

The authors used 10 ng of genomic DNA from formalin-fixed, paraffin-embedded human colorectal cancer tumors to sequence 46 cancer genes using the AmpliSeq platform. They developed an orthogonal resequencing approach, SimpliSeq, to evaluate the clinical application of AmpliSeq. The results suggest that AmpliSeq provides highly sensitive and quantitative mutation detection for most genes on its cancer panel; however, verification testing is critical for low-abundance variants and genes that are found to have recurrent false-positive variants.
Keywords: AmpliSeq, Ion Torrent, Next-generation sequencing, Mutation, Precision oncology, Molecular diagnostics
Abstract
Purpose.
The success of precision oncology relies on accurate and sensitive molecular profiling. The Ion AmpliSeq Cancer Panel, a targeted enrichment method for next-generation sequencing (NGS) using the Ion Torrent platform, provides a fast, easy, and cost-effective sequencing workflow for detecting genomic “hotspot” regions that are frequently mutated in human cancer genes. Most recently, the U.K. has launched the AmpliSeq sequencing test in its National Health Service. This study aimed to evaluate the clinical application of the AmpliSeq methodology.
Methods.
We used 10 ng of genomic DNA from formalin-fixed, paraffin-embedded human colorectal cancer (CRC) tumor specimens to sequence 46 cancer genes using the AmpliSeq platform. In a validation study, we developed an orthogonal NGS-based resequencing approach (SimpliSeq) to assess the AmpliSeq variant calls.
Results.
Validated mutational analyses revealed that AmpliSeq was effective in profiling gene mutations, and that the method correctly pinpointed “true-positive” gene mutations with variant frequency >5% and demonstrated high-level molecular heterogeneity in CRC. However, AmpliSeq enrichment and NGS also produced several recurrent “false-positive” calls in clinically druggable oncogenes such as PIK3CA.
Conclusion.
AmpliSeq provided highly sensitive and quantitative mutation detection for most of the genes on its cancer panel using limited DNA quantities from formalin-fixed, paraffin-embedded samples. For those genes with recurrent “false-positive” variant calls, caution should be used in data interpretation, and orthogonal verification of mutations is recommended for clinical decision making.
Implications for Practice:
Next-generation sequencing technologies permit deep sequencing of hundreds of cancer genes concurrently, offering an unprecedented opportunity to identify the clinically relevant mutations in personalized cancer care. The Ion AmpliSeq Cancer Panel provides a rapid and cost-effective sequencing workflow for single-tube preparation of amplicon libraries from genomic “hotspot” regions that are frequently mutated in human cancer genes. This study evaluates the advantages and disadvantages of the AmpliSeq platform for clinical applications, and describes how this new knowledge can be used to ensure the accuracy of mutation detection for precision oncology.
Introduction
Next-generation sequencing (NGS) technologies permit deep sequencing of hundreds of cancer genes concurrently, providing an unprecedented opportunity to identify the clinically actionable mutations relevant to personalized cancer care. Targeted NGS has emerged as a sensitive and efficient tool to detect complex and heterogeneous gene mutations, interrogating relevant gene content with a breadth that exceeds probe-based assays and conventional Sanger sequencing [1]. Several targeted NGS platforms exist, including cancer panels from Illumina and RainDance that require a minimum genomic DNA (gDNA) input of 50–250 ng and a practical turnaround time ranging from days to weeks. These limitations pose some real-world challenges, namely the clinical need for fast turnaround times as well as the obligatory requirement that assays must work with low DNA input given the typically poor yields achieved from formalin-fixed, paraffin-embedded (FFPE) specimens, fine-needle aspirates, rare circulating tumor cells, and microdissected tissue fragments.
Very recently, Ion Torrent has provided a potential solution for these problems by developing the Ion AmpliSeq Cancer Panel (henceforth abbreviated AmpliSeq). AmpliSeq is a targeted polymerase chain reaction (PCR)-based enrichment method performed upstream of NGS that reportedly enables efficient and cost-effective mutation screening of FFPE specimens [2, 3]. AmpliSeq is attractive for clinical applications for several reasons: (a) DNA input requirements are only 10 ng, compared with up to 1 μg of DNA required by other enrichment protocols and conventional sequencing methods; (b) the assay design targets relatively short gene segments for amplification (typically <120 bp), making it more compatible with heavily modified and degraded FFPE clinical specimens; (c) the turnaround time is only 10 hours (3.5 hours for library preparation, 4 hours for template preparation, 1.5 hours for sequencing, and 1 hour for data analysis), and is thus responsive to time-sensitive clinical applications; and (d) AmpliSeq enrichment targets 190 amplicons that encompass 739 known cancer-relevant mutations/variants (herein abbreviated variants) across 46 cancer-related genes [2, 3]. The AmpliSeq Cancer Panel was designed using a candidate pool of variants identified in large-scale sequencing studies, narrowing the content to critical genes and pathways important in the initiation and progression of human cancer. Currently, AmpliSeq is used in clinical cancer research with key applications for personalized cancer care. Most recently, the U.K. has launched an AmpliSeq-based 46-gene sequencing diagnostic test in its National Health Service [4].
Colorectal cancer (CRC) is the second leading cause of cancer-related deaths in the U.S. The pathogenesis of CRC is associated with mutations in several critical genes, including KRAS, BRAF, PIK3CA, SMAD4, PTEN, NRAS, and TGFBR2, all of which are covered by the Ion AmpliSeq Cancer Panel. In this study, we developed an orthogonal resequencing approach termed SimpliSeq to evaluate the clinical application of AmpliSeq. The new knowledge from this study will help inform the clinical applications of AmpliSeq by providing experimental evidence that addresses both the pros and cons of this approach for sequencing routine clinical tumor specimens.
Materials and Methods
Tumor Specimens, Cell Lines, and DNA Preparation
Twenty-two archival FFPE CRC tumor specimens at stage II to stage IV were obtained through an Institutional Review Board-approved research procurement protocol. Each specimen was microdissected as previously described [5]. Two different compartments were collected: epithelial tumor (labeled as xxxxx-E) and tumor-surrounding stroma (labeled as xxxxx-S), with a total area of 10 mm2 to 25 mm2 each. Briefly, FFPE sections (7-µM thick) were deparaffinized, briefly stained with hematoxylin and dehydrated in xylene, and then subjected to laser capture-assisted microdissection using the MMI CellCut Plus instrument (Molecular Machines & Industries, Glattbrugg, Switzerland, http://www.molecular-machines.com). In total, 44 individually microdissected tissues were collected from 22 CRC tumor specimens, and gDNA samples were prepared using the QIAamp DNA FFPE tissue kit (Catalog no. 56404, Qiagen, Hilden, Germany, http://www.qiagen.com). In separate studies, 8 additional archival CRC specimens were used for allele-specific PCR (AS-PCR) or TaqMan-based confirmation testing. The gDNA concentration and purity were measured using a NanoDrop spectrophotometer (NanoDrop, Wilmington, DE, http://www.nanodrop.com).
DNA from eight cancer cell lines with known mutation status was pooled at mass fractions ranging from 2% to 35% (assuming diploidy for these selected genes on the panel) as positive mutant controls (supplemental online Table 1). The cell line GP2d was obtained from Health Protection Agency Culture Collections (Salisbury, UK, http://phe-culturecollections.org.uk), and the remaining cell lines were obtained from ATCC (Manassas, VA, http://www.attc.org): MIA-PaCa-2, T-24, RKO, SK-Mel-2, HCT-116, SW1116, and A549.
AmpliSeq-Based Mutation Detection
AmpliSeq NGS analysis was run on an Ion Torrent Personal Genome Machine (PGM; Life Technologies, Carlsbad, CA, http://www.lifetechnologies.com) using the Ion AmpliSeq Cancer Panel (v1) with targeted coverage across 46 cancer genes [2]. Briefly, 10 ng of gDNA was used for single-tube preparation of 190-plex amplicon libraries. Each sample was bar- coded, amplified by emulsion polymerase chain reaction (emPCR), and sequenced on an Ion 316 chip. To optimize NGS experiment conditions, we compared three different gDNA inputs (5 ng, 10 ng, and 100 ng), two working PGM instruments, two independent analysis pipelines, and input titration of DNA template into emPCR with analyzed total reads, median coverage (across 739 hot spots), and variant frequency of KRAS G12D, which had been tested by other platforms. Within these different conditions, the increase of DNA input for emPCR from 48 million copies to 160 million copies was the most critical.
Orthogonal Mutation Validation
We developed SimpliSeq, an orthogonal resequencing approach featuring singleplex target enrichment and independent bioinformatic analysis, to validate the variant calls using the remaining gDNA from previous AmpliSeq runs. Briefly, 5 ng of gDNA was used to individually amplify genes of interest using validated gene-specific primers, covering 100 bp of genomic context sequence surrounding the mutant base. SimpliSeq primers were designed to be nonredundant with AmpliSeq primers and to minimize the sequence overlap with the corresponding AmpliSeq amplicon, providing an orthogonal PCR strategy that could be used to confirm putative variants identified in the primary AmpliSeq NGS screen. SimpliSeq amplicons were bar coded for standard library preparation, pooled and sequenced on an Ion Torrent PGM sequencer as described above.
Independent orthogonal mutation confirmation was also carried out using SuraSeq500, a multiplex PCR panel of 35 amplicons and targeted NGS on Illumina GAIIx platform using 40 ng of gDNA as described previously [6]. TaqMan or AS-PCR assays using 100 ng of gDNA [7] and Sanger sequencing using 100 ng of gDNA [8] were also used in confirmation studies.
Statistical Analysis
Variants were identified from AmpliSeq-targeted NGS data using the associated Torrent Variant Caller Plugin (version 2.0.1) and annotated manually per Human Genome Variation Society (HGSV) nomenclature. For reads with <100× coverage, variants were identified based on the version 2.0.1 Ion Torrent Variant Caller algorithm, which considers both base quality and coverage depth for defining a positive call. At very low coverage (<60×), a Bayesian algorithm is used.
Analysis of variants in SimpliSeq reads was accomplished using NextGENe version 2.18. Briefly, raw fastq files generated by the Torrent server were aligned to the reference genes downloaded from the National Center for Biotechnology Information with the criteria of at least 30 matching bases to the reference and at least 95% of all bases also matching the reference. Postalignment, variants were automatically called using the software default parameters and annotated per HGSV nomenclature. Pearson correlation coefficients were applied to measure the linear dependency or the strength of association between two variables.
Results
Evaluation of AmpliSeq-Based Variant Detection
To assess the feasibility of AmpliSeq with low DNA input, we examined the KRAS mutation status using the recommended amount of 10 ng of gDNA extracted from each of the two FFPE CRC tumor specimens with known KRAS genotypes previously identified by AS-PCR analysis. Following the optimized NGS workflow (Fig. 1), AmpliSeq correctly distinguished the mutant KRAS from wild-type allele, a result that was further validated by using both SuraSeq500 and Illumina GAIIx NGS platforms [6] (supplemental online Fig. 1).
Figure 1.

Targeted NGS workflow: AmpliSeq and SimpliSeq.
Abbreviations: emPCR, emulsion polymerase chain reaction; FFPE, formalin-fixed, paraffin-embedded; gDNA, genomic DNA; NGS, next-generation sequencing; PGM, Personal Genome Machine.
To evaluate the detection sensitivity of AmpliSeq, we screened gene mutations using eight pooled cancer cell lines [6] containing 18 well-characterized mutations with expected variant frequencies ranging from 1% to 35% (supplemental online Table 1). Of these 18 known mutations, 15 were covered by the AmpliSeq 790 hotspot mutation loci. Furthermore, AmpliSeq-targeted NGS reported 14 of 15 of these mutations to be consistent with the expected input abundance with variant frequency (VF) as low as 1% (supplemental online Fig. 1). The Pearson correlation coefficient between expected and detected mutation frequency was determined to be .94, indicating a strong linear relationship. However, it is important to note that 1 of the 14 mutations was a “false-negative mutation” as it was not in the automatic output made by the associated Ion Torrent Variant Caller and had to be mined from the raw data. Further analysis of the data revealed that JAK2 V617F was a systematic false-positive variant call in FFPE tissue and cell line-based studies (data not shown); we therefore excluded it from subsequent data analyses.
AmpliSeq-Based Variant Identification Using FFPE CRC Tissues
Next, we applied AmpliSeq to measure the mutation status of 44 microdissected FFPE tissues. Data analysis revealed an overall high yield of amplicons across 46 genes (supplemental online Table 2), achieving >100× coverage of 94.3% of targets with a mean read depth of 1241, comparable to those at higher coverage (250× and 500×) (supplemental online Table 3). Figure 2A shows the distribution of variants detected by AmpliSeq in these 44 microdissected FFPE samples. In total, 269 variants covering 97 mutational hotspots were detected in 31 of the 46 genes in the panel, with VFs ranging from 1% to 95%. Specifically, 61% (165/269) of variants have VF rates >5% and 39% (104/269) have VF <5%. In this study, we focused on the mutational profiling by AmpliSeq and did not study insertions and deletions because of the reported suboptimal utilities of AmpliSeq in studying genetic alterations such as these [2, 3].
Figure 2.

Mutation detection using formalin-fixed, paraffin-embedded (FFPE) colorectal cancer tumor specimens. Variants were detected by AmpliSeq (A) and SimpliSeq (B) in 44 FFPE genomic DNA samples. The red shade stands for variants with VF >5%; blue shade for variants with VF between 1% and 5%; and green shade for variants within a single gene containing multiple hotspot mutations, with VFs of both 1%–5% and >5%. Gray shade indicates no variants detected. The y-axis indicates the gene name, and the x-axis the identification of the matched pairings, i.e., the microdissected epithelial tumor and stroma tissues. False-negative variants in p53 and PTEN are marked with a circle (B). (C–E): Correlation of high-frequent variants (VF >5%) (C, D) and low-frequent variants (VF 1%–5%) (E) between AmpliSeq and SimpliSeq. Open circles within the marked area represent “true positive” and open circles on the x-axis represent “false positive.” Note that JAK2 was excluded from the above analysis and the outstanding “false-positive genes” (PIK3CA, NRAS, FGFR2) have been removed in (D, E). The percentage of bona fide variants (marked in red) was calculated based on SimpliSeq and AmpliSeq variant calls, and is provided in the upper right corner of each figure (C–E).
Abbreviation: VF, variant frequency.
Variant Validation by Orthogonal Platforms
To assess the potential effects of multiplex PCR-based target enrichment on AmpliSeq results, we selected genes of interest and resequenced them individually by SimpliSeq using amplicons that were individually amplified by nonredundant, target-specific singleplex PCR (Fig. 1). For AmpliSeq variants with VF >5%, SimpliSeq confirmed 68 of 101 (67%) AmpliSeq variant calls as true positives (Fig. 2B, 2C), covering 31 mutational hotspots in 14 genes across 44 FFPE gDNA samples. Notably, 33 of 101 (33%) AmpliSeq variants were identified as false positives, which was exemplified by PIK3CA, NRAS, and FGFR2 when a portion of their AmpliSeq data was evaluated by SimpliSeq (Table 1; supplemental online Tables 4 and 5; data not shown). In a separate confirmatory study of the SimpliSeq results, we achieved 100% accuracy for the PIK3CA, KRAS, and NRAS genes when compared with TaqMan or AS-PCR assays (supplemental online Tables 4, 5, and 6; data not shown), in addition to a complete concordance for KIT variants when compared with Sanger sequencing (supplemental online Table 7). After removal of these systematic “false-positive” genes (VF >5%), the rate of “true-positive” AmpliSeq variant calls increased from 65% to 92% (66/72) (Fig. 2D).
Table 1.
Representative “true-positive” and “false-positive” variant calls (variant frequency >5%) by AmpliSeq, verified by SimpliSeq
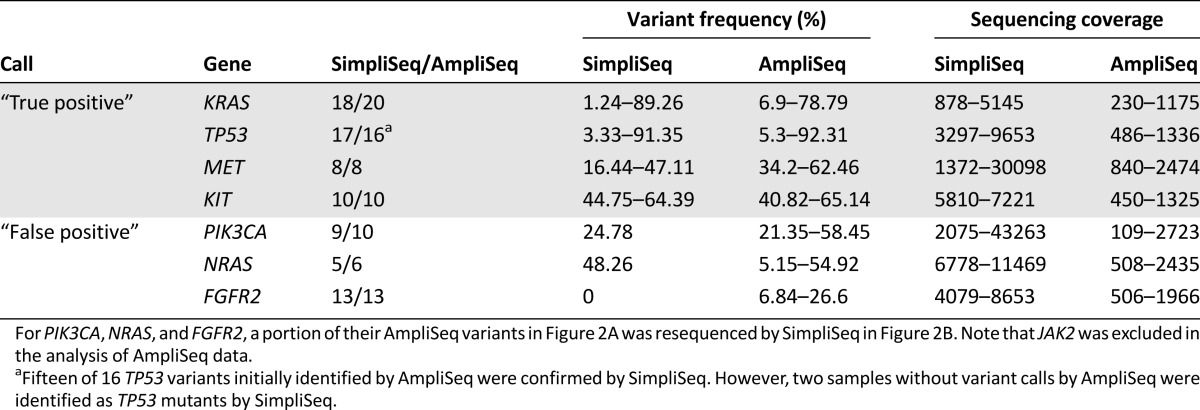
For AmpliSeq variants with low VF (1%–5%), our validation analysis demonstrated that 40 of 48 (83%) variants could not be confirmed and were likely false positives (Fig. 2E). This finding is consistent with the 5% detection threshold recommended for AmpliSeq. Furthermore, SimpliSeq analysis also revealed “false-negative” variant calls in TP53 and PTEN (Fig. 2B), in which 3 of 55 wild-type calls by AmpliSeq were actual TP53 R175H mutants (VF: 1.9% and 3.4%; coverage: 4778 and 5588) and PTEN Q214* mutant (VF: 1.01%; coverage: 6032).
Overall, 86.4% (38/44) of AmpliSeq variants with VF >5% and 100% (30/30) of AmpliSeq variants with VF <5% had at least one false-positive mutation. The read depth may not be an important factor impacting these “false-positive” calls. For instance, the average read depth of false positives was 1411.45 reads per target for variants with low VF (1%–5%) and 1155.74 for all variants (VF 1%–5% and VF > 5%), which is comparable to the average read depth of 1241.5 for all variant calls (“false positives” and “true positives,” VF 1%–5% and VF > 5%). Three types of variants were identified by SimpliSeq analysis: (a) tumor-specific variants (TP53 R273H in sample 40125-E [90.38% VF] vs. TP53 wild type in sample 40125-S); (b) stroma-specific variants (EGFR G810S in sample 41694-S [4.12% VF] vs. EGFR wild type in sample 41694-E); and (c) variants identified in both tumor and stroma samples (including somatic variants such as KRAS G12D in sample 40261-E [76.75% VF] vs. KRAS G12D in sample 40261-S (5.95% VF), which could have been caused by cross-contamination during the microdissection process. Furthermore, variants such as KIT M541L (VF ∼50%) and MET T1010I (VF ∼50%) were also identified in nine matched pairings of tumor and stroma samples, which is in agreement with the previously reported germline variations in human cancers [9, 10].
Genetic Landscape of Variants in 22 FFPE CRC Tumor Specimens
NGS analysis of the variant percentile indicated high-level molecular complexity of CRC at several levels. The number of variants varied considerably, ranging from one to five per CRC specimen, indicating intertumor molecular heterogeneity (Fig. 3A). Analysis of variants revealed considerable intratumor molecular heterogeneity, exemplified by a CRC specimen that carried mutations in TP53 R273H (90%), FBXW7 R465C (80%), SMAD4 R361H (62%), and SMAD4 R497H (13%) (Fig. 3B). Concurrent KRAS mutations with different VFs have been identified in a single CRC specimen, such as the mutually exclusive mutations in KRAS G12A (9%) and G13D (28%) on discrete reads (Fig. 3C), which were also coidentified by AS-PCR assays (supplemental online Table 5). The genomic mutational landscape of the 22 CRC tumor specimens is shown in Figure 3D. As anticipated, the most common mutations in CRC were KRAS (54.5%, 12/22) and TP53 (50%, 11/22), consistent with previous large-scale mutational analyses [11, 12] and the My Cancer Genome database (supplemental online Table 8).
Figure 3.

Genetic landscape of variants detected in CRC tumor specimens. (A): The number of variants verified in each of the 22 CRC tumor specimens. (B): Intratumor molecular heterogeneity revealed by variants with different VF (13%–90%) in a single CRC tumor specimen. (C): Sequencing snapshot of the KRAS mutations in a single CRC tumor specimen. (D): Distribution of gene mutations in 22 CRC tumor specimens.
Abbreviations: CRC, colorectal cancer; VF, variant frequency.
Taken together, the above studies suggest that AmpliSeq provides highly sensitive and quantitative mutation detection for most of the genes on its cancer panel; however, verification testing is critical for low-abundance variants and those genes that are found to have recurrent false-positive variants. As a result of this study, we suggest guidelines for implementing AmpliSeq NGS into routine clinical testing for precision oncology (Fig. 4A). Orthogonal validation is recommended for variants with VF <5% and variants (VF >5%) with recurrent “false positives” (PIK3CA, NRAS, FGFR2, and JAK2) (Fig. 4B).
Figure 4.

Clinical application of AmpliSeq. (A): Implementing AmpliSeq NGS into routine clinical testing for precision oncology. (B): Orthogonal validation is recommended for variants with VF <5% and variants (VF >5%) with recurrent “false positives” (PIK3CA, NRAS, FGFR2, and JAK2).
Abbreviations: gDNA, genomic DNA; NGS, next-generation sequencing; VF, variant frequency.
Discussion
AmpliSeq NGS is poised to enter the clinic as a new tool to advance precision medicine [3, 4]. We report here the mutational analyses of 44 FFPE samples using AmpliSeq for variant identification and SimpliSeq for orthogonal validation. This verification strategy offers nonredundant, target-specific singleplex PCR enrichment and independent NGS data analysis to confirm or deny variants identified in the primary AmpliSeq screen. Using as little as 10 ng of gDNA, AmpliSeq NGS detected variants with VF ranging from 1% to 95%, but at the expense of “false-positive” variant calls, including systematic and idiosyncratic mutation hotspots. In this work, we show the necessity of using orthogonal platforms to validate variant calls and eliminating the recurrent “false positives” from further analysis.
Our study clearly articulates the need for caution in interpreting AmpliSeq data. We found that “false-positive” mutations frequently occur in clinically relevant genes. As promising results have been recently shown in clinical trials using PI3K inhibitors [13], the false-positive PIK3CA variant calls raise serious concerns if physicians use the PIK3CA mutation status generated by AmpliSeq rather than an approved in vitro companion diagnostic assay to guide targeted cancer therapy [14–18]. AmpliSeq’s associated variant caller was also found to be inadequate and led to several “false-negative” gene mutations, and although not seen in this study, a sequencing bias has been reported in AT-rich genomes and long (>14-base) homopolymer tracts on the Ion Torrent PGM [19]. We cannot rule out the possibility that our results may have an overestimation of false-positive events given the caveats associated with small specimen cohorts (44 gDNA samples) and the quality of the FFPE samples, particularly samples with poor amplification.
At least one systematic false-positive error noted in the early stages of this study, JAK2 V617F, was specifically addressed in later versions of the Ion Torrent Server, in which an option was added to “turn off” detection of this variant. Of note, the “turning off” of JAK2 detection may have potentially large implications for the utilization of this platform in myeloproliferative neoplasms. Similar caveats are also associated with the “turning off” of other alleles (PIK3CA, FGFR2, and NRAS) [18, 20]. As each of these loci is a known hotspot, “dropping off” these genes would essentially render the assay incapable of making any calls and lead to the inability of the assay to offer sufficient negative predictive value for these loci. Additional versions of AmpliSeq may be redesigned and/or reformulated to minimize unreliable calls in the genes noted herein.
Interestingly, SimpliSeq validation also confirmed variants found in the stroma samples. This may be due to germline variants (KIT M541L and MET T1010I), resulting in the variants (VF ∼50%) detected in the matched pairings. Some stromal variants might be caused by cross-contamination during the repetitive microdissection procedure, as evidenced by a much lower variant frequency in stroma than in the matched epithelial tumor pairs. In two cases, stroma-specific variants were identified in EGFR (G810D), although the mechanism remains unclear. The current study does not exclude the possibility of stromal variants as reported in literature [21].
Although AmpliSeq NGS was associated with questionable or erroneous calls in such instances, we note that other enrichment methods and sequencing platforms are not immune to error. For example, sequencing errors in GGC sequences have been reported using the Illumina MiSeq [19]. In line with the “false-negative” variant calls in this study, we found that the built-in automatic variant calling was inadequate for AmpliSeq, a common problem shared by the Ion Torrent PGM and Illumina MiSeq platforms [19]. The findings presented here provide the basis for a suggestion that laboratories should strongly consider developing independent bioinformatics pipelines for AmpliSeq data acquisition and analyses.
Targeted NGS evaluates cancer-associated regions with a read depth and sensitivity that offer an unprecedented opportunity to decipher tumor heterogeneity and clinical therapeutic responses. A case in point is the KRAS oncogene, in which mutations in codons 12 and 13 are established biomarkers for lack of clinical benefit with anti-epidermal growth factor receptor therapies (cetuximab and panitumumab) in metastatic CRC. Although uncommon, two concurrent KRAS mutations were identified by AmpliSeq in a single specimen, suggesting intratumor heterogeneity. However, it cannot be determined on the basis of these separate reads whether the mutations are heterogeneously distributed in the same cells or in different subclones. The possibility exists that they may derive from a single clonal population in which the KRAS mutations are in trans. In targeted cancer therapies, the percentage of mutant tumor cells, indicated by the VF, in the drug-targetable mutations may help us to better understand the therapeutic response.
During the past decade, the identification of molecular biomarkers for clinically relevant mutations or other genetic abnormalities in cancer has improved the understanding of cancer pathogenesis and therapeutic response of cancer cells, setting the stage for a paradigm shift toward the adoption of treatments directed to the particular genetic makeup of the tumor [22–25]. With the advent of disruptive sequencing technologies, we envision the implementation of targeted NGS approaches into routine clinical practice, offering insights about oncology biomarkers for patient selection, therapeutic response, and prognostic predication in precision oncology. The findings from this study will inform the clinical application of AmpliSeq and support accurate mutation profiling toward biomarker-driven targeted therapies or investigational agents in clinical trials.
See http://www.TheOncologist.com for supplemental material available online.
Supplementary Material
Acknowledgments
We thank Adam Marko for assistance with NGS analysis, Paul Fielder and Rod Mathews for project support, and Kimberly Walter for critical reading of this article.
Author Contributions
Conception/design: Shidong Jia, Gary J. Latham, Priti Hegde, Liangxuan Zhang
Provision of study material or patients: Shidong Jia, Priti Hegde, Liangxuan Zhang, Gary J. Latham, Rajesh Patel, Rachel Tam, Erica Schleifman
Collection and/or assembly of data: Shidong Jia, Liangxuan Zhang, Liangjing Chen, Sachin Sah, Gary J. Latham, Haider Mashhedi, Sreedevi Chalasani, Ling Fu, Teiko Sumiyoshi
Data analysis and interpretation: Shidong Jia, Liangxuan Zhang, Liangjing Chen, Sachin Sah, Gary J. Latham, Qinghua Song, Hartmut Koeppen, Rajiv Raja, William Forrest, Garret M. Hampton, Mark R. Lackner, Priti Hegde
Manuscript writing: Shidong Jia, Liangxuan Zhang, Gary J. Latham
Final approval of manuscript: Liangxuan Zhang, Liangjing Chen, Sachin Sah, Gary J. Latham, Rajesh Patel, Qinghua Song, Hartmut Koeppen, Rachel Tam, Erica Schleifman, Haider Mashhedi, Sreedevi Chalasani, Ling Fu, Teiko Sumiyoshi, Rajiv Raja, William Forrest, Garret M. Hampton, Mark R. Lackner, Priti Hegde, Shidong Jia
Disclosures
Shidong Jia: Genentech (E); Mark Lackner: Genentech (E); Priti Hegde: Genentech (E); Ling Fu: Genentech (E); Teiko Sumiyoshi: Genentech (E); Nga Wan Rachel Tam: Genentech (E); Haider Mashhedi: Genentech (E); Sreedevi Chalasani: Genentech (E); Erica Schleifman: Genentech (E); Rajesh Patel: Genentech (E); Qinghua Song: Genentech (E); Liangxuan Zhang: Genentech (E); Liangjing Chen: Asuragen (E); Sachin Sah: Asuragen (E); Gary J. Latham: Asuragen (E, OI); Harmut Koeppen: Genentech (E); Rajiv Raja: Genentech (E), Roche (OI); William Forrest: Genentech, Roche (E, OI), Garret Hampton: Genentech, Roche (E, OI).
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1.Meldrum C, Doyle MA, Tothill RW. Next-generation sequencing for cancer diagnostics: A practical perspective. Clin Biochem Rev. 2011;32:177–195. [PMC free article] [PubMed] [Google Scholar]
- 2.Beadling C, Neff TL, Heinrich MC, et al. Combining highly multiplexed PCR with semiconductor-based sequencing for rapid cancer genotyping. J Mol Diagn. 2013;15:171–176. doi: 10.1016/j.jmoldx.2012.09.003. [DOI] [PubMed] [Google Scholar]
- 3.Singh RR, Patel KP, Routbort MJ, et al. Clinical validation of a next-generation sequencing screen for mutational hotspots in 46 cancer-related genes. J Mol Diagn. 2013;15:607–622. doi: 10.1016/j.jmoldx.2013.05.003. [DOI] [PubMed] [Google Scholar]
- 4.Next-generation screening goes national in UK Cancer Discov. 2013;3:OF6. [Google Scholar]
- 5.Jia S, Liu Z, Zhang S, et al. Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature. 2008;454:776–779. doi: 10.1038/nature07091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hadd AG, Houghton J, Choudhary A, et al. Targeted, high-depth, next-generation sequencing of cancer genes in formalin-fixed, paraffin-embedded and fine-needle aspiration tumor specimens. J Mol Diagn. 2013;15:234–247. doi: 10.1016/j.jmoldx.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 7.Patel R, Tsan A, Tam R, et al. Mutation scanning using MUT-MAP, a high-throughput, microfluidic chip-based, multi-analyte panel. PLoS One. 2012;7:e51153. doi: 10.1371/journal.pone.0051153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jia S, Gao X, Lee SH, et al. Opposing effects of androgen deprivation and targeted therapy on prostate cancer prevention. Cancer Discov. 2013;3:44–51. doi: 10.1158/2159-8290.CD-12-0262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tang X, Boxer M, Drummond A, et al. A germline mutation in KIT in familial diffuse cutaneous mastocytosis. J Med Genet. 2004;41:e88. doi: 10.1136/jmg.2003.015156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee JH, Han SU, Cho H, et al. A novel germ line juxtamembrane Met mutation in human gastric cancer. Oncogene. 2000;19:4947–4953. doi: 10.1038/sj.onc.1203874. [DOI] [PubMed] [Google Scholar]
- 11.Cancer Genome Atlas Network Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wood LD, Parsons DW, Jones S, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 13.Rodon J, Dienstmann R, Serra V, et al. Development of PI3K inhibitors: Lessons learned from early clinical trials. Nat Rev Clin Oncol. 2013;10:143–153. doi: 10.1038/nrclinonc.2013.10. [DOI] [PubMed] [Google Scholar]
- 14.Lackner MR. Prospects for personalized medicine with inhibitors targeting the RAS and PI3K pathways. Expert Rev Mol Diagn. 2010;10:75–87. doi: 10.1586/erm.09.78. [DOI] [PubMed] [Google Scholar]
- 15.Jia S, Roberts TM, Zhao JJ. Should individual PI3 kinase isoforms be targeted in cancer? Curr Opin Cell Biol. 2009;21:199–208. doi: 10.1016/j.ceb.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.She QB, Chandarlapaty S, Ye Q, et al. Breast tumor cells with PI3K mutation or HER2 amplification are selectively addicted to Akt signaling. PLoS One. 2008;3:e3065. doi: 10.1371/journal.pone.0003065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spoerke JM, O’Brien C, Huw L, et al. Phosphoinositide 3-kinase (PI3K) pathway alterations are associated with histologic subtypes and are predictive of sensitivity to PI3K inhibitors in lung cancer preclinical models. Clin Cancer Res. 2012;18:6771–6783. doi: 10.1158/1078-0432.CCR-12-2347. [DOI] [PubMed] [Google Scholar]
- 18.Zhang J, Roberts TM, Shivdasani RA. Targeting PI3K signaling as a therapeutic approach for colorectal cancer. Gastroenterology. 2011;141:50–61. doi: 10.1053/j.gastro.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 19.Quail MA, Smith M, Coupland P, et al. A tale of three next generation sequencing platforms: Comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genomics. 2012;13:341. doi: 10.1186/1471-2164-13-341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ilić N, Roberts TM. Comparing the roles of the p110α and p110β isoforms of PI3K in signaling and cancer. Curr Top Microbiol Immunol. 2010;347:55–77. doi: 10.1007/82_2010_63. [DOI] [PubMed] [Google Scholar]
- 21.Kurose K, Gilley K, Matsumoto S, et al. Frequent somatic mutations in PTEN and TP53 are mutually exclusive in the stroma of breast carcinomas. Nat Genet. 2002;32:355–357. doi: 10.1038/ng1013. [DOI] [PubMed] [Google Scholar]
- 22.Gandara DR, Li T, Lara PN, Jr, et al. Algorithm for codevelopment of new drug-predictive biomarker combinations: Accounting for inter- and intrapatient tumor heterogeneity. Clin Lung Cancer. 2012;13:321–325. doi: 10.1016/j.cllc.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Y, Hegde P, Zhang F, et al. Prostate cancer—A biomarker perspective. Front Endocrinol (Lausanne) 2012;3:72. doi: 10.3389/fendo.2012.00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yan M, Liu QQ. Targeted therapy: Tailoring cancer treatment. Chin J Cancer. 2013;32:363–364. doi: 10.5732/cjc.013.10114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aggarwal S. Targeted cancer therapies. Nat Rev Drug Discov. 2010;9:427–428. doi: 10.1038/nrd3186. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.