

Abstract
Background
Multiple endocrine neoplasia type 2 (MEN2) is an autosomal dominant hereditary cancer syndrome caused by mutations in the RET proto-oncogene. In patients with MEN 2A and 2B the penetrance of pheochromocytoma is variable and childhood pheochromocytoma in the setting of MEN 2 remains rare.
Patient and Intervention
We present the case of an 8-year-old female with known MEN 2A, C634Y RET mutation, found to have markedly elevated plasma normetanephrines and total metanephrines, and diagnosed with a 6 cm pheochromocytoma requiring laparoscopic unilateral adrenalectomy.
Conclusions
Given this patient’s age at diagnosis, current screening guidelines should recommend annual screening beginning at age 8 for patients with MEN 2B or for patients with MEN 2A with codons 630 or 634 RET mutations.
Keywords: Multiple endocrine neoplasia type 2, pheochromocytoma, screening, adrenalectomy
Introduction
Multiple endocrine neoplasia type 2 (MEN2) is an autosomal dominant hereditary cancer syndrome caused by mutations in the RET proto-oncogene. The syndrome is characterized by nearly complete penetrance of medullary thyroid cancer, however is further divided into three subtypes based upon the specific RET mutation and associated endocrinopathies, age at presentation, and clinical course. MEN 2A is characterized by multifocal medullary thyroid cancer, pheochromocytoma, and hyperparathyroidism. MEN 2B is characterized by medullary thyroid cancer with onset at a very young age, pheochromocytoma, and multiple mucosal neuromas and ganglioneuromatosis of the gastrointestinal tract. Familial medullary thyroid carcinoma (FMTC) is characterized by medullary thyroid cancer only. (1, 2).
In patients with MEN 2A and 2B the penetrance of pheochromocytoma is variable. Childhood pheochromocytoma in the setting of MEN 2 remains rare. However, given the dangers of catecholamine excess, especially in the peri-operative setting, appropriate screening for MEN 2 patients, based upon age and specific RET mutation, is warranted. In this case we present a patient with known MEN 2A, codon 634 RET mutation, who was diagnosed with a pheochromocytoma at the age of 8. Based upon extensive literature review, this represents the youngest documented MEN patient with a pheochromocytoma. The current screening guidelines of the American Thyroid Association for pheochromocytoma in MEN 2 are based upon this case (3).
Case
An 8-year-old female with known MEN 2A presented with occasional headaches. The patient had a known paternal family history of MEN 2A. All relatives with documented MEN had been diagnosed with pheochromocytoma and required adrenalectomy between the ages of 16 and 30.
The patient herself was diagnosed with the 634Y RET proto-oncogene heterozygous mutation via genetic testing at the age of 2. She had undergone prophylactic total thyroidectomy with parathyroid autotransplantation to the sternocleidomastoid muscle at the age of 4. At that time, routine Hematoxylin and Eosin (H&E) stained sections of the thyroid gland were histologically unremarkable. There was no evidence of medullary thyroid carcinoma. Scattered foci of minimal c-cell hyperplasia were detected by calcitonin immunohistochemistry. Following thyroidectomy the patient was maintained on thyroid hormone replacement therapy without additional complaints.
At the age of 8 the patient reported occasional headaches and was found to have biochemical evidence of pheochromocytoma with plasma metanephrines of 59 pg/mL (normal less than 57), elevated normetanephrines of 1858 pg/mL (normal less than 148), and elevated total metanephrines of 1917 pg/mL (normal less than 205). Calcitonin, parathyroid hormone, and thyroid hormone levels were all within normal limits. She denied palpitations or abdominal pain and was normotensive. Abdominal MRI revealed a 5.7 × 5.0 × 4.6 cm well circumscribed mass arising from the left adrenal gland, consistent with pheochromocytoma (figure 1).
Fig. 1.
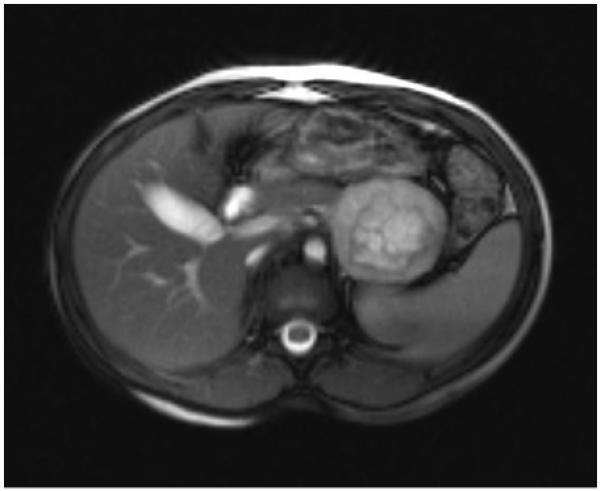
Abdominal MRI demonstrates a well-circumscribed heterogeneous 5.7 × 5 × 4.6 cm mass arising from the anterior limb of the left adrenal gland, consistent with a pheochromocytoma. Moderate post contrast enhancement is present with a large central non-enhancing area suggestive of necrosis.
The patient was placed on phenoxybenzamine alpha blockade preoperatively. A laparoscopic left adrenalectomy was performed. Pathology showed a pheochromocytoma (figure 2), which was confirmed by immunohistochemistry (figure 3). The patient tolerated the procedure well and was discharged home on post-operative day one. Her post-operative course was complicated by occasional headaches, however plasma metanephrine, normetanephrine, and total metanephrines evaluated two months after surgery were all within normal limits. The patient is now 52 months status post left adrenalectomy and shows no biochemical evidence of recurrent disease on biannual plasma screening.
Fig. 2.
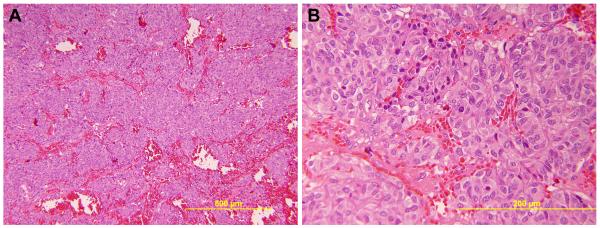
Routine H&E histologic evaluation of the adrenal gland shows a pheochromocytoma. The neoplasm has an organoid growth pattern with hemorrhage separating islands of tumor cells (A) [low-power, 100x]. The individual tumor cells have abundant eosinophilic cytoplasm (B) [high-power, 400x].
Fig. 3.

Immunohistochemical stains support the diagnosis of pheochromocytoma (200x). Chromogranin (A) and synaptophysin (B) are strongly and diffusely positive in the tumor cells, confirming neuroendocrine differentiation. A cytokeratin immunostain (C) is negative, excluding neuroendocrine carcinoma, as is an inhibin immunostain (D). The neuroendocrine differentiation and lack of inhibin expression also exclude an adrenocortical neoplasm.
Discussion
The penetrance of pheochromocytoma in MEN 2 patients is variable. Large case series report forty to fifty percent of MEN 2 patients will develop pheochromocytomas (4-6). Penetrance varies by family, with a reported range of six to one hundred percent penetrance in different kindreds (4). Patients typically present with palpitations, hypertensive attacks, or recurrent headaches (7-8). However, half of all patients may be asymptomatic at the time of diagnosis (8-9). The diagnosis of pheochromocytoma may be made before the diagnosis of medullary thyroid cancer (12.9 to 25.1 percent of patients), simultaneously (34.7 to 38.9 percent of patients), or after the diagnosis of medullary thyroid cancer (40.2 to 48.2 percent of patients) (9-10).
Childhood pheochromocytoma is rare. The average age at diagnosis of pheochromocytoma has been reported from 23 to 40 years of age, depending on the case series (4-14). Prior publications have documented cases of pheochromocytoma as young as 12 years of age (12, 15-16). This case represents the youngest documented case of pheochromocytoma in the setting of MEN 2. Based upon personal communication of this case, the 2009 American Thyroid Association guidelines recommend pheochromocytoma screening be initiated at the age of 8 for patients with MEN 2B or MEN 2A with codon 634 or 630 Ret mutations (3). Screening for patients with other MEN 2A Ret mutations should begin at the age of 20. Appropriate screening consists of annual plasma metanephrines and normetanephrines or 24-hour urine collection for metanephrines or normetanephrines (3). Elevations in plasma or urinary levels should prompt intra-abdominal imaging with a MRI or CT scan. Given the peri-operative risk of catecholamine excess, any known pheochromocytoma should be respected prior to surgical treatment for medullary thyroid cancer or hyperparathyroidism in an MEN 2 patient.
The majority of MEN 2 pheochromocytomas are benign and laparoscopic adrenalectomy is the treatment of choice (9, 11, 14). Patients with bilateral disease should undergo cortical-sparing adrenal surgery given the risk of Addisonian crisis following bilateral adrenalectomy (3). Patients undergoing unilateral adrenalectomy should continue to be screened for pheochromocytoma as approximately thirty percent of these patients will develop contralateral disease (11, 13).
Conclusion
We present the case of an 8-year-old female with known MEN 2A, C634Y Ret mutation, found to have markedly elevated plasma normetanephrines and total metanephrines, and diagnosed with a 6 cm pheochromocytoma requiring laparoscopic unilateral adrenalectomy. Given this patient’s age at diagnosis, current screening guidelines recommend annual screening beginning at age 8 for patients with MEN 2B or for patients with MEN 2A with codons 630 or 634 Ret mutations.
Acknowledgments
KR is supported by National Institutes of Health grant T32 CA009621.
Footnotes
DISCLOSURE STATEMENT: The authors have nothing to disclose.
References
- 1.Traugott AL, Moley JF. Multiple endocrine neoplasia type 2: clinical manifestations and management. Cancer Treat Res. 2010;153:321–337. doi: 10.1007/978-1-4419-0857-5_18. [DOI] [PubMed] [Google Scholar]
- 2.Moo-Young TA, Traugott AL, Moley JF. Sporadic and familial medullary thyroid carcinoma: state of the art. Surg Clin North Am. 2009;89(5):1193–1204. doi: 10.1016/j.suc.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.American Thyroid Association Guidelines Task Force. Kloos RT, Eng C, Evans DB, et al. Medullary thyroid cancer: management guidelines of the American Thyroid Association. Thyroid. 2009;19(6):565–612. doi: 10.1089/thy.2008.0403. [DOI] [PubMed] [Google Scholar]
- 4.Howe JR, Norton JA, Wells SA., Jr. Prevalence of pheochromocytoma and hyperparathyroidism in multiple endocrine neoplasia type 2A: results of long-term follow-up. Surgery. 1993;114(6):1070–1077. [PubMed] [Google Scholar]
- 5.Vasen HF, van der Feltz M, Raue F, et al. The natural course of multiple endocrine neoplasia type IIb. A study of 18 cases. Arch Intern Med. 1992;152(6):1250–1252. [PubMed] [Google Scholar]
- 6.Iihara M, Yamashita T, Okamoto T, et al. A nationwide clinical survey of patients with multiple endocrine neoplasia type 2 and familial medullary thyroid carcinoma in Japan. Jpn J Clin Oncol. 1997;27(3):128–134. doi: 10.1093/jjco/27.3.128. [DOI] [PubMed] [Google Scholar]
- 7.Shimotake T, Iwai N, Yanagihara J, et al. The natural history of multiple endocrine neoplasia type 2A – a clinical analysis. Jpn J Surg. 1990;20(3):290–293. doi: 10.1007/BF02470663. [DOI] [PubMed] [Google Scholar]
- 8.Pomares FJ, Canas R, Rodriguez JM, et al. Differences between sporadic and multiple endocrine neoplasia type 2A phaeochromocytoma. Clin Endocrinol (Oxf) 1998;48(2):195–200. doi: 10.1046/j.1365-2265.1998.3751208.x. [DOI] [PubMed] [Google Scholar]
- 9.Rodriguez JM, Balsalobre M, Ponce JL, et al. Pheochromocytoma in MEN 2A syndrome. Study of 54 patients. World J Surg. 2008;32(11):2520–2526. doi: 10.1007/s00268-008-9734-2. [DOI] [PubMed] [Google Scholar]
- 10.Modigliani E, Vasen HM, Raue K, et al. Pheochromocytoma in multiple endocrine neoplasia type 2: European study. The European Study Group. J Intern Med. 1995;238(4):363–367. doi: 10.1111/j.1365-2796.1995.tb01211.x. [DOI] [PubMed] [Google Scholar]
- 11.Brunt LM, Lairmore TC, Doherty GM, et al. Adrenalectomy for familial pheochromocytoma in the laparoscopic era. Ann Surg. 2002 May;235(5):713–720. doi: 10.1097/00000658-200205000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nguyen L, Niccoli-Sire P, Caron P, et al. Pheochromocytoma in multiple endocrine neoplasia type 2: a prospective study. Eur J Endocrinol. 2001;144(1):37–44. doi: 10.1530/eje.0.1440037. [DOI] [PubMed] [Google Scholar]
- 13.Yip L, Cote GJ, Shapiro SE, et al. Multiple endocrine neoplasia type 2: evaluation of the genotypephenotype relationship. Arch Surg. 2003;138(4):409–416. doi: 10.1001/archsurg.138.4.409. [DOI] [PubMed] [Google Scholar]
- 14.Wohllk N, Schweizer H, Erlic Z, et al. Multiple endocrine neoplasia type 2. Best Pract Res Clin Endocrinol Metab. 2010;24(3):371–387. doi: 10.1016/j.beem.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 15.Skinner MA, DeBenedetti MK, Moley JF, et al. Medullary thyroid carcinoma in children with multiple endocrine neoplasia types 2A and 2B. J Pediatr Surg. 1996;31(1):177–181. doi: 10.1016/s0022-3468(96)90343-7. [DOI] [PubMed] [Google Scholar]
- 16.Machens A, Brauckhoff M, Holzhaussen HJ, et al. Codon-specific development of pheochromocytoma in multiple endocrine neoplasia type 2. J Clin Endocrinol Metab. 2005;90(7):3999–4003. doi: 10.1210/jc.2005-0064. [DOI] [PubMed] [Google Scholar]