

Abstract
The cystic fibrosis transmembrane conductance regulator (CFTR) protein is the only known apical glutathione (GSH) transporter in the lung. The purpose of these studies was to determine whether oral GSH or glutathione disulfide (GSSG) treatment could increase lung epithelial lining fluid (ELF) GSH levels and whether CFTR plays a role in this process. The pharmacokinetic profile of an oral bolus dose of GSH (300 mg/kg) was determined in mice. Plasma, ELF, bronchoalveolar lavage (BAL) cells, and lung tissue were analyzed for GSH content. There was a rapid elevation in the GSH levels that peaked at 30 min in the plasma and 60 min in the lung, ELF, and BAL cells after oral GSH dosing. Oral GSH treatment produced a selective increase in the reduced and active form of GSH in all lung compartments examined. Oral GSSG treatment (300 mg/kg) resulted in a smaller increase of GSH levels. To evaluate the role of CFTR in this process, Cftr knockout (KO) mice and gut-corrected Cftr KO-transgenic (Tg) mice were given an oral bolus dose of GSH (300 mg/kg) and compared with wild-type mice for changes in GSH levels in plasma, lung, ELF, and BAL cells. There was a twofold increase in plasma, a twofold increase in lung, a fivefold increase in ELF, and a threefold increase in BAL cell GSH levels at 60 min in wild-type mice; however, GSH levels only increased by 40% in the plasma, 60% in the lung, 50% in the ELF, and twofold in the BAL cells within the gut-corrected Cftr KO-Tg mice. No change in GSH levels was observed in the uncorrected Cftr KO mice. These studies suggest that CFTR plays an important role in GSH uptake from the diet and transport processes in the lung.
Keywords: nutrition, transport, high-performance liquid chromatography, mice, cystic fibrosis, antioxidant, cystic fibrosis transmembrane conductance regulator
The respiratory tract is constantly challenged by a number of environmental agents including ozone, particulate matter, nitrogen oxides, cigarette smoke, and microbes. The epithelial lining fluid (ELF) is first to encounter these agents and serves as the body’s initial line of defense. ELF is well represented with water-soluble antioxidants including ascorbate, urate, and glutathione (GSH). GSH is a sulfhydryl containing tripeptide (L-γ-glutamyl-L-cysteinyl-glycine) that can scavenge oxidants either directly or indirectly, as a specific substrate for enzymes involved in detoxification pathways (31). GSH is unique among these airway antioxidants because it is selectively concentrated in the ELF compared with plasma (46).
The ELF GSH concentration is 100-fold greater than its circulating level in plasma (11). ELF GSH levels are elevated following primary lung infection with Pseudomonas aeruginosa (15), in asthmatics (14), in chronic beryllium disease (13), and in chronic smokers (11, 13). In many of these cases, the elevated GSH levels are thought to be a lung-adaptive response to oxidants. On the other hand, basal GSH levels in ELF are low in individuals who are acute smokers (42), in human immunodeficiency virus-positive subjects (36), and in a number of progressive lung disorders including idiopathic pulmonary fibrosis (IPF) (10), acute respiratory distress syndrome (ARDS) (5, 37), and cystic fibrosis (CF) (45). Decreased levels of ELF GSH may make the lung more susceptible to exogenous agents and contribute to chronic oxidative stress. The pathways by which the lung modulates ELF GSH levels are relatively unknown.
CF is a lethal hereditary disease caused by numerous autosomal recessive mutations of the 250-kb CF transmembrane conductance regulator (CFTR) protein located on chromosome 7 (41). It is estimated that nearly 30,000 Caucasians in North America and more than 200,000 individuals worldwide are affected by this disease (1). Studies suggest that the CFTR channel not only regulates chloride transport but also other anions including bicarbonate and GSH (34). Studies performed in our laboratory and by others have confirmed that CFTR is partly responsible for the transport of GSH across lung secretory epithelium (20, 32, 52). Thus it is hypothesized that the CFTR protein plays a prominent role in ELF GSH transport and that a defective CFTR protein would cause alterations in lung GSH distribution.
Previous studies have reported that GSH concentration in blood and lung can be increased following oral administration (2, 18). However, none of the previous oral studies have determined GSH changes associated with lung compartments such as the ELF and bronchoalveolar lavage (BAL) cells, nor the role of CFTR in mediating these changes. We hypothesized that oral GSH and/or GSSG treatments in mice could elevate GSH levels in ELF and BAL cells and that the CFTR protein plays a role in these processes. We tested this hypothesis using mouse models of CF: Cftr knockout (KO) and gut-corrected Cftr KO-transgenic (Tg) mice. Our laboratory reports that GSH levels can be increased in lung, ELF, and BAL cells and that intracellular GSH concentration in BAL cells may be dependent on ELF GSH levels. We also demonstrate the importance of lung and intestinal CFTR in these processes.
MATERIALS AND METHODS
Animals
Adult wild-type male C57BL/6J mice (24.6 ± 0.2 g) were purchased from Jackson Laboratories (Bar Harbor, ME). They were fed Purina mouse chow 5010 and autoclaved tap water ad libitum. C57BL/6J congenic Cftr KO (S489X) mice (22.3 ± 0.2 g) that possessed the S489X mutations in the murine equivalent to CFTR (47) and C57BL/6J congenic gut-corrected Cftr KO-Tg mice (23.7 ± 0.3 g) that had intestinal specific expression of normal human Cftr driven by the fatty acid binding promoter (57) were employed in these studies. These mice were originally obtained from Case Western Reserve University’s CF Animal Core, as previously described (49). The gut-corrected Cftr KO-Tg mice were fed Purina mouse chow 5010, and the Cftr KO were maintained on a liquid elemental diet (Peptamen; Nestle, Glendale, CA) to avoid lethal intestinal obstructions previously reported in these mice (24, 47). Autoclaved tap water in bottles with sipper tubes was provided. Mice were pathogen free and housed in microisolator cages with lights cycled 12 h on and 12 h off. Genotypes for each mouse were determined by PCR with DNA isolated from tail clips as described (47, 57). All mice were removed from food 8 h before dosing. Animal studies were approved by the National Jewish Medical and Research Center Animal Care and Use Committee.
Oral GSH administration
Wild-type mice were administered a single bolus dose (300 mg/kg) of GSH (Sigma, St. Louis, MO) by gavage using a flexible bead-tipped feeding tube 20 G × 1.5 inches (Braintree Scientific, Braintree, MA). Stock solution of GSH was prepared fresh daily in PBS at pH 7. Control mice received PBS (125 μl) as vehicle controls. Mice were killed at 0, 15, 30, 60, 120, or 240 min after treatment. Plasma, BAL fluid (BALF), and lung tissue were collected at all time points. Wild-type mice also received a single bolus dose (300 mg/kg) of GSSG (Sigma) by gavage as described above. Stock solution of GSSG was prepared fresh daily in PBS at pH 7. Control mice received PBS (125 μl) as vehicle control. Mice were killed 60 min after treatment. Plasma, BALF, and lung tissue were collected and analyzed for GSH levels. Groups of four to six Cftr KO mice (both gut-corrected and noncorrected) were dosed with GSH (300 mg/kg) by gavage and plasma, BALF, and lung tissue collected 60 min after treatment.
Isolation of BALF, plasma, and lung tissue
Mice were killed by administering pentobarbital (65 mg/kg ip), exsanguinated by cardiac puncture, and vascularly perfused with 0.9% saline. Blood was collected to measure GSH and perform urea analysis as described by the manufacturer (Sigma Diagnostics). Urea analysis was used to normalize data by accounting for the BAL dilution where ELF GSH concentrations were determined by multiplying the BALF concentrations by the dilution factor using a urea method (12). BAL was performed by cannulating the trachea in situ with a 22-gauge 0.5-inch blunt-tipped needle. Three aliquots of 1.0 ml each of sterile PBS, pH 7.4, were instilled and collected by gentle aspiration. The total lavage fluid was pooled together and centrifuged at 4,000 g for 10 min at 4°C to remove BALF cells. Supernatant and BAL cells were stored at −80°C until further analysis. The right and left lungs were then removed and snap-frozen in liquid nitrogen. The lungs were then ground into a fine powder in a liquid nitrogen-cooled mortar and pestle and then stored at −80°C until further analysis.
GSH analysis in BAL cells, BALF, plasma, and lung tissue
A 180-μl aliquot of BALF was acidified with 5% metaphosphoric acid and centrifuged at 16,000 g for 5 min at 4°C to remove proteins. BAL cells were resuspended in 200 μl of water and briefly sonicated. A 20-μl aliquot of cell lysate was removed for protein analysis, and the remaining sample was acidified with 5% metaphosphoric acid and centrifuged to remove precipitated proteins. To determine GSH concentrations in lung tissue, ~20 mg of the ground tissue was dissolved in 500 μl of water, acidified with 5% metaphosphoric acid, and centrifuged to remove precipitated proteins. Similarly, plasma was also treated with 5% metaphosphoric acid and centrifuged to remove precipitated proteins. GSH in BAL cells, BALF, lung tissue, and plasma was analyzed by high-performance liquid chromatography (HPLC) coupled with coulometric electrochemical detection (CoulArray model 5600; ESA, Chelmford, MA). Sample analysis was performed using a 7 × 53-mm C18 reverse phase (Platinum EPS C18 100A, 3 μm; Alltech, Deerfield, IL) and mobile phase consisting of 125 mM sodium acetate in 0.9% acetonitrile at pH 3.0. The electrode potentials for a channel 1, 2, 3, and 4 were set at 100, 215, 485, and 650 mV, respectively. Under these conditions, GSH exhibited retention time of 7.2 min. The GSH signals were distributed across electrodes 2, 3, and 4 with the signal on channel 3 being dominant. Concentrations of GSH were determined from a 25-μl injection and quantified from a five-point standard curve. These data were also confirmed with HPLC equipped with fluorometric detector as described below.
Determination of GSH and GSSG levels using HPLC coupled with fluorometric detection
To determine GSH levels, 75 μl of ELF was added to an equal volume of KPBS buffer (50 mM potassium phosphate buffer, 17.5 mM EDTA, 50 mM serine, and 50 mM boric acid at pH 7.4). To each sample, 10 μl of an internal standard was added (0.1 mM des-Gly-glutathione). The samples were then treated with 10 μl of monobromobimane (mBBr; 3 mM in acetonitrile) and incubated in the dark for 30 min at room temperature. The derivatization reaction was stopped by the addition of 10 μl of 70% perchloric acid. The samples were then centrifuged at 16,000 g for 10 min at 4°C to separate the protein pellet from the supernatant. The resulting supernatant was then transferred to HPLC vials to measure GSH. The samples were analyzed on a Hitachi HPLC (Elite LaChrom) coupled with a fluorometric detector (model L-2480). The samples were eluted with Synergi 4μ Hydro-RP 80A C18 column (150 × 4.6 mm; Phenomenex, Torrance, CA). The mobile phase consisted of 1% acetic acid and 7% acetonitrile, with a pH adjusted to 4.25 using ammonium hydroxide. An injection volume of 5 μl and a flow rate of 1.0 ml/min were used to detect GSH levels. The detector excitation and emission wavelengths were set at 390 and 480 nm, respectively. The ELF GSSG levels were measured using a recycling assay in which the samples were pretreated with a GSH reductase system before their derivatization with mBBr (25).
Statistical analysis
Data are expressed as means ± SE. Prizm version 5 (GraphPad, San Diego, CA) was used to carry out ANOVA statistical analysis with a Newman-Keuls multiple comparison test where a P < 0.05 was considered significant. Pharmacokinetic parameters were calculated using PKanalyst software using a two-compartment model (Micromath, San Diego, CA).
RESULTS
Pharmacokinetic profile of oral GSH administration in wild-type mice
Following oral administration of 300 mg/kg of GSH, plasma GSH levels peaked at 30 min posttreatment (Fig. 1A). The GSH peak corresponded to a threefold increase over control mice and returned to basal levels at 240 min posttreatment. The elimination half-life of plasma GSH was estimated to be 27 min, and the distribution half-life was estimated to be 22 min. A similar profile was observed for the lung except that peak GSH levels were delayed, peaking 60 min posttreatment (Fig. 1B). The change in lung GSH content corresponded to a twofold increase over basal levels. Lung tissue GSH levels returned to basal levels at 240 min posttreatment, with an elimination half-life estimated to be 54 min and a distribution half-life estimated to be 30 min.
Fig. 1.
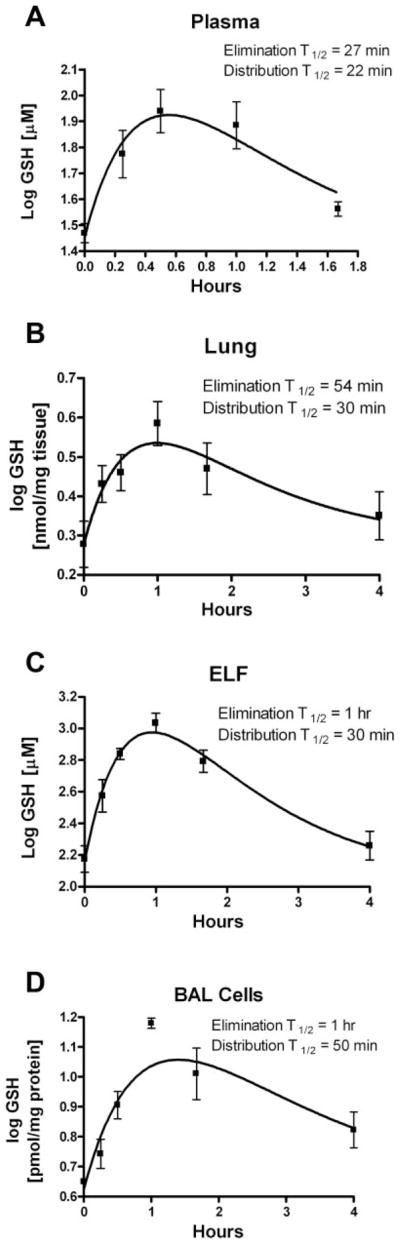
Pharmacokinetic profile of a single oral bolus dose of reduced glutathione (GSH, 300 mg/kg) in plasma (A), lung tissue (B), lung epithelial lining fluid (ELF; C), and lung bronchoalveolar lavage (BAL; D) cells in wild-type mice. Groups of 3–6 animals were used per time point. Pharmacokinetic values were derived from fitted curves to a 2-compartment model.
ELF represents the first line of defense against environmental agents. Hence, we were interested in determining the effects of oral GSH administration on ELF GSH levels. To our surprise, we saw a fivefold increase in ELF GSH levels over basal levels at 60 min (Fig. 1C). As seen with lung tissue, ELF GSH levels returned to basal levels by 240 min posttreatment with an estimated elimination half-life of 60 min and a distribution half-life estimated to be 30 min. Since BAL cells are present in ELF and play important roles in lung host defense, we also measured changes in their GSH levels. Following oral administration of GSH, we observed peak BAL cell GSH concentrations at 60 min posttreatment (Fig. 1D). This change in GSH corresponded to a threefold increase over basal levels, with an estimated elimination half-life of 60 min and a distribution half-life estimated to be 50 min. BAL cell GSH levels remained elevated at 240 min posttreatment. Pharmacokinetic modeling was used to estimate distribution and elimination half-lives and apparent volume of distribution based on the changes in plasma and lung GSH content (Table 1).
Table 1.
Pharmacokinetics of GSH in plasma and lung compartments
| ELF | BAL Cells | Lung | Plasma | |
|---|---|---|---|---|
| Cmax | 1,131 ± 319 μM | 15.1 ± 1 nmol/mg protein | 4.3 ± 0.3 nmol/mg tissue | 90 ± 31.0 μM |
| Tmax (h) | 1 | 1 | 1 | 0.5 |
| Apparent volume of distribution | 689 ml | |||
| Distribution t1/2 (min) | 30 | 50 | 30 | 22 |
| Elimination t1/2 (min) | 60 | 60 | 54 | 27 |
Data are expressed as means ± SE. ELF, epithelial lining fluid; BAL, bronchoalveolar lavage. Cmax is the maximum concentration reached; Tmax is the time at which the Cmax occurs.
Changes in plasma and lung GSH and GSSG levels after oral GSH administration
Wild-type mice were gavaged with 300 mg/kg of GSH, and 60 min later their GSH and GSSG levels were determined in plasma and lung using HPLC coupled with fluorometric detection (Table 2). Both HPLC GSH methods gave similar results. Oral GSH administration was associated with twofold increases in plasma GSH and GSSG levels; however, changes in GSSG did not reach statistical significance. ELF GSH levels increased fourfold without any significant increases in GSSG levels. Oral GSH administration produced similar elevations in lung tissue and BAL cell GSH levels without significant changes in GSSG levels. These data suggest that oral GSH administration can effectively elevate the reduced and active form of GSH in the lung. The significance of selectively raising the reduced form of GSH in fluids and tissues is a resulting change in redox state that renders the compartments more resistant to oxidative damage.
Table 2.
Plasma and lung GSH and GSSG changes following oral administration of GSH
| GSH
|
GSSG
|
GSH/GSSG
|
||||
|---|---|---|---|---|---|---|
| Basal | 60 min | Basal | 60 min | Basal | 60 min | |
| Plasma, μM | 22 ± 0.9 | 57 ± 2.5* | 5.0 ± 0.9 | 11.4 ± 2.2 | 4.4 ± 0.1 | 5.3 ± 0.7 |
| Lung tissue, nmol/mg tissue | 2.3 ± 0.2 | 4.3 ± 0.3* | 0.3 ± 0.04 | 0.5 ± 0.04 | 7.0 ± 0.2 | 7.8 ± 0.3 |
| ELF, μM | 233 ± 28.3 | 847 ± 25.3* | 36.4 ± 5.5 | 45.4 ± 11.0 | 6.7 ± 1.4 | 20.1 ± 4.4* |
| BAL cells, nmol/mg protein | 7.7 ± 0.5 | 11.7 ± 0.6* | 0.7 ± 0.09 | 0.9 ± 0.07 | 8.9 ± 0.9 | 12.8 ± 0.2* |
Values are means ± SE.
Significantly different from basal levels (P < 0.05). P values represent statistical significance using Student’s t-test.
Effect of oral GSSG administration in wild-type mice
Wild-type mice received 300 mg/kg of GSSG by gavage, and plasma, lung, ELF, and BAL cell GSH levels were measured 60 min later. There was a 40% increase in plasma GSH concentration in GSSG-treated animals compared with basal levels (Fig. 2A). There was a similar increase in lung GSH levels compared with basal levels (Fig. 2B). There was a 30% increase in ELF GSH levels in GSSG-treated wild-type animals compared with basal levels, although this increase was not as large as those seen with oral GSH treatment at the same time point (Fig. 2C). There was a 50% increase in BAL cell GSH level in GSSG-treated wild-type animals compared with basal levels (Fig. 2D). Overall, these increases in GSH levels from oral GSSG administration were not as great as that observed with oral GSH administration, suggesting that the reduced form is more easily utilized by the body.
Fig. 2.
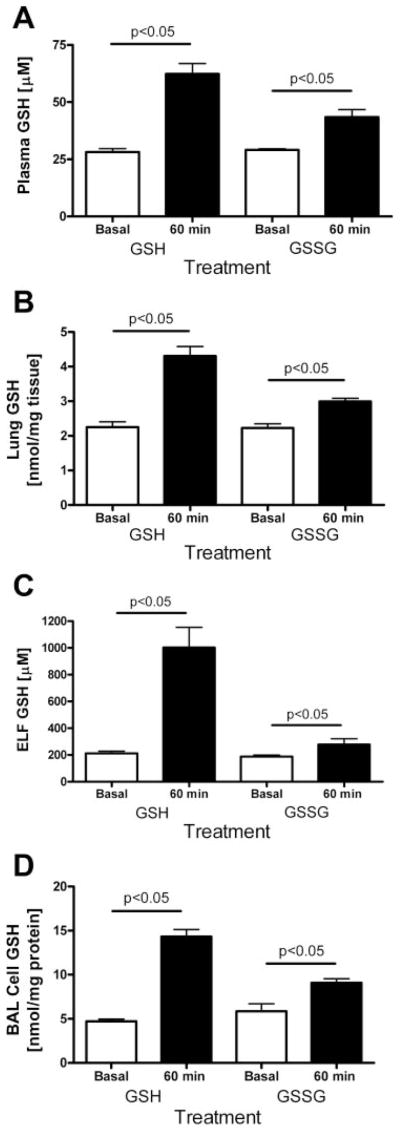
Effect of oral administration (300 mg/kg) of reduced (GSH) or oxidized (GSSG) glutathione on GSH levels in plasma (A), lung tissue (B), lung ELF (C), and lung BAL (D) cells in mice 60 min posttreatment compared with basal levels. Bars indicate statistical comparisons among groups of 3–6 mice.
Effect of oral GSH administration in gut-corrected Cftr KO-Tg and uncorrected Cftr KO mice
We report that oral administration of GSH in wild-type mice can raise GSH levels in different lung compartments, with maximum increases occurring as early as 60 min postdose (Table 1). Since CFTR dysfunction results in decreased steady-state levels of GSH in the ELF (45, 52), we were interested in determining whether oral GSH supplementation can increase GSH levels in different lung compartments of gut-corrected (Cftr-Tg) and uncorrected Cftr KO mice. We chose to examine this effect in both the uncorrected and the CFTR intestinal (gut) corrected mice to address any concerns with intestinal expression of CFTR on GSH bioavailability from an oral dose. Mice received a single bolus GSH dose (300 mg/kg) by gavage and were killed at 60 min posttreatment.
Basal plasma GSH levels were similar between wild-type and gut-corrected Cftr-Tg KO mice with the uncorrected Cftr KO mice having slightly lower basal plasma levels that did not reach statistical significance (Fig. 3). Wild-type mice had a twofold increase in plasma GSH levels 60 min postdose, whereas the gut-corrected Cftr KO-Tg mice had a much smaller 40% increase. The uncorrected Cftr KO mice showed no change in plasma GSH levels, suggesting an important role of CFTR in the bioavailability of GSH in the alimentary tract.
Fig. 3.
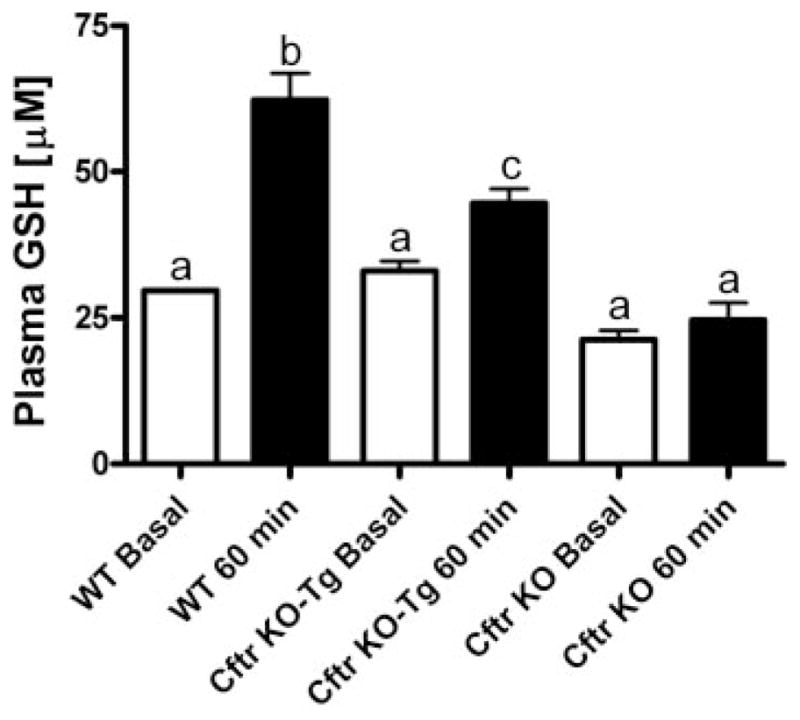
Effect of oral GSH (300 mg/kg) on plasma GSH levels in wild-type (WT), gut-corrected Cftr knockout-transgenic (KO-Tg), and uncorrected Cftr KO mice 60 min posttreatment (closed bars). Open bars are basal plasma GSH steady-state levels. Groups contained 3–6 animals. Bars with different letters (a–c) indicate statistical significant differences, P < 0.05.
Next, we examined changes in lung tissue GSH levels between wild-type, gut-corrected Cftr KO-Tg and uncorrected Cftr KO mice 60 min postoral GSH treatment. Basal lung GSH levels were similar between wild-type and both the gut-corrected Cftr KO-Tg and uncorrected Cftr KO mice (Fig. 4). There was a twofold increase in lung GSH levels in the wild-type lung content 60 min after GSH treatment. In comparison, the gut-corrected Cftr KO-Tg mice had only a 60% increase in lung GSH levels after GSH treatment. We did not see any significant increases in the lung GSH levels after GSH treatment in the uncorrected Cftr KO mice. These data are consistent with the lack of change in plasma GSH in the uncorrected Cftr KO mice.
Fig. 4.
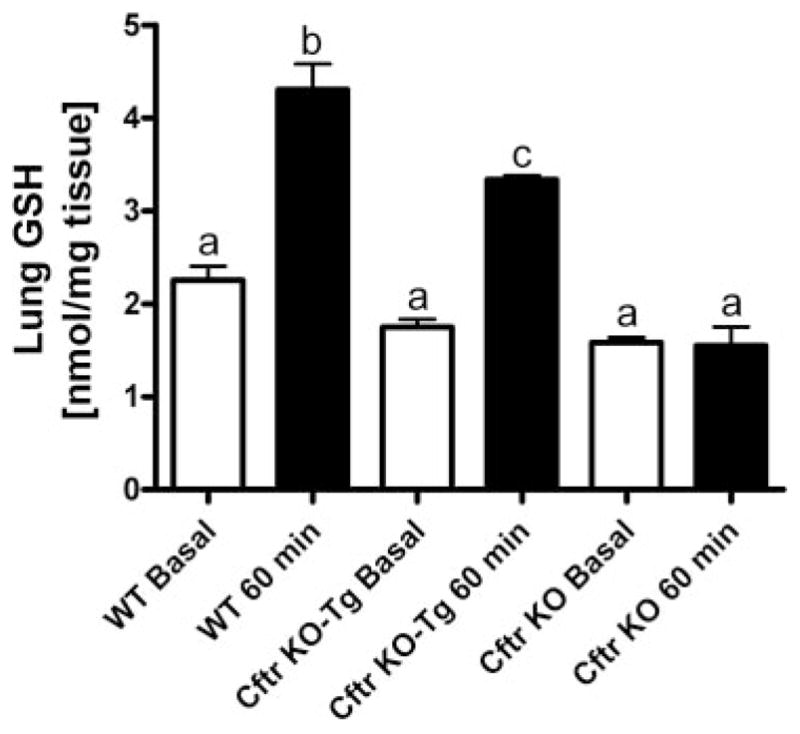
Effect of oral GSH (300 mg/kg) on lung GSH levels in WT, gut-corrected Cftr KO-Tg, and uncorrected Cftr KO mice 60 min posttreatment (closed bars). Open bars are basal lung GSH steady-state levels. Groups contained 3–6 animals. Bars with different letters (a–c) indicate statistical significant differences, P < 0.05.
Changes in the lung ELF GSH content were compared among wild-type, gut-corrected Cftr KO-Tg, and uncorrected Cftr KO mice 60 min following oral administration of GSH. Basal ELF GSH levels were 50% lower in both the gut-corrected and uncorrected Cftr KO mice compared with the wild-type mice (Fig. 5). Oral GSH treatment produced a nearly sixfold increase in ELF GSH concentrations in wild-type mice. In comparison, there was only a 50% increase in ELF GSH levels in gut-corrected Cftr KO-Tg compared with basal levels. Interestingly, the uncorrected Cftr KO mice failed to show any increases in ELF GSH levels after oral GSH treatment. To rule out possible loss of observed GSH from the artificial formation of S-nitrosoglutatione from acidified nitrite, we measured the basal ELF nitrite levels in wild-type, gut-corrected Cftr KO-Tg, and uncorrected Cftr KO mice. There were no significant changes in the ELF nitrite levels among different mice strains (data not shown).
Fig. 5.
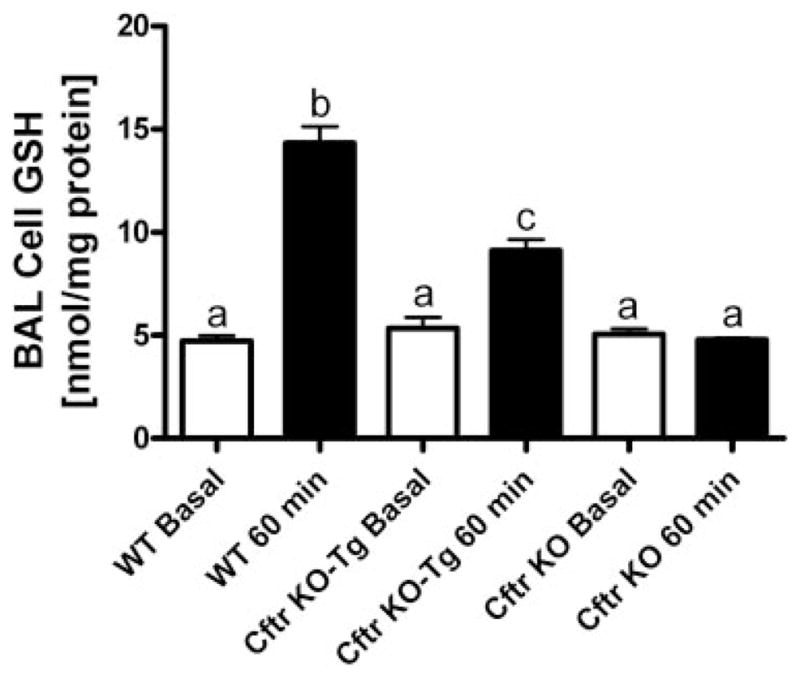
Effect of oral GSH (300 mg/kg) on lung BAL cell GSH levels in WT, gut-corrected Cftr KO-Tg, and uncorrected Cftr KO mice 60 min posttreatment (closed bars). Open bars are basal BAL cell GSH steady-state levels. Groups contained 3–6 animals. Bars with different letters (a–c) indicate statistical significant differences, P < 0.05.
Finally, we looked at changes in GSH levels in BAL cells among wild-type, gut-corrected, and uncorrected Cftr KO mice 60 min after GSH dosing. Basal BAL cell GSH levels were similar between the wild-type mice and both the gut-corrected and uncorrected Cftr KO mice (Fig. 6). Oral GSH treatment produced a threefold increase in BAL cell GSH levels in the wild-type mice but only a twofold increase in the gut-corrected Cftr KO-Tg mice. Again, no changes in BAL cell GSH content were observed in the uncorrected Cftr KO mice after oral GSH treatment.
Fig. 6.
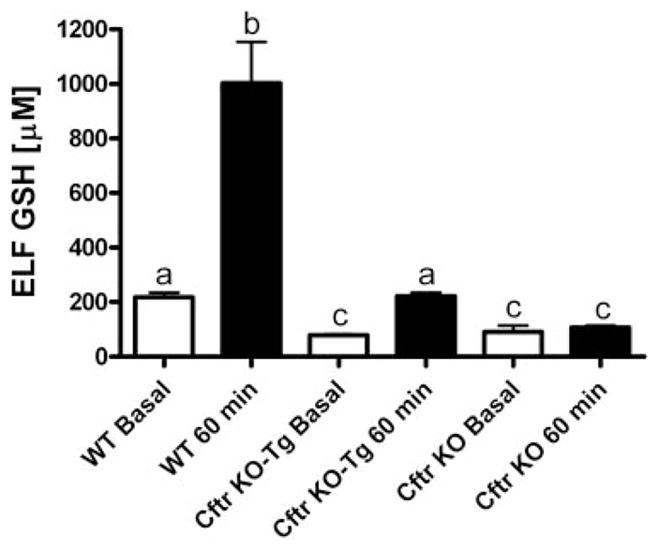
Effect of oral GSH (300 mg/kg) on lung ELF GSH levels in WT, gut-corrected Cftr KO-Tg, and uncorrected Cftr KO mice 60 min posttreatment (closed bars). Open bars are basal ELF GSH steady-state levels. Groups contained 3–6 animals. Bars with different letters (a–c) indicate statistical significant differences, P < 0.05.
DISCUSSION
It has been hypothesized that the ELF GSH deficiency plays an important role in pathophysiology of a number of a pulmonary disorders including IPF, CF, ARDS, and acquired immunodeficiency syndrome (28). Hence, it is logical to study whether oral administration of GSH would be beneficial in increasing GSH levels in lung compartments. Previous studies have shown that oral GSH administration can raise serum and lung tissue GSH levels in rodents (2, 18) but did not measure pharmacokinetic parameters or examine compartments within the lung. The present studies have created a pharmacokinetic profile for oral GSH administration in serum and lung compartments of wild-type mice and examined the role of CFTR in these processes. An important assumption one needs to make is that these changes in the GSH content are from the exogenously administered GSH. We acknowledge that the observed changes reported herein are steady-state levels and are likely a combination of GSH breakdown, resynthesis, and transport. The major finding of our current investigation is that GSH concentrations can be significantly increased in plasma, lung tissue, ELF, and BAL cells after oral administration, and this is dependent on functional lung and intestinal CFTR. Our studies also suggest that GSH levels in BAL cells may change in relationship to the GSH concentration in the ELF. Further studies with radiolabeled GSH will be required to tease apart the importance of these pathways to the overall observed changes in lung GSH reported herein.
There was a significant increase in plasma GSH levels in wild-type mice 1 h following the oral administration of both reduced and oxidized forms of GSH. This finding is in agreement with previous studies where oral GSH administration increased plasma GSH levels in mice and rats (2, 18). This is in contrast to the reported lack of an increase in plasma GSH levels from oral GSH administration in the human (55). Possible explanations for these discrepancies are the much lower dose of GSH used in the human studies compared with rodents (50 mg/kg vs. 100–300 mg/kg, respectively) and that there may be species specific differences in intestinal transport processes. The gut-corrected Cftr KO-Tg mice had about one-half of the increase in plasma GSH levels 1 h following oral GSH administration as seen in wild-type mice. The noncorrected Cftr KO mice were unable to raise plasma GSH levels after oral GSH administration. A potential explanation for only a partial increase of plasma GSH in the gut-corrected Cftr KO-Tg mice is that unlike endogenous CFTR, the Cftr transgene is not expressed in crypts, but in villi, of Cftr KO-Tg mice (57). Overall, these data imply that having a functional CFTR in the intestine is very important for the utilization of dietary GSH and that malabsorption and/or utilization of dietary antioxidants, including GSH, may set up for a systemic oxidative stress in CF.
Previous studies have shown that lung GSH levels are not significantly different in Cftr KOs from wild-type mice (52). We show a significant increase in lung GSH levels in wild-type animals and in gut-corrected Cftr KO-Tg mice, but not in uncorrected Cftr KO after oral GSH administration. The CF lung is under chronic oxidative stress due to exogenous and endogenous generation of free radicals (8, 16). Previous studies in our laboratory have shown that Cftr KO mice have some basal oxidative stress as indicated by an increase in the oxidation of DNA and the increased activities of antioxidant enzymes, such as glutathione peroxidases and glutathione disulfide reductase (52). This increased demand on GSH supply may limit GSH availability to maintain steady-state and adaptive GSH levels in the ELF.
ELF GSH and other antioxidants serve as a first point of defense against free radicals and help to maintain the cellular redox balance. The redox status of a cell is an important determinant in modulation of inflammatory responses (38). Any deficiency in GSH can increase the steady-state level of oxidants that may lead to tissue destruction. Previous studies have shown that ELF GSH levels are low in Cftr KO mice compared with wild-type mice, and this deficiency is associated with increased oxidative stress (51). P. aeruginosa infections are associated with lung neutrophilia that upon activation release large amounts of myleoperoxidase (35, 48, 54). Myleoperoxidase has antimicrobial effects due to its formation of strong oxidants such as hypochlorous acid. GSH is a very effective scavenger of hypochlorous acid (39). Our laboratory has shown that infections with P. aeruginosa can increase ELF GSH levels in wild-type animals to counter the oxidative stress induced by this pathogen. However, this adaptive response is severely blunted in Cftr KO animals, resulting in oxidative stress (15) and increased mortality (26). Overall, these studies point out the importance of ELF GSH in maintaining normal lung epithelium homeostasis and the possible benefits of methods to augment ELF GSH levels.
It is known that GSH plays a prominent role in immune responses (17, 23, 29, 38), extracellular matrix remodeling (50, 56), and in inflammatory responses (43). Chronic bronchopulmonary infection is a hallmark feature in CF patients. A number of investigators have implicated a defective host defense response in CF (19, 40, 53). To clear microorganisms, neutrophils are recruited, which can release reactive oxygen species causing further depletion of antioxidants and oxidative stress. Previous studies have shown that GSH plays an important role in modulating inflammatory and immune responses (42). Inhalation of GSH has been shown to decrease the levels of superoxide generated in BAL cells from CF subjects (44). GSH also helps in T cell activation and proliferation, important immune responses needed to adequately clear microbial infections. Also, the literature suggests that lymphocyte activation is dependent on GSH levels (29). Interestingly, in our study, we observed that BAL cell GSH levels could be raised following oral GSH administration in wild-type and gut-corrected Cftr KO-Tg mice. This increase correlated well with increases in ELF GSH levels in both strains. However, uncorrected Cftr KO mice failed to raise their ELF cell GSH levels. This novel observation suggests that functional CFTR not only plays an important role in transporting GSH from the lung into the ELF but also may indirectly affect the GSH levels in BAL cells.
Given the number of lung disorders that have low basal ELF GSH levels, the rationale of increasing GSH is logical. Numerous studies have been performed to increase the lung GSH levels using GSH or its precursors, including N-acetyl cysteine or cysteine esters (6, 7, 33). N-acetyl cysteine did not increase the GSH concentrations significantly in the plasma or in the lung compartment of chronic obstructive pulmonary disorder patients compared with controls even at the high dose of 600 mg three times daily for 5 days (7). Attempts have also been made to increase the ELF GSH by means of aerosol delivery (3, 4, 9, 22, 27, 44), intravenous administration (9, 30), and oral administration of GSH (2, 18, 55). Inhalation of GSH increased the ELF GSH levels, but it also increased the levels of GSSG (44). Interestingly, we observed that oral administration of GSH could effectively elevate ELF GSH levels without associated increased GSSG levels. The increase in the levels of GSSG could be problematic in asthmatics, because GSSG can exacerbate the bronchiole constriction. In addition, aerosolized GSH was not found to change the levels of oxidized proteins in BAL from CF (22). We observed a nearly sixfold increase in ELF GSH levels in wild-type animals at 60 min posttreatment, suggesting that it is possible to raise the ELF GSH levels via oral administration. It was also possible to increase the GSH levels in ELF of Cftr KO-Tg animals, but to a lesser extent. In contrast, the uncorrected Cftr KO mice failed to show any increase in ELF GSH over wild-type animals. Nutritional status of CF patients has long been known to affect mortality and morbidity. Investigators have shown that dietary supplementation with whey products can affect GSH status in CF subjects (21). Both the reduced and oxidized forms of GSH when given orally increase plasma and BAL cell GSH levels. GSH is the most abundant thiol in plants and animals and would make up a large amount of dietary cysteine in our daily diet. Studies have also shown that GSH levels have diurnal fluctuations that may be linked to rodent feeding patterns (30). It is tempting to speculate that diet may contribute to one’s GSH status and that GSH could be utilized directly or indirectly from our diet. These findings also raise a question on whether CF subjects can be effectively treated by only correcting lung CFTR deficiency.
In summary, this study indicates that oral GSH administration can increase plasma and lung compartment GSH levels in wild-type mice and to a lesser extent in gut-corrected Cftr KO-Tg animals. It also suggests that oral GSH treatment can boost BAL cell GSH levels. However, since this study failed to show significant increases in serum and lung compartment GSH levels in uncorrected Cftr KO mice, it is questionable whether oral GSH administration to CF patients with intestinal malabsorption would benefit from this therapy. We also show that GSH is rapidly distributed to the serum and lung compartments. We speculate that in addition to CFTR, there are other transporter(s) responsible for transporting GSH and probably other important dietary molecules to the lung, which may be responsible for dietary deficiencies observed in various lung diseases.
Acknowledgments
GRANTS
These studies were supported in part by funding from a Cystic Fibrosis Foundation grant (B. J. Day) and National Institutes of Health Grants RO1-HL-075523 (B. J. Day) and P30-DK-027651 (A. VH).
References
- 1.Accurso FJ. Update in cystic fibrosis 2005. Am J Respir Crit Care Med. 2006;173:944–947. doi: 10.1164/rccm.2601006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aw TY, Wierzbicka G, Jones DP. Oral glutathione increases tissue glutathione in vivo. Chem Biol Interact. 1991;80:89–97. doi: 10.1016/0009-2797(91)90033-4. [DOI] [PubMed] [Google Scholar]
- 3.Bishop C, Hudson VM, Hilton SC, Wilde C. A pilot study of the effect of inhaled buffered reduced glutathione on the clinical status of patients with cystic fibrosis. Chest. 2005;127:308–317. doi: 10.1378/chest.127.1.308. [DOI] [PubMed] [Google Scholar]
- 4.Borok Z, Buhl R, Grimes GJ, Bokser AD, Hubbard RC, Holroyd KJ, Roum JH, Czerski DB, Cantin AM, Crystal RG. Effect of glutathione aerosol on oxidant-antioxidant imbalance in idiopathic pulmonary fibrosis. Lancet. 1991;338:215–216. doi: 10.1016/0140-6736(91)90350-x. [DOI] [PubMed] [Google Scholar]
- 5.Bowler RP, Velsor LW, Duda B, Chan ED, Abraham E, Ware LB, Matthay MA, Day BJ. Pulmonary edema fluid antioxidants are depressed in acute lung injury. Crit Care Med. 2003;31:2309–2315. doi: 10.1097/01.CCM.0000085090.06078.8C. [DOI] [PubMed] [Google Scholar]
- 6.Bridgeman MM, Marsden M, MacNee W, Flenley DC, Ryle AP. Cysteine and glutathione concentrations in plasma and bronchoalveolar lavage fluid after treatment with N-acetylcysteine. Thorax. 1991;46:39–42. doi: 10.1136/thx.46.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bridgeman MM, Marsden M, Selby C, Morrison D, MacNee W. Effect of N-acetyl cysteine on the concentrations of thiols in plasma, bronchoalveolar lavage fluid, and lung tissue. Thorax. 1994;49:670–675. doi: 10.1136/thx.49.7.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown RK, Kelly FJ. Evidence for increased oxidative damage in patients with cystic fibrosis. Pediatr Res. 1994;36:487–493. doi: 10.1203/00006450-199410000-00013. [DOI] [PubMed] [Google Scholar]
- 9.Buhl R, Vogelmeier C, Critenden M, Hubbard RC, Hoyt RF, Jr, Wilson EM, Cantin AM, Crystal RG. Augmentation of glutathione in the fluid lining the epithelium of the lower respiratory tract by directly administering glutathione aerosol. Proc Natl Acad Sci USA. 1990;87:4063–4067. doi: 10.1073/pnas.87.11.4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cantin AM, Hubbard RC, Crystal RG. Glutathione deficiency in the epithelial lining fluid of the lower respiratory tract in idiopathic pulmonary fibrosis. Am Rev Respir Dis. 1989;139:370–372. doi: 10.1164/ajrccm/139.2.370. [DOI] [PubMed] [Google Scholar]
- 11.Cantin AM, North SL, Hubbard RC, Crystal RG. Normal alveolar epithelial lining fluid contains high levels of glutathione. J Appl Physiol. 1987;63:152–157. doi: 10.1152/jappl.1987.63.1.152. [DOI] [PubMed] [Google Scholar]
- 12.Chinard FP. Quantitative assessment of epithelial lining fluid in the lung. Am J Physiol Lung Cell Mol Physiol. 1992;263:L617–L618. doi: 10.1152/ajplung.1992.263.6.L617. [DOI] [PubMed] [Google Scholar]
- 13.Comhair SA, Lewis MJ, Bhathena PR, Hammel JP, Erzurum SC. Increased glutathione and glutathione peroxidase in lungs of individuals with chronic beryllium disease. Am J Respir Crit Care Med. 1999;159:1824–1829. doi: 10.1164/ajrccm.159.6.9810044. [DOI] [PubMed] [Google Scholar]
- 14.Dauletbaev N, Viel K, Buhl R, Wagner TO, Bargon J. Glutathione and glutathione peroxidase in sputum samples of adult patients with cystic fibrosis. J Cyst Fibros. 2004;3:119–124. doi: 10.1016/j.jcf.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 15.Day BJ, van Heeckeren AM, Min E, Velsor LW. Role for cystic fibrosis transmembrane conductance regulator protein in a glutathione response to bronchopulmonary Pseudomonas infection. Infect Immun. 2004;72:2045–2051. doi: 10.1128/IAI.72.4.2045-2051.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dominguez C, Gartner S, Linan S, Cobos N, Moreno A. Enhanced oxidative damage in cystic fibrosis patients. Biofactors. 1998;8:149–153. doi: 10.1002/biof.5520080125. [DOI] [PubMed] [Google Scholar]
- 17.Droge W, Breitkreutz R. Glutathione and immune function. Proc Nutr Soc. 2000;59:595–600. doi: 10.1017/s0029665100000847. [DOI] [PubMed] [Google Scholar]
- 18.Favilli F, Marraccini P, Iantomasi T, Vincenzini MT. Effect of orally administered glutathione on glutathione levels in some organs of rats: role of specific transporters. Br J Nutr. 1997;78:293–300. doi: 10.1079/bjn19970147. [DOI] [PubMed] [Google Scholar]
- 19.Fick RB, Jr, Sonoda F, Hornick DB. Emergence and persistence of Pseudomonas aeruginosa in the cystic fibrosis airway. Semin Respir Infect. 1992;7:168–178. [PubMed] [Google Scholar]
- 20.Gao L, Kim KJ, Yankaskas JR, Forman HJ. Abnormal glutathione transport in cystic fibrosis airway epithelia. Am J Physiol Lung Cell Mol Physiol. 1999;277:L113–L118. doi: 10.1152/ajplung.1999.277.1.L113. [DOI] [PubMed] [Google Scholar]
- 21.Grey V, Mohammed SR, Smountas AA, Bahlool R, Lands LC. Improved glutathione status in young adult patients with cystic fibrosis supplemented with whey protein. J Cyst Fibros. 2003;2:195–198. doi: 10.1016/S1569-1993(03)00097-3. [DOI] [PubMed] [Google Scholar]
- 22.Griese M, Ramakers J, Krasselt A, Starosta V, Van Koningsbruggen S, Fischer R, Ratjen F, Mullinger B, Huber RM, Maier K, Rietschel E, Scheuch G. Improvement of alveolar glutathione and lung function but not oxidative state in cystic fibrosis. Am J Respir Crit Care Med. 2004;169:822–828. doi: 10.1164/rccm.200308-1104OC. [DOI] [PubMed] [Google Scholar]
- 23.Grimble RF. The effects of sulfur amino acid intake on immune function in humans. J Nutr. 2006;136:1660S–1665S. doi: 10.1093/jn/136.6.1660S. [DOI] [PubMed] [Google Scholar]
- 24.Guilbault C, Saeed Z, Downey GP, Radzioch D. Cystic fibrosis mouse models. Am J Respir Cell Mol Biol. 2007;36:1–7. doi: 10.1165/rcmb.2006-0184TR. [DOI] [PubMed] [Google Scholar]
- 25.Gupta S, Rogers LK, Taylor SK, Smith CV. Inhibition of carbamyl phosphate synthetase-I and glutamine synthetase by hepatotoxic doses of acetaminophen in mice. Toxicol Appl Pharmacol. 1997;146:317–327. doi: 10.1006/taap.1997.8228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heeckeren A, Walenga R, Konstan MW, Bonfield T, Davis PB, Ferkol T. Excessive inflammatory response of cystic fibrosis mice to bronchopulmonary infection with Pseudomonas aeruginosa. J Clin Invest. 1997;100:2810–2815. doi: 10.1172/JCI119828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holroyd KJ, Buhl R, Borok Z, Roum JH, Bokser AD, Grimes GJ, Czerski D, Cantin AM, Crystal RG. Correction of glutathione deficiency in the lower respiratory tract of HIV seropositive individuals by glutathione aerosol treatment. Thorax. 1993;48:985–989. doi: 10.1136/thx.48.10.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hudson VM. Rethinking cystic fibrosis pathology: the critical role of abnormal reduced glutathione (GSH) transport caused by CFTR mutation. Free Radic Biol Med. 2001;30:1440–1461. doi: 10.1016/s0891-5849(01)00530-5. [DOI] [PubMed] [Google Scholar]
- 29.Iwata S, Hori T, Sato N, Ueda-Taniguchi Y, Yamabe T, Nakamura H, Masutani H, Yodoi J. Thiol-mediated redox regulation of lymphocyte proliferation. Possible involvement of adult T cell leukemia-derived factor and glutathione in transferrin receptor expression. J Immunol. 1994;152:5633–5642. [PubMed] [Google Scholar]
- 30.Jaeschke H, Wendel A. Diurnal fluctuation and pharmacological alteration of mouse organ glutathione content. Biochem Pharmacol. 1985;34:1029–1033. doi: 10.1016/0006-2952(85)90606-9. [DOI] [PubMed] [Google Scholar]
- 31.Kelly FJ. Gluthathione: in defence of the lung. Food Chem Toxicol. 1999;37:963–966. doi: 10.1016/s0278-6915(99)00087-3. [DOI] [PubMed] [Google Scholar]
- 32.Kogan I, Ramjeesingh M, Li C, Kidd JF, Wang Y, Leslie EM, Cole SP, Bear CE. CFTR directly mediates nucleotide-regulated glutathione flux. EMBO J. 2003;22:1981–1989. doi: 10.1093/emboj/cdg194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lailey AF, Upshall DG. Thiol levels in rat bronchioalveolar lavage fluid after administration of cysteine esters. Hum Exp Toxicol. 1994;13:776–780. doi: 10.1177/096032719401301106. [DOI] [PubMed] [Google Scholar]
- 34.Linsdell P, Hanrahan JW. Glutathione permeability of CFTR. Am J Physiol Cell Physiol. 1998;275:C323–C326. doi: 10.1152/ajpcell.1998.275.1.C323. [DOI] [PubMed] [Google Scholar]
- 35.Meyer KC, Zimmerman J. Neutrophil mediators, Pseudomonas, and pulmonary dysfunction in cystic fibrosis. J Lab Clin Med. 1993;121:654–661. [PubMed] [Google Scholar]
- 36.Pacht ER, Diaz P, Clanton T, Hart J, Gadek JE. Alveolar fluid glutathione decreases in asymptomatic HIV-seropositive subjects over time. Chest. 1997;112:785–788. doi: 10.1378/chest.112.3.785. [DOI] [PubMed] [Google Scholar]
- 37.Pacht ER, Timerman AP, Lykens MG, Merola AJ. Deficiency of alveolar fluid glutathione in patients with sepsis and the adult respiratory distress syndrome. Chest. 1991;100:1397–1403. doi: 10.1378/chest.100.5.1397. [DOI] [PubMed] [Google Scholar]
- 38.Paolicchi A, Dominici S, Pieri L, Maellaro E, Pompella A. Glutathione catabolism as a signaling mechanism. Biochem Pharmacol. 2002;64:1027–1035. doi: 10.1016/s0006-2952(02)01173-5. [DOI] [PubMed] [Google Scholar]
- 39.Peskin AV, Winterbourn CC. Kinetics of the reactions of hypochlorous acid and amino acid chloramines with thiols, methionine, and ascorbate. Free Radic Biol Med. 2001;30:572–579. doi: 10.1016/s0891-5849(00)00506-2. [DOI] [PubMed] [Google Scholar]
- 40.Pier GB, Grout M, Zaidi TS, Olsen JC, Johnson LG, Yankaskas JR, Goldberg JB. Role of mutant CFTR in hypersusceptibility of cystic fibrosis patients to lung infections. Science. 1996;271:64–67. doi: 10.1126/science.271.5245.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pilewski JM, Frizzell RA. Role of CFTR in airway disease. Physiol Rev. 1999;79:S215–S255. doi: 10.1152/physrev.1999.79.1.S215. [DOI] [PubMed] [Google Scholar]
- 42.Rahman I, MacNee W. Lung glutathione and oxidative stress: implications in cigarette smoke-induced airway disease. Am J Physiol Lung Cell Mol Physiol. 1999;277:L1067–L1088. doi: 10.1152/ajplung.1999.277.6.L1067. [DOI] [PubMed] [Google Scholar]
- 43.Rahman I, MacNee W. Oxidative stress and regulation of glutathione in lung inflammation. Eur Respir J. 2000;16:534–554. doi: 10.1034/j.1399-3003.2000.016003534.x. [DOI] [PubMed] [Google Scholar]
- 44.Roum JH, Borok Z, McElvaney NG, Grimes GJ, Bokser AD, Buhl R, Crystal RG. Glutathione aerosol suppresses lung epithelial surface inflammatory cell-derived oxidants in cystic fibrosis. J Appl Physiol. 1999;87:438–443. doi: 10.1152/jappl.1999.87.1.438. [DOI] [PubMed] [Google Scholar]
- 45.Roum JH, Buhl R, McElvaney NG, Borok Z, Crystal RG. Systemic deficiency of glutathione in cystic fibrosis. J Appl Physiol. 1993;75:2419–2424. doi: 10.1152/jappl.1993.75.6.2419. [DOI] [PubMed] [Google Scholar]
- 46.Slade R, Crissman K, Norwood J, Hatch G. Comparison of antioxidant substances in bronchoalveolar lavage cells and fluid from humans, guinea pigs, and rats. Exp Lung Res. 1993;19:469–484. doi: 10.3109/01902149309064358. [DOI] [PubMed] [Google Scholar]
- 47.Snouwaert JN, Brigman KK, Latour AM, Malouf NN, Boucher RC, Smithies O, Koller BH. An animal model for cystic fibrosis made by gene targeting. Science. 1992;257:1083–1088. doi: 10.1126/science.257.5073.1083. [DOI] [PubMed] [Google Scholar]
- 48.Suntres ZE, Omri A, Shek PN. Pseudomonas aeruginosa-induced lung injury: role of oxidative stress. Microb Pathog. 2002;32:27–34. doi: 10.1006/mpat.2001.0475. [DOI] [PubMed] [Google Scholar]
- 49.van Heeckeren AM, Schluchter M, Xue L, Alvarez J, Freedman S, St George J, Davis PB. Nutritional effects on host response to lung infections with mucoid Pseudomonas aeruginosa in mice. Infect Immun. 2004;72:1479–1486. doi: 10.1128/IAI.72.3.1479-1486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vayalil PK, Olman M, Murphy-Ullrich JE, Postlethwait EM, Liu RM. Glutathione restores collagen degradation in TGF-β-treated fibroblasts by blocking plasminogen activator inhibitor-1 expression and activating plasminogen. Am J Physiol Lung Cell Mol Physiol. 2005;289:L937–L945. doi: 10.1152/ajplung.00150.2005. [DOI] [PubMed] [Google Scholar]
- 51.Velsor LW, Kariya C, Kachadourian R, Day BJ. Mitochondrial oxidative stress in the lungs of cystic fibrosis transmembrane conductance regulator protein mutant mice. Am J Respir Cell Mol Biol. 2006;35:579–586. doi: 10.1165/rcmb.2005-0473OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Velsor LW, van Heeckeren A, Day BJ. Antioxidant imbalance in the lungs of cystic fibrosis transmembrane conductance regulator protein mutant mice. Am J Physiol Lung Cell Mol Physiol. 2001;281:L31–L38. doi: 10.1152/ajplung.2001.281.1.L31. [DOI] [PubMed] [Google Scholar]
- 53.Wilson GB, Fudenberg HH. Does a primary host defense abnormality involving monocytes-macrophages underlie the pathogenesis of lung disease in cystic fibrosis? Med Hypotheses. 1982;8:527–542. doi: 10.1016/0306-9877(82)90014-7. [DOI] [PubMed] [Google Scholar]
- 54.Witko-Sarsat V, Delacourt C, Rabier D, Bardet J, Nguyen AT, Descamps-Latscha B. Neutrophil-derived long-lived oxidants in cystic fibrosis sputum. Am J Respir Crit Care Med. 1995;152:1910–1916. doi: 10.1164/ajrccm.152.6.8520754. [DOI] [PubMed] [Google Scholar]
- 55.Witschi A, Reddy S, Stofer B, Lauterburg BH. The systemic availability of oral glutathione. Eur J Clin Pharmacol. 1992;43:667–669. doi: 10.1007/BF02284971. [DOI] [PubMed] [Google Scholar]
- 56.Yumei F, Zhou Y, Zheng S, Chen A. The antifibrogenic effect of (−)–epigallocatechin gallate results from the induction of de novo synthesis of glutathione in passaged rat hepatic stellate cells. Lab Invest. 2006;86:697–709. doi: 10.1038/labinvest.3700425. [DOI] [PubMed] [Google Scholar]
- 57.Zhou L, Dey CR, Wert SE, DuVall MD, Frizzell RA, Whitsett JA. Correction of lethal intestinal defect in a mouse model of cystic fibrosis by human CFTR. Science. 1994;266:1705–1708. doi: 10.1126/science.7527588. [DOI] [PubMed] [Google Scholar]