fraction of premalignant lesions and in the vast majority of human colorectal carcinomas, we hypothesized that 1CT cells cultured under defined, serumdepleted conditions acquired a third copy of chromosome 7 and upregulated EGFR levels, supporting a clonal expansion of ‘EGFR-addicted’ 1CT + 7 cells. Therefore, a more potent cytostatic effect is observed when inhibiting EGFR in oncogene-addicted cells32 (that is, the effect of cetuximab on 1CT + 7 cells compared to 1CT). Interestingly, ectopic overexpression of EGFR in both normal human bronchial epithelial cells33 or in HCEC 1CT is insufficient to sensitize cells to AICAR (Supplementary Figure S7). These results suggest that AICAR is more effective against EGFR-dependent cells and/or perhaps those that endogenously overexpress EGFR.
Cells typically respond to AICAR through phosphorylation and subsequent activation of AMPK, the best-known target of AICAR.34 Although Tang et al.21 provided evidence of an AMPK-dependent cytostatic effect in aneuploid mouse embryonic fibroblasts, they did not observe similar specificity when activating AMPK through an alternative method, such as treatment with the compound metformin, an indirect AMPK activator. We also observed that metformin did not specifically sensitize 1CT + 7 cells (Supplementary Figure S8a), suggesting that the specificity of AICAR may be independent of AMPK function. Furthermore, both HCEC lines were similarly responsive to mTOR repressionmediated growth inhibition using the compound Torin1 (Supplementary Figure S8b), indicating that the sensitivity of trisomy 7 cells to AICAR is also not regulated through the canonical mTOR pathway. EGFR degradation induced by AICAR could be either dependent on AMPK (treatment with AICAR leads to AMPK activation by LKB1 leading to EGFR depletion) or independent of AMPK (treatment with AICAR affects another pathway that leads to EGFR depletion). Although we have no direct experimental evidence whether the effects of AICAR on EGFR are dependent on AMPK, we hypothesize that AMPK, like LKB1, is not required for EGFR degradation as T172 activation of AMPK is dependent on LKB1.35 Because AMPK activation requires LKB1 and our data indicate that LKB1 is not required for EGFR degradation (Supplementary Figure S3), these results altogether suggest that AICAR-induced EGFR depletion is independent of AMPK activation. Although activation of AMPK may play a large role in reducing cell proliferation induced by AICAR in some cells, our data indicate that EGFR depletion may be an alternative mechanism of growth inhibition. AMPK activation and targeted proteolysis of EGFR could also be working collectively to reduce the growth of aneuploid cells.
Our findings describe AICAR as a novel compound in regulating EGFR protein levels in both non-malignant and malignant genomically unstable cells, a potential therapeutic strategy in the chemoprevention of aneuploidy-driven CRC. AICAR selectively inhibits proliferation of 1CT + 7 HCECs, whereas diploid cells are largely unaffected. Furthermore, these effects are reversible (Figure 3c) and EGFR levels are restored upon 24-h washout. These results indicate that AICAR may be effective in treating patients with colonic adenomas driven by aneuploidy with minimal toxicity to normal epithelial tissue. As the human colon constantly undergoes rapid turnover, AICAR may be used to prevent the spontaneous growth of aneuploid cells until they are shedded at the apex of the colonic crypt.36 Upon turnover-mediated abolishment of aneuploid cells from the colonic epithelium, AICAR treatment may be discontinued and EGFR levels will presumably be restored in normal tissue. Also worth noting is that AICAR efficacy is dependent on the presence of EGF ligand, thus only affecting cycling cells in a mitogen-containing environment.
To conclude, we have identified AICAR as a novel regulator of EGFR signaling that potently prevents proliferation of a minimally aneuploid (trisomy 7) HCEC line. The downregulation of EGFR protein levels can be attributed to increased ubiquitination and proteasomal-mediated degradation in an EGF-dependent manner. We propose that this negative regulation of EGFR (in possible combination with AMPK activation) inhibits the growth of both malignant and non-malignant human colonic cells with unstable chromosome numbers. Future studies involve testing the effects of AICAR on a diverse set of aneuploid cancer cell lines, further elucidating the mechanism of preferential sensitivity to cells with genomic instability, and to determine critical pathways involved in AICAR-mediated EGFR regulation. Further analyses on a broad panel of aneuploid, non-transformed human cell lines are also unquestionably needed, although the field is currently limited by the lack of these cell-based models. Therefore, the development of new aneuploid systems using human cells or mouse models is of utmost importance towards therapeutic discovery in exploiting the aneuploid state for clinical benefit. This study provides further rationale for the potential clinical utility of AICAR as a chemopreventive compound to patients predisposed or recurring from sporadic CRC or as a therapeutic agent to those currently diagnosed with CRC.
Materials and Methods
Cell culture and reagents
Culture conditions of HCECs have been previously described elsewhere.10 Briefly, HCECs are maintained in 2% oxygen/5% carbon dioxide conditions on Primaria dishes (BD Biosciences, San Jose, CA, USA). The 4:1 DMEM:Medium 199 (Hyclone, Thermo Scientific, Waltham, MA, USA) is supplemented with 2% cosmic calf serum (Hyclone, Thermo Scientific) and 20 ng/ml EGF (PeproTech, Rocky Hill, NJ, USA). HCEC experiments are performed in serum-free, defined conditions in the presence of EGF, unless otherwise stated. Human CRC cell lines are cultured with 10% serum. The identity of all cell lines were verified by DNA fingerprinting.
AICAR (Toronto Research Chemicals, Toronto, ON, Canada), metformin (Sigma, St Louis, MO, USA) and MG132 (Torcris Biosciences, Bristol, UK) were dissolved in dimethylsulfoxide. Reverse transcription-polymerase chain reaction primers for EGFR-tyrosine kinase domains were previously described elsewhere.9 siGENOME siRNAs targeting LKB1 or non-targeting controls were purchased from Dharmacon (Lafayette, CO, USA) and reverse transfections were performed using RNAiMAX transfection reagent (Invitrogen, Grand Island, NY, USA) under the manufacturer's instructions.
Growth assays
Cells (2 × 103) were seeded in 96-well clear-bottom plates in serum-free medium for 24 h before the addition of vehicle or drug-containing medium. At 5 days post-drug treatment, CellTiter-Glo (Promega, Madison, WI, USA) reagent is added to each well as per the manufacturer's instructions and shaken. Luminescent ATP levels were detected by an Envision plate reader (Perkin-Elmer, Waltham, MA, USA) and cell numbers are normalized to control wells.
For colony formation assays, cells were plated at clonal density (200–400 cells per 10cm2 dish) in 2% cosmic calf serum to promote low-density cell adhesion to plates. After 24 h, cells were washed with PBS and replaced with either vehicle or 0.25 mm AICAR in serum-free medium. At 15 days post-treatment, colonies were washed with PBS and stained with 6% glutaraldehyde/0.5% crystal violet solution for 30 min and counted.
Metabolic analysis
HCECs were seeded in six-well dishes at various densities (1–5 × 103 cells) in 2 ml serum-free medium and replaced with 1 ml vehicle or AICAR-containing medium 24 h later. Cell culture supernatants were collected 48 h post-treatment and centrifuged to exclude debris. Supernatants were analyzed using a BioProfile Basic-4 Automated Analyzer (Nova Biomedical, Waltham, MA, USA) and values were normalized to cell-free medium incubated alongside drug-treated cells. Glucose consumption and lactate production is then normalized to the relative sum of total cell counts per hour over a 48-h treatment period.
Western blot
Whole cell lysates were collected in Laemmli sample buffer and boiled. Equal amount of lysates were resolved on 4–15% polyacrylamide gels (Bio-Rad, Hercules, CA, USA) and transferred to polyvinylidene difluoride. Membranes were then probed with the following primary antibodies: anti-EGFR (sc-03; Santa Cruz, Santa Cruz, CA, USA), anti-Met (L41G3; Cell Signaling, Danvers, MA, USA), anti-RalA (610221; BD Biosciences), anti-LKB1 (27D10; Cell Signaling) and anti-HA (C29F4; Cell Signaling). Anti-phospho-AMPK, phospho-ACC and phospho-mTOR are from the AMPK and ACC sampler kit (9957; Cell Signaling). Horse radish peroxidase-conjugated goat anti-mouse or anti-rabbit secondary antibodies (Jackson ImmunoResearch, West Grove, PA, USA) were detected by SuperSignal West Femto Chemiluminescent Substrate kit (Thermo Scientific) and imaged on a G:Box (Syngene, Frederick, MD, USA) gel documentation system. Bands were quantified using the GeneTools (Syngene) software and normalized to loading controls.
Immunofluorescence and immunohistochemistry
For immunofluorescence, cells cultured in chamberslides were fixed in 4% paraformaldehyde for 10 min, permeabilized with 0.5% Triton X and blocked (10% goat serum, 3% bovine serum albumin) for 5 min. Anti-EGFR primary antibody (sc-03) was diluted 1:50 in blocking solution and applied for 1 h, followed by goat-anti-rabbit fluorescein isothiocyanate secondary for 1 h. Cells were then stained for 4′,6-diamidino-2-phenylindole, mounted in Mowiol 4-88 (Calbiochem, Darmstadt, Germany), and observed at × 100 magnification.
Immunohistochemistry was performed on 5-μm paraffin-embedded sections of Bouin-fixed HT29 and LoVo xenografted tumors following heat-induced antigen retrieval with 0.01 M sodium citrate buffer (pH 6.0). Tumor formation and vehicle/AICAR treatment of immunocompromised mice was previously described elsewhere.21 Anti-EGFR primary antibody (sc-03) at a 1:200 dilution was applied overnight at 4°C. Sections were then blocked using the Avidin/Biotin Blocking Kit (Vector Labs, Burlingame, CA, USA) and incubated with biotinylated secondary antibody, streptavidin-conjugated horse radish peroxidase and DAB reagents as described and supplied in the Peroxidase Detection System (Novocastra/Leica, Buffalo Grove, IL, USA).
Ubiquitination assay
HCEC 1CT + 7 (106) were reversed transfected with 1μg HA-tagged ubiquitin plasmid using Effectene Transfection Reagent (Qiagen, Valencia, CA, USA) according to the manufacturer's instructions in 2% serum medium. The next day, cells were washed with PBS and replaced with serum-free medium containing 0.25 mm AICAR and 10μm MG132. After 24 h, cells were washed with ice-cold PBS, collected in IP buffer (50 mm Tris, 150 mm NaCl, 10% glycerol, 1% Tween-20, protease/phosphatase inhibitors) and lysed for 30 min at 4°C, followed by passage through a 27.5 G needle. Lysates were then immunoprecipitated with EGFR agaroseconjugated antibody (sc120AC; Santa Cruz) for 2 h at 4°C, followed by three washes. Samples were then boiled in the presence of SDS and resolved by 4–15% SDS–PAGE. Membranes were probed using anti-EGFR and anti-HA antibodies.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank RY Quach for figure artwork and U Eskiocak, G Stadler and AR Mullen (UT Southwestern) for technical assistance. We also express gratitude to YC Tang and A Amon (MIT) for providing xenograft tumors for IHC analysis and for critical reading of the manuscript. HA-tagged ubiquitin plasmids and anti-HA antibodies were a kind gift from LJ Huang (UT Southwestern). EGFR-V5 expression constructs and human bronchial epithelial cell-EGFR cell lines were provided by C Nirodi (UT Southwestern). This work was supported by CPRIT Training Grant RP101496 to PL, NASA Grants NNX09AU95G, NNX11AC15G, and NNX11AC54G and NCI SPORE CA70907 to JWS.
Footnotes
Conflict Of Interest: The authors declare no conflict of interest.
Disclaimer: The content of this manuscript is original and has not been published or accepted for publication, either in whole or in part, in any form (other than as an abstract or other preliminary publication). The authors declare that no part of the manuscript is currently under consideration for publication elsewhere.
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
References
- 1.Gordon DJ, Resio B, Pellman D. Causes and consequences of aneuploidy in cancer. Nat Rev Genet. 2012;13:189–203. doi: 10.1038/nrg3123. [DOI] [PubMed] [Google Scholar]
- 2.Sheltzer JM, Amon A. The aneuploidy paradox: costs and benefits of an incorrect karyotype. Trends Genet. 2011;27:446–453. doi: 10.1016/j.tig.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bakhoum SF, Compton DA. Chromosomal instability and cancer: a complex relationship with therapeutic potential. J Clin Invest. 2012;122:1138–1143. doi: 10.1172/JCI59954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McGranahan N, Burrell RA, Endesfelder D, Novelli MR, Swanton C. Cancer chromosomal instability: therapeutic and diagnostic challenges. EMBO Rep. 2012;13:528–538. doi: 10.1038/embor.2012.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weaver BA, Silk AD, Montagna C, Verdier-Pinard P, Cleveland DW. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell. 2007;11:25–36. doi: 10.1016/j.ccr.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 6.Habermann JK, Paulsen U, Roblick UJ, Upender MB, McShane LM, Korn EL, et al. Stage-specific alterations of the genome, transcriptome, and proteome during colorectal carcinogenesis. Genes Chromosomes Cancer. 2007;46:10–26. doi: 10.1002/gcc.20382. [DOI] [PubMed] [Google Scholar]
- 7.Bomme L, Bardi G, Pandis N, Fenger C, Kronborg O, Heim S. Clonal karyotypic abnormalities in colorectal adenomas: clues to the early genetic events in the adenoma–carcinoma sequence. Genes Chromosomes Cancer. 1994;10:190–196. doi: 10.1002/gcc.2870100307. [DOI] [PubMed] [Google Scholar]
- 8.Manchado E, Malumbres M. Targeting aneuploidy for cancer therapy. Cell. 2011;144:465–466. doi: 10.1016/j.cell.2011.01.037. [DOI] [PubMed] [Google Scholar]
- 9.Ly P, Eskiocak U, Kim SB, Roig AI, Hight SK, Lulla DR, et al. Characterization of aneuploid populations with trisomy 7 and 20 derived from diploid human colonic epithelial cells. Neoplasia. 2011;13:348–357. doi: 10.1593/neo.101580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roig AI, Eskiocak U, Hight SK, Kim SB, Delgado O, Souza RF, et al. Immortalized epithelial cells derived from human colon biopsies express stem cell markers and differentiate in vitro. Gastroenterology. 2010;138:e1–e5. doi: 10.1053/j.gastro.2009.11.052. [DOI] [PubMed] [Google Scholar]
- 11.Briand P, Nielsen KV, Madsen MW, Petersen OW. Trisomy 7p and malignant transformation of human breast epithelial cells following epidermal growth factor withdrawal. Cancer Res. 1996;56:2039–2044. [PubMed] [Google Scholar]
- 12.Sareen D, McMillan E, Ebert AD, Shelley BC, Johnson JA, Meisner LF, et al. Chromosome 7 and 19 trisomy in cultured human neural progenitor cells. PLoS One. 2009;4:e7630. doi: 10.1371/journal.pone.0007630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garewal H, Meltzer P, Trent J, Prabhala R, Sampliner R, Korc M. Epidermal growth factor receptor overexpression and trisomy 7 in a case of Barrett's esophagus. Dig Dis Sci. 1990;35:1115–1120. doi: 10.1007/BF01537584. [DOI] [PubMed] [Google Scholar]
- 14.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 15.Longva KE, Blystad FD, Stang E, Larsen AM, Johannessen LE, Madshus IH. Ubiquitination and proteasomal activity is required for transport of the EGF receptor to inner membranes of multivesicular bodies. J Cell Biol. 2002;156:843–854. doi: 10.1083/jcb.200106056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dikic I. Mechanisms controlling EGF receptor endocytosis and degradation. Biochem Soc Trans. 2003;31:1178–1181. doi: 10.1042/bst0311178. [DOI] [PubMed] [Google Scholar]
- 17.Levkowitz G, Waterman H, Ettenberg SA, Katz M, Tsygankov AY, Alroy I, et al. Ubiquitin ligase activity and tyrosine phosphorylation underlie suppression of growth factor signaling by c-Cbl/Sli-1. Mol Cell. 1999;4:1029–1040. doi: 10.1016/s1097-2765(00)80231-2. [DOI] [PubMed] [Google Scholar]
- 18.Levkowitz G, Waterman H, Zamir E, Kam Z, Oved S, Langdon WY, et al. c-Cbl/Sli-1 regulates endocytic sorting and ubiquitination of the epidermal growth factor receptor. Genes Dev. 1998;12:3663–3674. doi: 10.1101/gad.12.23.3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cohen G, Mustafi R, Chumsangsri A, Little N, Nathanson J, Cerda S, et al. Epidermal growth factor receptor signaling is up-regulated in human colonic aberrant crypt foci. Cancer Res. 2006;66:5656–5664. doi: 10.1158/0008-5472.CAN-05-0308. [DOI] [PubMed] [Google Scholar]
- 20.Messersmith WA, Ahnen DJ. Targeting EGFR in colorectal cancer. N Engl J Med. 2008;359:1834–1836. doi: 10.1056/NEJMe0806778. [DOI] [PubMed] [Google Scholar]
- 21.Tang YC, Williams BR, Siegel JJ, Amon A. Identification of aneuploidy-selective antiproliferation compounds. Cell. 2011;144:499–512. doi: 10.1016/j.cell.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo D, Hildebrandt IJ, Prins RM, Soto H, Mazzotta MM, Dang J, et al. The AMPK agonist AICAR inhibits the growth of EGFRvIII-expressing glioblastomas by inhibiting lipogenesis. Proc Natl Acad Sci USA. 2009;106:12932–12937. doi: 10.1073/pnas.0906606106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9:563–575. doi: 10.1038/nrc2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aschenbach WG, Hirshman MF, Fujii N, Sakamoto K, Howlett KF, Goodyear LJ. Effect of AICAR treatment on glycogen metabolism in skeletal muscle. Diabetes. 2002;51:567–573. doi: 10.2337/diabetes.51.3.567. [DOI] [PubMed] [Google Scholar]
- 25.Williams BR, Prabhu VR, Hunter KE, Glazier CM, Whittaker CA, Housman DE, et al. Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science. 2008;322:703–709. doi: 10.1126/science.1160058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang F, Goh LK, Sorkin A. EGF receptor ubiquitination is not necessary for its internalization. Proc Natl Acad Sci USA. 2007;104:16904–16909. doi: 10.1073/pnas.0707416104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meltzer SJ, Yin J, Manin B, Rhyu MG, Cottrell J, Hudson E, et al. Microsatellite instability occurs frequently and in both diploid and aneuploid cell populations of Barrett's-associated esophageal adenocarcinomas. Cancer Res. 1994;54:3379–3382. [PubMed] [Google Scholar]
- 28.Torres EM, Sokolsky T, Tucker CM, Chan LY, Boselli M, Dunham MJ, et al. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science. 2007;317:916–924. doi: 10.1126/science.1142210. [DOI] [PubMed] [Google Scholar]
- 29.Chen G, Bradford WD, Seidel CW, Li R. Hsp90 stress potentiates rapid cellular adaptation through induction of aneuploidy. Nature. 2012;482:246–250. doi: 10.1038/nature10795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pavelka N, Rancati G, Zhu J, Bradford WD, Saraf A, Florens L, et al. Aneuploidy confers quantitative proteome changes and phenotypic variation in budding yeast. Nature. 2010;468:321–325. doi: 10.1038/nature09529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Torres EM, Dephoure N, Panneerselvam A, Tucker CM, Whittaker CA, Gygi SP, et al. Identification of aneuploidy-tolerating mutations. Cell. 2010;143:71–83. doi: 10.1016/j.cell.2010.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gazdar AF, Shigematsu H, Herz J, Minna JD. Mutations and addiction to EGFR: the Achilles ‘heal’ of lung cancers? Trends Mol Med. 2004;10:481–486. doi: 10.1016/j.molmed.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 33.Das AK, Chen BP, Story MD, Sato M, Minna JD, Chen DJ, et al. Somatic mutations in the tyrosine kinase domain of epidermal growth factor receptor (EGFR) abrogate EGFR-mediated radioprotection in non-small cell lung carcinoma. Cancer Res. 2007;67:5267–5274. doi: 10.1158/0008-5472.CAN-07-0242. [DOI] [PubMed] [Google Scholar]
- 34.Rattan R, Giri S, Singh AK, Singh I. 5-Aminoimidazole-4-carboxamide-1-beta-d-ribofuranoside inhibits cancer cell proliferation in vitro and in vivo via AMP-activated protein kinase. J Biol Chem. 2005;280:39582–39593. doi: 10.1074/jbc.M507443200. [DOI] [PubMed] [Google Scholar]
- 35.Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, et al. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci USA. 2004;101:3329–3335. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lamprecht SA, Lipkin M. Migrating colonic crypt epithelial cells: primary targets for transformation. Carcinogenesis. 2002;23:1777–1780. doi: 10.1093/carcin/23.11.1777. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.