

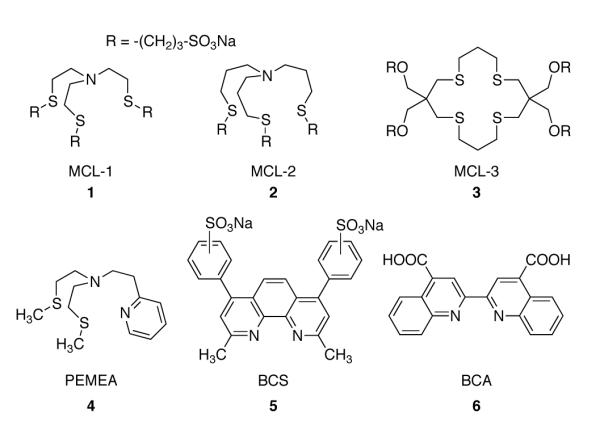
Abstract
The measurement of reliable Cu(I) protein binding affinities requires competing reference ligands with similar binding strengths; however, the literature on such reference ligands is not only sparse but often conflicting. To address this deficiency, we have created and characterized a series of water-soluble monovalent copper ligands, MCL-1, MCL-2, and MCL-3, that form well-defined, air-stable, and colorless complexes with Cu(I) in aqueous solution. Concluding from X-ray structural data, electrochemical measurements, and an extensive network of equilibrium titrations, all three ligands form discrete Cu(I) complexes with 1:1 stoichiometry and are capable of buffering Cu(I) concentrations between 10−10 and 10−17 M. As most Cu(I) protein affinities have been obtained from competition experiments with bathocuproine disulfonate (BCS) or 2,2′-bicinchoninic acid (BCA), we further calibrated their Cu(I) stability constants against the MCL-series. To demonstrate the application of these reagents, we determined the Cu(I) binding affinity of CusF (logK = 14.3±0.1), a periplasmic metalloprotein required for the detoxification of elevated copper levels in E. coli. Altogether, this interconnected set of affinity standards establishes a reliable foundation that will facilitate the precise determination of Cu(I) binding affinities of proteins and small molecule ligands.
Keywords: copper, metallochaperone, stability constant, metal buffer
Introduction
Copper is an essential trace element that is central to a broad range of biological processes, including cellular respiration, connective tissue formation, pigment synthesis, antioxidant defense, and photosynthesis in plants and bacteria.1 Cellular copper levels are controlled by an intricate network of membrane transporters and metallochaperones, which escort the metal ion to specific subcellular locations for incorporation into metalloproteins or for export through secretory pathways.2 To rationalize the directivity of these trafficking routes, a detailed knowledge of the copper binding affinities of all involved ligands is essential.3,4 Although a host of copper transporting proteins have been characterized, the reported affinity constants are often conflicting.4,5,6-11 These discrepancies were likely caused by the difficulties associated with determining the stability constants for complexes of Cu(I), the predominant oxidation state of copper in the reducing intracellular environment. Aqueous Cu(I) is not only highly reactive towards dioxygen but also prone to disproportionation at acidic pH or precipitation as Cu2O at neutral or basic pH. Recently, Xiao et al. pointed out common pitfalls in stability constant measurements and offered a set of guidelines for the determination of Cu(I) protein binding affinities.8,9 Foremost, the choice of suitable equilibrium conditions is key for obtaining reliable binding affinities, irrespective of the analytical method applied. As a general rule, the degree of complex formation should not exceed 90% at equimolar concentrations of metal ion and protein to avoid large errors during data analysis.12 Given the high affinity of Cu(I) transporting proteins,10 direct addition of Cu(I) would result in nearly quantitative binding. For this reason, the stability constants of high-affinity ligands are best obtained through competition experiments with an affinity standard whose stability constant and solution chemistry have been well characterized. At present, only a few such reference ligands exist for Cu(I) and similar to the reports on protein affinities, published values are often conflicting. Due to the limited stability of Cu(I) in aqueous solution, the majority of Cu(I) stability constants were obtained from experimental Cu(II) affinities and the standard reduction potentials of the free and ligand-bound Cu(II/I) couple using the Nernst equation. Although this approach eliminates the challenges associated with the redox instability of aqueous Cu(I), uncertainties remain due to the ionic strength dependence of the reduction potentials and the often large structural differences between the mono- and divalent forms of the copper-ligand complexes, which may lead to irreversible redox processes.
To address the scarcity of reliable Cu(I) affinity standards, we report here a set of three new water-soluble Monovalent Copper Ligands, MCL-1, MCL-2, and MCL-3 (1-3, Chart 1), which we rigorously characterized in terms of their Cu(I) binding affinities and coordination chemistry in aqueous buffer. Rather than relying solely on the Nernst relationship for determination of Cu(I) affinities, we also identified the dibasic ligand PEMEA13 (4) as a pH-modulated intermediary to anchor the Cu(I) affinities of the strongly binding MCL-series chelators to that of the much weaker ligand acetonitrile, whose Cu(I)-complex stability constants have been determined via a kinetic method.14 As the majority of Cu(I) protein affinities have been obtained from competition experiments with the chromogenic ligands bathocuproine disulfonate (BCS, 5) and 2,2′-bicinchoninic acid (BCA, 6), we additionally determined their Cu(I) stability constants by competition titrations with the MCL-series of compounds. The colorless MCL-series of affinity standards complement the chromogenic reagents by allowing direct monitoring of the degree of Cu(I) saturation of the target ligand in spectrophotometric or fluorimetric titrations. To illustrate this capability, we measured the Cu(I) affinity of CusF, a tryptophan-containing periplasmic protein important to copper resistance in Escherichia coli. Altogether, we have established a set of reliable Cu(I) affinity standards that should be broadly applicable for the determination of Cu(I) affinities in aqueous buffer at physiological pH.
Chart 1.
Experimental Section
Synthesis
The procedures and analytical data for the synthesis of ligands 1-3, their Cu(I) complexes, and all intermediates are provided in the Supporting Information. The ligands BCS (disodium bathocuproine disulfonate hydrate, 97%) and BCA (2,2′-bicinchoninic acid, free acid, 98%) were purchased from Acros Organics and MP Biomedicals, respectively; PEMEA and DHEAMP were synthesized according to published procedures13,15 and purified by recrystallization as the phosphate (PEMEA) and hydrochloride (DHEAMP) salts.
Protonation Constants
All protonation constants (logKH) were derived based on measurements of the hydronium ion concentration −log[H3O+] or p[H], not activity.16 For this purpose, a combination glass electrode was calibrated by titrating a 5 mM solution of HCl in 0.1 M KCl (ionic background) with a standardized solution of 0.1 M KOH at 25°C. The endpoint, electrode potential and slope were determined using Gran’s method as implemented in the GLEE software package.17 The protonation constants of PEMEA were measured by spectrophotometric titrations at 25°C with p[H] ranging between 1.9 and 8.2 (0.1 M KCl), followed by non-linear least-squares fitting over the entire spectral range using the SPECFIT software package.18 The logKH of BCA was analogously determined by varying p[H] between 3.7 and 5.5. All other protonation constants were obtained through potentiometric titrations by stepwise addition (motorized burette) of a standardized solution of 0.1 M KOH to a solution of the ligand in 0.1 M KCl at 25°C (ligand concentrations were 1 mM for BCS and 5 mM for MCL-1, MCL-2, and DHEAMP•2HCl). In the case of BCS, MCL-1, and MCL-2, at least one full molar equivalent of HCl was added to protonate the basic sites prior to titration with KOH. The protonation constants were then determined from the potentiometric data based on non-linear least-squares fitting using the Hyperquad software package.19
Electrochemistry
Cyclic voltammograms were acquired in 10 mM PIPBS buffer, pH 5, containing 0.1 M KClO4 as the electrolyte using a CH-Instruments potentiostat (model 600A). Experiments were carried out under an atmosphere of argon in a single compartment cell with a glassy carbon working electrode, a Pt counter-electrode, and an aqueous Ag/AgCl reference electrode (1 M KCl). The half-wave potentials (E1/2) were referenced to ferrocenium (0.40 vs. SHE)20,21 or ferroin (1.112 vs. SHE)13,22 as external standards. Measurements were typically performed with a scan rate of 20 - 50 mV s−1.
Stability Constants
Titrations involving Cu(I) were carried out under an atmosphere of argon with thorough exclusion of oxygen. All solutions were degassed immediately before use, and reagents were supplied from freshly prepared stock solutions with a gas-tight syringe. All spectrophotometric competition equilibrium titrations were carried out in a 1 cm path length quartz cuvette, which was sealed with a PTFE-lined rubber septum. Equilibration times were determined based on separate kinetic experiments. Stability constants were obtained from the titration data by non-linear least-squares fitting over the relevant spectral range using the SPECFIT software package.18 Detailed experimental conditions for each equilibrium titration are provided in the Supporting Information.
X-ray Structural Determinations
Crystals were coated with ParatoneN oil, suspended in a small fiber loop, and placed in a cooled nitrogen gas stream at 173 K on a Bruker D8 APEX II CCD sealed tube diffractometer with graphite monochromated Mo-Kα (0.71073 Å) radiation. Data collection, indexing, and initial cell refinements were carried out using APEX II software.23 Frame integration and final cell refinements were done using the SAINT software.24 All structures were solved using direct methods and difference Fourier techniques (SHELXTL, V6.12).25 Hydrogen atoms were placed in their expected chemical positions and were included in the final cycles of least-squares with isotropic Uij’s related to the atom’s ridden upon. All nonhydrogen atoms were refined anisotropically except for the disordered oxygen and fluorine atoms of the ClO4 and PF6 anions, respectively. Additional details of data collection and structure refinement are given in Tables S1-8 (Supporting Information). Supplementary crystallographic data (CCDC 948257 (13a), 948431 (14a), and 949729 (16)) can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request.cif.
Expression and Purification of CusF
E. coli BL21(DE3) cells hosting the pASK-IBA3 plasmid with the gene encoding for the CusF residues 6-88 were grown at 37°C to an optical density of 0.6 – 1.1 (600 nm) in LB medium containing 100 mg/L ampicillin.26 Protein expression was induced with anhydrotetracycline (0.8 mg/L) and the cells were harvested after 5 hours by centrifugation. The cell pellet was resuspended in MOPS buffer (50 mM MOPS, 0.15 M NaCl, pH 7) and the protein was extracted by three freeze-thaw cycles. After agitation at 4°C for 1 h, the cell suspension was centrifuged at 6000×g for 15 min at 4°C, and the supernatant was concentrated with a 3K microsep centrifugal device (Pall Life Sciences). The protein extract was purified by two sequential gel filtrations (GE Äkta) using a Superdex 75 column equilibrated with MOPS buffer. Fractions containing pure protein were confirmed by Laemmli-SDS-PAGE, pooled, and concentrated. The protein was stored at −20°C in the elution buffer for up to two days. Trace elemental analysis by TXRF (Bruker S2 Picofox Spectrometer) revealed that purified CusF was present in the apo-form (Figure S27, Supporting Information).
Cu(I)-Affinity of CusF
A) Fluorescence Equilibrium Titration with MCL-3
A 20 μM solution of holo-CusF in MOPS buffer (50 mM MOPS, 0.15 M NaCl, pH 7.0) was titrated with MCL-3 (up to a concentration of 280 μM). After addition of each aliquot, a fluorescence emission spectrum was acquired over the range of 305-500 nm by excitation at 290 nm. The precise concentration of CusF was determined from a molar-ratio titration of the apo-protein with Cu(I), which was generated in situ by reduction of CuSO4 with sodium ascorbate. To ensure quantitative loading with Cu(I), the holo-protein was prepared from apo-CusF by addition of 1.0 molar equivalent of Cu(I). The Cu(I) stability constant of CusF was then determined by non-linear least-squares fitting of the fluorescence emission traces over the entire spectral range using the SPECFIT18 software package.
B) Spectrophotometric Equilibrium Titration with BCA
The equilibrium titrations were carried out by the following two approaches: Either the Cu(I)-complex of BCA was first formed in situ by addition of [Cu(I)(CH3CN)4]PF6 (16 μM) to a solution of BCA (50 μM) in MOPS buffer at pH 7 (50 mM MOPS, 150 mM NaCl, 25°C) followed by titration with apo-CusF (0-72 μM) at pH 7, or apo-CusF (65 μM) was titrated with [Cu(I)(CH3CN)4]PF6 (0-87 μM) in the presence of 80 μM BCA as a competing ligand. In either case, the Cu(I) stability constant of CusF was obtained by non-linear least-squares fitting (SPECFIT)18 of the UV-vis traces over 450-700 nm.
Results and Discussion
Ligand Design
For biochemical applications, Cu(I)-affinity standards should be water-soluble, discourage heteroleptic complex formation, and ideally form a redox-stable Cu(I) complex, even under aerobic conditions. The latter requires a substantially positive reduction potential of the ligand-bound Cu(II/I) couple and discourages the use of redox-labile donors such as thiols or phosphines in the ligand design. Various aliphatic thioether-based macrocyclic or tripodal ligands with S4 or NS3 donor sets form stable 1:1 Cu(I)-complexes under aerobic conditions and offer the additional advantage of transparency in the visible to near-UV range, thus minimizing spectral overlap with a competing ligand during spectrophotometric or fluorimetric titrations. While linear or macrocyclic ligands with an aliphatic S4 or NS3 donor set provide affinities in the range of logKCu(I) = 12-14,27,28 N-centered tripodal ligands such as tris(2-methylthioethyl)amine (TMMEA) provide stability constants 2-3 orders of magnitude larger and LCu(II/I) reduction potentials approximately 300 mV higher than their macrocyclic counterparts.13 Building upon the TMMEA architecture, we designed the water-soluble derivative MCL-1 by replacing the terminal methyl groups with 3-sulfopropyl moieties (Chart 1). In the structurally related ligand MCL-2, each of the three N-S bridges was elongated by a methylene spacer, a change that was found to lower the apparent affinity at pH 7 by approximately 5 orders of magnitude compared to MCL-1 (vide infra). Finally, the thiocrown ligand MCL-3 was devised to bridge the affinity gap between the two tripodal ligands. While the parent macrocycle [16]aneS4 exhibits a logKCu(I) of 12.0,27 we anticipated that the conformational preorganization imposed by the bulky solubilizing groups should substantially increase the affinity. A similar effect has been reported for [14]aneS4, whose Ni(II) affinity increased 49-fold in nitromethane solution (1.7 logarithmic units) upon geminal substitution with two pairs of methyl groups.29
Ligand Synthesis
Both MCL-1 and MCL-2 were prepared in good yield from the corresponding tris(ω-chloroalkyl)amines 7 and 8, respectively, via reaction with excess sodium 3-mercaptopropanesulfonate in the presence of sodium hydroxide followed by recrystallization from methanol-water mixtures (Scheme 1). The byproduct NaCl is efficiently removed under these conditions due to the negative temperature dependence of its solubility in methanol.30 After rigorous drying, both ligands were obtained as the anhydrous trisodium salts. The macrocyclic ligand MCL-3 was constructed from the versatile thietane building block 9 previously utilized in our syntheses of water-soluble Cu(I) selective fluorescent probes.31 As outlined in Scheme 1, double ring-opening with 1,3-diiodopropane gave diiodide 10, which was cyclized with 1,3-propanedithiol under Kellogg conditions32 and deprotected with aqueous HCl to give thiocrown 12. Alkylation of the corresponding lithium alkoxide with 1,3-propanesultone followed by precipitation with ethanolic NaI provided MCL-3 as the tetrasodium salt.
Scheme 1.
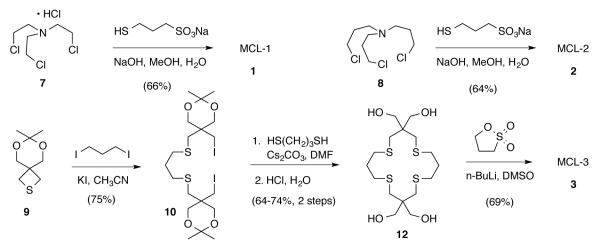
Synthesis of the water-soluble Cu(I)-ligands MCL-1 (1), MCL-2 (2), and MCL-3 (3).
Synthesis and X-ray Crystal Structures of Cu(I) Complexes
Ligands MCL-1, MCL-2, and MCL-3 were each reacted with stoichiometric quantities of [Cu(I)(CH3CN)4]PF6 to produce the respective Cu(I) complexes 13-15. All three complexes were isolated as hexafluorophosphate adducts containing equal numbers of sodium cations and sulfonate groups, corresponding to the formulas of [Na3LCu(I)]PF6 for MCL-1 and MCL-2 and [Na4LCu(I)]PF6 for MCL-3. Crystallization of the MCL-2-Cu(I) complex (14) from ethanol-water yielded the hydrate Na6(Cu(I)L)2(PF6)2•15H2O (14a) as colorless tablets suitable for X-ray diffraction. Under the same condition, the PF6− adduct of MCL-1-Cu(I) produced only very thin acicular crystals; however, in the presence of excess NaClO4 the complex formed the perchlorate adduct Na7(Cu(I)L)2(ClO4)3•4H2O (13a), which crystallized as colorless triclinic prisms. Because crystallization of MCL-3-Cu(I) (15) under a wide variety of conditions yielded only fibrous acicular crystals or powders, we selected compound 12 (Scheme 1) as a truncated analog of MCL-3 (abbreviated as trMCL-3 hereafter) for crystal structure determination. The corresponding Cu(I) complex crystallized from ethanol as clear prisms with the formula [(trMCL-3)-Cu(I)]PF6 • EtOH (16). To gauge the molecular preorganization of the tetrasubstituted thiocrown, we also determined the structure of free trMCL-3, which was crystallized from DMSO-water. A list of selected bond distances and angles for each structure are compiled in Tables 1 and 2. Additional data concerning the X-ray structural determination are provided in the Supporting Information.
TABLE 1.
Selected bond distances (Å) and angles (deg) describing the Cu(I)-coordination geometries in the crystal structures of [Na7(Cu(I)(MCL-1))2](ClO4)3•4H2O (13a) and [Na6(Cu(I)MCL-2)2](PF6)2•15H2O (14a).
| 13a | 14a (I) | 14a (II) | |
|---|---|---|---|
| Cu(1)-S(1) | 2.2486(7) | 2.2868(6) | 2.2874(6) |
| Cu(1)-S(2) | 2.2886(7) | 2.2870(6) | 2.2941(6) |
| Cu(1)-S(3) | 2.2680(7) | 2.2889(6) | 2.2910(6) |
| Cu(1)-N(1) | 2.1610(17) | 2.1261(17) | 2.1242(17) |
| S(1)-Cu(1)-N(1) | 91.91(6) | 98.78(5) | 98.85(5) |
| S(2)-Cu(1)-N(1) | 89.88(5) | 97.86(5) | 98.90(5) |
| S(3)-Cu(1)-N(1) | 91.32(5) | 98.51(5) | 97.98(5) |
| S(1)-Cu(1)-S(2) | 121.41(2) | 117.60(2) | 117.33(2) |
| S(1)-Cu(1)-S(3) | 125.49(2) | 118.36(2) | 118.53(2) |
| S(2)-Cu(1)-S(3) | 112.99(3) | 117.77(2) | 117.60(2) |
TABLE 2.
Selected bond distances (Å) and angles (deg) for the two cationic units in the crystal structure of [Cu(I)-(trMCL-3)]PF6 •EtOH (16).
| 16 (I) | 16 (II) | |
|---|---|---|
| Cu(1)-S(1) | 2.2558(2) | 2.2729(3) |
| S(2)-Cu(1) | 2.2577(2) | 2.2576(2) |
| S(3)-Cu(1) | 2.3024(2) | 2.3339(2) |
| S(4)-Cu(1) | 2.3108(2) | 2.3088(3) |
| S(1)-Cu(1)-S(3) | 118.896(9) | 116.703(10) |
| S(1)-Cu(1)-S(4) | 110.019(9) | 104.482(9) |
| S(1)-Cu(1)-S(2) | 106.648(9) | 106.517(9) |
| S(2)-Cu(1)-S(3) | 106.772(9) | 109.358(9) |
| S(2)-Cu(1)-S(4) | 117.871(9) | 123.669(10) |
| S(3)-Cu(1)-S(4) | 97.026(8) | 96.580(9) |
Complexes with tripodal ligands
The Cu(I) adducts of the tripodal ligands MCL-1 and MCL-2 crystallized as discrete monomeric complexes, in which the Cu(I) center is coordinated by the apical nitrogen and all three thioether donors (Figure 1). In both structures, the sulfopropyl arms adopt a helical conformation and do not participate in copper coordination. Instead, the anionic sulfonate groups are bridged by sodium cations and water molecules, forming a hydrophilic layer that links two Cu(I)-complexes of opposing helicity within a single unit cell (Figure S1, Supporting Information). In the case of MCL-1 (13a), the two complexes contained within a unit cell are exact mirror images, whereas for MCL-2 (14a), slight structural differences produce a pair of pseudo-enantiomeric geometries (I) and (II) (Table 1).
Figure 1.

ORTEP drawing and atomic numbering scheme for the crystal structures of the Cu(I) complexes of A) MCL-1 (13a) and B) MCL-2 (14a, I). Ellipsoids shown represent 50% probability. Hydrogen atoms and counter ions have been omitted for clarity. A packing diagram for each structure is provided in the Supporting Information (Figure S1).
As illustrated with the ORTEP plot in Figure 1A, the Cu(I)-complex of MCL-1 (13a) adopts a nearly trigonal pyramidal coordination geometry with an S-Cu-S bond angle sum of 359.9°. Despite the open coordination space below the basal plane of the trigonal pyramid, no additional donors are present within reasonable bonding distance in the crystal structure, rendering the coordination geometry of MCL-1-Cu(I) essentially identical to that previously reported for Cu(I)-TMMEA.13 In contrast to MCL-1, the longer N-S bridges of MCL-2 might be expected to accommodate a near-tetrahedral Cu(I)-center; however, the coordination geometry of complex 14a is still strongly distorted towards a trigonal pyramidal arrangement (Figure 1B). The sum of the S-Cu-S bond angles (353.7°) is substantially greater than expected for an ideal tetrahedral geometry (328.4°) and approaches a trigonal arrangement as in MCL-1. For comparison, the Cu(I) complex of the structurally related ligand tris(3-(benzylthio)propan-1-yl)amine (18), containing benzyl groups in place of the sulfopropyl moieties crystallized in four distinct conformers (Z = 4) with S-Cu-S bond angle sums ranging from 331.7 to 348.3°,33 suggesting that the modest structural differences between 13a and 14a are not associated with a significant energetic penalty.
As evident from Table 1, a comparison of the bond distances reveals a similar picture compared to the trends in bond angles. The Cu-N and average Cu-S bond lengths of 2.161(2) and 2.27(2) Å, respectively observed for 13a are almost identical to the values of 2.17(1) and 2.26(1) Å previously reported for TMMEA-Cu(I),13 indicating that the pendant sulfopropyl groups have little effect on the Cu(I)-S interaction. The average Cu-N bond in 14a is shorter by 0.036 Å compared to 13a, a difference that may be attributed to the greater basicity of the nitrogen in MCL-2 compared to MCL-1 (vide infra); however, the magnitude of the effect is rather modest in view of the Cu-N bond variations observed for the Cu(I) complex of ligand 18, which differed by up to 0.030 Å within the same unit cell.33
Macrocyclic Ligand and Complex
The macrocyclic ligand trMCL-3 forms with Cu(I) a discrete complex (16) with 1:1 stoichiometry (Figure 2A). Similar to the crystal structure of complex 14a, the unit cell contains two conformers with notable structural differences (Table 2). The average S-Cu bond distance in complex 16 (2.287±0.030 Å over both conformers) is essentially identical to that of the MCL-2 complex 14a (2.289 Å), and the overall coordination geometry is best described as flattened tetrahedral, with transannular S-Cu-S angles of 116.7-123.7° vs. 109.5° for an ideal tetrahedral arrangement. Overall, the crystallographic features of complex 16 are consistent with strong coordination between trMCL-3 and Cu(I). Furthermore, all hydroxyl groups point away from the Cu(I) center, suggesting that complex 16 is a viable structural analog for the Cu(I)-MCL-3 complex (15).
Figure 2.
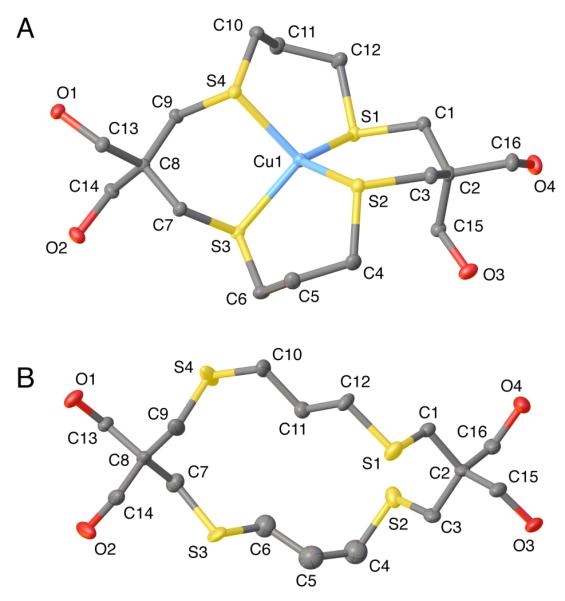
ORTEP drawing and atomic numbering scheme for the crystal structures of A) [Cu(I)-(trMCL-3)]PF6 (16, I) and B) the free ligand trMCL-3 (12). Ellipsoids shown represent 50% probability. Hydrogen atoms have been omitted for clarity.
The crystal structure of free trMCL-3 (Figure 2B) indicates significant preorganization toward Cu(I) coordination, especially when compared to the previously reported crystal structure of the unsubstituted parent ligand [16]aneS4 (Figure 3).34 In the latter, two of the thioether donors are in exo-positions and point away from the macrocyclic cavity with an S-S separation of 8.48 Å. By contrast, the corresponding S-S distance in trMCL-3 is only 6.13 Å, thus bringing the thioether donors much closer to the 4.03 Å transannular separation observed for the Cu(I)-complex 16. Underscoring the importance of this preorganization, no discrete monomeric [16]aneS4-Cu(I) complex has been previously observed in the solid state; in fact, the crystal structure of a [16]aneS4-copper(I) iodide adduct revealed a coordination polymer with the thiocrown in an exo-dentate conformation.36 A dihydroxy-derivative of [16]aneS4 also failed to yield a discrete Cu(I) complex, and the Cu(I)-perchlorate adduct showed evidence of oxidation to Cu(II) even under an argon atmosphere.37 In stark contrast, both trMCL-3 and MCL-3 itself yield Cu(I)-complexes that are air-stable even in solution. Based on these results, it appears that the preorganization imposed by the placement of the bulky solubilizing groups in MCL-3 not only increases the Cu(I)-affinity relative to [16]aneS4 (vide infra), but may actually be essential for selective formation of a discrete 1:1 Cu(I) complex.
Figure 3.
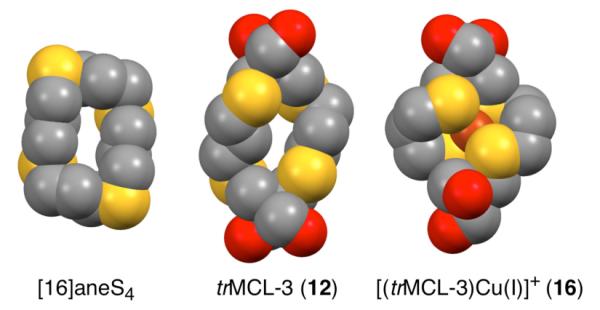
Space-filling model illustrating the molecular preorganization of trMCL-3 (12) vs. [16]aneS434 in comparison to the geometry of [Cu(I)-(trMCL-3)]PF6 (16) (representations generated with Mercury CSD35 from crystal structure atomic coordinates).
Protonation Constants
As the metal ion affinity of ligands with one or more basic donor atoms is pH dependent, we determined for each compound the corresponding protonation constants (logKH). A compilation of these results is provided in Table 3. All logKH values were determined based on the hydronium ion concentration, −log[H3O+] or p[H]. Following the recommendation of Martell and Smith,38 the tabulated concentration constants must be corrected upward by 0.11 (I = 0.1) to derive the corresponding mixed-mode protonation constants where hydronium ions are expressed in terms of their activity (pH) (see Supporting Information). Because neutral PEMEA and BCA were not soluble at millimolar concentrations in aqueous solution, the protonation constants for these ligands were determined by spectrophotometric titrations at micromolar concentrations (Figure S2-5, Supporting Information). The protonation constant of BCS was measured at 1 mM. All other compounds were titrated at 5 mM followed by data fitting with Hyperquad.19
TABLE 3.
Protonation Constants for ligands 1-6 and DHEAMP15 (17) in Aqueous Solution at 25°C (0.1 M KCl).a
| ligand | logKH1 | logKH2 |
|---|---|---|
| MCL-1 (1) | 7.00(2) | |
| MCL-2 (2) | 8.98(1) | |
| MCL-3 (3) | – | |
| PEMEA (4) | 7.24(4) | 3.23(4) |
| BCS (5) | 5.70(2) | |
| BCA (6) | 3.80(2) | |
| DHEAMP (17) | 6.94(1) |
the standard deviation of the mean of three independent titrations is listed in parentheses. The digits refer to the significant figures furthest to the right (e.g. 7.00(2) means 7.00±0.02).
Potentiometric titrations of MCL-1 at 0.1 M ionic strength yielded a protonation constant of 7.00±0.02 for the aliphatic amino group of the ligand. The corresponding mixed-mode protonation constant of 7.11 is lower compared to those reported for TMMEA (8.36) and its ethyl analogue TEMEA (8.32) in aqueous solution,13 but closely matches the value of 7.1 for TEMEA extrapolated from data acquired in 80% water-methanol.39 The differences between the extrapolated and measured protonation constants reported in the literature might be due to hydrophobic effects. Elongation of the N-S bridge in MCL-2 increases the basicity of the amino group by two orders of magnitude to 8.98, a trend that was also observed for tris(3-methylthiopropyl)amine (TMMPA) in 80% methanol when compared to TEMEA under the same conditions.39 Finally, the previously reported values for the protonation constants of BCS (5.7),9 BCA (3.74±0.04; 0.2 M ionic strength),40 and PEMEA (logKH1 = 7.33 and logKH2 =3.26, reported as mixed mode protonation constants)13 corroborated well with the values determined in this work (Table 3).
Stability Constants of Cu(I) Complexes
The simplest method for the determination of complex stability constants involves the direct titration of a ligand with the metal ion; however, this approach only yields reliable values if the fractional complex formation resides below 90%, a condition that is difficult to satisfy with tight binding ligands. To arrive at a set of reliable thermodynamic stability constants for the Cu(I)-complexes of ligands 1-6, we instead performed a series of equilibrium titrations, each involving a pair of ligands that compete for Cu(I) binding (Scheme 2). Because the concentration of aqueous Cu(I) is very low under these conditions, the precipitation of Cu2O or disproportion to Cu(II) and Cu(0) are thermodynamically unfavorable and therefore not of concern. Even the reactivity towards dioxygen is greatly reduced in the presence of a Cu(I)-stabilizing ligand, though all titrations were still carried out under deoxygenated conditions as a standard cautionary measure. For ligands that formed Cu(I)-complexes with a reversible or quasi-reversible redox reaction, we derived the corresponding Cu(I) stability constants from the half-wave potentials Ef and the Cu(II) complex stability constant based on the Nernst relationship (1)
| (1) |
where E°aq represents the reduction potential for the aqueous Cu(II/I) couple expressed in terms of concentrations. To account for the differential activity at 0.1 M ionic strength, the IUPAC standard potential of E° = 0.153 V20 was adjusted to E°aq = 0.13 V as recommended by Rorabacher (see also Supporting Information).28 Combined with the titration data, we established an interconnected network of thermodynamic equilibria from which we were able to arrive at a set of consistent Cu(I) stability constants for all involved ligands (Scheme 2). To outline the relative dependence and cross validations within this extensive data set, the following section describes each of the thermodynamic equilibria in more detail. A compilation of the pertinent thermodynamic data is given in Table 4. Stability constants which were determined based on multiple thermodynamic equilibria are reported as the cumulative average.
Scheme 2.

TABLE 4.
Reduction Potentials and Average Thermodynamic and Conditional Stability Constants for Cu(I)/Cu(II) Complexes of ligands 1-6 in Aqueous Solution at 25°C (0.1 M KCl).
| ligand |
Ef
vs. SHE [V]a |
log KCu(II)L |
log KCu(I)L |
log K’Cu(I) Lb |
pCu c |
|---|---|---|---|---|---|
| MCL-1 (1) | 0.716(1) | 6.42(2) | 16.33(2) | 16.0 | 16.9 |
| MCL-2 (2) | n/a | - | 13.08(13) | 11.0 | 11.9 |
| MCL-3 (3) | 0.729(3) | 3.47(4) | 13.80(3) | 13.8 | 14.8 |
| PEMEA (4) | 0.595(2) | 7.85(8) | 15.71(2) | 15.2 | 16.2 |
| BCS (5)f | 0.626(1) | 12.42(7) d | 20.81(2)d | 20.8e | 16.6 |
| BCA (6) | n/a | - | 17.66(3)d | 17.7e | 13.5 |
Half-wave potential at pH 5.0;
apparent affinity at pH 7.0 (0.1 M KClO4);
pCu = −log [Cu(I)aq] at 10 μM ligand and 1 μM Cu(I) concentration;
logβ2 of the 2:1 complex;
logβ2’.
mixture of the m-m’, m-p’, and p-p’ isomers (60:32:8).
Ligand MCL-1
Slow-scan cyclic voltammetry studies of MCL-1 at pH 5 in the presence of excess Cu(II) revealed a quasi-reversible one-electron redox process at Ef = 0.716 V (vs. SHE) with a peak separation of 74 mV (Figure 4A), thus offering the opportunity to determine the corresponding Cu(I) stability constant from the Nernst relationship (1). The Cu(II) stability constant of MCL-1 was obtained by spectrophotometric titration of the ligand with Cu(II)SO4 under the same conditions (pH 5.0, 10 mM PIPBS, 0.1 M KClO4, Figure S9, Supporting Information). Non-linear least squares fitting over the entire spectral range yielded logKCu(II)L = 6.42±0.02. Based on the above data, we derived a Cu(I) stability constant of logKCu(I)L = 16.33±0.02. Taking into account the protonation constant of MCL-1, we calculated an apparent affinity of logK’Cu(I)L = 16.0 at pH 7.0.
Figure 4.
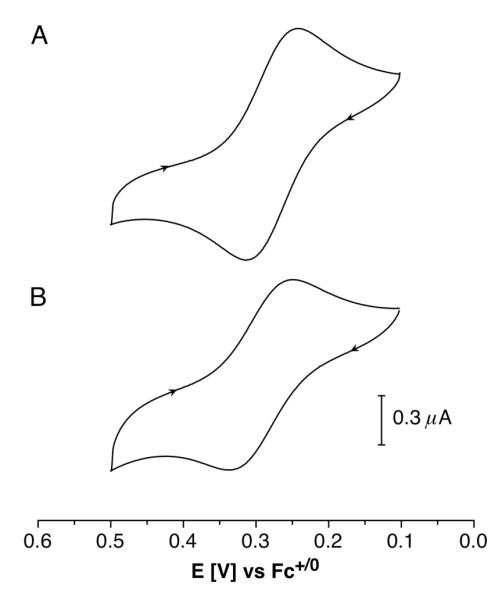
Cyclic voltammograms for (A) MCL-1 (150 μM) and (B) MCL-3 (150 μM) in the presence of CuSO4 (1 mM) at pH 5.0 (10 mM PIPBS, 0.1 M KClO4; glassy-carbon electrode, scan rate 20 mV/s).
2,2′-Bicinchoninic Acid (BCA)
In the presence of Cu(I), this ligand forms a purple 2:1 complex with an absorption maximum at 562 nm (ε = 7900 M−1cm−1).41 Cyclic voltammetry experiments revealed an irreversible redox behavior, thus precluding the application of the Nernst relationship to determine its Cu(I) stability constant. Taking advantage of the optical transparency of Cu(I)-MCL-1 in the visible range, we determined the stability constant of BCA by spectrophotometric competition titration between the two ligands (Figure 5A). For this purpose, equimolar quantities of BCA and the Cu(I) complex of MCL-1 were pre-equilibrated in 10 mM PIPBS (pH 5.0, 0.1 M KClO4). Under these conditions, less than 50% of the total BCA is bound to Cu(I), thus excluding the presence of a significant amount of the 1:1 complex. Titration with MCL-1 up to 4 molar equivalents followed by non-linear least squares fitting yielded a stability constant of logβ2 = 17.67±0.03. This value is 0.5 logarithmic units higher compared to the Cu(I) affinity estimated by Xiao et al.;9 however, the difference is consistent with the selected value of E°aq,42 which is 34 mV higher compared to the ionic strength adjusted potential of 0.13 V used throughout this work.
Figure 5.

Spectrophotometric titrations of (A) 2,2′-bicinchoninic acid (BCA, 6; 100 μM) at pH 5.0 (10 mM PIPBS, 0.1 M KClO4) and (B) bathocuproine disulfonate (BCS, 5; 50 μM) at pH 7.0 (10 mM PIPES, 0.1 M KClO4) with MCL-1 in the presence of Cu(I).
Ligand MCL-2
Despite its structural similarity with MCL-1, we were not able to detect the formation of a complex with Cu(II), even at high millimolar concentrations. Consistent with this observation, cyclic voltammetry experiments with the Cu(I)-complex of MCL-2 showed an irreversible redox process, suggesting rapid dissociation of the copper ion upon oxidation. For this reason, we determined the Cu(I) affinity of MCL-2 only by spectrophotometric competition titrations with BCA (Figure S16, Supporting Information). The data fit well to a 1:1 binding model and yielded a logKCu(I)L of 13.08±0.13, corresponding to an apparent affinity of 11.0 at pH 7.0. Compared to MCL-1, the Cu(I) affinity of MCL-2 is remarkably lower, a result that would not be predicted based on the crystal structure data which indicated a similarly strong Cu(I)-coordination for both ligands. The dramatically lower Cu(I) affinity of MCL-2 is presumably due to a greater entropic penalty for conformational restriction of the longer N-S bridges upon chelation. While 6-membered chelate rings generally result in less stable metal complexes compared to the analogous 5-membered rings, Cu(I)-complexes often exhibit little or no such effect.43
Bathocuproine disulfonate (BCS, disodium salt)
Similar to BCA, this heterocyclic ligand forms with Cu(I) a colored 2:1 complex with an absorption maximum at 483 nm (ε = 13,300 M−1 cm−1).44 Commercially available BCS is supplied as a mixture of three regioisomers bearing the sulfonate groups either in the meta or para positions of the 4- and 7-aryl rings (Chart 1).45 Taking advantage of the resonance assignments previously established by multidimensional NMR techniques,45 we determined for our batch a 60:32:8 composition of the m-m’, m-p’, and p-p’ isomers, respectively. The following data should therefore be interpreted as a statistical average for this composition. Competition titrations with MCL-1, performed in the same fashion as described for BCA, yielded a stability constant of logβ2 = 20.80±0.03 (Figure 5B, Figure S17, Supporting Information). Slow-scan cyclic voltammetry experiments with BCS in the presence of either [Cu(I)(CH3CN)4]PF6 or Cu(II)SO4 revealed a quasi-reversible one-electron redox process with Ef = 0.626 V (vs. SHE) with a peak separation of 73 mV (Figure S7, Supporting Information), allowing independent determination of the Cu(I)-complex stability constant via Equation (1). Although the Cu(II)-complex stability constant of BCS was previously assessed via a potentiometric method requiring millimolar ligand concentrations,9 we relied instead on spectrophotometric titration at micromolar concentrations to avoid interference from the reported self-association of BCS in aqueous solution.46 For this purpose, we identified N,N-bis(2-hydroxyethyl)-N-(2-pyridylmethyl)amine (DHEAMP, 17) as suitable competing ligand, which according to Damu et al. forms a 1:1 Cu(II) complex with a stability constant of logKCu(II)L = 9.2±0.1.15 On the basis of a molar ratio titration of DHEAMP with Cu(II) at low pH combined with the experimental protonation constant of 6.94±0.01 (lit. 6.92±0.01), we obtained a logKCu(II)L of 9.21±0.02 in good agreement with the above literature value (Figure S11, Supporting Information). Finally, competition titration of BCS with Cu(II)SO4 in the presence of DHEAMP yielded a stability constant of logβ2 = 12.42±0.07 for the 2:1 Cu(II) complex of BCS (Figure S12, Supporting Information). Together with the half-wave potential of the Cu(II/I) complex, we calculated a Cu(I) stability constant of logβ2 = 20.81±0.08, which is in excellent agreement with the data obtained by above competition titration with MCL-1. As in the case of BCA, the above result is higher than the previously reported logβ2 of 19.9;9 although the deviation is here somewhat larger than the 0.6 logarithmic units expected for a 34 mV difference in E°aq.
Ligand MCL-3
In the presence of Cu(II), this ligand forms a complex with an absorption maximum at 577 nm and a strong charge-transfer band centered around 443 nm. A direct spec-trophotometric molar ratio titration at pH 5.0 yielded a Cu(II) stability constant of logKCu(II)L = 3.47±0.04 (Figure S10, Supporting Information). Because MCL-3 does not contain any basic sites, its stability constants are invariant towards pH in aqueous solution. Slow-scan cyclic voltammetry studies of MCL-3 in the presence of Cu(II) showed a one-electron process at Ef = 0.729 V vs. SHE (Figure 4B); however, with a peak separation of 100 mV, even at scan rates as low as 2 mV/s (Figure S8, Supporting Information), the redox process cannot be considered reversible and is therefore not suitable to derive a Cu(I) stability constant based on equation (1). Instead, we performed spectrophotometric competition titrations with BCS, from which we derived a Cu(I) stability constant of 13.80±0.04 (Figure S19, Supporting Information). Furthermore, we were able to corroborate this value by competition titration with BCA, which yielded a stability constant of 13.78±0.06 (Figure S20, Supporting Information). It is noteworthy that the Cu(I) affinity of MCL-3 is significantly higher compared to the parent macrocycle [16]aneS4 (logKCu(I)L = 12.0),27 an observation that is likely due to the conformational preorganization of the free ligand. In fact, the 65-fold gain in Cu(I) affinity is very similar to the previously reported 49-fold increase in Ni(II) affinity upon methylation of [14]aneS4 as mentioned above,29 despite the different coordination preferences of Cu(I) and Ni(II).
Although the combination of competition titrations and thermodynamic cycles (Equation 1) yielded a consistent set of cross-validated stability constants, the entire data set hinges on a single value, the standard reduction potential of the aqueous Cu(II/I) couple used in Equation (1). When considering the spread of E°aq values applied in the literature,20,28,42 the calculated Cu(I) stability constant may vary up to 0.6 logarithmic units. To address this uncertainty, we sought to measure at least one of the stability constants by a method that is not dependent on the value of E°aq. At present, there are only few ligands for which Cu(I) stability constants have been reported without the use of electrochemical methods. At first, we explored the utility of cyanide as competing ligand in spectrophotometric titrations with BCS; however, careful data analysis indicated the presence of heteroleptic ternary complexes, which prevented us from deriving a reliable stability constant. We next explored the suitability of acetonitrile, which may not only act as a competing ligand but also stabilize Cu(I) towards disproportionation.43 Given the rather low stability constants of logβ1 = 2.63, logβ2 = 4.02, and logβ3 = 4.29,14 acetonitrile cannot compete for Cu(I) binding with either BCS or BCA. We therefore tried to identify a spectrophotometrically active dibasic Cu(I) ligand whose apparent affinity could be adjusted over several orders of magnitude simply by changing the pH. At low pH, acetonitrile would be able to compete for Cu(I) binding, while at neutral pH the higher apparent affinity would match that of BCS or BCA.
With the reported Cu(I) stability constant of logKCu(I)L = 15.76 and protonation constants of 3.26±0.08 and 7.33±0.15, the dibasic pyridine derivative PEMEA (4) emerged as the most promising candidate.13 Slow-scan cyclic voltammetry measurements in the presence of Cu(II) revealed a quasi-reversible one-electron redox process with Ef = 0.595 V (vs. SHE) and a peak separation of 68 mV (Figure S6, Supporting Information), thus allowing independent verification of the PEMEA Cu(I) stability constant through a thermodynamic cycle. Spectrophotometric titration of PEMEA with Cu(II) at pH 5.0 gave rise to a new charge-transfer absorption band centered around 350 nm due to formation of the corresponding 1:1 complex. Non-linear least squares fitting over the spectral range of 300-450 nm provided a Cu(II) stability constant of logKCu(II)L = 7.85±0.08 (Figure S13, Supporting Information). Combined with the electrochemical data, we calculated a Cu(I) stability constant of logKCu(I)L = 15.71±0.08, which agrees well with the value reported by Rorabacher and coworkers (15.76).13 As anticipated, acetonitrile was able to compete for Cu(I) binding in the presence of PEMEA at low pH. Based on a molar ratio titration of PEMEA with [Cu(I)(CH3CN)4]PF6, in aqueous buffer at pH 2.0 (14 mM HClO4, 100 μM sodium ascorbate, 3.9% CH3CN, 0.1 M KClO4, 25°C), we determined a Cu(I) stability constant of logKCu(I)L = 15.75±0.02 (Figure S21, Supporting Information). This value is in excellent agreement with the data obtained based on Equation (1) with E°aq = 0.13 V (vs. SHE). Finally, we used PEMEA to perform two additional competition titrations with MCL-1 (Figure S22, Supporting Information) and BCA (Figure S23, Supporting Information) to further cross-validate the above equilibrium network. Based on spectrophotometric titrations followed by non-linear least-squares fitting we determined a Cu(I) stability constant of logβ2 = 17.63±0.05 for BCA and logKCu(I)L = 16.33±0.07 for MCL-1. Both results agree well with the values from the initial series of competition titrations (17.67±0.03 and 16.33±0.02 for BCA and MCL-1, respectively).
Based on all of the above data, we present recommended values for the Cu(I)-complex stability constants of the MCL-series as well as BCA, BCS, and PEMEA in Table 4. All standard deviations are based on independent repeats with n = 3 or higher. When additionally taking into account the uncertainty associated with the measurement of formal potentials, the listed stability constants are expected to be accurate within ±0.1 logarithmic units. In addition to the Cu(I) stability constants (logKCu(I)L), we also listed the corresponding conditional or apparent stability constants (logK’Cu(I)L) at neutral pH for each ligand. Originally proposed by Schwarzenbach et al.,47 the apparent affinity takes into account the protonation equilibria of the ligand and metal ion at a given pH (Supporting Information). Due to competing protonation equilibria, the stability constants of ligands with basic donor atoms are strongly pH dependent. For example, at neutral pH the apparent Cu(I) affinity of MCL-2 (pKa 9.09, I = 0.1) is lowered by two orders of magnitude, whereas the less basic ligand MCL-1 (pKa 7.11, I = 0.1) exhibits a much smaller change of 0.33 logarithmic units (Table 4). Because BCS and BCA form Cu(I) complexes with a 2:1 stoichiometry, their stability constants cannot be directly compared to the MCL series of ligands. For this reason, we also listed in Table 4 the corresponding pCu values, which equate to −log([Cu(I)aq]) for a solution containing a total concentration of 10 μM of ligand and 1 μM of Cu(I) at pH 7.0 (I = 0.1).48 Under these conditions, BCS and BCA exhibit a similar chelating ability compared to MCL-1 and MCL-3, respectively. Due to the 2:1 complex stoichiometry, the solution equilibrium depends not only on the ligand/Cu(I) ratio but also the total ligand concentration (Figure 6A). For example, if the BCS and Cu(I) concentrations are increased by 10-fold, the pCu(I) value increases from 16.6 to 17.6.
Figure 6.

Dynamic range for buffering free Cu(I)aq with ligands exhibiting 2:1 (A) and 1:1 (B) complex stoichiometries. The fractional saturation of the corresponding ligand was calculated as a function of −log([Cu(I)aq]) for a buffer with constant total ligand concentration and varying [Cu(I)]total at pH 7.0 (I = 0.1).
Concluding from the apparent affinities at pH 7, the MCL series is suitable to buffer aqueous Cu(I) concentrations from 10−10 to 10−17 M, a range that is well matched with the affinity of many Cu(I) metalloproteins.9,11 The three ligands exhibit complementary buffer ranges with almost equal spacing (Figure 6B). As the corresponding Cu(I) complexes are all crystal-line and air-stable, Cu(I)-buffer solutions can be readily prepared without the need of an additional Cu(I) source. Furthermore, the ligands and the corresponding Cu(I) complexes are optically transparent down to 300 nm (Figure S14), permitting interference-free monitoring of Cu(I)-binding to targets with an intrinsic spectroscopic signature, thus complementing the chromogenic reagents BCS and BCA.
The MCL series may also be applied for the calibration of Cu(I)-responsive fluorescent probes, the majority of which have been characterized via competition with low-affinity mono-dentate ligands amenable to heteroleptic complex formation. Given that most fluorescent probes reported to date exhibit dissociation constants in the picomolar range,49 MCL-2 would be well suited as a reference standard. Compared to CH3CN and two other low-affinity ligands recently proposed as quantitative probes for Cu(I),50 MCL-2 forms a Cu(I) complex with improved redox stability and reduced reactivity towards dioxygen. The Cu(I) complex of MCL-2 would also be well suited for the metallation of copper proteins, which are typically isolated in their apo-forms upon overexpression.9 Similarly, the Cu(I) complexes of MCL-1 and MCL-3 offer superior redox stability compared to those of PEMEA, BCS, or BCA.
To illustrate the application of the MCL-series as affinity standards, we describe in the last section the determination of a Cu(I) protein complex stability constant based on fluorimetric titrations. For this purpose, we chose a tryptophan-containing protein whose affinity has been previously characterized based on competition with a chromogenic ligand.
Determination of the Cu(I) Stability Constant of CusF
As a component of the CusCFBA efflux complex,51 the periplasmic protein CusF plays an important role in the detoxification of elevated levels of copper and silver in E. coli.52 Besides coordination to a Met2His motif, the protein utilizes a tryptophan residue to stabilize Cu(I) via a strong cation-π interaction.6,7,53 In the apo-protein, the tryptophan residue fluoresces at 343 nm and is quenched upon binding of Cu(I), thus offering a convenient spectroscopic handle to monitor the degree of Cu(I) binding in an equilibrium titration with a competing ligand. Concluding from preliminary titrations, we found that MCL-3 matched best the Cu(I) affinity of CusF. As illustrated in Figure 7, gradual addition of MCL-3 to a solution of holo-CusF in MOPS buffer (pH 7.0) restored the tryptophan fluorescence. Non-linear least-squares fitting of the emission response revealed a CusF-Cu(I) stability constant of log KCu(I)L = 14.29±0.11.
Figure 7.
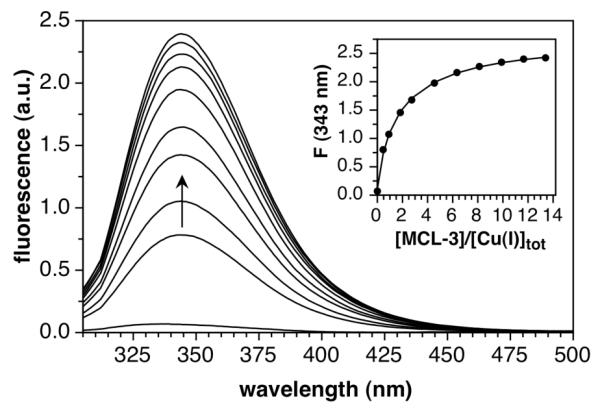
Fluorescence titration of CusF (20 μM) with MCL-3 at pH 7.0 (50 mM MOPS, 0.15 M NaCl, 25°C). Inset: Change of the fluorescence intensity at 343 nm (excitation at 290 nm).
This value is 0.5 log units higher compared to an earlier report, in which the Cu(I) stability constant of CusF was determined relative to BCA (Kd (CusF) × β2 (BCA) = 7.3 × 103; or logK (CusF) = 13.8 assuming logβ2 (BCA) = 17.7),6 but agrees well with the data of a more recent independent re-evaluation using the same competitor ligand (logK (CusF) = 14.3 assuming logβ2 (BCA) = 17.7).7 Due to this discrepancy, we also reexamined the Cu(I)-affinity of CusF by spectrophotometric equilibrium titrations using BCA as a competing ligand. Regardless whether the Cu(I)-BCA complex was preformed and then titrated with apo-CusF (Figure S25, Supporting Information), or whether a mixture of apo-CusF and BCA was titrated with Cu(I) supplied from a stock solution in acetonitrile (Figure S26, Supporting Information), the stability constants converged at a value of logKCu(I)L = 14.21±0.03, thus confirming within experimental error above data from competition titration with MCL-3.
Conclusions
To address the pressing need for robust affinity standards in Cu(I) biochemistry, we have created and characterized a series of three water-soluble ligands that form well-defined, colorless, and air-stable Cu(I) complexes in aqueous solution. All three ligands and their Cu(I) complexes can be isolated and recrystallized in multigram quantities, thus allowing for the preparation of precisely defined Cu(I)-buffer solutions simply by mixing the free ligand and corresponding Cu(I) complex in the appropriate ratios. Grounded in an extensive network of equilibrium titrations and electrochemical measurements, we determined reliable acidity and Cu(I) stability constants, not only for the colorless MCL series of ligands, but also for two chromogenic reagents, bathocuproine disulfonate 5 (BCS) and 2,2′-bicinchoninic acid 6 (BCA), frequently used for the determination of Cu(I) protein stability constants. Altogether, these ligands represent a cohesive set of optically transparent and chromogenic affinity standards, which are capable of buffering Cu(I) concentrations from 10−10 to 10−18 M. Despite the high binding affinities, rapid equilibration was observed for all ligand competition experiments, presumably due to an associative Cu(I)-ligand exchange mechanism. The fast exchange kinetics also raises the possibility of conducting competition experiments by isothermal titration calorimetry,54 which would allow the measurement of reliable Cu(I) binding affinities of proteins and ligands regardless of their spectroscopic signatures. Furthermore, the Cu(I)-complex of the lowest affinity ligand MCL-2 may be used for the quantitative metallation of Cu(I)-binding proteins, thus offering an alternative Cu(I) source in place of the air- and moisture-sensitive tetrakis(acetonitrile)-Cu(I) salts. Conversely, the chelation of copper with high affinity ligands might offer avenues for the treatment of diseases associated with copper overload such as Alzheimer’s and Wilson’s disease.55 Most importantly, we anticipate that these reagents will facilitate the reliable measurement of Cu(I) binding affinities of metalloproteins and other biologically relevant Cu(I)ligands.
Supplementary Material
ACKNOWLEDGMENT
We thank Prof. M.M. McEvoy for the generous gift of the CusF expression vector, and K. Odegaard for assistance with the TXRF measurements. Financial support from the National Institutes of Health (R01GM067169) is gratefully acknowledged.
Footnotes
Supporting Information. Synthetic procedures, compound characterization, crystal structure data, supporting information for the determination of protonation constants and Cu(I) stability constants. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions The manuscript was written through contributions of all authors, and all authors have given approval to the final version of the manuscript.
Notes The authors declare no competing financial interest.
REFERENCES
- (1).Linder MC. Biochemistry of Copper. Springer; New York: 1991. [Google Scholar]
- (2).Kim B-E, Nevitt T, Thiele DJ. Nat. Chem. Biol. 2008;4:176. doi: 10.1038/nchembio.72. [DOI] [PubMed] [Google Scholar]; Lutsenko S. Curr. Opin. Chem. Biol. 2010;14:211. doi: 10.1016/j.cbpa.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Huffman DL, O’Halloran TV. J. Biol. Chem. 2000;275:18611. doi: 10.1074/jbc.C000172200. [DOI] [PubMed] [Google Scholar]; Xiao ZG, Wedd AG. Chem. Commun. 2002:588. doi: 10.1039/b111180a. [DOI] [PubMed] [Google Scholar]; Badarau A, Dennison C. Proc. Natl. Acad. Sci. USA. 2011;108:13007. doi: 10.1073/pnas.1101448108. [DOI] [PMC free article] [PubMed] [Google Scholar]; Niemiec MS, Weise CF, Wittung-Stafshede P. PLoS One. 2012;7:e36102. doi: 10.1371/journal.pone.0036102. [DOI] [PMC free article] [PubMed] [Google Scholar]; Fu Y, Tsui H-CT, Bruce KE, Sham L-T, Higgins KA, Lisher JP, Kazmierczak KM, Maroney MJ, Dann CE, Winkler ME, Giedroc DP. Nat. Chem. Biol. 2013;9:177. doi: 10.1038/nchembio.1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Banci L, Bertini I, Ciofi-Baffoni S, Kozyreva T, Zovo K, Palumaa P. Nature. 2010;465:645. doi: 10.1038/nature09018. [DOI] [PubMed] [Google Scholar]
- (5).Jensen PY, Bonander N, Møller LB, Farver O. Biochim. Biophys. Acta. 1999;1434:103. doi: 10.1016/s0167-4838(99)00161-2. [DOI] [PubMed] [Google Scholar]; Wernimont AK, Yatsunyk LA, Rosenzweig AC. J. Biol. Chem. 2004;279:12269. doi: 10.1074/jbc.M311213200. [DOI] [PubMed] [Google Scholar]; Kittleson JT, Loftin IR, Hausrath AC, Engelhardt KP, Rensing C, McEvoy MM. Biochemistry. 2006;45:11096. doi: 10.1021/bi0612622. [DOI] [PubMed] [Google Scholar]; Yatsunyk LA, Rosenzweig AC. J. Biol. Chem. 2007;282:8622. doi: 10.1074/jbc.M609533200. [DOI] [PubMed] [Google Scholar]
- (6).Xue Y, Davis AV, Balakrishnan G, Stasser JP, Staehlin BM, Focia P, Spiro TG, Penner-Hahn JE, O’Halloran TV. Nat. Chem. Biol. 2008;4:107. doi: 10.1038/nchembio.2007.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Loftin IR, Blackburn NJ, McEvoy MM. J. Biol. Inorg. Chem. 2009;14:905. doi: 10.1007/s00775-009-0503-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Xiao ZG, Wedd AG. Nat. Prod. Rep. 2010;27:768. doi: 10.1039/b906690j. [DOI] [PubMed] [Google Scholar]
- (9).Xiao Z, Brose J, Schimo S, Ackland SM, La Fontaine S, Wedd AG. J. Biol. Chem. 2011;286:11047. doi: 10.1074/jbc.M110.213074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Badarau A, Dennison C. J. Am. Chem. Soc. 2011;133:2983. doi: 10.1021/ja1091547. [DOI] [PubMed] [Google Scholar]
- (11).Allen S, Badarau A, Dennison C. Biochemistry. 2012;51:1439. doi: 10.1021/bi201370r. [DOI] [PubMed] [Google Scholar]
- (12).Hansen LD, Fellingham GW, Russell DJ. Anal. Biochem. 2011;409:220. doi: 10.1016/j.ab.2010.11.002. [DOI] [PubMed] [Google Scholar]
- (13).Ambundo EA, Deydier MV, Grall AJ, Aguera-Vega N, Dressel LT, Cooper TH, Heeg MJ, Ochrymowycz LA, Rorabacher DB. Inorg. Chem. 1999;38:4233. [Google Scholar]
- (14).Kamau P, Jordan RB. Inorg. Chem. 2001;40:3879. doi: 10.1021/ic001447b. [DOI] [PubMed] [Google Scholar]
- (15).Damu KV, Shaikjee MS, Michael JP, Howard AS, Hancock RD. Inorg. Chem. 1986;25:3879. [Google Scholar]
- (16).Martell AE, Motekaitis RJ. The Determination and Use of Stability Constants. Wiley; New York: 1992. [Google Scholar]
- (17).Gans P, O’Sullivan B. Talanta. 2000;51:33. doi: 10.1016/s0039-9140(99)00245-3. [DOI] [PubMed] [Google Scholar]
- (18).SPECFIT Global Analysis System. Spectrum Software Associates . Marlborough MA: 2001. p. 01752. [Google Scholar]
- (19).Gans P, Sabatini A, Vacca A. Talanta. 1996;43:1739. doi: 10.1016/0039-9140(96)01958-3. [DOI] [PubMed] [Google Scholar]
- (20).Milazzo G, Caroli S. Tables of Standard Electrode Potentials. Wiley; New York: 1978. [Google Scholar]
- (21).Koepp H-M, Wendt H, Stkehlow HZ. Elektrochem. 1960;64:483. [Google Scholar]
- (22).Yee EL, Cave RJ, Guyer KL, Tyma PD, Weaver MJ. J. Am. Chem. Soc. 1979;101:1131. [Google Scholar]
- (23).APEX II, 2005. Bruker AXS, Inc.; Madison, WI: 2005. [Google Scholar]
- (24).SAINT, v. 6.45A. Bruker AXS, Inc.; Madison, WI: 2003. [Google Scholar]
- (25).Sheldrick GM. Acta Crystallogr., Sect. A. 2008;A64:112. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- (26).Loftin IR, Franke S, Roberts SA, Weichsel A, Héroux A, Montfort WR, Rensing C, McEvoy MM. Biochemistry. 2005;44:10533. doi: 10.1021/bi050827b. [DOI] [PubMed] [Google Scholar]
- (27).Bernardo MM, Schroeder RR, Rorabacher DB. Inorg. Chem. 1991;30:1241. [Google Scholar]
- (28).Bernardo MM, Heeg MJ, Schroeder RR, Ochrymowycz LA, Rorabacher DB. Inorg. Chem. 1992;31:191. [Google Scholar]
- (29).Desper JM, Gellman SH, Wolf RE, Jr, Cooper SR. J. Am. Chem. Soc. 1991;113:8663. [Google Scholar]
- (30).Pinho SP, Macedo EA. J. Chem. Eng. Data. 2005;50:29. [Google Scholar]
- (31).Morgan MT, Bagchi P, Fahrni CJ. J. Am. Chem. Soc. 2011;133:15906. doi: 10.1021/ja207004v. [DOI] [PMC free article] [PubMed] [Google Scholar]; Morgan MT, Bagchi P, Fahrni CJ. Dalton Trans. 2013;42:3240. doi: 10.1039/c2dt31985c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Buter J, Kellogg RM. J. Org. Chem. 1981;46:4481. [Google Scholar]
- (33).Blomenkemper M, Schröder H, Pape T, Hahn FE. Inorg. Chim. Acta. 2011;366:76. [Google Scholar]
- (34).Blake AJ, Gould RO, Halcrow MA, Schroder M. Acta Crystallogr., Sect. B. 1993;49:773. [Google Scholar]
- (35).Macrae CF, Bruno IJ, Chisholm JA, Edgington PR, McCabe P, Pidcock E, Rodriguez-Monge L, Taylor R, Streek J. v., Wood PA. J. Appl. Crystallogr. 2008;41:466. [Google Scholar]
- (36).Brooks NR, Blake AJ, Champness NR, Cooke PA, Hubberstey P, Proserpio DM, Wilson C, Schröder M. J. Chem. Soc., Dalton Trans. 2001:456. [Google Scholar]
- (37).Munakata M, Wu LP, Yamamoto M, Kuroda-Sowa T, Maekawa M. J. Chem. Soc., Dalton Trans. 1995:3215. [Google Scholar]
- (38).Martell AE, Smith RM. Critical Stability Constants. Plenum Press; New York: 1974. [Google Scholar]
- (39).Cooper TH, Mayer MJ, Leung KH, Ochrymowycz LA, Rorabacher DB. Inorg. Chem. 1992;31:3796. [Google Scholar]
- (40).Gershuns AL, Koval VL. Izv. Vyssh. Uchebn. Zaved., Khim. Khim. T. 1975;18:1543. [Google Scholar]
- (41).Xiao Z, Donnelly PS, Zimmermann M, Wedd AG. Inorg. Chem. 2008;47:4338. doi: 10.1021/ic702440e. [DOI] [PubMed] [Google Scholar]
- (42).Hawkins CJ, Perrin DD. J. Chem. Soc. 1963:2996. [Google Scholar]
- (43).Balakrishnan KP, Kaden TA, Siegfried L, Zuberbühler AD. Helv. Chim. Acta. 1984;67:1060. [Google Scholar]
- (44).Xiao Z, Loughlin F, George GN, Howlett GJ, Wedd AG. J. Am. Chem. Soc. 2004;126:3081. doi: 10.1021/ja0390350. [DOI] [PubMed] [Google Scholar]
- (45).De Pascali SA, Migoni D, Papadia P, Muscella A, Marsigliante S, Ciccarese A, Fanizzi FP. Dalton Trans. 2006:5077. doi: 10.1039/b610945d. [DOI] [PubMed] [Google Scholar]
- (46).Yao S, Cherny RA, Bush AI, Masters CL, Barnham KJ. J. Pept. Sci. 2004;10:210. doi: 10.1002/psc.539. [DOI] [PubMed] [Google Scholar]
- (47).Schwarzenbach G. Complexometric Titrations. Interscience Publisher; New York: 1957. [Google Scholar]
- (48).Harris WH, Raymond KN. J. Am. Chem. Soc. 1979;101:6534. [Google Scholar]
- (49).Fahrni CJ. Curr. Opin. Chem. Biol. 2013;17:656–662. doi: 10.1016/j.cbpa.2013.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]; Morgan MT, Bagchi P, Fahrni CJ. Fluorescent probes for monovalent copper. In: Culotta V, Scott RS, editors. Metals in Cells. John Wiley & Sons, Ltd; Chichester, UK: 2013. pp. 65–83. [Google Scholar]; Hyman LM, Franz KJ. Coord. Chem. Rev. 2012;256:2333–2356. doi: 10.1016/j.ccr.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Xiao Z, Gottschlich L, van der Meulen R, Udagedara SR, Wedd AG. Metallomics. 2013;5:501. doi: 10.1039/c3mt00032j. [DOI] [PubMed] [Google Scholar]
- (51).Franke S, Grass G, Rensing C, Nies DH. J. Bacteriol. 2003;185:3804. doi: 10.1128/JB.185.13.3804-3812.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Mealman TD, Blackburn NJ, McEvoy MM. Curr. Top. Membr. 2012;69:163. doi: 10.1016/B978-0-12-394390-3.00007-0. [DOI] [PubMed] [Google Scholar]
- (53).Chakravorty DK, Wang B, Ucisik MN, Merz KM. J. Am. Chem. Soc. 2011;133:19330. doi: 10.1021/ja208662z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Trapaidze A, Hureau C, Bal W, Winterhalter M, Faller P. J. Biol. Inorg. Chem. 2012;17:37. doi: 10.1007/s00775-011-0824-5. [DOI] [PubMed] [Google Scholar]
- (55).Delangle P, Mintz E. Dalton Trans. 2012;41:6359. doi: 10.1039/c2dt12188c. [DOI] [PubMed] [Google Scholar]; Pujol AM, Cuillel M, Jullien A-S, Lebrun C, Cassio D, Mintz E, Gateau C, Delangle P. Angew. Chem. Int. Ed. Engl. 2012;51:7445. doi: 10.1002/anie.201203255. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.