

This article reviews current management strategies for low-grade gliomas (LGG), including surgery, radiotherapy, and chemotherapy. In addition, the importance of profiling the genetic and molecular properties of LGGs in the development of targeted anticancer therapies is also reviewed. Finally, given the prevalence of these tumors in otherwise healthy young patients, the impact of treatment on neurocognitive function and quality of life is also evaluated.
Keywords: Glioma, Astrocytoma, Oligodendroglioma, Surgery, Radiation
Abstract
Low-grade gliomas (LGGs) are a diverse group of primary brain tumors that often arise in young, otherwise healthy patients and generally have an indolent course with longer-term survival in comparison with high-grade gliomas. Treatment options include observation, surgery, radiation, chemotherapy, or a combined approach, and management is individualized based on tumor location, histology, molecular profile, and patient characteristics. Moreover, in this type of brain tumor with a relatively good prognosis and prolonged survival, the potential benefits of treatment must be carefully weighed against potential treatment-related risks.
We review in this article current management strategies for LGG, including surgery, radiotherapy, and chemotherapy. In addition, the importance of profiling the genetic and molecular properties of LGGs in the development of targeted anticancer therapies is also reviewed. Finally, given the prevalence of these tumors in otherwise healthy young patients, the impact of treatment on neurocognitive function and quality of life is also evaluated.
Implications for Practice:
This review summarizes the epidemiology, presentation, diagnosis, and treatment of low-grade gliomas. The article discusses recent advances in genetic characterization of these tumors, which has become particularly important in guiding tumor identification and classification, and may in some cases offer information about prognosis and/or expected response to treatment. The major studies regarding the use of radiation, chemotherapy, and surgical approaches in the treatment of these tumors are discussed. This information will aid medical oncologists in understanding the challenges inherent in diagnosing and treating patients with low-grade gliomas, and in recognizing the important factors to consider in devising treatment plans.
Introduction
Central nervous system tumors are defined by their cell of origin and their histopathological characteristics, which predict their behavior [1]. Gliomas are neuroepithelial tumors originating from the supporting glial cells of the central nervous system (CNS). Glial tumors consist of astrocytomas, oligodendrogliomas, mixed oligo-astrocytic, and mixed glioneuronal tumors, which arise from astrocytic, oligodendroglial, mixed oligoastrocytic, or neuronal-glial cells, respectively. The World Health Organization (WHO) classification system categorizes gliomas from grade 1 (lowest grade) through grade 4 (highest grade), based upon histopathologic characteristics such as cytological atypia, anaplasia, mitotic activity, microvascular proliferation, and necrosis. Low-grade gliomas (LGGs) consist of grade I tumors, which contain none of the aforementioned histologic features, and grade II tumors, characterized by the presence of cytologic atypia alone [1]. Low-grade astrocytic tumors include diffuse astrocytoma, pilomyxoid astrocytoma, and pleomorphic xanthoastrocytoma (WHO grade II), as well as subependymal giant cell astrocytoma (SEGA) and pilocytic astrocytoma (WHO grade I tumors). Low-grade oligodendroglial tumors include oligodendrogliomas and oligoastrocytomas (WHO grade II tumors) [1]. Low-grade glioneuronal tumors include the following WHO grade I tumors: ganglioglioma, desmoplastic infantile astrocytoma and ganglioglioma, dysembryoplastic neuroepithelial tumor, papillary glioneuronal tumor, and rosette-forming glioneuronal tumor of the fourth ventricle [1]. In this review, we discuss the epidemiology, clinical, and diagnostic characteristics, histopathologic and molecular features, prognosis, and treatment of LGG. For the purposes of this review, we will focus on supratentorial nonpilocytic astrocytomas, oligodendrogliomas, and oligoastrocytomas. Selected other LGG subtypes, including subependymal giant cell astrocytoma (SEGA), pleomorphic xanthoastrocytoma, brainstem glioma, and pilocytic astrocytoma, will be discussed briefly.
Epidemiology
In 2012, more than 66,000 primary CNS tumors were diagnosed in the U.S., 30% (approximately 20,000) of which were gliomas [2]. In young adults (20–34 years of age), gliomas account for 32% of all primary CNS tumors, 17% of which are astrocytic tumors; 28% of these are glioblastomas [2]. Available data do not separate high-grade versus low-grade tumors; thus, the annual incidence of LGG is difficult to determine. Incidence rates for oligodendrogliomas, anaplastic astrocytomas, glioblastomas, and mixed gliomas are more than two times higher in whites than in blacks [2]. The reason for this racial discrepancy is uncertain. It may represent detection bias, a genetic difference, or another as yet unidentified explanation. Various environmental risk factors have been examined for evidence of a link between environmental exposures and an increased risk of brain tumor formation. The only factor definitively shown to be correlated with an increased risk of secondary brain tumors is CNS exposure to therapeutic or high-dose radiation [3]. Other environmental exposures have been investigated, without compelling evidence to support their role in brain tumor formation. Numerous genetic mutations conferring increased glioma risk have been described, including NF1 and NF2 mutations in neurofibromatosis types 1 and 2, respectively; TSC1 and TSC2 mutations in tuberous sclerosis; TP53 mutations in the Li-Fraumeni syndrome; and a number of gene mutations associated with Turcot’s syndrome and multiple hamartomas, including APC, hMLH1, hMLH2, PMS2, and PTEN mutations [3]. However, these genetic conditions are found in only a very small percentage of patients diagnosed with LGG each year in the U.S.
Presentation
LGGs present most commonly in the second through fourth decades of life, with peak incidence in the third and fourth decades of life. Clinical signs and symptoms vary and are largely attributed to mass effect from invasion into surrounding parenchyma or obstructive hydrocephalus [4]. Seizure is the presenting symptom in up to 80% of patients [4]. Others may present with cognitive or behavioral changes, focal neurologic deficits, or clinical signs or symptoms of increased intracranial pressure, such as headache or papilledema. However, patients may also be asymptomatic, without evident abnormalities on neurologic examination.
Diagnosis
Diagnosis of LGGs is made through a combination of imaging, histopathology, and molecular diagnostic methods. On computed tomography scan, low-grade gliomas appear as diffuse areas of low attenuation. On conventional magnetic resonance imaging (MRI), which is currently the imaging modality of choice, LGGs are often homogeneous with low signal intensity on T1-weighted sequences and hyperintensity on T2-weighted and Fluid-Attenuated Inversion Recovery (FLAIR) sequences (Fig. 1). Calcifications may be evident as areas of T2 hyperintensity/T1 hypointensity in up to 20% of lesions, including oligodendrogliomas and astrocytomas, and are particularly suggestive of oligodendrogliomas [5]. Gliomas, in general, infiltrate the surrounding parenchyma despite apparent radiographic margins observed on T2/FLAIR sequences [5, 6]. Contrast enhancement, if present, is minimal, and is more likely to be seen with oligodendrogliomas [5]. Although contrast enhancement has been classically associated with a higher degree of malignancy, some degree of contrast enhancement may be seen in up to 60% of LGG [4]. LGGs differ from grade III and IV gliomas, as the latter often demonstrate a higher degree of tumor heterogeneity and contrast enhancement, restricted diffusion on diffusion-weighted imaging magnetic resonance (MR) sequences, and increased relative cerebral blood volume on perfusion-weighted MRI [7, 8]. Despite characteristic radiographic findings, tumor grade cannot be determined by imaging alone. Newer imaging techniques, such as MR spectroscopy (MRS) and positron emission tomography (PET) imaging, may improve the diagnostic potential; however, at this time, histopathologic examination of tissue remains the gold standard for diagnosis and grading of LGG.
Figure 1.

Imaging features of low-grade glioma. The grade 2 oligoastrocytoma pictured in these magnetic resonance images appears as a relatively homogeneous region of high signal intensity on T2/FLAIR-weighted images (A) and low signal intensity on T1 precontrast images (B). There is faint contrast enhancement on the T1 postcontrast images (C).
Surgery
The main goal of surgery is to obtain pathological diagnosis and, when feasible, to achieve a gross total resection. Advances such as preoperative functional MRI and tractography, as well as intraoperative neurophysiological monitoring, allow surgeons to safely maximize resection of T2/FLAIR abnormalities on MRI often involving eloquent areas. Patients with tumors that cannot be safely resected, or who have lesions of uncertain etiology, may undergo stereotactic biopsy using preoperative or intraoperative MRI imaging to obtain tissue for histopathological analysis. Surgeons target the potentially higher grade component of the lesion (for example, contrast enhancement) for biopsy. The yield of such biopsies is as high as 90%–95%; however, because of the potential heterogeneity of these tumors, biopsy may not reflect the highest grade for diagnosis, with reported accuracy rates ranging from 51% to 83% [4].
Histopathology
The tissue sample is stained using hematoxylin and eosin, which allows for identification and classification of tumor type. Diffuse astrocytomas consist of well-differentiated fibrillary or gemistocytic neoplastic astrocytes on a loose matrix. Oligoastrocytomas are diffusely infiltrating tumors with a mixture of oligodendroglial and astrocytic cell types (Fig. 2) [1]. Oligodendrogliomas are infiltrating tumors containing cells with uniform-appearing nuclei and perinuclear clearing, often described as having a “fried egg” appearance.
Figure 2.

Histopathologic features of low-grade glioma. (A): Oligodendrogliomas are characterized by uniform-appearing, infiltrating cells with perinuclear clearing in a honeycomb pattern. Hematoxylin and eosin stain. Magnification, 400×. (B): Astrocytoma, consisting of fibrillary neoplastic astrocytes on a loose tumor matrix background. Hematoxylin and eosin stain. Magnification, 400×. (C): Oligoastrocytoma, containing a mixture of both tumor cell types. Hematoxylin and eosin stain. Magnification, 400×. Images courtesy of Declan McGuone, neuropathology fellow, Department of Neuropathology, Massachusetts General Hospital.
Molecular Pathology
In the last decade, genetic characterization has become paramount in tumor identification and classification and is often predictive of tumor behavior, by providing information about prognosis and/or expected response to treatment. Deletion of selected regions on chromosomes 1p and 19q is of particular importance in low-grade gliomas, as it has a strong association with the oligodendroglioma tumor subtype. Loss of the 1p36 region has been noted in 18% of astrocytomas and 73% of oligodendrogliomas; loss of the 19q13.3 region is described in 38% of astrocytomas and 73% of oligodendrogliomas [9]. The 1p36 and 19q13.3 regions are codeleted in 11% of astrocytomas and 64% of oligodendrogliomas [9].
Isocitrate dehydrogenase 1 and 2 gene mutations (IDH1 and IDH2) have been reported in LGGs. Mutations in amino acid 132 of the IDH1 gene are observed in the majority (>70%) of WHO grade II and III astrocytomas and oligodendrogliomas, as well as in secondary glioblastomas originating from these lesions [10, 11]. In contrast, IDH2 mutations are rare, found in only about 6% of LGG [12]. Molecular diagnostic methods are typically used to identify the presence of such mutations following tumor tissue sampling (via biopsy or resection). There are also emerging noninvasive methods of detecting such mutations. The commonly identified arginine 132 (R132) mutation in IDH1 results in excess production of the 2-hydroxyglutarate metabolite, which can be detected using optimized in vivo spectral editing and one- and two-dimensional brain MRS (Fig. 3) [11].
Figure 3.

Noninvasive detection of genetic mutations. One-dimensional magnetic resonance spectroscopy with use of specialized spectral-editing sequence for the detection of 2HG as demonstrated in a secondary glioblastoma patient with IDH1R132H mutation (A), in comparison with the spectra from a healthy volunteer with wt IDH1 (B). Figure reprinted (adapted) with permission from Andronesi OC, Kim GS, Gerstner E et al. Detection of 2HG in IDH-mutated glioma patients by in vivo spectral editing and two-dimensional correlation magnetic resonance spectroscopy. Sci Transl Med 2012;4:116ra4.
Abbreviations: a.u., arbitrary units; GABA, γ-aminobutyric acid; Gln, glutamine; Glu, glutamate; 2HG, 2-hydroxyglutarate; I, signal intensity; MM, denotes contamination of GABA signal with macromolecule signal; wt, wild-type.
There has been recent interest in the role of the CIC gene, a homolog of the Drosophila gene capicua, located on chromosome 19q. Mutations in this gene have been associated with oligodendroglioma and oligoastrocytomas [13, 14]. CIC gene mutations were observed in 69% of oligodendrogliomas in one series and have been described as highly associated with isocitrate dehydrogenase 1 and 2 (IDH1 and IDH2) mutations and 1p/19q codeletion, with clinical significance as yet unclear [15]. Mutations in the FUBP1 gene on chromosome 1p have also been associated with oligodendrogliomas [13].
Mutation and overexpression of tumor protein 53 (TP53) are said to be the “genetic hallmark” of low-grade astrocytomas (>60%), particularly gemistocytic astrocytomas, of which >80% carry a TP53 mutation [1, 16].
In cases in which pathologic diagnosis is difficult, such as distinguishing between gliosis and tumor, the presence of TP53 and IDH1 expression by immunohistochemistry can be very helpful in establishing the diagnosis of tumor.
Prognosis
Immunohistochemical markers of tumor cell proliferation, molecular and genetic characteristics of the tumor cells, and patient characteristics can all be useful in predicting tumor behavior, response to treatment, and patient prognosis.
Proliferative Indices
Immunohistochemical labeling of Ki-67 or MIB-1 monoclonal antibodies against the Ki-67 nuclear proliferation-related protein is used to evaluate the mitotic activity of the glioma cells. In LGG, these indices are generally low, suggesting low mitotic activity: <4% in diffuse astrocytoma, 3%–5% in oligodendrogliomas, and <6% in oligoastrocytomas [17]. Higher mitotic indices by immunohistochemistry are typically associated with more aggressive LGG behavior.
Molecular Pathology
The molecular pathology of LGG is playing an increasingly important role in the prediction of tumor response to treatment and prognosis. The 1p-19q codeletion has been identified as a significant marker of prolonged survival in oligodendroglioma, regardless of tumor grade; such favorable association between 1p-19q status and prognosis was not demonstrated in patients with astrocytoma or oligoastrocytoma [18]. IDH1 and IDH2 gene mutations are also associated with prolonged survival and enhanced sensitivity to treatment. In a study of 132 LGG patients with IDH gene mutations, the mutations were associated with prolonged overall survival [12]. The authors also demonstrated a significant increase in response to the oral alkylating agent temozolomide in the patients with IDH-mutated tumors [12]. The DNA repair protein O6-methylguanine-methyltransferase (MGMT) has also been shown to play a key role in treatment-related prognosis, as this protein confers some degree of resistance to alkylating agents [19]. Methylation of the MGMT promoter, which thereby silences the gene, is associated with improved response to treatment and prolonged progression-free survival in temozolomide-treated patients [12, 19]. Combinations of two or more of these molecular aberrations also have significant prognostic power. There is longer survival in patients with combined IDH mutant/MGMT methylation status versus patients with IDH wild-type tumors and even more favorable prognosis in those patients with 1p-19q codeletion [20]. The prognostic significance of isolated TP53 mutation and overexpression is not well-established, although some studies have suggested its role as a poor prognostic marker with respect to survival [16, 17]. The combination of nuclear TP53 immunopositivity with IDH gene mutation and MGMT methylation is associated with a significant risk of malignant transformation [20]. With all of these molecular markers, some question remains as to whether they are truly prognostic indicators on their own, or are merely predictors of survival in the setting of chemotherapy and/or radiation treatment regimens [4, 21].
Additional Prognostic Factors
Several studies have identified patient and tumor characteristics that together portend poor outcomes. The European Organisation for Research and Treatment of Cancer (EORTC) conducted a multivariate analysis of prognostic factors and created a prognostic scoring system based on characteristics of LGG patients enrolled in the EORTC 22844 and 22845 trials. The investigators identified the following poor prognostic indicators in patients with LGG: age ≥40, astrocytic tumor type (vs. oligodendroglioma or oligo-dominant), tumor size ≥6 cm, tumor crossing the midline, and presence of neurologic deficit(s) at diagnosis (before surgery) [17, 22, 23].
Treatment
There are significant challenges in designing and evaluating therapeutic trials for LGG treatment. Some limitations of these studies include the incorporation of multiple histological types of LGG without distinguishing between subtypes, lack of molecular diagnostics in several studies, absence of consensus on the definition of radiographic response, failure to account for the possibility of pseudoprogression in patients treated with radiotherapy, and limited incorporation of measures regarding quality of life (QoL), neurocognitive outcomes, and neurotoxicity. A summary of LGG treatment modalities is provided in Table 1.
Table 1.
Treatment of low-grade gliomas
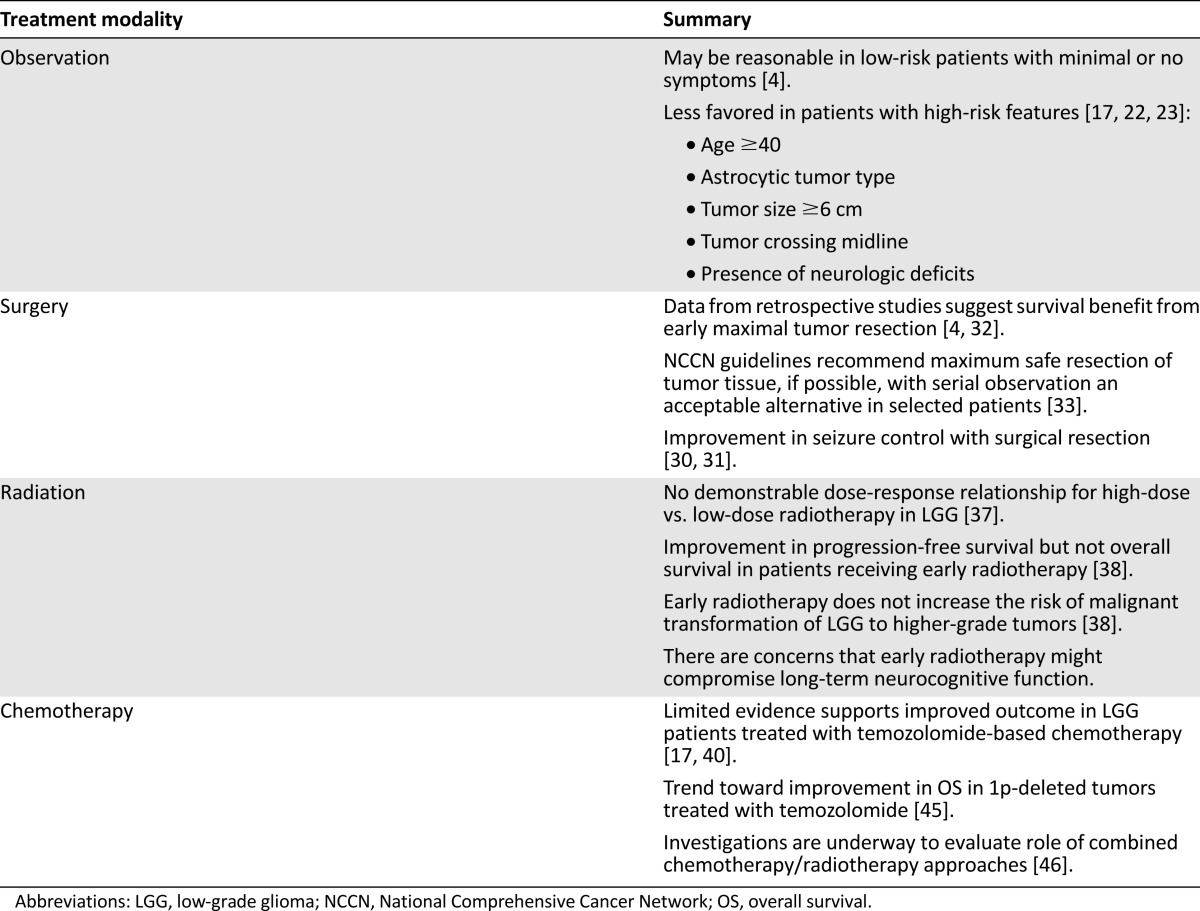
Surgery
Increasingly, studies have supported surgical resection rather than observation to improve overall survival [24, 25]. Additionally, some studies suggest a benefit of extent of resection on progression-free survival [26–29]. Whether gliomas are incidentally found or symptomatic, surgery has been reported to improve seizure control [30, 31].
In one review of the surgical management of LGG, the authors noted the historical arguments in favor of watchful waiting in selected patients with minimal or medically controlled symptoms, with one of the primary arguments based on data suggesting that such an approach did not worsen patients’ QoL, nor did it negatively impact overall survival, although the value of such data is limited by its retrospective nature [4]. Of nine retrospective surgical studies, six demonstrated significant overall survival benefit with extensive surgical resection. Two prospective trials evaluating resection and postoperative radiation therapy demonstrated a significant survival benefit with more aggressive resection on univariate analysis, but not on multivariate analysis. These studies are limited by unblinded assessment of resection (i.e., in many cases, the surgeon determined the extent of resection), as well as patient and treatment selection biases [4]. In another review, the authors examined all major publications since 1990 addressing the effect of extent of surgical resection on glioma outcome. They concluded that there was a trend toward improvement in survival with more extensive surgical resection. In univariate and multivariate analyses of these LGG studies, they noted that extent of resection had significant prognostic value in 7 of the 10 studies [32].
In one review of the surgical management of LGG, the authors noted the historical arguments in favor of watchful waiting in selected patients with minimal or medically controlled symptoms, with one of the primary arguments based on data suggesting that such an approach did not worsen patients’ QoL, nor did it negatively impact overall survival, although the value of such data is limited by its retrospective nature.
The National Comprehensive Cancer Network guidelines for the management of low-grade infiltrative supratentorial astrocytoma/oligodendroglioma in adult patients recommend maximum safe resection of tumor tissue, if possible, with the caveat that serial observation may be appropriate for selected patients [33].
A number of surgical advances have allowed for improvement in the surgeon’s ability to maximize the degree of tumor resection, while sparing eloquent brain. The use of functional MRI and magnetic source imaging allows the surgeon to map functional brain areas such as motor and language cortices, in relation to the tumor [34]. Diffusion tensor imaging may be helpful to identify functional anatomical tracts that are as important as the functioning areas themselves; this technique allows for careful surgical planning to minimize risk of deficits and distinguish between tumor cells and peritumoral edema [7]. Intraoperative MRI and MRS may be used to evaluate the degree of tumor resection during the surgical procedure and more clearly identify residual tumor [35, 36].
Radiation
Several prospective clinical trials have examined the utility of high-dose versus low-dose radiation and the costs versus benefits of early versus delayed radiotherapy. In EORTC 22844, investigators assessed the overall effectiveness of radiotherapy and the potential of a dose-response relationship. A total of 379 adult patients with LGG was randomized to receive radiotherapy postoperatively (or postbiopsy) with 45 Gy in 5 weeks versus 59.4 Gy in 6.6 weeks. At a median follow-up of 74 months, there was no significant difference in overall survival (58% in the low-dose group and 59% in the high-dose group) or progression-free survival (47% in the low-dose group and 50% in the high-dose group), and there was no demonstrable dose-response relationship for radiotherapy in LGG [37].
Similar results were observed in the North Central Cancer Treatment Group/Radiation Therapy Oncology Group (RTOG)/Eastern Cooperative Oncology Group (ECOG) study, in which survival and toxicity were evaluated in low- and high-dose radiation arms. In this study, 203 patients with supratentorial LGG from 1986 to 1994 were randomized to either the low-dose (50.4 Gy in 28 fractions) or high-dose (64.8 Gy in 36 fractions) treatment group. There was no advantage of higher-dose radiation therapy observed in this study. In fact, there was a trend toward improved survival at 2 and 5 years with low-dose therapy (2- and 5-year survival of 94% and 72%, respectively, in comparison with 85% and 64% with high-dose therapy). However, this difference did not reach statistical significance. In addition, there was a higher incidence of radiation neurotoxicity (radiation necrosis) in the high-dose radiotherapy group (5% vs. 2.5% in the low-dose radiotherapy group) [28].
Timing of radiotherapy was addressed in the EORTC 22845 study, in which early versus delayed radiotherapy was assessed. This study, initiated in 1986, included 314 patients with LGG from 24 European centers, who were randomized to early postoperative radiotherapy versus deferred radiotherapy (postponed until the time of disease progression). In this study, there was a significant improvement in progression-free survival in the early radiotherapy group, with a median progression-free survival of 5.3 years in the early radiotherapy group versus 3.4 years in the delayed radiotherapy group. There was no significant difference, however, in overall survival, which was 7.4 years in the early group versus 7.2 years in the delayed group (p = .872) [38]. In the EORTC 22845 study, it was also noted that early radiation did not increase the risk of malignant transformation of LGG to higher-grade tumors. The investigators noted that in study patients undergoing a second surgery at the time of tumor recurrence, the proportion of subjects diagnosed with high-grade glioma was no different in the early versus delayed radiotherapy groups [38]. Quality of life measures were not evaluated in the EORTC 22845 study; thus, the clinical significance of the improvement in progression-free survival in the early radiotherapy group remains uncertain [38].
Newer radiation techniques allow for more precisely directed radiation, improving the ability to target the tumor while sparing healthy surrounding brain tissue, thus minimizing radiation toxicity. Examples include intensity-modulated radiation therapy and stereotactic radiosurgery. However, at this point, neither of these techniques has demonstrated superior efficacy in the treatment of LGG. Proton radiation is another technique that allows for targeted radiation with sparing of surrounding tissues. There is an ongoing trial investigating the efficacy of proton radiation therapy on patients with LGG (ClinicalTrials.gov Identifier: NCT01358058). In this trial, the effectiveness of proton therapy with a more restricted radiation field will be compared with photon therapy, with particular attention paid to treatment-related side effects [39]. As this is a noncomparative, single arm trial, it will not directly address the efficacy and toxicity of proton radiation in comparison with conventional radiotherapy; this question could ideally be addressed by future investigational studies.
Chemotherapy
Although chemotherapy is often used in high-grade gliomas, its role, with or without radiation therapy, in the treatment of patients with LGG remains a topic of investigation. Given its demonstrated efficacy in the high-grade glioma population, temozolomide has been of particular interest. In one phase II study, there was demonstrated activity of temozolomide in LGG patients with progressive disease. In this cohort of 46 patients, 61% of subjects achieved radiographic responses—24% having achieved complete response and 37% having achieved partial response. The median progression-free survival (PFS) was 22 months, with a 6-month PFS of 98% and a 12-month PFS of 76% [40].
In a review of 7 trials evaluating postoperative temozolomide in LGG, with or without prior chemotherapy and/or radiation therapy, the authors concluded that, whereas these trials demonstrated tumor “shrinkage” in response to temozolomide, it is unclear whether this shrinkage is adequate to meet formal criteria for a partial response to treatment [17].
The RTOG 9802 trial assessed treatment of LGG patients with radiation alone versus radiation followed by 6 weeks of chemotherapy with procarbazine, lomustine, and vincristine (PCV). The study enrolled 251 patients from 1998 to 2002, and there was a statistically significant improvement in progression-free survival, but not overall survival, in the radiotherapy plus PCV group. In the first 2 years of treatment, overall survival and progression-free survival were similar. The utility of this analysis was limited by the short 2-year follow-up time before data analysis. However, subsequent analysis with longer follow-up demonstrated possible delayed benefits of chemotherapy; 2-year survivors in the PCV treatment arm had a significantly increased likelihood of surviving an additional 3 years and 5 years versus nonchemotherapy recipients [41].
The RTOG 0424 trial (ClinicalTrials.gov Identifier: NCT00114140) was a phase II study of temozolomide-based chemotherapy in high-risk low-grade glioma, which compared 3-year survival of patients with high-risk LGG treated with temozolomide alone versus those enrolled in EORTC 22844 and 22845. In this nonrandomized, multicenter study, patients received concurrent radiation and daily temozolomide for 6 weeks, followed by postradiation temozolomide for up to 12 additional months. Three-year survival was assessed and compared with patients enrolled on clinical trials EORTC 22844 and EORTC 22845. Additional primary outcome measures include PFS, toxicity, association of survival and progression-free survival with MGMT methylation status, quality of life, and neurocognitive function [42]. Patients enrolled in the RTOG 0424 trial had a 3-year overall survival rate of 73.1%, which exceeded that of historical controls [43]. This noncontrolled study is limited by its reliance upon a comparison group using data from trials that had been conducted 20 years earlier.
The EORTC 22033-26033 trial (ClinicalTrials.gov Identifier: NCT00182819) was a phase III randomized, multicenter study comparing progression-free survival of patients with LGG treated with radiotherapy versus temozolomide. In this study, patients were stratified according to participating center, chromosome 1p status (deleted vs. normal vs. undeterminable), contrast enhancement on MRI (yes vs. no), age (<40 years vs. ≥40 years), and WHO performance status (0 or 1 vs. 2). Subjects were then randomized to one of two treatment arms—a radiotherapy group, which underwent radiotherapy 5 days per week for a total of 28 fractions, and a chemotherapy arm, in which patients received oral temozolomide daily for 21 days of each 28-day cycle, for up to 12 treatment cycles. Outcome measures included progression-free survival, overall survival, and quality of life [44]. In the overall study population, there was no difference in progression-free survival or overall survival between the two groups. In patients with chromosome 1p maintained who received temozolomide, there was a trend toward inferior PFS. In those patients with chromosome 1p deleted who were treated with temozolomide, there was a trend toward improvement in OS [45]. Further follow-up is required before the final results of this trial can be assessed.
There are other ongoing clinical trials that are seeking to further define the ideal treatment regimen for patients with LGG, including ECOG-E3F05 (ClinicalTrials.gov Identifier: NCT00978458), a phase III randomized study of radiotherapy with or without temozolomide in patients with symptomatic or progressive LGG. The primary objectives of this study are to determine whether the addition of temozolomide to fractionated radiotherapy improves progression-free survival and/or median overall survival. This study is currently recruiting participants [46].
Monitoring Response to Treatment
The optimal method of assessing treatment response in LGG remains an active area of investigation. Currently, MRI (T2/FLAIR sequence), with or without contrast enhancement, is used to identify tumor size and associated peritumoral edema. Some authors suggest that treatment outcomes might be more reliably evaluated using advanced imaging techniques designed to assess specific biological aspects of the tumor, including amino acid PET, MRS, and/or cerebral blood volume assessment with perfusion-weighted MRI [47]. However, none of these alternative imaging markers have been validated for use in LGG clinical trials or in clinical practice.
In addition, the challenges for assessing tumor response as described by Macdonald et al. in 1990 have been highlighted, including the use of cross-sectional rather than volumetric area to measure tumor size, failure to account for neurologic deterioration or increasing steroid usage in assessing disease status, and limitations of the imaging itself, including difficulty distinguishing between tumor borders and new lesions in gliomas, which often have satellite lesions, as well as the challenge of identifying tumor mimics such as pseudoprogression, in which increased contrast enhancement in response to treatment does not equate to true tumor progression [6, 48, 49].
The Response Assessment in Neuro -Oncology defines a set of criteria for assessing outcome in trials of diffuse LGG. This includes specific guidelines for using tumor size and appearance on T2/FLAIR MRI sequences to define complete response, partial response, and minor response to treatment, as well as stable disease and progression. The criteria take into account stability of corticosteroid dosing, clinical status, and differentiation between new T2 or FLAIR abnormalities related to tumor spread in comparison with those attributable to radiation effects [6]. These consensus guidelines await validation in future randomized studies.
Treatment-Related Complications
An important consideration in determining the optimal treatment approach in patients with LGG is weighing the potential benefits of various treatment regimens against treatment-related side effects, which may limit treatment intensity and/or duration and have a significant impact on the patient’s quality of life. For example, neurosurgeons plan surgical approaches to maximize resection (when feasible) while minimizing neurological deficit.
Of particular concern in brain-directed treatment is the cognitive impact on patients, particularly related to radiation therapy. Although radiation therapy may prolong progression-free survival, this may come at some cost of cognitive performance. This issue was addressed in a retrospective evaluation of LGG patients who had received radiotherapy in comparison with those who had not received radiotherapy. This study was a follow-up of long-term survivors (mean of 12 years) from an earlier study on cognitive outcomes with radiation therapy. The original study had assessed 195 patients with LGG at a mean of 6 years from diagnosis, and it was concluded that LGG patients had worse cognitive performance when compared with healthy controls or patients with hematologic malignancies, regardless of radiation treatment status [50]. Within the LGG patients, there was a nonsignificant trend toward inferior cognitive functioning in patients who received radiotherapy versus those who did not (p = .145). The authors determined that the tumor itself, not the radiation treatment, had the most significant adverse impact on cognitive functioning, although high-dose radiation (>2 Gy) had an additional significant impact on cognitive decline [50, 51]. In the follow-up study, 65 long-term survivors were evaluated at a mean of 12 years from the time of diagnosis; 32 of these patients received radiation therapy. The authors reported significant cognitive deficits in patients who had received prior radiation (17 patients; 53%) versus those who did not receive radiation therapy (4 patients; 27%). These deficits were evident in at least 5 of 18 neuropsychological test parameters. The authors concluded that with long-term follow-up, radiation doses less than 2 Gy caused significant long-term cognitive deficits in patients who underwent radiotherapy [50, 51]. The EORTC 22844 study included a 47-item QoL questionnaire evaluating psychological, physical, social, and symptom domains over time. The authors concluded that, whereas no major differences in QoL between the high-dose versus low-dose radiation groups were evident, significantly higher levels of fatigue/malaise and insomnia immediately after radiation therapy in the high-dose group were detected. In addition, the high-dose group experienced worse emotional functioning and reported decreased leisure time, with these effects persisting for 7–15 months after randomization [52].
Of particular concern in brain-directed treatment is the cognitive impact on patients, particularly related to radiation therapy. Although radiation therapy may prolong progression-free survival, this may come at some cost of cognitive performance.
In a study of LGG patients receiving temozolomide, patients were assessed with respect to QoL measures before chemotherapy and at 2-month intervals while on temozolomide therapy. In comparison with a normal population, patients with LGG had lower social and emotional well-being. There were no significant changes in QoL scores with each chemotherapy cycle in comparison with baseline scores, and some scores improved with treatment (p = .01) [53].
In addition to the effects of treatment described in these QoL studies, there are medication-specific side effects, which play a role in the patient’s quality of life and ability to tolerate ongoing chemotherapy administration. Temozolomide may cause anorexia, nausea, vomiting, fatigue, and hematologic toxicity, including leukopenia and thrombocytopenia. These side effects may negatively impact QoL for this patient population.
Special Topics—Selected LGG Subtypes
Brainstem Glioma
Most prevalent in children, brainstem gliomas are a heterogeneous group of tumors, which includes diffuse intrinsic pontine glioma, exophytic medullary glioma, and tectal gliomas (Fig. 4) [54]. These tumors are often diffusely infiltrative and may have cystic components. Patients often present with hydrocephalus and signs of elevated intracranial pressure (e.g., vomiting, headaches), ataxia, cranial nerve abnormalities, or other signs of brainstem dysfunction. Diffuse intrinsic pontine glioma has a median age of onset of 6.5 years and an extremely poor prognosis, with median survival of less than 1 year, although adult patients with diffuse intrinsic pontine glioma tend to have longer survival [54]. Treatment of these tumors generally involves radiation with or without chemotherapy. Surgery is not feasible in most patients because of the location of the tumors. Exophytic medullary glioma and tectal gliomas are more indolent forms of brainstem gliomas with better prognoses and may be managed conservatively with serial radiographic and clinical evaluation [54].
Figure 4.
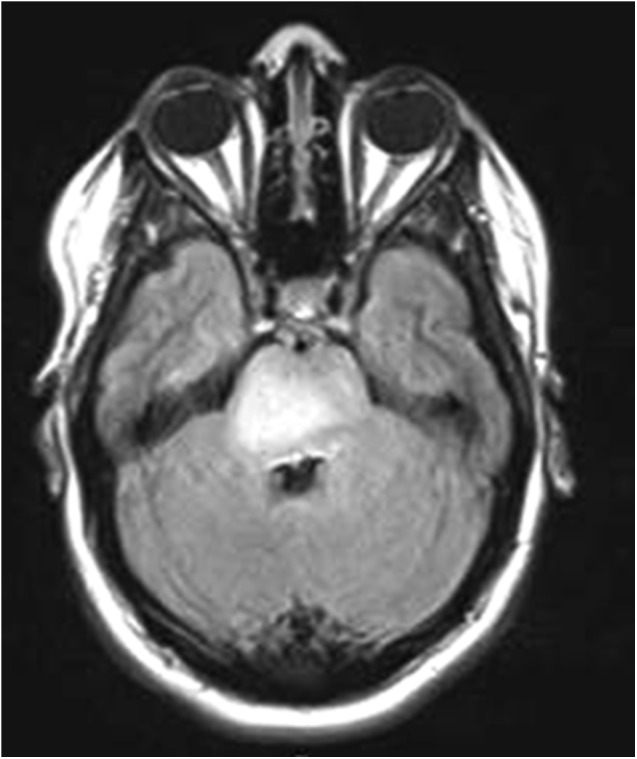
Brainstem glioma. The diffuse pontine glioma pictured in this T2/FLAIR-weighted magnetic resonance image appears as an expansile, hyperintense lesion centered within the pons.
Pilocytic Astrocytoma
Pilocytic astrocytomas (PAs) are WHO grade I, well-circumscribed, indolent, often cystic tumors that represent 5%–6% of all gliomas (Fig. 5) [1]. Genetic abnormalities implicated in pilocytic astrocytoma formation include tandem duplication of the BRAF proto-oncogene locus at 7q34, as well as activating point mutations of BRAF, including BRAF V600E mutations [55]. PAs are the most common glioma subtype in children, commonly referred to as juvenile pilocytic astrocytoma, and are most often located in the cerebellum. Although PAs are more often found in an infratentorial location in children, they may also be observed in either children or adults in the optic nerves, optic chiasm, hypothalamus, thalamus/basal ganglia, or cerebral hemispheres, with presenting symptoms and signs depending largely on tumor location [1]. PAs may spread into the subarachnoid space and/or periventricular spaces and may disseminate along the craniospinal axis [1]. Treatment typically consists of surgical resection, with long-term survival possible after gross total resection; in cases of residual postoperative tumor, chemotherapy and/or radiotherapy might be used as adjuvant treatment [56].
Figure 5.

Juvenile pilocytic astrocytoma. The juvenile pilocytic astrocytoma pictured in these magnetic resonance images appears as a mainly cystic neoplasm centered in the cerebellar vermis. There is a dominant cyst and smaller mural solid component, as seen on T1 precontrast images (A), with small areas of nodular enhancement within the mural component, as seen on T1 postcontrast images (B). The tumor fills and obstructs the fourth ventricle, causing noncommunicating hydrocephalus.
Subependymal Giant Cell Astrocytoma
Subependymal giant cell astrocytomas (SEGAs) are benign, indolent, well-circumscribed, and often calcified tumors, generally arising from the wall of the lateral ventricles [1]. These tumors are associated with tuberous sclerosis complex (TSC), an autosomal dominant neurocutaneous syndrome whose characteristic features include cognitive impairment, cutaneous angiofibromas, cardiac rhabdomyomas, and renal angiomyolipomas [1]. These WHO grade I tumors often present in the first two decades of life with seizures or with signs of increased intracranial pressure [1]. Treatment may include surgery or observation. Everolimus, an inhibitor of the mammalian target of rapamycin (mTOR) complex, was studied in a nonrandomized trial of TSC patients with subependymal giant cell astrocytomas. In TSC patients, mutations in either the TSC1 or TSC2 gene lead to constitutive upregulation of mTOR complex 1, which promotes abnormal cell growth and proliferation. In this study, the administration of everolimus was associated with tumor responses and reduction in seizure frequency. The drug was subsequently approved by the Food and Drug Administration for this indication [57]. This represents an important step forward in the search for novel targeted agents in the treatment of LGG and may help guide future investigation into similarly tailored therapy in other LGG subtypes.
Pleomorphic Xanthoastrocytoma
Pleomorphic xanthoastrocytomas (PXAs) are WHO grade II tumors comprising less than 1% of all astrocytic tumors and generally occur in children and young adults, with two thirds of patients under the age of 18 years [1]. These tumors are generally adherent to the meninges and may be cystic with a mural nodule; microscopic features include nuclear and cytoplasmic pleomorphism and large xanthomatous cells containing lipid droplets [1]. BRAF V600E mutations have been described in this type of LGG [58]. PXAs are generally located superficially in the cerebral hemispheres, with 98% occurring supratentorially, and often involve the meninges, hence their description as meningocerebral neoplasms [1]. As a result of this superficial location, patients with this type of tumor often present with seizures. Prognosis is largely favorable, with estimated 81% 5-year and 70% 10-year survival [1]. The majority of patients undergo surgical resection, which is generally possible because of the superficial location of these tumors, and there is a trend toward improved outcomes with greater extent of resection [59].
Footnotes
For Further Reading: Marco C. Pinho, Pavlina Polaskova, Jayashree Kalpathy-Cramer et al. Low Incidence of Pseudoprogression by Imaging in Newly Diagnosed Glioblastoma Patients Treated With Cediranib in Combination With Chemoradiation. The Oncologist 2014:19;75–81.
Implications for Practice: Pseudoprogression is an unsolved clinical dilemma during postchemoradiation surveillance for malignant gliomas. It has received substantial attention in the era of temozolomide-based regimens, mimicking early disease progression on imaging studies and challenging patient management and interpretation of clinical trials. This study suggests that the incidence of pseudoprogression in patients receiving antiangiogenic therapy is low and that enlarging lesions in this patient population are more likely to represent true tumor progression than transient post-treatment effects.
Author Contributions
Conception/Design: Tracy T. Batchelor
Collection and/or assembly of data: Deborah A. Forst
Data analysis and interpretation: Brian V. Nahed, Tracy T. Batchelor, Deborah A. Forst
Manuscript writing: Jay S. Loeffler, Brian V. Nahed, Tracy T. Batchelor, Deborah A. Forst
Final approval of manuscript: Jay S. Loeffler, Brian V. Nahed, Tracy T. Batchelor, Deborah A. Forst
Disclosures
Tracy T. Batchelor: Merck, Roche, and Novartis (C/A); Robert Michael Educational Institute, Educational Concepts Group, Research to Practice, and Oakstone (H); Pfizer, Millennium, AstraZeneca, UpToDate, Imedex, Advance Medical, and Champions Biotech (RF). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1.et al. In: WHO Classification of Tumours of the Central Nervous System. Louis DN, Ohgaki H, Wiestler OD, editors; Lyon, France: IARC Press; 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Central Brain Tumor Registry of the United States . Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2004-2008 (March 23, 2012 Revision) Hinsdale, IL: CBTRUS; 2012. [Google Scholar]
- 3.Schwartzbaum JA, Fisher JL, Aldape KD, et al. Epidemiology and molecular pathology of glioma. Nat Clin Pract Neurol. 2006;2:494–503; quiz 1, 516. doi: 10.1038/ncpneuro0289. [DOI] [PubMed] [Google Scholar]
- 4.Pouratian N, Asthagiri A, Jagannathan J, et al. Surgery insight: The role of surgery in the management of low-grade gliomas. Nat Clin Pract Neurol. 2007;3:628–639. doi: 10.1038/ncpneuro0634. [DOI] [PubMed] [Google Scholar]
- 5.Pirzkall A, Nelson SJ, McKnight TR, et al. Metabolic imaging of low-grade gliomas with three-dimensional magnetic resonance spectroscopy. Int J Radiat Oncol Biol Phys. 2002;53:1254–1264. doi: 10.1016/s0360-3016(02)02869-9. [DOI] [PubMed] [Google Scholar]
- 6.van den Bent MJ, Wefel JS, Schiff D, et al. Response assessment in neuro-oncology (a report of the RANO group): Assessment of outcome in trials of diffuse low-grade gliomas. Lancet Oncol. 2011;12:583–593. doi: 10.1016/S1470-2045(11)70057-2. [DOI] [PubMed] [Google Scholar]
- 7.Fan GG, Deng QL, Wu ZH, et al. Usefulness of diffusion/perfusion-weighted MRI in patients with non-enhancing supratentorial brain gliomas: A valuable tool to predict tumour grading? Br J Radiol. 2006;79:652–658. doi: 10.1259/bjr/25349497. [DOI] [PubMed] [Google Scholar]
- 8.Baehring JM, Bi WL, Bannykh S, et al. Diffusion MRI in the early diagnosis of malignant glioma. J Neurooncol. 2007;82:221–225. doi: 10.1007/s11060-006-9273-3. [DOI] [PubMed] [Google Scholar]
- 9.Smith JS, Alderete B, Minn Y, et al. Localization of common deletion regions on 1p and 19q in human gliomas and their association with histological subtype. Oncogene. 1999;18:4144–4152. doi: 10.1038/sj.onc.1202759. [DOI] [PubMed] [Google Scholar]
- 10.Yan H, Parsons DW, Jin G, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Andronesi OC, Kim GS, Gerstner E, et al. Detection of 2-hydroxyglutarate in IDH-mutated glioma patients by in vivo spectral-editing and 2D correlation magnetic resonance spectroscopy. Sci Transl Med. 2012;4:116ra4. doi: 10.1126/scitranslmed.3002693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Houillier C, Wang X, Kaloshi G, et al. IDH1 or IDH2 mutations predict longer survival and response to temozolomide in low-grade gliomas. Neurology. 2010;75:1560–1566. doi: 10.1212/WNL.0b013e3181f96282. [DOI] [PubMed] [Google Scholar]
- 13.Bettegowda C, Agrawal N, Jiao Y, et al. Mutations in CIC and FUBP1 contribute to human oligodendroglioma. Science. 2011;333:1453–1455. doi: 10.1126/science.1210557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sahm F, Koelsche C, Meyer J, et al. CIC and FUBP1 mutations in oligodendrogliomas, oligoastrocytomas and astrocytomas. Acta Neuropathol. 2012;123:853–860. doi: 10.1007/s00401-012-0993-5. [DOI] [PubMed] [Google Scholar]
- 15.Yip S, Butterfield YS, Morozova O, et al. Concurrent CIC mutations, IDH mutations, and 1p/19q loss distinguish oligodendrogliomas from other cancers. J Pathol. 2012;226:7–16. doi: 10.1002/path.2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ricard D, Idbaih A, Ducray F, et al. Primary brain tumours in adults. Lancet. 2012;379:1984–1996. doi: 10.1016/S0140-6736(11)61346-9. [DOI] [PubMed] [Google Scholar]
- 17.Schiff D, Brown PD, Giannini C. Outcome in adult low-grade glioma: The impact of prognostic factors and treatment. Neurology. 2007;69:1366–1373. doi: 10.1212/01.wnl.0000277271.47601.a1. [DOI] [PubMed] [Google Scholar]
- 18.Smith JS, Perry A, Borell TJ, et al. Alterations of chromosome arms 1p and 19q as predictors of survival in oligodendrogliomas, astrocytomas, and mixed oligoastrocytomas. J Clin Oncol. 2000;18:636–645. doi: 10.1200/JCO.2000.18.3.636. [DOI] [PubMed] [Google Scholar]
- 19.Everhard S, Kaloshi G, Crinière E, et al. MGMT methylation: A marker of response to temozolomide in low-grade gliomas. Ann Neurol. 2006;60:740–743. doi: 10.1002/ana.21044. [DOI] [PubMed] [Google Scholar]
- 20.Leu S, von Felten S, Frank S, et al. IDH/MGMT-driven molecular classification of low-grade glioma is a strong predictor for long-term survival. Neuro-oncol. 2013;15:469–479. doi: 10.1093/neuonc/nos317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hartmann C, Hentschel B, Tatagiba M, et al. German Glioma Network Molecular markers in low-grade gliomas: Predictive or prognostic? Clin Cancer Res. 2011;17:4588–4599. doi: 10.1158/1078-0432.CCR-10-3194. [DOI] [PubMed] [Google Scholar]
- 22.Baumert BG, Stupp R, European Organization for Research and Treatment of Cancer (EORTC) Radiation Oncology Group. European Organization for Research and Treatment of Cancer (EORTC) Brain Tumor Group Low-grade glioma: A challenge in therapeutic options: The role of radiotherapy. Ann Oncol. 2008;19(suppl 7):vii217–vii222. doi: 10.1093/annonc/mdn434. [DOI] [PubMed] [Google Scholar]
- 23.Pignatti F, van den Bent M, Curran D, et al. European Organization for Research and Treatment of Cancer Brain Tumor Cooperative Group. European Organization for Research and Treatment of Cancer Radiotherapy Cooperative Group Prognostic factors for survival in adult patients with cerebral low-grade glioma. J Clin Oncol. 2002;20:2076–2084. doi: 10.1200/JCO.2002.08.121. [DOI] [PubMed] [Google Scholar]
- 24.Afra D, Osztie E, Sipos L, et al. Preoperative history and postoperative survival of supratentorial low-grade astrocytomas. Br J Neurosurg. 1999;13:299–305. doi: 10.1080/02688699943727. [DOI] [PubMed] [Google Scholar]
- 25.Jakola AS, Myrmel KS, Kloster R, et al. Comparison of a strategy favoring early surgical resection vs a strategy favoring watchful waiting in low-grade gliomas. JAMA. 2012;308:1881–1888. doi: 10.1001/jama.2012.12807. [DOI] [PubMed] [Google Scholar]
- 26.Youland RS, Brown PD, Giannini C, et al. Adult low-grade glioma: 19-year experience at a single institution. Am J Clin Oncol. 2013;36:612–619. doi: 10.1097/COC.0b013e31825d580a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Claus EB, Horlacher A, Hsu L, et al. Survival rates in patients with low-grade glioma after intraoperative magnetic resonance image guidance. Cancer. 2005;103:1227–1233. doi: 10.1002/cncr.20867. [DOI] [PubMed] [Google Scholar]
- 28.Shaw E, Arusell R, Scheithauer B, et al. Prospective randomized trial of low- versus high-dose radiation therapy in adults with supratentorial low-grade glioma: Initial report of a North Central Cancer Treatment Group/Radiation Therapy Oncology Group/Eastern Cooperative Oncology Group study. J Clin Oncol. 2002;20:2267–2276. doi: 10.1200/JCO.2002.09.126. [DOI] [PubMed] [Google Scholar]
- 29.Peraud A, Ansari H, Bise K, et al. Clinical outcome of supratentorial astrocytoma WHO grade II. Acta Neurochir (Wien) 1998;140:1213–1222. doi: 10.1007/s007010050241. [DOI] [PubMed] [Google Scholar]
- 30.Chang EF, Potts MB, Keles GE, et al. Seizure characteristics and control following resection in 332 patients with low-grade gliomas. J Neurosurg. 2008;108:227–235. doi: 10.3171/JNS/2008/108/2/0227. [DOI] [PubMed] [Google Scholar]
- 31.Englot DJ, Han SJ, Berger MS, et al. Extent of surgical resection predicts seizure freedom in low-grade temporal lobe brain tumors. Neurosurgery. 2012;70:921–928; discussion 928. doi: 10.1227/NEU.0b013e31823c3a30. [DOI] [PubMed] [Google Scholar]
- 32.Sanai N, Berger MS. Glioma extent of resection and its impact on patient outcome. Neurosurgery. 2008;62:753–764; discussion 264–266. doi: 10.1227/01.neu.0000318159.21731.cf. [DOI] [PubMed] [Google Scholar]
- 33.National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology. Central Nervous System Cancers. Version 2.2013. Available at http://www.nccn.org/professionals/physician_gls/f_guidelines.asp Accessed July 7, 2013.
- 34.Pouratian N, Schiff D. Management of low-grade glioma. Curr Neurol Neurosci Rep. 2010;10:224–231. doi: 10.1007/s11910-010-0105-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pamir MN, Özduman K, Yıldız E, et al. Intraoperative magnetic resonance spectroscopy for identification of residual tumor during low-grade glioma surgery: Clinical article. J Neurosurg. 2013;118:1191–1198. doi: 10.3171/2013.1.JNS111561. [DOI] [PubMed] [Google Scholar]
- 36.Pamir MN, Ozduman K. 3-T ultrahigh-field intraoperative MRI for low-grade glioma resection. Expert Rev Anticancer Ther. 2009;9:1537–1539. doi: 10.1586/era.09.134. [DOI] [PubMed] [Google Scholar]
- 37.Karim AB, Maat B, Hatlevoll R, et al. A randomized trial on dose-response in radiation therapy of low-grade cerebral glioma: European Organization for Research and Treatment of Cancer (EORTC) Study 22844. Int J Radiat Oncol Biol Phys. 1996;36:549–556. doi: 10.1016/s0360-3016(96)00352-5. [DOI] [PubMed] [Google Scholar]
- 38.van den Bent MJ, Afra D, de Witte O, et al. EORTC Radiotherapy and Brain Tumor Groups and the UK Medical Research Council Long-term efficacy of early versus delayed radiotherapy for low-grade astrocytoma and oligodendroglioma in adults: The EORTC 22845 randomised trial. Lancet. 2005;366:985–990. doi: 10.1016/S0140-6736(05)67070-5. [DOI] [PubMed] [Google Scholar]
- 39.U.S. National Institutes of Health. Proton Radiation Therapy for Low Grade Gliomas. ClinicalTrials.gov Identifier: NCT01358058. Available at http://clinicaltrials.gov/show/NCT01358058 Accessed July 18, 2013.
- 40.Quinn JA, Reardon DA, Friedman AH, et al. Phase II trial of temozolomide in patients with progressive low-grade glioma. J Clin Oncol. 2003;21:646–651. doi: 10.1200/JCO.2003.01.009. [DOI] [PubMed] [Google Scholar]
- 41.Shaw EG, Wang M, Coons SW, et al. Randomized trial of radiation therapy plus procarbazine, lomustine, and vincristine chemotherapy for supratentorial adult low-grade glioma: Initial results of RTOG 9802. J Clin Oncol. 2012;30:3065–3070. doi: 10.1200/JCO.2011.35.8598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.U.S. National Institutes of Health. Temozolomide and Radiation Therapy in Treating Patients With Gliomas. ClinicalTrials.gov Identifier: NCT00114140. Available at http://clinicaltrials.gov/ct2/show/NCT00114140 Accessed July 18, 2013.
- 43.Fisher BJ, Lui J, Macdonald DR, et al. A phase II study of a temozolomide-based chemoradiotherapy regimen for high-risk LGG: preliminary results of RTOG 0424. Proc Am Soc Clin Oncol. 2013 [Abstract 2008] [Google Scholar]
- 44.National Cancer Institute. Radiation Therapy or Temozolomide in Treating Patients With Gliomas. ClinicalTrials.gov Identifier: NCT00182819. Available at http://www.cancer.gov/clinicaltrials/search/view?cdrid=442397&version=HealthProfessional Accessed July 18, 2013.
- 45.Baumert BG, Mason WP, Ryan G, et al. Temozolomide chemotherapy versus radiotherapy in molecularly characterized (1p loss) low-grade glioma: a randomized phase III intergroup study by the EORTC/NCIC-CTG/TROG/MRC-CTU (EORTC 22033-26033) Proc Am Soc Clin Oncol. 2013 [Google Scholar]
- 46.National Cancer Institute. Radiation Therapy With or Without Temozolomide in Treating Patients With Low-Grade Glioma. ClinicalTrials.gov Identifier: NCT00978458. Available at http://www.cancer.gov/clinicaltrials/search/view?cdrid=654697&version=healthprofessional Accessed July 18, 2013.
- 47.Dhermain FG, Hau P, Lanfermann H, et al. Advanced MRI and PET imaging for assessment of treatment response in patients with gliomas. Lancet Neurol. 2010;9:906–920. doi: 10.1016/S1474-4422(10)70181-2. [DOI] [PubMed] [Google Scholar]
- 48.Sorensen AG, Batchelor TT, Wen PY, et al. Response criteria for glioma. Nat Clin Pract Oncol. 2008;5:634–644. doi: 10.1038/ncponc1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Macdonald DR, Cascino TL, Schold SC, Jr, et al. Response criteria for phase II studies of supratentorial malignant glioma. J Clin Oncol. 1990;8:1277–1280. doi: 10.1200/JCO.1990.8.7.1277. [DOI] [PubMed] [Google Scholar]
- 50.Khasraw M, Lassman AB. Neuro-oncology: Late neurocognitive decline after radiotherapy for low-grade glioma. Nat Rev Neurol. 2009;5:646–647. doi: 10.1038/nrneurol.2009.194. [DOI] [PubMed] [Google Scholar]
- 51.Douw L, Klein M, Fagel SS, et al. Cognitive and radiological effects of radiotherapy in patients with low-grade glioma: Long-term follow-up. Lancet Neurol. 2009;8:810–818. doi: 10.1016/S1474-4422(09)70204-2. [DOI] [PubMed] [Google Scholar]
- 52.Kiebert GM, Curran D, Aaronson NK, et al. EORTC Radiotherapy Co-operative Group Quality of life after radiation therapy of cerebral low-grade gliomas of the adult: Results of a randomised phase III trial on dose response (EORTC trial 22844) Eur J Cancer. 1998;34:1902–1909. doi: 10.1016/s0959-8049(98)00268-8. [DOI] [PubMed] [Google Scholar]
- 53.Liu R, Solheim K, Polley MY, et al. Quality of life in low-grade glioma patients receiving temozolomide. Neuro-oncol. 2009;11:59–68. doi: 10.1215/15228517-2008-063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grimm SA, Chamberlain MC. Brainstem glioma: A review. Curr Neurol Neurosci Rep. 2013;13:346. doi: 10.1007/s11910-013-0346-3. [DOI] [PubMed] [Google Scholar]
- 55.Sadighi Z, Slopis J. Pilocytic astrocytoma: A disease with evolving molecular heterogeneity. J Child Neurol. 2013;28:625–632. doi: 10.1177/0883073813476141. [DOI] [PubMed] [Google Scholar]
- 56.Aryan HE, Meltzer HS, Lu DC, et al. Management of pilocytic astrocytoma with diffuse leptomeningeal spread: Two cases and review of the literature. Childs Nerv Syst. 2005;21:477–481. doi: 10.1007/s00381-004-1002-7. [DOI] [PubMed] [Google Scholar]
- 57.Krueger DA, Care MM, Holland K, et al. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med. 2010;363:1801–1811. doi: 10.1056/NEJMoa1001671. [DOI] [PubMed] [Google Scholar]
- 58.Dias-Santagata D, Lam Q, Vernovsky K, et al. BRAF V600E mutations are common in pleomorphic xanthoastrocytoma: Diagnostic and therapeutic implications. PLoS One. 2011;6:e17948. doi: 10.1371/journal.pone.0017948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Perkins SM, Mitra N, Fei W, et al. Patterns of care and outcomes of patients with pleomorphic xanthoastrocytoma: A SEER analysis. J Neurooncol. 2012;110:99–104. doi: 10.1007/s11060-012-0939-8. [DOI] [PubMed] [Google Scholar]