

Abstract
This unit describes the process of gene shuffling (also known as sexual PCR). Gene shuffling is a facile method for the generation of sequence libraries containing the information from a family of related genes. Essentially, related genes are fragmented by DNase I digestion and reassembled by primerless PCR. The resulting chimeric genes can then be screened or selected for a desired function.
Keywords: directed evolution, recombination, PCR
Introduction
Proteins serve as the catalytic, structural, and signaling backbone of cellular life and biotechnology. The ability to evolve proteins with novel function, then, has implications for medicine, industry, and research and can provide insight into the fundamental processes of evolution and protein function. In the search for proteins with novel function, researchers are confronted with a vast landscape of all possible proteins. Improvements in high throughput screening and selection of functional proteins have allowed for greater exploration of the sequence space, but this still falls orders of magnitude short of full coverage. Thus, the generation of “smart libraries” (those that explore functional sequence space more efficiently) remains important to increase the likelihood of finding proteins with a desired function.
It is often prudent to begin the search for new protein functionality by starting with a known protein with similar features. By querying the sequence space immediately adjacent to the starting point (by random mutagenesis), it may be possible to stumble upon the desired functionality. However, “smart libraries” formed by neutral drift mutation (Bershtein et al., 2008), targeted saturation mutagenesis (Miyazaki and Arnold, 1999), or gene shuffling (Stemmer, 1994) allows for more efficient exploration of functional sequence space.
The genes found in nature today are the result of countless, iterative rounds of mutation and selection over billions of years. Each extant gene has been selected to perform a particular function beneficial to its host organism. Sequence variability among members of a gene family is often acquired in the evolution of new functionality. However, not all mutations confer a specific advantage, and most variation is due to the accumulation of seemingly neutral mutations over time. The collective variation in the gene family may possess latent functional potential. By shuffling genes, it is possible to tap this potential and uncover new functionality (Figure 1).
Figure 1.
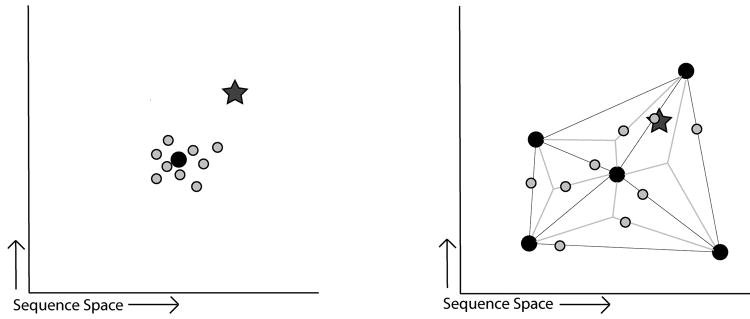
The sequence space of all possible proteins is depicted as a 2-dimentional plane. The x- and y-axis represent genotypic distance, such that neighboring points have a similar genotype and distant points are more dissimilar. Left) A starting parental sequence (black dot) is randomly mutagenized, resulting in a typical random mutagenesis library (gray dots). This library explores the sequence space near the parental sequence, but does not contain the sequence exhibiting a new function (star). Right) Multiple, divergent parental sequences (black dots) are recombined, resulting in a gene-shuffled library (gray dots). This library explores the larger, intervening area, and does contain the sequence exhibiting a new function (star). Thus, the latent evolutionary potential of a gene family can be tapped to find new functionality.
The following protocol allows for the random shuffling of related genes (Figure 2). In brief, related genes with identical flanking sequences are digested with DNase I. The resulting fragments are then thermal cycled in a primerless PCR, in which fragments anneal to one another and mutually serve as primer and template. The products are then amplified in a standard PCR, yielding a shuffled library.
Figure 2.
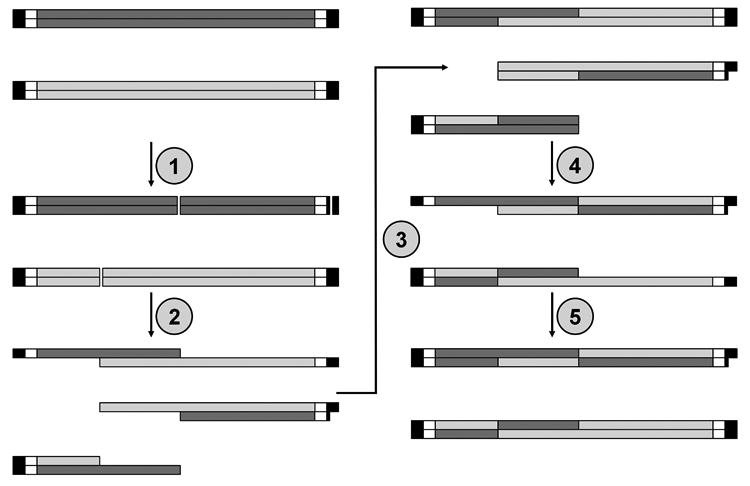
The parental Linear Input DNA sequences are composed of a library region of related genes (shades of gray) flanked by regions for outer primer annealing (black) and inner primer annealing (white). 1) Random double-stranded breaks are introduced by DNase I. 2) The fragments are denatured and anneal to each other at regions of homology. 3) The annealed fragments mutually serve as templates for extension, resulting in chimeric sequences. 4) Another cycle of denaturation and annealing 5) further extension resulting in full length products.
The products are amplified by PCR using inner primers that anneal proximal to the library region (white boxes) because the ends (black boxes) are often recessed. Some fragments are not shown for clarity.
Under certain circumstances, it may be advantageous to shuffle genes at predefined cross-over points. This is discussed further in Alternatives-Defined Cross-over Points. A detailed method for this alternative can be found in Current Protocols in Protein Science, UNIT 26.2 (Farrow and Arnold, 2010).
Protocol for the Generation of Libraries by Gene Shuffling
Materials
Parental Plasmid DNA (See Critical Parameters-Generation of Parental Plasmid DNA)
20 μM outer primers or restriction enzymes (See Critical Parameters-Primer Design and Figure 2)
20 μM inner primers (See Critical Parameter-Primer Design and Figure 2)
Thermostable, proofreading DNA polymerase (e.g. Pfu DNA polymerase or KOD DNA polymerase) and associated buffer. (See Critical Parameters-Polymerase Choices)
DNA Polymerase Family A and Family B Blend (See Critical Parameters-Polymerase Choices)
10 μl 600 mM Tris-SO4 (pH 8.9), 180 mM Ammonium Sulfate
4 mM dNTPs
50 mM MgSO4
Gel Purification kit
PCR Clean-Up kit
1 M Tris-HCl pH 7.4
200 mM MnCl2
DNase I
Thermal cycler (see UNIT 15.1)
Additional reagents and equipment for PCR amplification (UNIT 15.1) and agarose gel electrophoresis (UNIT 2.7)
Preparation of Linear Input DNA
- Generate linear, double stranded DNA versions of target region for each gene of interest. This may be accomplished by restriction digests of Parental Plasmid DNA or by PCR amplification of Parental Plasmid DNA. Plasmid preparation, DNA endonuclease digestion, and PCR are described in UNIT 1.6, UNIT 3.1, and UNIT 15.1 respectively.Digestion of a plasmid is marginally less error-prone than PCR amplification of the target region (Zhao and Arnold, 1997) but it requires that the genes to be shuffled are flanked by the same sequence in their respective plasmids.PCR potentially introduces point mutations, but allows flexibility in starting material because the initial primers can append appropriate flanking sequences. To minimize error and maximize yield, use of a thermostable, proofreading DNA polymerase (e.g. Pfu DNA polymerase or KOD DNA polymerase) is recommended (See Critical Parameters-Polymerase Choices). 20 ng Parental Plasmid DNA and 20 thermal cycles are routinely employed.
- Purify this “Linear Input DNA” by agarose gel purification. Gel purification is described in UNIT 2.6.2 μg total Linear Input DNA is required for the shuffling process as described. Gel purification is recommended to eliminate residual primers, starting template, and protein.
Fragmentation and purification of fragments
-
3Combine equal volumes 1 M Tris-HCl pH 7.4 and 200 mM MnCl2 to generate 10X DNase I buffer10X DNase I buffer should be made fresh before use. See Critical Parameters-Digestion Buffer.
-
4Combine the following in a 0.2 ml PCR tube:
- 5 μl 10X DNase I buffer
- 2 μg Linear Input DNA
- Water to bring total volume to 50 μlThe amount of Linear Input DNA from each parental gene depends on the desired results. For example, a two-parent library may start with 1 μg of each parent, but it is acceptable to use unequal amounts of each parental sequence in order to bias the outcome.
-
5Equilibrate the above reaction at 15°C for 5 minutes in a thermal cycler.
-
6Add 0.5 μl DNase I (diluted to 1 U/ μl in 1X DNase I buffer) to the mixtureThe dilution of DNase I in 1X DNase I buffer should be done immediately prior to use
-
7Incubate the reaction at 15°C for 3 minutes
-
8Stop the reaction by incubating at 80°C for 10 minutes
-
9Perform a PCR clean-up.As 2 μg of Linear Input DNA is fragmented, recovery of at least 1 μg of fragments is common. Only 200 ng of fragments are required for the reassembly reaction as described.Gel purification can be employed to specify the size range of DNA fragments recovered. This can be done by cutting a large band over the desired range. Fragments of intermediate size (400-1000 base pairs) are most often chosen (Joern et al., 2002). Larger fragments can reduce diversity and smaller fragments don't anneal properly. However, gel purification is not essential because the PCR-clean up purification disfavors the retention of fragments below 100 base pairs.
Reassembly
-
10Combine the following in a 0.2 ml PCR tube:
- 200 ng DNA fragments
- 2 units DNA Polymerase Family A and Family B Blend (See Critical Parameters-Polymerase Choices)
- 10 μl 600 mM Tris-SO4 (pH 8.9), 180 mM Ammonium Sulfate
- 5 μl 4 mM dNTPs
- 4 μl 50 mM MgSO4
- Water to bring the total volume to 100 μl
-
11Thermal cycle above reaction as follows: 94°C for 2 minutes; 35 cycles of (94°C for 30 seconds, 65°C for 90 seconds, 62°C for 90 seconds, 59°C for 90 seconds, 56°C for 90 seconds, 53°C for 90 seconds, 50°C for 90 seconds, 47°C for 90 seconds, 44°C for 90 seconds, 41°C for 90 seconds, 68°C for 90 seconds per kb) 68°C for 2 minutes per kb.The long, multi-step annealing step is termed “progressive hybridization” and is thought to favor low-stringency annealing, thus enabling recombination at regions of moderate homology (Abécassis et al., 2000).Difficult (e.g. G-C rich) sequences are often problematic. Careful choice of polymerases can help mitigate this problem. See Critical Parameters-Polymerase Choices.Reaction may incubate at 10°C for several hours thereafter.
-
12
Perform a PCR clean-up.
Reamplification
-
13PCR amplify 5 μl (one tenth) of the elution reassembly product using a thermostable, proofreading DNA polymerase and the inner set of primers. PCR is described in UNIT 15.1.20 cycles of PCR are routinely performed. See Critical Parameters-Polymerase Choices
-
14Purify amplified library by agarose gel purification. Gel purification is described in UNIT 2.6.The purified product will become the input library for selection or screening, and greater yield at this stage makes for a larger library. The actual desired size of the library depends on the nature of the screen or selection to be performed. Manual in vivo screens generally assess 103 variants, while in vitro selections can handle 1010 or more variants.If the genes are to be screened or selected in vivo, then digestion, ligation, and transformation/transfection may follow. For screens or selections utilizing cell-free lysate the DNA library can remain in linear form. In either case, the library should be flanked by appropriate sequences (e.g. promoters, terminators, etc.).
Alternatives
Defined Cross-over Points
As described, this protocol introduces random cross-over between related sequences. However, it is possible to predefine the regions of cross-over. Predefined cross-over points may allow for shuffling of less closely related genes and allow for the retention of functional domains. However, this approach may bias the selection against novel combinations needed to achieve new functionality.
Predefined shuffling can be accomplished by the PCR amplification of defined regions of the gene with primers that anneal to one family member, and append sequences suitable for homologous recombination to another family member (Jézéquel et al., 2008). Alternatively, Type IIB restriction endonuclease sites may be introduced into predefined cross-over points. When the template is digested, each domain is left with unique overhangs that allow for scar-less ligation of fragments in the correct order (Hiraga and Arnold, 2003; Farrow and Arnold, 2010).
Commentary
Background Information
The utility of gene shuffling was first demonstrated in the evolution of the TEM-1 Beta lactamase by Willem PC Stemmer (Stemmer, 1994). The lactamase gene was shuffled, and cloned and expressed in E. coli. The E. coli were plated with the antibiotic cefotaxime at concentration twenty fold higher than the minimum inhibitory concentration of E. coli harboring wild-type TEM-1. Colonies able to withstand this higher concentration of antibiotic served as the starting template for the next round of shuffling and selection. After several rounds of selection on increasing levels of cefotaxime, variants of TEM-1 conferring 32,000-fold greater resistance to cefotaxime than wild-type TEM-1 were isolated.
Shuffling has since been employed in the directed evolution of enzyme function, such as the ability to work at higher temperatures (Giver et al., 1998) or recognize new substrates (Brühlmann and Chen, 1999) and of viral coat proteins that expand host range (Pekrun et al., 2002) and enable enhanced gene delivery (Maheshri et al., 2006).
Critical Parameters
Generation of Parental Plasmid DNA
The first step in gene shuffling is decided upon the genes to shuffle and preparing the parental sequences. The genes are usually amplified from a genome or existing plasmid or they are made through gene synthesis. Regardless of the original source of the genes to be shuffled, it is recommended that they first be cloned into a plasmid and sequence verified.
The number of genes to shuffle is widely variable. Iterative shuffling of the same gene (relying on random point mutations for the introduction of diversity) has proven successful in altering the phenotype (Stemmer, 1994). In the later rounds of this process, the Linear Input DNA to be shuffled was quite variable, suggesting there is no upper limit to the number of distinct species that can be shuffled.
There is no minimum sequence homology between two genes to allow shuffling per se, but the frequency of crossover events is largely determined by the number and size of stretches of identity. Most crossover events occur at regions of least 10 consecutive, identical nucleotides (Joern et al., 2002).
As the ability to custom-build a gene becomes increasingly routine, it may be advisable to synthesize the starting genes using codons that facilitate shuffling. Due to the degeneracy of the genetic code, two genes may be very similar at the amino acid level, but quite divergent at the nucleotide level. By careful choice of the codon used at each position, two wild-type genes with insufficient sequence identity for efficient recombination can be optimized for shuffling. Traditional codon optimization (the use of only the most frequently used codon for each residue) ensures that genes with identical amino acids in a given position will be identical at the nucleotide level. This can vastly increase the sequence identity between gene family members, thus facilitating shuffling.
At non-identical amino acid residues, codons can be chosen that minimize the number of nucleotide mismatches. For example, CGT is the optimal arginine codon in E. coli and AGC the optimal serine codon. These share only one nucleotide in common. However, CGC is an acceptable choice for arginine, and shares two nucleotides with the optimal serine codon. Thus, if a protein has an arginine residue and its shuffling partner has a serine residue, the use of the CGC arginine codon and AGC serine codon will make genes more amenable to shuffling.
Primer Design
The fragmentation and reassembly process tends to leave the ends of the products recessed (see Figure 2). Thus, the primers used to generate the Linear Input DNA are unsuitable for the final reamplification step. The use of “outer” primers that leave 60 base pairs on either side of the target region is recommended for the generation of Linear Input DNA. This can be achieved using short (20 base pairs) primers that anneal 40 to 60 base pairs from the target region or by using long (60 base pairs) primers that anneal proximal to the target region. “Inner” primers that anneal proximal to the target region should be used for the final reamplification step. These primers may contain additional sequence (e.g. restriction sites) to facilitate subsequent reactions. Primers should be designed to be 20-25 base pairs, with about 50% GC content.
Polymerase Choices
For the reassembly reaction, a blend of family A DNA polymerase (e.g. Taq DNA polymerase) and proofreading family B DNA polymerase (e.g. Pfu DNA polymerase or KOD DNA polymerase) is recommended. The high activity and robustness of family A DNA polymerase allows for the amplification of even difficult sequences while the proof-reading activity of the family B DNA polymerase offers relatively high-fidelity amplification. Platinum Taq DNA Polymerase High Fidelity (Life Technologies) is a commercially available example.
For all other PCR amplifications, a thermostable, proofreading DNA polymerase (e.g. Pfu DNA polymerase or KOD DNA polymerase) is recommended to reduce the accumulation of point mutations. Other, low fidelity polymerases (e.g. Taq DNA polymerase) may be used to achieve a higher mutation rate. The increase in mutational load may unlock new functionality, but also increases the proportion of non-functional variants.
Digestion Buffer
This protocol calls for the use of MnCl2 rather than MgCl2 during the fragmentation step. This favors double-strand breaks rather than nicks. This is preferable, as heavily nicked strands are retained in the purification steps, but then upon heat denaturation yield small fragments that are unsuitable for use as primers in the reassembly reaction (Lorimer and Pastan, 1995). The 10X DNase I buffer should be made immediately prior to use as it may form precipitates, become discolored, and lose efficacy over time.
Troubleshooting
The most common problem is a failure of the reamplification reaction. The most likely culprit is improper primer design, as discussed above. Primers should be tested prior to use. The amount of reassembly product added to the reamplification reaction may need to be optimized. The number of reamplification PCR cycles can also be reduced.
Another possible problem is a preponderance of genes with no crossover events. This can be mitigated by gel-purification of the DNA fragments prior to reassembly, but may be a result of insufficient homology (discussed above).
Anticipated Results
The expected result is several micrograms of shuffled product. 2-4 cross-over events per kb are routinely observed, but this depends on the homology of the shuffling partners. Mutations tend to be rare, roughly 0.1 point mutations per kb. The percentage of functional variants created depends greatly on the homology and the function under consideration.
In an excellent study of this topic, Joern et al. (2002) shuffled a family of dioxygenases. In one experiment, two dioxygenases (85% identical) were recombined. In another, a third dioxygenase (63-64% identical to the other two) was added to the recombination. The recombined libraries were probed with parent-specific oligonucleotides at several positions and were also sequenced. The two-parent library contained about 2.5 cross-overs per kb, while the more divergent three-membered library contained about 2 cross-overs per kb. The sequencing revealed about 0.1 point mutations (all transitions in this study) per kb.
Time Considerations
Once starting material has been purified, the fragmentation and purification steps take 1 hour. The reassembly reaction takes 10-12 hours depending on the target size and is usually performed overnight. The clean-up, reamplification, and purification takes roughly 5 hours to perform.
Acknowledgments
This work was supported by the Welch Foundation (F-1654), National Institutes of Health (5 R21 HG005763-01,02 & 5 R01 AI092839-01,02), National Science Foundation (MCB-0943383), and the Office of Naval, Research (N00014-09-1-1087).
Literature Cited
- Abécassis V, Pompon D, Truan G. High efficiency family shuffling based on multi-step PCR and in vivo DNA recombination in yeast: statistical and functional analysis of a combinatorial library between human cytochrome P450 1A1 and 1A2. Nucleic Acids Research. 2000;28:E88. doi: 10.1093/nar/28.20.e88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bershtein S, Goldin K, Tawfik DS. Intense neutral drifts yield robust and evolvable consensus proteins. Journal of Molecular Biology. 2008;379:1029–44. doi: 10.1016/j.jmb.2008.04.024. [DOI] [PubMed] [Google Scholar]
- Brühlmann F, Chen W. Tuning biphenyl dioxygenase for extended substrate specificity. Biotechnology and Bioengineering. 1999;63:544–551. doi: 10.1002/(sici)1097-0290(19990605)63:5<544::aid-bit4>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Farrow MF, Arnold FH. Combinatorial Recombination of Gene Fragments to Construct a Library of Chimeras. Current Protocols in Protein Science. 2010:26.2.1–26.2.20. doi: 10.1002/0471140864.ps2602s61. [DOI] [PubMed] [Google Scholar]
- Giver L, Gershenson A, Freskgard PO, Arnold FH. Directed evolution of a thermostable esterase. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:12809–13. doi: 10.1073/pnas.95.22.12809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraga K, Arnold FH. General Method for Sequence-independent Site-directed Chimeragenesis. Journal of Molecular Biology. 2003;330:287–296. doi: 10.1016/s0022-2836(03)00590-4. [DOI] [PubMed] [Google Scholar]
- Jézéquel L, Loeper J, Pompon D. Sequence-independent construction of ordered combinatorial libraries with predefined crossover points. BioTechniques. 2008;45:523–32. doi: 10.2144/000112954. [DOI] [PubMed] [Google Scholar]
- Joern JM, Meinhold P, Arnold FH. Analysis of shuffled gene libraries. Journal of Molecular Biology. 2002;316:643–56. doi: 10.1006/jmbi.2001.5349. [DOI] [PubMed] [Google Scholar]
- Lorimer IA, Pastan I. Random recombination of antibody single chain Fv sequences after fragmentation with DNaseI in the presence of Mn2+ Nucleic Acids Research. 1995;23:3067–8. doi: 10.1093/nar/23.15.3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maheshri N, Koerber JT, Kaspar BK, Schaffer DV. Directed evolution of adeno-associated virus yields enhanced gene delivery vectors. Nature Biotechnology. 2006;24:198–204. doi: 10.1038/nbt1182. [DOI] [PubMed] [Google Scholar]
- Miyazaki K, Arnold FH. Exploring nonnatural evolutionary pathways by saturation mutagenesis: rapid improvement of protein function. Journal of Molecular Evolution. 1999;49:716–20. doi: 10.1007/pl00006593. [DOI] [PubMed] [Google Scholar]
- Pekrun K, Shibata R, Igarashi T, Reed M, Sheppard L, Patten PA, Stemmer WPC, Martin MA, Soong NW. Evolution of a Human Immunodeficiency Virus Type 1 Variant with Enhanced Replication in Pig-Tailed Macaque Cells by DNA Shuffling. Journal of Virology. 2002;76:2924–2935. doi: 10.1128/JVI.76.6.2924-2935.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stemmer WP. Rapid evolution of a protein in vitro by DNA shuffling. Nature. 1994;370:389–91. doi: 10.1038/370389a0. [DOI] [PubMed] [Google Scholar]
- Zhao H, Arnold FH. Optimization of DNA shuffling for high fidelity recombination. Nucleic Acids Research. 1997;25:1307–8. doi: 10.1093/nar/25.6.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]