

Abstract
Ultrafast two-dimensional infrared (2D-IR) spectroscopy reveals picosecond protein and hydration dynamics of crowded hen egg white lysozyme (HEWL) labeled with a metal-carbonyl vibrational probe covalently attached to a solvent accessible His residue. HEWL is systematically crowded alternatively with polyethylene glycol (PEG) or excess lysozyme in order to distinguish the chemically inert polymer from the complex electrostatic profile of the protein crowder. The results are threefold: (1) A sharp dynamical jamming-like transition is observed in the picosecond protein and hydration dynamics that is attributed to an independent-to-collective hydration transition induced by macromolecular crowding that slows the hydration dynamics up to an order of magnitude relative to bulk water; (2) The interprotein distance at which the transition occurs suggests collective hydration of proteins over distances of 30-40 Å; and (3) Comparing the crowding effects of PEG400 to our previously reported experiments using glycerol exposes fundamental differences between small and macromolecular crowding agents.
Introduction
The hydrophobic effect is a powerful driving force crucial in biological systems,1 playing a key role in protein folding2-4 and membrane formation,5 as well as directing surface association processes.6,7 It has been predicted8,9 and experimentally observed10,11 that the energetic balance of hydrophobic hydration depends on the size of the hydrated molecule. For small solutes, the cost of hydration is largely entropic as the water enhances its local structure to minimize hydrogen bond loses, while the cost of hydrating larger molecules is largely borne by enthalpic contributions as the solute forces the disruption of water's hydrogen bonding network.9 The corresponding dynamics of the surrounding water has been more difficult to access, though experiments and simulations are converging on a view where small hydrophobes exert negligible influence over the dynamics of the surrounding water molecules when in dilute concentrations,12-14 while large hydrophobic solutes can constrain and hinder the surrounding water by limiting the ability of hydrogen bond exchange.13-15 The crossover occurs on the nanometer length scale, which is characteristic of proteins, lipids and other biomolecules.
The perturbation of water by hydrophobic structures can have significant implications in cellular environments, where the structural and dynamic correlation lengths may extend beyond the space available from interstitial water. Crowding effects are generally considered in terms of energetics focusing on protein stability and refolding kinetics,16-23 where entropic forces arising from hard-core repulsions between macromolecules compete with enthalpic forces arising from weak attractions. Due to the challenging nature of experiments, dynamic aspects of crowding are more elusive, though progress in new methods of spectroscopy, including time-resolved fluorescence,24 terahertz absorption,25,26 NMR27,28 and 2D-IR,14 have allowed for the interfacial region of hydrated proteins to be studied directly. In particular, studies using THz absorption spectroscopy, coupled with molecular dynamics (MD) simulations, have found evidence of a dynamic hydration shell surrounding proteins ranging from 10-30 Å, depending on the protein.25,26 As a striking example, antifreeze proteins were found to have a hydration environment that can extend upwards of 30 Å.26 Additionally, photon echo experiments of hemoglobin in erythrocytes29 and optical Kerr effect (OKE) spectroscopy,30,31 which measures the low-frequency Raman response, have been used to observe a general slowing of the system dynamics with increasing concentrations, though no dynamic transition was apparent from the data.
Within the context of crowding, there is a dichotomy between what can broadly be classified as “chemical” and “physical” effects. For instance, studies comparing monomeric and polymeric sucrose (Ficoll 70) arrive at different conclusions. Pielak et al.19 observe no difference in protein stability (chymotrypsin inhibitor 2), whereas Gruebele et al.20 find pronounced differences in folding kinetics (phosphoglycerate). Our work focuses on dynamics using a similar comparison. If the differences in chemical interactions are minimal, is there a fundamental difference between macromolecular and small molecule crowding? In order to make progress, we have discovered that it is essential to perform experiments over a wide range of additive concentrations, as will be detailed below.
Questions remain regarding the relevant length and time scales associated with crowding. While ultrafast spectroscopic studies have uncovered the strong coupling between hydration water and protein flexibility, it is still unclear over what distances this coupling can persist, and whether the disruption of water upon crowding has a structural component or if it is a purely dynamic phenomenon. If there is a crowding dependence to the hydration structure, basic statistical mechanics tells us that there will be an energetic contribution due to the altered water-water and water-protein pair correlation functions. In the absence of a structural change, however, only dynamical measurements will be able to discern a detailed microscopic picture, as is the case, for example, with studies on the glass transition. In addition, measurements of diffusion in cellular environments show a general increase in diffusion constants upon crowding,23,32 but it is difficult to directly relate macroscopic diffusion constants to microscopic properties of the solvent, namely local solvent friction.
To address these issues, we use ultrafast two-dimensional infrared (2D-IR) to study the picosecond dynamics of HEWL labeled with a transition metal carbonyl vibrational probe covalently attached to the surface exposed His15 residue (the labeled protein is referred to as HEWL-RC).33 Metal carbonyls offer ideal vibrational probes for biological molecules due to the inherent strength of the transition and the frequency of the vibrational modes, giving strong signal in a region of the IR spectrum that is free from the protein and water background.13,14 Additionally, lysozymes are robust proteins that maintain structural integrity in crowded solutions.34 The x-ray crystal structure of HEWL-RC is shown in Figure 1, as well as a linear FTIR spectrum of the C≡O modes of the vibrational probe. We study the dynamics of the system through the frequency-frequency correlation function (FFCF), a powerful observable unique to 2DIR that reports on the equilibrium structural fluctuations that modulate the transition frequency of a probe molecule. The surface location of the vibrational probe used here allows us to study both the hydration dynamics and the protein dynamics simultaneously. The FFCFs exhibit rapid initial picosecond decays due to motion of the hydration water, followed by a significant static offset arising from fluctuations that are too slow to be fully sampled within the experimental window.13 We attribute the static offset of the correlation function to slow protein fluctuations, though other work looking at similar correlation functions have suggested that the slow dynamics could arise from very slow exchange between surface water and bulk water.24 While these contributions are difficult to distinguish experimentally, we believe that the spectral signatures between surface and bulk water are not as significant as the inhomogeneity arising from protein fluctuations. Since the region of the protein we probe experimentally is not located on the cleft region, but rather on an open, flat region of the protein, we do not expect idiosyncratically slow exchange of hydration water with the bulk. Simulations by Laage et al.15 have used site-specific analysis around a protein surface and have found that the majority of the water molecules experience only a mild slowdown due to the protein surface, while a handful of water molecules located in cleft regions of the protein or in the interior, experience significant slowdown upwards of 100 ps. Thus, distinct populations of hydration water can lead to mean residence times that are significantly longer than what the majority of the water experiences. Recent work on biomolecule hydration has highlighted the importance of considering metrics other than averages in describing interfacial water structure and thermodynamics.35 In addition, slow translational motion of water from the surface to the bulk is more apparent through techniques such as NMR27,28 and Overhauser dynamic nuclear polarization (ODNP),36 whereas experiments that measure ultrafast correlation functions tend to be predominantly sensitive to local dynamics.
Fig 1.
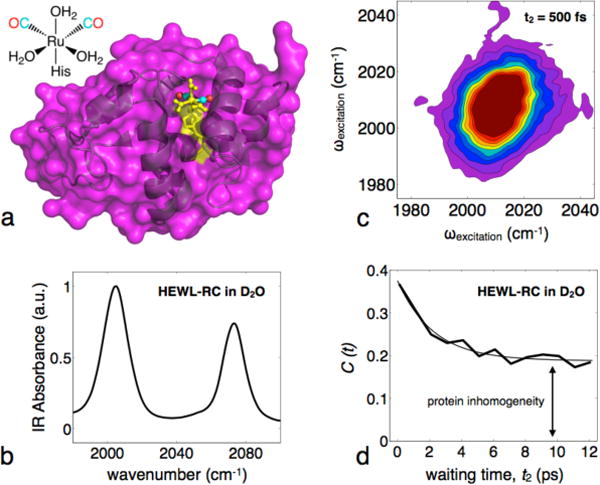
Crystal structure of HEWL-RC, linear and 2D-IR spectra, example FFCF. a, Structure of the metal-carbonyl vibrational probe and the crystal structure of the His 15 labeled HEWL carbonyl complex (probe site highlighted in yellow). b, Linear FTIR spectrum and (c) 2DIR spectrum shown for the metal-carbonyl CO region. d, Example of a typical frequency-frequency correlation function, showing an initial decay on the order of a few picoseconds corresponding to the hydration dynamics, followed by a static offset due to protein inhomogeneity that is not sampled within the experimental window.
Though the vibrational relaxation of the probe precludes time resolving the protein motion, the magnitude of the static offset can be used as a proxy for the protein dynamics. Hence, a single probe's FFCF is sensitive to both the hydration and protein dynamics separately, offering a perspective that is generally not available from THz or OKE spectroscopy, where the two contributions are mixed. We measure the protein-hydration dynamics of HEWL-RC in aqueous (D2O) solutions of PEG400 (8-9mer) ranging from 0 to 80% PEG400 by volume, and compare these results to previously reported experiments using glycerol.13 In addition, we carry out a parallel experiment of HEWL-RC in varying concentrations of excess lysozyme ranging from 20 to 160 mg/mL, which acts to self-crowd the labeled protein with a complex electrostatic surface, which contrasts starkly with that presented by the uncharged polymer crowder.
We present a comprehensive picture of the picosecond protein and hydration dynamics under crowding conditions. We find an abrupt dynamical transition of the protein and hydration dynamics induced by crowding, which is unique from the temperature dependent transition that is observed in hydrated proteins.37,38 The results suggests a dynamic hydration shell around the protein extending 15-20 Å, resulting in collective hydration for interprotein separations of 30-40 Å. We also find that the collective water dynamics can be up to an order of magnitude slower than bulk water. In addition, we find that the presence of this transition seems to be due to the macromolecular nature of the crowding agent since it is absent in the case of solvation by glycerol/water solutions. The existence of two distinct regimes, each of which is largely dynamically decoupled from the fine details of the surrounding solvent fluctuations, suggests the partitioning of biomacromolecules into “undercrowded” and “overcrowded” conditions. Based on our measurements, many cellular environments can be classified as being “overcrowded.”
Results
Polymer Crowding
There is experimental evidence that PEG400 adopts a compact structure when in dilute aqueous solution.39,40 For example, small angle neutron scattering results show that the radius of gyration of PEG400 measured at 1% (v/v) in D2O is 2 nm,39,40 which is similar in size to a typical protein. The structure of PEG400 at high concentrations, however, remains unclear, though it has been proposed that the short polymer adopts an entangled structure. Nevertheless, the effect of PEG on protein and hydration dynamics should be largely due to the volume it excludes and the associated perturbation of its hydration environment, where the protein and hydration water reside largely in the pores of the entangled polymer solution.
The protein and hydration dynamics were studied in D2O/PEG400 solvent mixtures of 0, 20, 40, 60 and 80% PEG400 v/v. Figure 2 shows the FFCFs for each solution and the experimental fits, consisting of a single exponential decay (due to hydration water) and a static offset (due to slow protein dynamics). In pure D2O the hydration dynamics occur with a 2.7 ps time constant, which is slower than bulk D2O by a factor of 2. This observation has previously been reported13 and is in quantitative agreement with MD simulations of Laage and coworkers that specifically investigated the influence of the protein on extended hydrogen bond jumps of the hydration water.15 At high PEG400 (80% v/v) concentration the hydration dynamics slow by nearly a factor of 4, and the protein contribution increases by about 75% relative to pure D2O. Surprisingly, a dynamic transition is observed around 50% D2O where there is a significant, abrupt slowing of the protein-hydration dynamics. On either side of this transition, the protein and hydration dynamics are only weakly coupled to the polymer concentration, though the protein dynamics and the hydration dynamics stay strongly coupled to each other at all solvent compositions (evident from the correlation between τhydration and C(t=∞) in Figure 2b). To ensure the protein is not dehydrated by PEG, at least in the local region of the probe molecule, we use the vibrational lifetime, which we have shown to be a unique observable capable of reporting on local hydration levels.14 The lifetimes shown in Figure 4c exhibit decay times consistent with water-assisted relaxation at all PEG400 concentrations, ensuring that the local area of the probe remains fully hydrated.
Fig. 2.
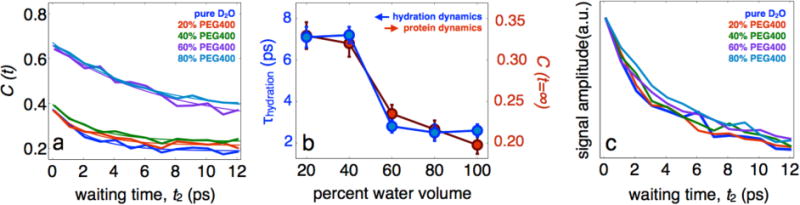
Interfacial water and protein dynamics of HEWL-RC in D2O/glycerol mixtures. a, FFCFs for HEWL-RC in D2O/PEG mixtures, ranging from pure D2O to 80% PEG by volume. b, Hydration timescale, obtained by the initial decay of the correlation function, and the protein dynamics, estimated by the static offset of the correlation function, plotted as a function of solvent composition. A strong coupling is clear from the data, with both the hydration and protein dynamics slowing down as glycerol is added to the system. There is also a sharp dynamic transition occurring at roughly 60% PEG. We suggest this transition results from the extended protein hydration environment overlapping with the PEG hydration environment. c, The vibrational relaxation—estimated from the rephasing signal amplitude—lacks any PEG400 dependence suggesting that the protein remains fully hydrated in the region around the probe.
Fig. 4.
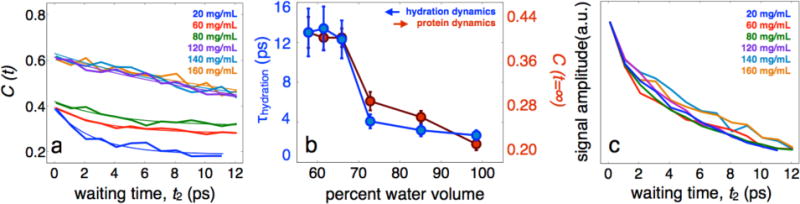
Interfacial water and protein dynamics of HEWL-RC in the presence of excess lysozyme. a, FFCFs for HEWL-RC in self-crowding conditions, ranging from 20 mg/mL to 160 mg/mL. b, Hydration timescale, obtained by the initial decay of the correlation function, and the protein dynamics, estimated by the static offset of the correlation function, plotted as a function of solvent composition. A strong coupling is clear from the data, with both the hydration and protein dynamics slowing down as excess lysozyme is added to the system. Similar to the PEG400 crowding, a dynamical transition is observed at sufficient crowding, though this transition occurs at lower concentrations of HEWL because of the more significant constraining effect that HEWL has on surrounding waters. c, Vibrational lifetimes estimated through the signal amplitude of the rephasing spectrum again show a consistently short lifetime, consistent with a lack of protein-protein interactions that would result in surface dehydration and increased lifetimes.
These results are fundamentally different from previous observations made on HEWL-RC in D2O/glycerol solutions.13 In those experiments we observed a gradual, uniform slowdown of the protein-hydration dynamics as a function of glycerol concentration, with no clear signs of a dynamical transition. Additionally, the slowdown in hydration dynamics was significantly more mild than what would be expected for the viscosity increase, demonstrating a weak coupling between interfacial water and the bulk solution. It is noteworthy that similar nonlinear scaling of interfacial water around liposomes has recently been observed using an NMR-based technique, Ovehauser dynamic nuclear polarization, which measures hydration water through the incorporation of a free-radical probe.36 For our current and previous results, a comparison of the interfacial water dynamics is shown in Figure 3. The influence of either glycerol or PEG400 on the hydration dynamics has similarities and differences. While the magnitude of the slowdown induced by high concentrations of either cosolvent is similar, and thus the coupling between the interfacial water and bulk solvent remains weak,13,36 the presence of a dynamic transition is observed only with the macromolecular crowding agent.
Fig. 3.
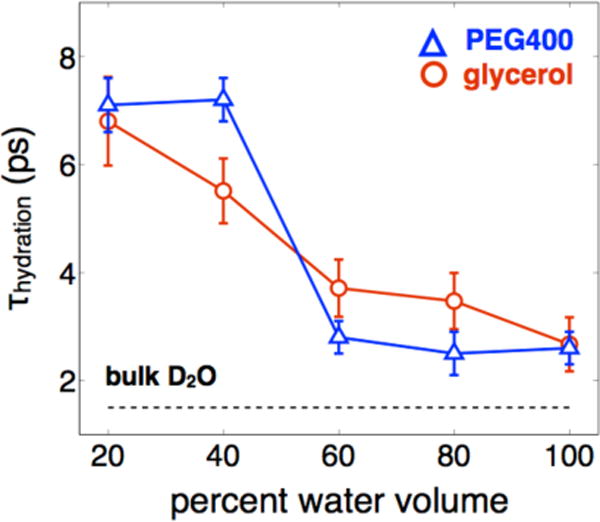
Comparison of interfacial water dynamics of HEWL-RC in solutions of glycerol and PEG400. While the magnitude of the hydration dynamics slowdown induced by each cosolvent is similar at high concentrations, the dynamic transition is observed only in the presence of the macromolecular crowding agent.
Self-Crowding
The presence of surface charges, site-specific interactions and an intricate surface topology make proteins a more complex and biologically relevant crowding agent than a simple polymer. Additionally, proteins have well-defined structures that are often not significantly perturbed by concentration, which is not necessarily the case for PEG400. Here, we use unlabeled lysozyme to serve as the crowding agent to determine if the presence of a critical crowding level could exist in cell-like environments. In addition to providing a more realistic crowding agent, the well-defined shape and structure of lysozyme allows for the protein-protein distances to be estimated for a given concentration of protein.
A starting solution of HEWL-RC was prepared at 20 mg/mL, then excess lysozyme was added to concentrations up to 160 mg/mL. As before, we use the vibrational lifetime (Figure 4c) to ensure that no protein-protein contacts alter the hydration of the protein surface. Similar to the PEG400 data, the vibrational lifetimes exhibit negligible lysozyme concentration dependence, suggesting that the protein remains fully hydrated.
The FFCFs and fit parameters are shown in Figure 4. As with PEG400, there is a clear dynamic transition. The transition occurs at a higher water composition (˜70%) than with the PEG400 crowding agent, which is attributed to the more significant constraining effect of HEWL on the surrounding waters. This view is supported by the fact that lysozyme is a highly charged (pI = 11) protein at neutral pH, and the dynamical constraints placed on the hydration water reduces the local dielectric, effectively extending the electrostatic footprint of the protein.41 Assuming a homogeneous mixture42 and a spherical approximation to the volume (computed using the van der Waals surface) to approximate the size of lysozyme, we estimated the typical protein-protein distances (surface-to-surface) at each crowding concentration. This distance is only an idealized estimate assuming homogeneous protein solution, and should be viewed as an estimated upper limit (see Supporting Information, SI, for details). Plotting the protein-hydration dynamics in terms of our estimated protein-protein distance (Figure 5) reveals that this transition occurs at distances around 30-40 Å, suggesting a dynamical influence of the hydration water extending upwards of 15-20 Å extending from each surface.
Fig. 5.
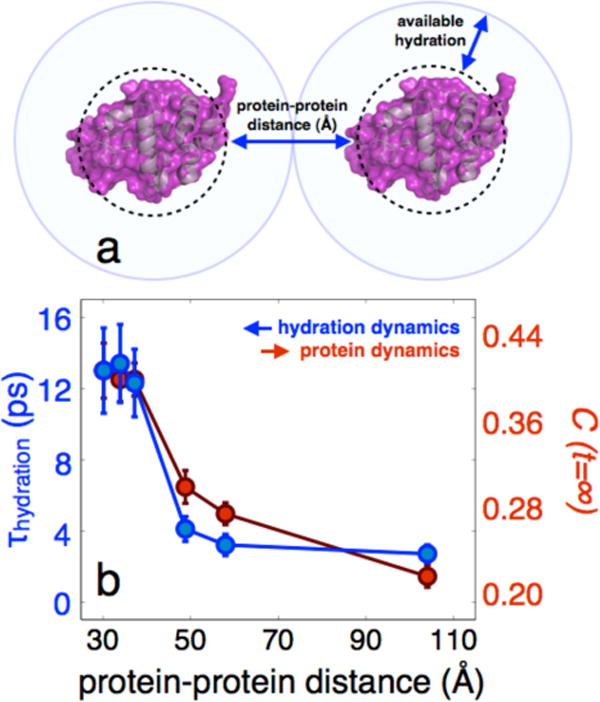
Hydration and protein dynamics of HEWL-RC in crowding conditions plotted as a function of protein-protein distance. a, The protein-protein distance is defined as the average surface-to-surface distance between proteins using a spherical approximation, which can be estimated for each concentration. b, Assuming a homogeneous mixture, the average surface-to-surface distance between proteins can be estimated, revealing that the transition occurs at a protein-protein distance of 30-40 Å.
Molecular Dynamics Simulations
The experimental results motivated efforts to simulate hydration dynamics of proteins under crowding conditions. In one case, two proteins are separated by a variable distance, and in the other case, four proteins are arranged tetrahedrally, with surfaces separated by a variable distance (Figure 6a,b). Each configuration (two- and four-protein geometries) was replicated in six individual simulations with average surface-to-surface distances ranging from 5-30 Å (see SI for more details). The water between the protein structures was selected for analysis of both the average hydrogen bond number (Figure 6c) as well as the hydrogen bond correlation time (Figure 6d). The hydrogen bond correlation time is reported as the 1/e time, alleviating complications of fitting a non-exponential relaxation. For the hydrogen bond number, there is a slight decrease at small interprotein distances reflecting the proportional increase in interfacial water, which exhibits reduced hydrogen bonding relative to bulk water. There is no observed threshold behavior in the extent of hydrogen bonding, suggesting a lack of significant structural changes of the water upon crowding. The dynamics of the water, however, show a very strong dependence on the crowding, including a dynamic transition that occurs at a critical protein-protein separation. In the two-protein simulation this critical distance was found to be 10-15 Å, whereas in the four-protein simulation we observed this transition at 20-25 Å. This distance is consistent with what was observed experimentally (30-40 Å), though there is a clear dependence of the dynamic transition on the configuration and geometry of crowding. The decoupling of the dynamics from crowding above and below the dynamic transition observed experimentally is also evident in the simulations.
Fig. 6.
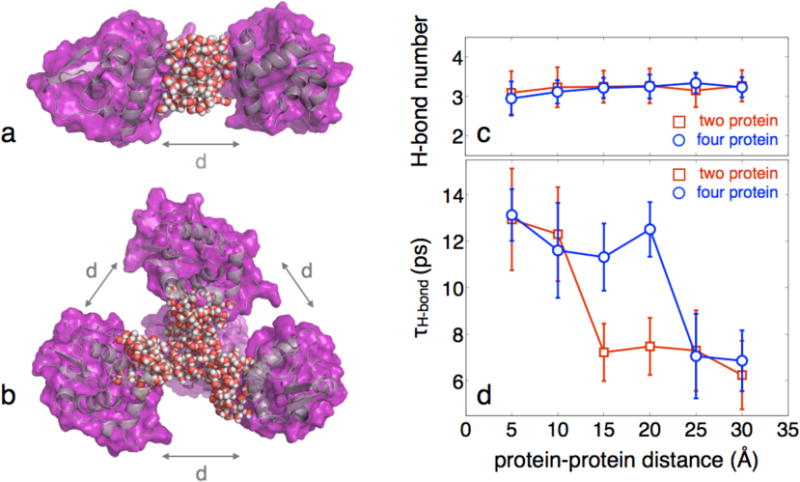
Example of the simulation analysis where (a) two proteins are separated by a set distance d and the bridging water is selected for analysis and (b) four proteins are arranged tetrahedrally, all of which are separated by the same variable distance. The water that was selected for analysis is shown. c, Hydrogen bond number of the crowded water as a function of protein-protein distance. In each case there is no clear transition in the average hydrogen bonds per water molecule, suggesting no significant change in structure. A slight downward trend is observed as the interprotein distance is reduced, though this is the result of a higher relative contribution from the interfacial water, which has fewer hydrogen bonds than bulk water. d, Hydrogen bond correlation times of the crowded water as a function of protein-protein distance. The occurrence of a dynamic transition is found between 10-15 Å for two proteins and 20-25 Å for the four protein simulation. In each case, only a weak coupling is observed before and after the dynamic transition. The results not only demonstrate a percolation-like transition of water dynamics upon crowding, but also show that the distance of this transition is a function of the degree and geometry of crowding.
Surprisingly, the dynamic transition is accompanied by no significant net structural changes, as seen in the average hydrogen bonding number of the interfacial water remaining constant (Figure 6c). The lack of any clear structural signature accompanying the dynamical transition is similar to glassy43 and jammed systems.44 Our observations are the first examples of a purely dynamical transition induced by macromolecular crowding. The lack of a significant change in the degree of hydrogen bonding differs qualitatively from previous studies based largely on inelastic neutron scattering experiments. At hydration levels over an order of magnitude lower than what we consider here, there is clear evidence for a pronounced change in water structure.45-47 Using comparisons with simulation, several workers have identified percolation transitions, where at a threshold hydration level, there is a significant increase in the size of the largest hydrogen bonded cluster solvating the protein.45 Such abrupt structural changes leading to hydrogen bonding networks that span large areas of the protein-water interface have been interpreted in terms of percolation theory. The new dynamical transition that we have identified here is distinct from these previous observations of hydrogen bond percolation.32 Most importantly, our highest protein concentration is 160 mg/mL, which corresponds to a hydration level (h = mass of D2O/mass of lysozyme) of h = 6. Neutron scattering experiments and accompanying simulations are carried out at hydration levels less than h = 1. These studies on hydrated protein powders represent an extreme case of crowding, while here the studies were performed on more dilute aqueous solutions. The dynamical transition that we observe occurs at comparably much larger values of protein hydration, highlighting the subtle nature of the collective hydration leading to a transition of the hydration water dynamics without significantly distorting its structure.
Discussion
Water is capable of forming extensive hydrogen bonding networks that reorganize in a collective manner through an angular jump mechanism.48,49 Furthermore, the barrier to hydrogen bond jumps is dominated by entropic contributions arising from the availability of hydrogen bonding accepting partners.50 Hydrogen bond exchange dynamics can be stifled by limiting the configuration space available for accepting waters, and thus larger hydrophobic molecules are capable of hindering hydrogen bond dynamics while small hydrophobes have a negligible effect.12 The collective nature of hydrogen bond motion can lead to spatially extended dynamic perturbations, inducing long-range coupling effects in crowded environments.25 Extended collective motion of water over distances of 30-40 Å has been observed not only in crowded protein solutions,26 but also in water pools confined within reverse micelles.51 In each case, the transition to collective water motion is found to be abrupt.
The measured retardation factors of crowded water are roughly 5 and 10 for PEG400 and lysozyme, respectively, relative to bulk D2O. Given that the expected concentrations of macro-molecules inside of cells is on the order of 300 mg/mL,52 the experimental results suggest that the majority of water within cells is involved in slow, collective hydration, with only trace amounts of “bulk-like” water present, despite 50-70% water content by volume. The long-range disruption of water dynamics around macromolecules is likely to be a general property of compact proteins, and particular proteins, such as antifreeze proteins,26 may leverage the perturbation to carry out a function.
Since this study primarily investigates dynamics as sensed on the picosecond time scale, there is no straightforward link to biological function, which spans a vast range of time scales.53 However, recent work has suggested fast fluctuations of proteins have significant implications on longer timescale dynamics, such as conformational sampling54 and possibly enzyme activity.53 Due to the strong coupling between the low-frequency fluctuations of proteins and the hydration water,13,55-57 the observed jamming-like transition of the water is accompanied by a transition in the fast protein dynamics. In crowded environments these low-frequency modes are significantly slowed from what they would be in solutions with excess water (Figure 2b and 4b). The collective hydration environment in crowded conditions effectively increases the viscosity felt by the protein, and thus the protein undergoes pronounced slowing at a critical crowding concentration. Based on our estimated macromolecular crowder concentration threshold, it would appear that most, if not all, regions of the cell are “overcrowded.”
Conclusions
We carried out two parallel experiments measuring the protein-hydration dynamics of HEWL-RC in both solutions of D2O/PEG400 and solutions of excess lysozyme to act as crowding agents. From the experimental results we draw three conclusions. 1. Both PEG and protein crowders induce a dynamical transition, where the coupled protein-hydration dynamics exhibit a sharp slowdown above a critical degree of crowding indicative of an independent-to-collective hydration transition. It is observed that water in sufficiently crowded environments is roughly an order of magnitude slower than bulk water. 2. Using the results from self-crowding, we estimate that the distance between protein surfaces at which this transition occurs is 30-40 Å, which is a striking manifestation of the collective and coordinated behavior of strongly hydrogen bonding environments. 3. The macromolecular nature of the crowder is essential as demonstrated through comparisons between PEG400 crowding and previously reported glycerol/water solutions. While similar degrees of slowing are found at high concentrations of both, the presence of a dynamical transition is observed only in the PEG400 experiments. Simulation results confirm the experimental findings, while introducing an additional observation. In contrast to previous studies of protein hydration, where hydrogen bonding in the hydrating water is perturbed by the protein, our simulations indicate no significant changes in hydrogen bonding. Rather, the observed and simulated abrupt transition is purely dynamical in nature, and reflects the long-range influence of protein surface-induced constraints on water's orientational flexibility.
These results suggest that little to no “bulk-like” water is present within cellular environments. Instead, biological macromolecules are hydrated by significantly constrained water that that in turn can strongly modulate the flexibility and dynamics of the biomolecules. Future work will be dedicated to studying the connection between the picosecond dynamics of the hydration water, which we suggest to be the origin of dynamical crowding effects, on much longer processes, such as protein folding and catalytic activity. The partitioning of hydration dynamics into two apparent regimes suggests that large scale implicit solvent simulations of biomolecules may be able to produce realistic dynamics by adopting distance-dependent frictional damping. Based on our observations of distinct under- and overcrowded regimes, perhaps as few as two macromolecule-specific friction values are needed to capture the essential dynamical contrast between isolated and crowded macromolecules. Macromolecule-modified hydration dynamics has also been related to a change in the local dielectric constant41,58, a quantity which enters both the generalized Born model of solvation,59 as well as the accurate estimates of donor-acceptor distances in Förster resonant energy transfer experiments.60 In both cases, the distance dependent solvation dynamics may produce qualitative deviations from conventional models based on a homogeneous dielectric continuum. With new methods such as site-specific 2DIR and other techniques, it is becoming clear that the complexity of biomolecule hydration can be addressed experimentally and linked directly to simulation, likely providing insight into the active nature of water in mediating biological processes.
Methods
Protein labeling
Hen egg white lysozyme (HEWL) was purchased from Sigma Aldirich (bioultra, > 98%). No further purification steps were taken. HEWL (approximately 2 mg/mL) was then combined in a 1:1 ratio with tricarbonylchloro(glycinato)ruthenium (II) in D2O (Sigma) and stirred at room temperature for 1 hour. The resulting labeled protein we refer to as HEWL-RC. The resulting product was purified in a desalting column (GE Healthcare, PD-10 Disposable Desalting Column), which removes unreacted tricarbonylchloro(glycinato)ruthenium (II). The reaction was carried out on the morning of the experiments, and no HEWL-RC was stored to be used at a later date33.
2D-IR spectroscopy
Mid-IR pulses are generated through two home-built dual stage optical parametric amplifiers (OPAs) coupled with difference frequency generation (DFGs) which are pumped with an regeneratively amplified Ti:Sapphire laser. The mid-IR pulses are then split into fields E1, E2, E3, and ELO with respected wavevectors k1, k2, k3, kLO (75 fs, 150 cm-1 bandwidth, 400 nJ/pulse), where the first three pulses are focused onto the sample in a box geometry to generate a third-order nonlinear signal, and the final pulse is used for heterodyne detection. We implement an upconversion detection technique that mixes a highly chirped pulse centered at 800 nm and fwhm = 160 ps with the mid-IR signal and local oscillator in a sum-frequency crystal (MgO doped LiNbO3) to allow for detection in the visible with a silicon CCD camera. The detection frequency of the 2D-IR spectrum is provided by the spectrometer. The excitation frequency is measured by scanning the time delay between the first two pulses and then Fourier transforming over the generated coherence period. A series of 2D spectra are then acquired as a function of waiting time between the excitation pulse pair and the detection pulse, which is stepped from 0-12 ps.
Molecular Dynamics simulations
Full details of the molecular dynamics simulations can be found in the Supporting Information. Protein crowding was simulated by analyzing the interstitial water of two protein configurations: 2 proteins near each other, and 4 proteins in a packed tetrahedral configuration. Six replicas of each configuration were made by varying the average separation between protein surfaces into a gradient of distances: 5, 10, 15, 20, 25, and 30 Å.
Hydrogen-bond (HB) auto-correlation functions and the average number of HB partners per water were calculated for interstitial water. Cutoffs for HB partners were defined as acceptor-donor distances of less than 3.5 Å (O-O distance), and acceptor-donor-hydrogen angles of less than 30° as outlined by Skinner et al61. The center of each protein was calculated as the mean position of all protein atoms. For each protein in each replica, spheres of water each with a radius of 10 Å were selected around the protein atom closest to the overall center. Hydrogen-bond auto-correlation functions were calculated at 15 ps intervals using the g_hbond utility from GROMACS. These functions were averaged to obtain the mean 1/e time. The average number of hydrogen bonds per water were calculated for each saved frame using in-house MATLAB code and the cutoff criteria detailed previously.
Supplementary Material
Acknowledgments
This work was supported by the National Science Foundation (CHE-0748501), the National Institutes of Health (RR012255), and the Camille & Henry Dreyfus Foundation.
Footnotes
Additional Information: Supporting Information detailing the estimation of protein-protein distance and molecular dynamics simulations are available. This information is available free of charge via the internet at http://pubs.acs.org.
Notes: The authors declare no competing financial interest.
References
- 1.Ball P. Chem Rev. 2008;108:74–108. doi: 10.1021/cr068037a. [DOI] [PubMed] [Google Scholar]
- 2.Nicholls A, Sharp KA, Honig B. Proteins:Struct Funct Genet. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
- 3.Liu P, Huang XH, Zhou RH, Berne BJ. Nature. 2005;437:159–162. doi: 10.1038/nature03926. [DOI] [PubMed] [Google Scholar]
- 4.Cheung MS, Garcia AE, Onuchic JN. Proc Natl Acad Sci USA. 2002;99:685–690. doi: 10.1073/pnas.022387699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hamill OP, Martinac B. Physiol Rev. 2001;81:685–740. doi: 10.1152/physrev.2001.81.2.685. [DOI] [PubMed] [Google Scholar]
- 6.Bhat TN, Bentley GA, Boulot G, Greene MI, Tello D, Dallacqua W, Souchon H, Schwarz FP, Mariuzza RA, Poljak RJ. Proc Natl Acad Sci USA. 1994;91:1089–1093. doi: 10.1073/pnas.91.3.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsai CJ, Lin SL, Wolfson HJ, Nussinov R. Protein Sci. 1997;6:53–64. doi: 10.1002/pro.5560060106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lum K, Chandler D, Weeks JD. J Phys Chem B. 1999;103:4570–4577. [Google Scholar]
- 9.Chandler D. Nature. 2005;437:640–647. doi: 10.1038/nature04162. [DOI] [PubMed] [Google Scholar]
- 10.Li ITS, Walker GC. Proc Natl Acad Sci USA. 2011;108:16527–16532. doi: 10.1073/pnas.1105450108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davis JG, Gierszal KP, Wang P, Ben-Amotz D. Nature. 2012;491:582–585. doi: 10.1038/nature11570. [DOI] [PubMed] [Google Scholar]
- 12.Laage D, Stirnemann G, Hynes JT. J Phys Chem B. 2009;113:2428–2435. doi: 10.1021/jp809521t. [DOI] [PubMed] [Google Scholar]
- 13.King JT, Kubarych KJ. J Am Chem Soc. 2012;134:18705–18712. doi: 10.1021/ja307401r. [DOI] [PubMed] [Google Scholar]
- 14.King JT, Arthur EJ, Brooks CL, III, Kubarych KJ. J Phys Chem B. 2012;116:5604–5611. doi: 10.1021/jp300835k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sterpone F, Stirnemann G, Laage D. J Am Chem Soc. 2012;134:4116–4119. doi: 10.1021/ja3007897. [DOI] [PubMed] [Google Scholar]
- 16.Minton AP. Biopolymers. 1981;20:2093–2120. [Google Scholar]
- 17.Zimmerman SB, Minton AP. Annu Rev Biophys Biomol Struct. 1993;22:27–65. doi: 10.1146/annurev.bb.22.060193.000331. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y, Sarkar M, Smith AE, Krois AS, Pielak GJ. J Am Chem Soc. 2012;134:16614–16618. doi: 10.1021/ja305300m. [DOI] [PubMed] [Google Scholar]
- 19.Benton LA, Smith AE, Young GB, Pielak GJ. Biochemistry. 2012;51:9773–9775. doi: 10.1021/bi300909q. [DOI] [PubMed] [Google Scholar]
- 20.Dhar A, Samiotakis A, Ebbinghaus S, Nienhaus L, Homouz D, Gruebele M, Cheung MS. Proc Natl Acad Sci USA. 2010;107:17586–17591. doi: 10.1073/pnas.1006760107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheung MS, Klimov D, Thirumalai D. Proc Natl Acad Sci USA. 2005;102:4753–4758. doi: 10.1073/pnas.0409630102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feig M, Sugita Y. J Phys Chem B. 2012;116:599–605. doi: 10.1021/jp209302e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McGuffee SR, Elcock AH. Plos Comput Biol. 2010;6:e1000694. doi: 10.1371/journal.pcbi.1000694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qiu W, Kao YT, Zhang L, Yang Y, Wang L, Stites WE, Zhong D, Zewail AH. Proc Natl Acad Sci USA. 2006;103:13979–13984. doi: 10.1073/pnas.0606235103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ebbinghaus S, Kim SJ, Heyden M, Yu X, Heugen U, Gruebele M, Leitner DM, Havenith M. Proc Natl Acad Sci USA. 2007;104:20749–20752. doi: 10.1073/pnas.0709207104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meister K, Ebbinghaus S, Xu Y, Duman JG, DeVries A, Gruebele M, Leitner DM, Havenith M. Proc Natl Acad Sci USA. 2012 doi: 10.1073/pnas.1214911110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nucci NV, Pometun MS, Wand AJ. Nat Struct Mol Biol. 2011;18:245–U315. doi: 10.1038/nsmb.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nucci NV, Pometun MS, Wand AJ. J Am Chem Soc. 2011;133:12326–12329. doi: 10.1021/ja202033k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McClain BL, Finkelstein IJ, Fayer MD. J Am Chem Soc. 2004;126:15702–15710. doi: 10.1021/ja0454790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mazur K, Heisler IA, Meech SR. J Phys Chem A. 2012;116:2678–2685. doi: 10.1021/jp2074539. [DOI] [PubMed] [Google Scholar]
- 31.Hunt NT, Kattner L, Shanks RP, Wynne K. J Am Chem Soc. 2007;129:3168–3172. doi: 10.1021/ja066289n. [DOI] [PubMed] [Google Scholar]
- 32.Doster W, Cusack S, Petry W. Nature. 1989;6209:754–756. doi: 10.1038/337754a0. [DOI] [PubMed] [Google Scholar]
- 33.Santos-Silva T, Mukhopadhyay A, Seixas JD, Bernardes GJL, Romao CC, Romao MJ. J Am Chem Soc. 2011;133:1192–1195. doi: 10.1021/ja108820s. [DOI] [PubMed] [Google Scholar]
- 34.Huang L, Jin R, Li J, Luo K, Huang T, Wu D, Wang W, Chen R, Xiao G. Faseb J. 2010;24:3536–3543. doi: 10.1096/fj.09-150987. [DOI] [PubMed] [Google Scholar]
- 35.Patel AJ, Varilly P, Jamadagni SN, Hagan MF, Chandler D, Garde S. J Phys Chem B. 2012;116:2498–2503. doi: 10.1021/jp2107523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Franck JM, Scott JA, Han S. J Am Chem Soc. 2013;135:4175–4178. doi: 10.1021/ja3112912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tarek M, Tobias D. J Phys Rev Lett. 2002;88:138101. doi: 10.1103/PhysRevLett.88.138101. [DOI] [PubMed] [Google Scholar]
- 38.Doster W, Cusack S, Petry W. Nature. 1989;337:754–756. doi: 10.1038/337754a0. [DOI] [PubMed] [Google Scholar]
- 39.Linegar KL, Adeniran AE, Kostko AF, Anisimov MA. Colloid J. 2010;72:279–281. [Google Scholar]
- 40.Lancz G, Avdeev MV, Petrenko VI, Garamus MV, Koneracka M, Kopcansky P. Acta Phys Pol A. 2010;118:980–982. [Google Scholar]
- 41.Despa F, Fernandez A, Berry RS. Phys Rev Lett. 2004;93:228104. doi: 10.1103/PhysRevLett.93.228104. [DOI] [PubMed] [Google Scholar]
- 42.Shukla A, Mylonas E, Di Cola E, Finet S, Timmins P, Narayanan T, Svergun DI. Proc Natl Acad Sci USA. 2008;105:5075–5080. doi: 10.1073/pnas.0711928105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Angell CA. Science. 1995;267:1924–1935. doi: 10.1126/science.267.5206.1924. [DOI] [PubMed] [Google Scholar]
- 44.Trappe V, Prasad V, Cipelletti L, Segre PN, Weitz DA. Nature. 2001;411:772–775. doi: 10.1038/35081021. [DOI] [PubMed] [Google Scholar]
- 45.Oleinikova A, Brovchenko I, Smolin N, Krukau A, Geiger A, Winter R. Phys Rev Lett. 2005;95:247802. doi: 10.1103/PhysRevLett.95.247802. [DOI] [PubMed] [Google Scholar]
- 46.Smolin N, Oleinikova A, Brovchenko I, Geiger A, Winter R. J Phys Chem B. 2005;109:10995–11005. doi: 10.1021/jp050153e. [DOI] [PubMed] [Google Scholar]
- 47.Nakagawa H, Kataoka M. J Phys Soc Japan. 2010;79:083801. [Google Scholar]
- 48.Eaves JD, Loparo JJ, Fecko CJ, Roberts ST, Tokmakoff A, Geissler PL. Proc Natl Acad Sci USA. 2005;102:13019–13022. doi: 10.1073/pnas.0505125102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Laage D, Hynes JT. Science. 2006;311:832–835. doi: 10.1126/science.1122154. [DOI] [PubMed] [Google Scholar]
- 50.Laage D, Hynes JT. Chem Phys Lett. 2006;433:80–85. [Google Scholar]
- 51.Moilanen DE, Fenn EE, Wong D, Fayer MD. J Chem Phys. 2009;131:014704. doi: 10.1063/1.3159779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zimmerman SB, Trach SO. J Mol Biol. 1991;222:599–620. doi: 10.1016/0022-2836(91)90499-v. [DOI] [PubMed] [Google Scholar]
- 53.Henzler-Wildman KA, Lei M, Thai V, Kerns SJ, Karplus M, Kern D. Nature. 2007;450:913–U927. doi: 10.1038/nature06407. [DOI] [PubMed] [Google Scholar]
- 54.Vashisth H, Brooks CL., III J Phys Chem Lett. 2012:3379–3384. doi: 10.1021/jz301650q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lubchenko V, Wolynes PG, Frauenfelder H. J Phys Chem B. 2005;109:7488–7499. doi: 10.1021/jp045205z. [DOI] [PubMed] [Google Scholar]
- 56.Frauenfelder H, Chen G, Berendzen J, Fenimore PW, Jansson H, McMahon BH, Stroe IR, Swenson J, Young RD. Proc Natl Acad Sci USA. 2009;106:5129–5134. doi: 10.1073/pnas.0900336106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vinh NQ, Allen SJ, Plaxco KW. J Am Chem Soc. 2011;133:8942–8947. doi: 10.1021/ja200566u. [DOI] [PubMed] [Google Scholar]
- 58.Harada R, Sugita Y, Feig M. J Am Chem Soc. 2012;134:4842–4849. doi: 10.1021/ja211115q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bashford D, Case DA. Annu Rev Phys Chem. 2000;51:129–152. doi: 10.1146/annurev.physchem.51.1.129. [DOI] [PubMed] [Google Scholar]
- 60.Beljonne D, Curutchet C, Scholes GD, Silbey RJ. J Phys Chem B. 2009;113:6583–6599. doi: 10.1021/jp900708f. [DOI] [PubMed] [Google Scholar]
- 61.Kumar R, Schmidt JR, Skinner JL. J Chem Phys. 2007;126:204107. doi: 10.1063/1.2742385. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.