

Abstract
We have previously shown that 1,2,3-triazole ureas (1,2,3-TUs) act as versatile class of irreversible serine hydrolase inhibitors that can be tuned to create selective probes for diverse members of this large enzyme class, including diacylglycerol lipase-β (DAGLβ), a principal biosynthetic enzyme for the endocannabinoid 2-arachidonoylglycerol (2-AG). Here, we provide a detailed account of the discovery, synthesis, and structure-activity relationship (SAR) of (2-substituted)-piperidyl-1,2,3-TUs that selectively inactivate DAGLβ in living systems. Key to success was the use of activity-based protein profiling (ABPP) with broad-spectrum and tailored activity-based probes to guide our medicinal chemistry efforts. We also describe an expanded repertoire of DAGL-tailored activity-based probes that includes biotinylated and alkyne agents for enzyme enrichment coupled with mass spectrometry-based proteomics and assessment of proteome-wide selectivity. Our findings highlight the broad utility of 1,2,3-TUs for serine hydrolase inhibitor development and their application to create selective probes of endocannabinoid biosynthetic pathways.
Introduction
Serine hydrolases (SHs) represent one of the largest and most diverse enzyme families in Nature. The 200+ human members of this enzyme class catalyze the hydrolysis of small-molecule transmitters, lipids, peptides, and proteins1, 2 and have emerged as therapeutic targets for several clinically approved drugs that treat obesity,3 type 2 diabetes,3, 4 and cognitive disorders.5 Despite their pervasive roles in biology, most mammalian SHs remain poorly characterized with respect to their biochemical and physiological functions. The development of selective inhibitors to probe the function of individual SHs in living systems would be of great value, but this goal has only been accomplished for a limited number of SH targets.6-12
We have shown that 1,2,3-triazole ureas (1,2,3-TUs) serve as a versatile scaffold for developing selective inhibitors of SHs.8 1,2,3-TUs inhibit SHs by an irreversible mechanism involving carbamylation of the active-site serine nucleophile (Supplementary Figure 1). We recently reported the development of potent and selective inhibitors of diacylglycerol lipase-β (DAGLβ) based on a (2-substituted)-piperidyl (Pip)-1,2,3-TU scaffold.13, 14 DAGLβ and DAGLα are sequence-related SHs that produce the endocannabinoid, 2-arachidonoylglycerol (2-AG).15-17 The development of selective, in vivo-active inhibitors targeting DAGLβ enabled the functional analysis of this enzyme in peritoneal macrophages where it regulates an endocannabinoideicosanoid network involved in inflammatory responses.13
Here, we provide a full account of the discovery, synthesis, structure-activity relationship (SAR), and inhibitory activities of (2-substituted)-Pip-1,2,3-TUs that culminated in the discovery of the DAGLβ-selective inhibitor 11 (KT109) and the dual DAGLα/β inhibitor 27 (KT172).13 We also describe the synthesis of alkyne and biotin-coupled 1,2,3-TUs and their use as tailored activity-based probes for DAGL enzymes. Finally, we show that structurally related (2-phenyl)-Pip-1,2,3-TUs do not inhibit DAGLβ, but maintain potency against the off-target enzyme ABHD6, thus serving as useful negative-control probes for DAGLβ-related studies, as well as a novel scaffold for developing in vivo-active inhibitors of ABHD6.18
Results
Discovery and characterization of (2-substituted)-Pip-1,2,3-TUs as DAGLβ inhibitors
As described in our previous studies,13 we found that the 1,4-regioisomer of (2-substituted)-Pip-1,2,3-TUs, such as 2, show enhanced potency for DAGLβ compared with the corresponding 2,4-regisomer (3) as measured by competitive activity-based protein profiling (ABPP) (Figure 1). ABPP is a chemoproteomic technology that utilizes small-molecule probes containing: 1) a reactive group that binds and covalently modifies the active sites of a large number of enzymes that share conserved mechanistic and/or structural features; and 2) a reporter tag, such as a fluorophore or biotin, to facilitate detection, enrichment, and identification of probe-labeled enzymes by SDS-PAGE (gel-based ABPP19) and/or quantitative mass spectrometry-based proteomic methods (ABPP-SILAC8, 13). The regioisomers of (2-substituted)-Pip-1,2,3-TUs arise due to acylation at the N1 or N2 position of the triazole ring during triphosgene-mediated urea formation (Scheme 1). Here, we wanted to determine whether enantiomers of 2 show differences in DAGLβ potency and selectivity. We resolved racemic 2 into its enantiomers (2a and b) using chiral-HPLC and found that 2a showed ∼100-fold greater potency than 2b against DAGLβ (Figure 1). We also observed a similar increase in activity against the off-target ABHD6, demonstrating that 2a provides a similar overall selectivity profile as the racemate. We therefore concluded that the racemate is suitable for biological studies, given that the active enantiomer displayed a similar selectivity profile and only a two-fold increase in potency over the racemic mixture.
Figure 1.

Characterization of (2-benzyl)-Pip-1,2,3-TU isomers as DAGLβ inhibitors. The activity of regio- (2 and 3) and stereo- (2a and b) isomers was tested against recombinant DAGLβ expressed in HEK293T cells and endogenous ABHD6 in mouse Neuro2A neuroblastoma cell proteome using a gel-based competitive ABPP assay. Compounds were preincubated with HEK293T-DAGLβ membrane (0.3 mg/mL) or Neuro2A (1 mg/mL) proteomes for 30 min at 37 °C prior to treatment with the activity based probes 38 (HT-01)13 or fluorophosphonate-rhodamine (FP-Rh), respectively (1 μM probe, 30 min) and analysis by gel-based ABPP, where compound activity was measured by reductions in probe-labeling of target SHs. This gel and subsequent fluorescent gels are shown in gray scale.
Scheme 1.

General synthesis of (2-benzyl)-Pip-1,2,3-TUs.
In order to evaluate whether replacement of the triazole ring with other nitrogenous heterocycles would affect activity against DAGLβ, we synthesized the triazole (5), pyrazole (8), and imidazole (9) derivatives of the 2-piperidyl-heterocyclic urea scaffold (Scheme 2). We synthesized compound 5 by direct coupling of 4-(4-(trifluoromethoxy)phenyl)-triazole to 2-benzylpiperidine and purified the 1,4-regioisomer as described above. The heterocyclic derivatives were synthesized by triphosgene-mediated coupling of 2-benzylpiperidine with either 4-bromopyrazole or 4-bromoimidazole followed by Suzuki-coupling with (4-(trifluoromethoxy)phenyl)boronic acid to generate 8 or 9 as single regioisomers. The activities of compounds 5, 8, and 9 against recombinant DAGLβ and other SHs in the mouse brain proteome demonstrated that the triazole (5) showed potent activity against DAGLβ and also inhibited other SHs at higher concentrations, whereas the pyrazole (8) and imidazole (9) derivatives were largely inactive as determined by gel-based ABPP (Table 1). These results indicate that the triazole ring is important for reactivity of (2-benzyl)-Pip-1,2,3-TUs with DAGLβ.
Scheme 2.

Synthesis of (2-benzyl)-Pip-heterocyclic urea derivatives.
Table 1.
Comparison of activity of (2-benzyl)-Pip-heterocyclic urea derivatives.
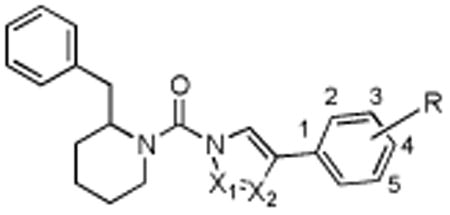
| |||
|---|---|---|---|
|
| |||
| Structure | Compound | DAGLβ % inhibition (100 nM) | Off-targets (10 μM) |
| X1 = N, X2 = N, R = 4-OCF3 | 5 | 52 | FAAH, ABHD6, ABHD11, PLA2G7 |
| X1 = N, X2 = C, R = 4-OCF3 | 8 | 0 | none |
| X1 = C, X2 = N, R = 4-OCF3 | 9 | 0 | none |
| X1 = N, X2 = N, R = 3-5 diF | 10 | 55.4 | FAAH, KIAA1363, ABHD6, LYPLA1, LYPLA2, PLA2G7, ABHD11 |
| X1 = N, X2 = N, R = 4-Ph | 11 (KT109) | 60.8 | ABHD6, PLA2G7 |
| X1 = N, X2 = N, R = 3-Ph | 14 | 27.4 | FAAH, MGLL, ABHD6, 80 kDa SH, ABHD11 |
| X1 = N, X2 = N, R = 2-Ph | 15 | 17.6 | FAAH, MGLL, ABHD6, ABHD11 |
| X1 = N, X2 = N, R = 4-piperidineamide | 19 | 40.9 | FAAH, ABHD6, ABHD11 |
Modifications to the triazole ring enhance potency and selectivity of DAGLβ inhibitors
We next evaluated substitutions to the 4-position of the triazole ring of DAGLβ inhibitors. 4-phenyl-triazoles were synthesized from commercially-available alkynes by copper-catalyzed azide-alkyne cycloaddition reaction20 (CuACC or click chemistry), purified using silica gel chromatography, and coupled to 2-benzyl piperidine to yield compounds 10 – 13 (Scheme 3). Further coupling of compounds 12 and 13 with phenylboronic acid afforded the biphenyl derivatives 14 and 15. Compound 19 was synthesized using commercially available 4-bromo-1,2,3-triazole coupled to 2-benzyl piperidine under standard conditions to generate a TU intermediate that was then treated with 4-(piperidine-1-carbonyl)-phenylboronic acid in the presence of Pd(II) and K2CO3 to provide compound 19 (Scheme 3). Competitive ABPP revealed that compounds 5, 10, and 19 were equipotent against DAGLβ (Figure 2A), but 5 and 19 showed better selectivity compared to 10 (Figure 2B, C). We next synthesized the biphenyl compound 11 and found that it maintained potency against DAGLβ and exhibited improved selectivity compared to 5 and 19 (Figure 2A and Table 1). We also compared the activity of isomers of compound 11 that varied in the position of the distal phenyl ring and found that substitutions at the 3- (14) or 2-position (15) reduced potency against DAGLβ (Figure 2A) or increased off-target activity (Figure 2 and Table 1).
Scheme 3. Synthesis of (2-benzyl)-Pip-1,2,3-TUs with 4-substituted triazoles.

Figure 2.

Potency and selectivity of (2-benzyl)-Pip-1,2,3-TU inhibitors of DAGLβ. (A) Inhibitory activity of compounds (0.1 μM or 10 μM) against recombinant DAGLβ expressed by transient transfection in HEK293T cells (DAGLβ-HEK293T lysates) or recombinant mouse ABHD11 (50 nM) as determined by gel-based competitive ABPP using 38 or FP-Rh, respectively. (B, C) Selectivity of compounds (10 or 1 μM) against mouse brain SHs as measured by gel-based competitive ABPP using FP-Rh (B) or 38 (C). The gel-based ABPP assays were performed as described in Figure 1. Assignment of serine hydrolase enzyme activities in competitive ABPP gels are based on gel migration patterns consistent with past studies.13, 21
Compound 11 still showed potent activity against ABHD6, as well as low-level cross-reactivity with lipoprotein-associated phospholipase A2 (PLA2G7) at higher concentrations (Figure 2C). To improve the selectivity of compound 11 further, we synthesized derivatives with various substituents added to the distal phenyl ring (Scheme 4). All compounds were synthesized using 2 as a common intermediate for subsequent Suzuki coupling reactions. We tested derivatives with a trifluoromethyl substituent at para- (24), meta- (25), and ortho-positions (26) and found that 26 showed the best potency and selectivity (Figure 3 and Table 2). Substitutions at the para-position of compounds (20 and 24) increased PLA2G7 off-target activity with the exception of larger polar groups (21 and 23). However, these larger polar substitutions also reduced potency against DAGLβ (Figure 3 and Table 2), and we therefore focused on smaller substituents and found that addition of a hydroxymethyl group at the ortho-(29) but not meta-position (22) improved potency against DAGLβ (Figure 3). Replacement of the hydroxymethyl with a methoxy substituent (27) eliminated FAAH as an off-target and attenuated PLA2G7 off-target activity while largely retaining potency against DAGLβ (Figure 3 and Table 2). Low-level cross-reactivity with monoacylglycerol lipase (MGLL) was, however, observed with compound 27 when tested at 10 μM (Figure 3B). These results demonstrate that ortho-substitutions of the biphenyl-triazole ring with small, moderately polar groups generate DAGLβ inhibitors with excellent activity and selectivity.
Scheme 4.
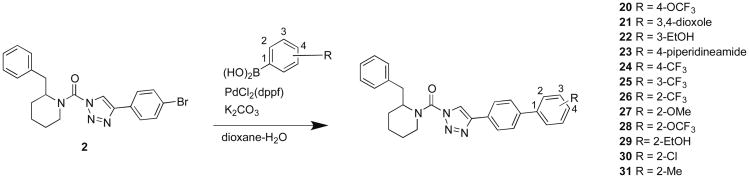
Synthesis of biphenyl derivatives of (2-benzyl)-Pip-1,2,3-TUs.
Figure 3.

Potency and selectivity of biphenyl derivatives of (2-benzyl)-Pip-1,2,3-TUs. (A) Inhibitory activity of compounds (0.1 μM or 10 μM) against recombinant DAGLβ expressed by transient transfection in HEK293T cells (DAGLβ-HEK293T lysates) or recombinant mouse ABHD11 (50 nM) as determined by gel-based competitive ABPP using 38 or FP-Rh, respectively. (B, C) The selectivity of compounds (10 or 1 μM) against mouse brain SHs was measured using gel-based competitive ABPP with FP-Rh (B) or 38 (C). The gel-based ABPP assays were performed as described in Figure 1. Serine hydrolase activities in gels were assigned as described in Figure 2.
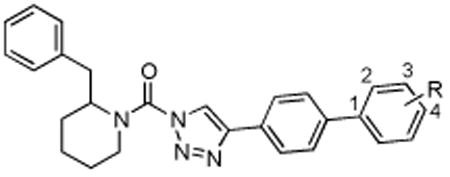
Table 2.
Activity of biphenyl derivatives of 2-benzyl Pip-1,2,3-TUs.
| |||
|---|---|---|---|
|
| |||
| Structure | Compound | DAGLβ % Inhibition (100 nM) | Off-targets (10 μM) |
| 4-OCF3 | 20 | 38.1 | KIAA1363, ABHD6, PLA2G7 |
| 3,4-dioxole | 21 | 17.8 | FAAH, ABHD6 |
| 3-EtOH | 22 | 11.4 | ABHD6 |
| 4-piperidineamide | 23 | 9.8 | ABHD6 |
| 4-CF3 | 24 | 31.1 | FAAH, KIAA1363, ABHD6,PLA2G7 |
| 3-CF3 | 25 | 44.4 | FAAH, ABHD6 |
| 2-CF3 | 26 | 59 | ABHD6 |
| 2-OMe | 27 (KT172) | 55.3 | MGLL, ABHD6 |
| 2-OCF3 | 28 | 59 | MGLL, ABHD6, PLA2G7 |
| 2-EtOH | 29 | 67 | FAAH, MGLL, ABHD6 |
| 2-Cl | 30 | 66.2 | ABHD6, PLA2G7, ABHD11 |
| 2-Me | 31 | 63.2 | ABHD6, PLA2G7, ABHD11 |
In summary, our SAR studies designated compounds 11 and 27 as potent and selective DAGLβ inhibitors. Both compounds inhibited DAGLβ in vitro with IC50 values of 50-80 nM as measured by gel-based ABPP and LC-MS substrate assays.13 Compounds 11 and 27 showed good selectivity with minimal and complementary cross-reactivity against other SHs with only a single shared off-target, ABHD6 (Figure 2B and 3B). Further studies showed that, while compound 27 was equipotent against DAGLα and DAGLβ, 11 displayed enhanced potency for DAGLβ over DAGLα (∼60-fold selectivity) as measured by gel-based ABPP,13 designating these compounds as dual DAGLα/β and DAGLβ-selective inhibitors, respectively. Both compounds displayed excellent potency and selectivity in situ, with near-complete inactivation of DAGLβ (and ABHD6) in Neuro2A cells at concentrations < 50 nM (in situ IC50 values of 11-14 nM) and negligible cross-reactivity with the 45+ additional SHs detected in this cell line by ABPP-SILAC.13 Finally, both compounds inactivated DAGLβ in peritoneal macrophages from mice (1-5 mg/kg of compound, i.p.).13
A clickable analog of compound 27 confirms proteome-wide selectivity for DAGLβ/ABHD6
Our previous competitive ABPP results (both gel- and MS-based analyses)13 showed that the DAGL inhibitors 11 and 27 exhibit excellent selectivity across the SH class, but did not address the possibility that these inhibitors may react with other proteins in the proteome. To determine proteome-wide selectivity, we synthesized analogs of 27 (32 and 33; Figure 4A) that bear an alkyne group to serve as a latent affinity handle suitable for modification by reporter tags using copper-catalyzed azide-alkyne cycloaddition chemistry22 (click chemistry). First, we confirmed that both 32 and 33 retain good inhibitory activity against DAGLβ and ABHD6 as measured by gel-based competitive ABPP in Neuro2A proteomes (Figure 4B). Next, we treated Neuro2A cells with varying concentrations of 32 or 33 for 1 hr. Cells were then lysed and the membrane proteomes conjugated by click chemistry with an azide-Rh tag,23 separated by SDS-PAGE, and probe-labeled proteins visualized by in-gel fluorescence scanning (Figure 4C). This analysis identified two major protein targets of ∼70 and 35 kDa, matching the molecular weights of DAGLβ and ABHD6, respectively, that could be detected at concentrations of 32 or 33 as low as 10 nM (Figure 4C). Good selectivity for DAGLβ and ABHD6 was maintained up to ∼600 nM of the probes, at which point, a handful of additional probe-labeled proteins were detected. Considering that the parent inhibitors 11 and 27 exhibit in situ activities in the 25-50 nM range,13 these data argue that both inhibitors maintain good proteome-wide specificity at concentrations required to inhibit DAGLβ and ABHD6 in cells.
Figure 4.

Structure and activity of clickable analogs of compound 27. (A) Chemical structures of clickable probes 32 and 33. (B) In vitro potency of clickable probes against DAGLβ and ABHD6 in Neuro2A proteome as measured by gel-based competitive ABPP using 38. Neuro2A lysates (1 mg/mL) were incubated with the indicated concentration of compounds (30 min, 37 °C) followed by labeling with 1 μM 38 (30 min, 37 °C). (C) Click chemistry ABPP of Neuro2A cells treated in situ with 32 and 33. Neuro2A cells were treated with the indicated concentrations of compound (1 hr, 37 °C), lysed and labeled proteins in the membrane fraction were visualized by click chemistry reaction with azide-Rh followed by SDS PAGE and in-gel fluorescence scanning. Protein bands corresponding to DAGLβ and ABHD6 bands are labeled. Serine hydrolase activities in gels were assigned as described in Figure 2.
Acyclic phenethyl-1,2,3-TUs as a scaffold for creating DAGL-tailored activity-based probes
In order to determine whether modifications to the carbamoylating portion of (2-substituted)-Pip-1,2,3-TUs would affect potency and selectivity towards DAGLβ, we synthesized acyclic phenethyl-derivatives following the procedure outlined in Scheme 5. Reductive amination of 2-phenylethanamine with aldehydes resulted in secondary amines that were subsequently coupled with 4-(4-(trifluoromethoxy)phenyl)-triazole to yield compounds 35 – 37 (Scheme 5). The acyclic phenethyl-1,2,3-TUs still retained potent activity against recombinant DAGLβ (Figure 5A) with reasonable selectivity against other SHs in the mouse brain proteome as judged by gel-based ABPP (Figure 5B). Depending on the compound, off-target activity was observed for a subset of SHs, including FAAH, KIAA1363, PLA2G7, ABHD6, and an additional unidentified FP-reactive protein (∼80 kDa) (Figure 5C and Table 3). These studies demonstrate that acyclic phenethyl-1,2,3-TUs serve as a suitable scaffold to incorporate diverse substituents, including a bulky Boc-group (compound 37), into the DAGLβ inhibitor scaffold.
Scheme 5.

Synthesis of acyclic phenethyl 1,2,3-TUs.
Figure 5.

Potency and selectivity of acyclic phenethyl-1,2,3-TUs. (A) Inhibitory activity of compounds (0.1 μM) against recombinant DAGLβ expressed by transient transfect in HEK293T cells (DAGLβ-HEK293T lysates) as determined by gel-based competitive ABPP using 38 probe. (B, C) Selectivity of compounds against mouse brain SHs as measured by gel-based competitive ABPP using FP-Rh (B) or 38 (C). For the gel-based ABPP assays, proteomes were incubated with compound (0.1, 1, or 10 μM) for 30 min at 37 °C followed by reaction with fluorescent ABPP probes (38 or FP-Rh, 1 μM, 30 min, 37 °C). Fluorescent gel images are shown in gray scale. Serine hydrolase activities in gels were assigned as described in Figure 2.
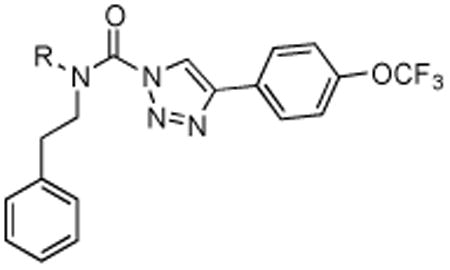
Table 3. Activity of acyclic phenethyl-1,2,3-TUs.
| |||
|---|---|---|---|
|
| |||
| Structure | Compound | DAGLB % Inhibition (100 nM) | Off-targets (10 μM) |
|
35 | 69.9 | ABHD6, 80 kDa SH, PLA2G7 |
|
36 | 70.5 | FAAH, KIAA1363, ABHD6, 80 kDa SH, PLA2G7 |
|
|
37 | 73 | ABHD6, 80 kDa SH, PLA2G7 |
We found that 37 served as a versatile intermediate for the design of DAGL-tailored activity-based probes. Initially, we substituted a BODIPY-dye in place of the Boc- group of 37 resulting in the development of the activity-based probe, 38 (Figure 6a) that could detect native DAGL activity in gel-based formats.13 Compound 38 also provided a complementary probe for other SHs, including PLA2G7, that are difficult to visualize in gel-based assays with the more broad-spectrum agent FP-Rh.21 Here, we wanted to determine the full spectrum of SHs targeted by compound 38 in Neuro2A proteomes using the quantitative mass-spectrometry (MS)-based proteomic method ABPP-SILAC.7, 8, 13 In brief, light and heavy Neuro2A proteomes (membrane and soluble fractions) were treated with DMSO or 38 (20 μM, 30 min), respectively, followed by enrichment of SHs using FP-biotin (10 μM, 2 hr). As shown in Figure 6B, we observed substantial blockade of FP-biotin labeling for ABHD6, DAGLβ, DDHD2, and PAFAH2, identifying these SHs as principal targets of 38. The MS-based results helped to assign identities to compound 38-labeled SH targets observed in our gel-based analysis, indicating that the ∼80-90 kDa doublet observed in both Neuro2A (Figure 6C) and mouse brain proteomes with 38 (Figure 2C, 3C, and 5C) is likely the poorly-characterized phospholipase, DDHD2.24, 25 Collectively, these results underscore the value of compound 38 as an activity-based probe for the focused visualization of low-abundance SHs in mammalian proteomes.
Figure 6.
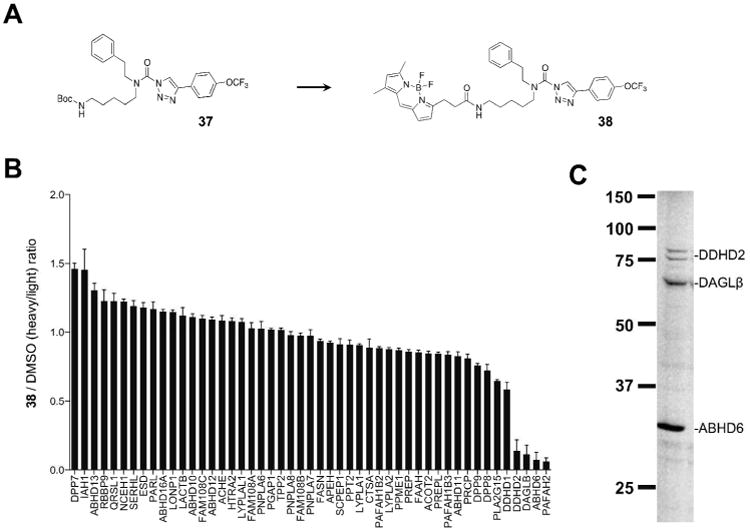
Development of the DAGL-tailored activity-based probe, compound 38. (A) 37 served as a precursor for coupling of an amine-reactive BODIPY fluorophore to generate 38. (B) Competitive ABPP-SILAC of SH activities from Neuro2A cells treated in situ with 20 μM of 38 for 4 hr. Compound 38 inhibited DDHD2, DAGLβ, ABHD6, and PAFAH2 by ≥ 80%, but did not inhibit any of the other detected SHs. Error bars represent mean ± s.e.m. of heavy/light ratios for the multiple peptides observed for each enzyme (minimum of 3 unique peptides per enzyme) in both soluble and membrane fractions. The ABPP-SILAC experiments confirmed (for DAGLβ and ABHD6) and revealed (for DDHD2) the identity of probe-labeled bands detected by gel-based ABPP in compound 38-treated Neuro2A membrane proteome (C; experiment performed as described in Figure 1). Note that PAFAH2 is a soluble enzyme and its signals are likely too low for detection by compound 38 in gel-based ABPP experiments of Neuro2A membrane proteome.
Expansion of DAGL-tailored probes to include biotin and alkyne derivatives of compound 38
We next synthesized biotin and alkyne analogs of compound 38 that can be used to directly enrich probe-labeled targets. We incorporated alkyne (39) and biotin (40) functional groups in the 37 precursor using simple amine coupling reactions. Both 39 and 40 showed moderate potency against recombinant DAGLα and DAGLβ with IC50 values in the range of 0.4 – 2 μM range (Figure 7). Next, we compared the specificity of 40 with its more broad-spectrum counterpart, FP-biotin26 using SILAC-ABPP, where probe-treated (heavy) Neuro2A proteomes were compared to DMSO-treated (light) Neuro2A proteomes for SH activities enriched by probes (heavy/light ratios > 20). More than 45 SH activities were enriched by FP-biotin (Table 4), which matches previous experiments that inventoried the full-range of SH activities in Neuro2A cells.13 In contrast, compound 40-enriched samples only contained 16 SHs (Table 4), including all of the established targets of 38 – DAGLβ, ABHD6, DDHD2, and PAFAH2 (Figure 6B). Several additional SH targets were also enriched by compound 40, including ABHD11, ESD, and LYPLAL1 (Table 4). That these enzymes were not identified in competitive ABPP studies with compound 38 (Figure 6B) suggests they are likely lower potency targets of compounds 38 and 40 (or exhibit preferential reactivity with 40).
Figure 7.
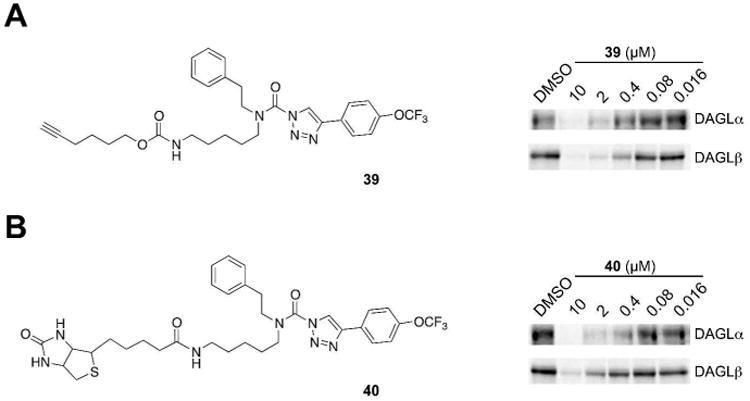
Inhibition of recombinant DAGLα and β by 39 and 40. Inhibitory activity of the alkyne (A) and biotin (B) probes, 39 and 40, respectively, was measured by competitive ABPP using DAGL-HEK293T membrane lysates. Proteomes were preincubated with varying concentrations of 39 or 40 for 30 min at 37 °C prior to treatment with 38 (1 μM, 30 min, 37 °C) and analysis by gel-based ABPP. Both probes blocked 38 labeling of DAGLα and β with IC50 values ranging from 0.4 – 2 μM.
Table 4.
Comparison of SHs enriched by FP-biotin or 40. Light and heavy proteomes were treated with DMSO or probe (10 μM of FP-biotin or 40) for 2 hr at room temperature and analyzed by ABPP-SILAC as described in Supporting Information. SHs shown in the table were detected with ratio values > 20 in probe/DMSO comparisons (minimum of 3 unique peptides per protein), denoting selective enrichment in probe-treated proteomes. We also performed a control experiment where we heat-denatured heavy proteomes prior to enrichment with FP-biotin to demonstrate that the detected SHs are enriched in an activity-dependent manner in ABPP-SILAC experiments (Supplementary Table 1).
| Serine hydrolases enriched in probe vs. no-probe treated samples | ||
|---|---|---|
|
| ||
| FP-biotin | 40 | |
| 1 | ABHD10 | ABHD11 |
| 2 | ABHD11 | ABHD6 |
| 3 | ABHD12 | CTSA |
| 4 | ABHD13 | DAGLβ |
| 5 | ABHD6 | DDHD2 |
| 6 | ACHE | ESD |
| 7 | ACOT2 | FASN |
| 8 | APEH | LYPLA1 |
| 9 | BAT5 | LYPLA2 |
| 10 | CTSA | LYPLAL1 |
| 11 | DAGLβ | PAFAH2 |
| 12 | DDHD2 | PLA2G15 |
| 13 | DPP7 | PLA2G6 |
| 14 | DPP8 | PNPLA2 |
| 15 | DPP9 | PPME1 |
| 16 | ESD | PREP |
| 17 | FAAH | |
| 18 | FAM108B1 | |
| 19 | FASN | |
| 20 | HTRA2 | |
| 21 | IAH1 | |
| 22 | LACTB | |
| 23 | LIPE | |
| 24 | LYPLA1 | |
| 25 | LYPLA2 | |
| 26 | LYPLAL1 | |
| 27 | NCEH1 | |
| 28 | PAFAH1B2 | |
| 29 | PAFAH1B3 | |
| 30 | PAFAH2 | |
| 31 | PARL | |
| 32 | PGAP1 | |
| 33 | PLA2G15 | |
| 34 | PLA2G6 | |
| 35 | PLAT | |
| 36 | PNPLA6 | |
| 37 | PNPLA7 | |
| 38 | PNPLA8 | |
| 39 | PPME1 | |
| 40 | PPT2 | |
| 41 | PRCP | |
| 42 | PREP | |
| 43 | PREPL | |
| 44 | QRSL1 | |
| 45 | RBBP9 | |
| 46 | SERHL | |
We next compared the subset of SHs enriched by compound 40 and FP-biotin and asked whether low or high-abundance targets were preferentially identified by each probe, where SH abundance was estimated by spectral counting (Figure 8). Certain high-abundance SHs, such as FASN, are enriched to an equal degree in both sample sets (ratio of log spectral counts ∼ 1; Figure 8). However, other high-abundance SHs, such as PREP, show much lower signals in 40 versus FP-biotin datasets (log spectral count ratio ∼ 0.5; Figure 8). Several additional medium-high abundance SHs (e.g. LYPLA1 and LYPLA2) were also preferentially enriched in FP-biotin-experiments. Conversely, some of the lower abundance targets, like DDHD2, showed substantially higher signals in 40- compared with FP-biotin-enriched samples (log spectral count ratio ∼ 2; Figure 8). Taken together, these results show that 40 functions as a tailored activity-based probe by providing both enhanced reactivity with low-abundance SHs (DDHD2, PLA2G6, PAFAH2) and attenuated labeling of higher abundance targets (PREP, LYPLA1, and LYPLA2).
Figure 8.

Comparative analysis of 40- and FP-biotin-enriched SHs in ABPP-SILAC experiments of Neuro2A proteomes. The signals (log spectral counts) for SH activities that were detected with ratios > 20 in probe/DMSO experiments (denoting selective enrichment in probe-treated proteomes; see Table 4 for complete list) and enriched in both FP-biotin and compound 40 datasets are plotted. SHs that show equal enrichment in both probe datasets have log spectral count ratios ∼1 (denoted by dashed line). SHs that show preferential enrichment by FP-biotin (red) or 40 (blue) deviate from the slope (log spectral count ratios < or > 1, respectively). The low-abundance SHs DDHD2, PLA2G6, and PAFAH2 show preferential enrichment by compound 40.
Development of a DAGLβ-inactive control probe that also provides a novel scaffold for ABHD6 inhibitors
The ABHD6 cross-reactivity displayed by compounds 11 and 27 proved difficult to eliminate with medicinal chemistry. We therefore addressed this issue by synthesizing ABHD6-selective inhibitors that function as negative-control probes for our biological studies. Four separate piperidine analogs were coupled to a 4-bromophenyl-triazole to generate TU intermediates that were then further coupled with substituted phenylboronic acids to produce compounds 46 – 52 (Scheme 6). We tested the activities of these derivatives using competitive ABPP and found that changing the location of the benzyl group on the piperidine ring from the 2- (27) to 3- (47) or 4-position (49) increased off-target activity (Figure 9A). After determining that the 2-position was optimal, we found that substitution of the 2-benzyl- for a 2-phenyl- (46) or 2-phenethyl-piperidine group (48) dramatically attenuated activity against DAGLβ in comparison with the potent 2-benzyl-derivative 27 (Figure 3A, Table 2). To further explore 2-phenyl- and 2-phenethyl-scaffolds, we synthesized 50 – 52 and found that both the 2-phenyl (50) and 2-phenethyl (51) derivatives that lack substituents on the biphenyl-triazole group still retained some activity against DAGLβ (Figure 9B). In contrast, addition of a para-methoxy group produced compound 52 (KT195),13 which showed a slight reduction in potency for ABHD6 (IC50 value of 10 nM13), but negligible cross-reactivity with DAGLβ (Figure 9B) or other brain SHs (Figure 9A), and was thus designated as a suitable negative-control probe that could be utilized at concentrations similar to those used for compounds 11 and 27 to selectively inhibit ABHD6.
Scheme 6.

Synthesis of (2-substituted)-Pip-1,2,3-TUs as candidate negative-control probes that inhibit ABHD6.
Figure 9.

Inhibitory activity and selectivity of candidate (2-substituted)-Pip-1,2,3-TU negative-control probes that target ABHD6. (A) Selectivity of control probes against mouse brain SHs as measured by gel-based competitive ABPP using FP-Rh. (B) Inhibitory activity of candidate negative-control probes against DAGLβ-HEK293T lysates as measured by gel-based competitive ABPP using 38. Previous studies showed concentration-dependent inhibition of ABHD6 in mouse brain proteomes by 52 (IC50 = 10 nM) as measured by competitive ABPP.13 For the gel-based ABPP assays, proteomes were incubated with compound at the indicated concentrations for 30 min at 37 °C followed by reaction with fluorescent ABPP probes (38 or FP-Rh, 1 μM, 30 min, 37 °C). Serine hydrolase activities in gels were assigned as described in Figure 2.
Conclusion
In summary, we describe the synthesis and SAR of a series of (2-substituted)-Pip-1,2,3-TUs that culminated in discovery of potent, selective, and in vivo-active inhibitors of DAGLβ.13 The successful development of DAGLβ-selective inhibitors highlights the utility of ABPP as not only a universal activity assay for the SH class of enzymes, but also adaptable to create tailored activity-based probes to facilitate detection of low-abundance enzymes like DAGLβ, PLA2G7, and DDHD2. Key to the development of these specialized activity-based probes was the tolerance that DAGLβ displays for acyclic analogs of the piperidine agents 11 and 27, which permitted the incorporation of diverse reporter tags into the DAGLβ inhibitor scaffold. Utilizing this synthetic strategy, we were able to generate a suite of fluorescent (38), alkyne (39), and biotin (40) probes that should find broad utility in both gel- and MS-based ABPP studies.
A major finding in our SAR studies was that substitution of mono- for bi-phenyl triazoles enhanced the selectivity of (2-benzyl)-Pip-1,2,3-TUs for DAGLβ, leading to the development of the optimized DAGLβ inhibitors, 11 and 27. We went on to design clickable analogs (32 and 33) of optimized DAGLβ inhibitors, which exhibited good proteome-wide selectivity at concentrations matching those used for the parent inhibitors to inactivate DAGLβ and ABHD6 in Neuro2A cells. We also discovered from our SAR studies key structural modifications to the DAGL inhibitor scaffold that attenuate DAGLβ activity while maintaining potency against ABHD6. These studies culminated in the discovery of a (2-phenyl)-Pip-1,2,3-TU, compound 52, that can serve as a negative-control probe for DAGLβ-related studies, as well as a novel chemotype for developing next-generation ABHD6-selective inhibitors for functional studies in vivo.18
Projecting forward, we believe that 1,2,3-TUs will continue to serve as a useful scaffold for developing selective probes targeting endocannabinoid biosynthetic pathways. With a modest number of analogs, we were able to develop potent and isoform-selective inhibitors capable of inactivating DAGLβ in vitro and in vivo. Future work will focus on optimizing DAGL inhibitors for improved brain penetrance and CNS activity. Another important goal is the development of DAGLα-selective inhibitors, which should benefit from the availability of gel-based ABPP assays using DAGL-tailored probes (38). Achieving these objectives would create a near-complete set of selective chemical probes for studying 2-AG-mediated endocannabinoid pathways in vivo.
Experimental Section
General Synthetic Methods
All chemicals and reagents that were commercially-available were purchased from Sigma-Aldrich, Matrix Scientific, BioBlocks, Combi-Blocks, Maybridge, Fisher, Acros, ChemExper, or Capot Chemicals and used without further purification. Commercial solvents (pre-dried, oxygen-free formulations) were passed through activated alumina columns to obtain dry solvents. Unless noted otherwise, all reactions were performed under a nitrogen atmosphere using oven-baked glassware. Flash chromatography was performed using mesh silica gel (230-400). Analytical thin-layer chromatography (TLC) on glass backed silica gel 60 F254 plates was used to monitor reactions. pTLC on silica gel 60 F254 plates or flash chromatography on 40-60 MYM mesh silica gel were used to purify reactions. 1H-NMR and spectra were recorded in CDCl3 on a Varian Mercury-300 spectrometer, a Varian Inova-400, Bruker DRX-500, or a Bruker DRX-600 spectrometer, and referenced to trimethylsilane (TMS). Chemical shifts were reported in ppm relative to TMS and J values were reported in Hz. High resolution mass spectrometry (HRMS) experiments were performed at The Scripps Research Institute Mass Spectrometry Core on an Agilent mass spectrometer using electrospray ionization-time of flight (ESI-TOF). The purity of final compounds were determined to be ≥95% by HPLC analysis (Agilent 1100 LC/MS).
Hex-5-yn-1-yl (5-(N-phenethyl-4-(4-(trifluoromethoxy)phenyl)-1H-1,2,3-triazole-1-carboxamido)pentyl)carbamate (39)
A solution of 5-hexyn-1-ol (10 mg, 0.10 mmol) in CH2Cl2 (2 mL) was treated with triphosgene (30 mg, 0.10 mmol, 1.0 equiv) and pyridine (8 μL, 0.10 mmol, 1.0 equiv), and the mixture was stirred for 30 min at 4 °C. The mixture was poured into H2O and extracted with ethyl acetate. The organic layer was washed with H2O and brine, dried over Na2SO4 and concentrated under reduced pressure. The residue was dissolved in THF (1 mL), and Boc-deprotected urea 3713 (7 mg, 14 μmol) and iPr2NEt (10 μL) were added to the solution. After stirring for 1 hr at room temperature, the mixture was poured into H2O and extracted with ethyl acetate. The organic layer was washed with H2O and brine, dried over Na2SO4 and concentrated under reduced pressure. pTLC (ethyl acetate:hexane=1:3) afforded 39 (8 mg, 98%).
1H NMR (CDCl3, 300 MHz) δ 8.42-7.80 (m, 3H), 7.40-7.10 (m, 7H), 4.70 (br, 1H), 4.15-3.50 (m, 6H), 3.25-2.95 (m, 4H), 2.22 (td, 2H, J = 7.0, 2.6 Hz), 1.95 (t, 1H, J = 2.6 Hz), 1.65-1.20 (m, 10H). HRMS calculated for C30H35F3N5O4 [M+H]+ 586.2636, found 586.2633.
N-(5-(5-(2-Oxohexahydro-1H-thieno[3,4-d]imidazol-4-yl)pentanamido)pentyl)-N-phenethyl-4-(4-(trifluoromethoxy)phenyl)-1H-1,2,3-triazole-1-carboxamide (40)
A solution of the urea 37 (13 mg, 23 μmol) in CH2Cl2 (0.6 mL) was treated with 4N HCl-dioxane (0.6 mL), and the mixture was stirred for 3 hr at room temperature. The solvent was evaporated and the residue was dissolved in DMF (1 mL). iPr2NEt (12 μL, 69 μmol, 3.0 equiv) and biotin-NHS (8 mg, 23 μmol, 1.0 equiv) was added to the solution, and the mixture was stirred for 2 hr at room temperature. The mixture was poured into H2O and extracted with ethyl acetate. The organic layer was washed with H2O and brine, dried over Na2SO4 and concentrated under reduced pressure. Et2O (1 mL) was added and the mixture was sonicated. The supernatant was removed and this procedure was repeated twice to afford 40 (10 mg, 63%).
1H NMR (CDCl3, 300 MHz) δ 8.43-7.80 (m, 3H), 7.35-7.05 (m, 7H), 6.25-6.03 (m, 2H), 5.30 (s, 1H), 4.48 (m, 1H), 4.29 (m, 1H), 4.00-3.50 (m, 4H), 3.30-2.80 (m, 6H), 2.70 (d, 1H, J = 13.0 Hz), 2.18 (t, J = 7.4 Hz), 1.80-1.20 (m, 10H). HRMS calculated for C33H41F3N7O4S [M+H]+ 688.2887, found 688.2883.
Materials for Biological Experiments
FP-rhodamine,26 FP-biotin,26, 27 and 3813 were synthesized as previously described. Neuro2A and HEK293T cells were purchased from ATCC.
Cell culture and preparation of cell line proteomes
Neuro2A and HEK293T cells were grown in DMEM supplemented with 10% fetal bovine serum at 37 °C with 5% CO2. For in vitro experiments, cells were grown to 80-90% confluency, washed twice with cold PBS (pH 7.5) and scraped. Cell pellets were then isolated by centrifugation at 1,400 × g for 3 min at 4 °C. The pellets were resuspended in 500 μL of cold PBS (pH 7.5), sonicated, and centrifuged at 100,000 × g for 45 min to generate soluble and membrane fractions. Mouse DAGLα and DAGLβ were transiently overexpressed in HEK293T cells as previously described.13 Total protein concentration of membrane and soluble fractions was determined using a protein assay kit (Bio-Rad). Samples were stored at -80 °C until use.
Preparation of mouse brain and liver proteomes
Brains and livers from C57Bl/6 mice were Dounce-homogenized in PBS, pH 7.5, followed by a low-speed spin (1,400 × g, 5 min) to remove debris. The supernatant was then centrifuged (100,000 × g, 45 min) to generate the cytosolic fraction in the supernatant and the membrane fraction as a pellet. The pellet was washed and resuspended in PBS buffer by sonication. The total protein concentration in each fraction was determined using a protein assay kit (Bio-Rad). Samples were stored at -80 °C until further use.
Gel-based competitive ABPP
Gel-based competitive ABPP experiments were performed following previously described protocols.13, 28, 29 Proteomes (1 mg/mL) were pretreated with compounds at indicated concentrations (30 min, 37 °C) followed by labeling with either FP-rhodamine or 38 (1 μM final concentration) in a 50 μL total reaction volume. After 30 min or 1 hr at 37 °C, the FP-rhodamine- or 38-labeled reactions, respectively, were quenched with SDS-PAGE loading buffer. After separation by SDS-PAGE (10% acrylamide), samples were visualized by in-gel fluorescence scanning using a flatbed fluorescent scanner (Hitachi FMBio IIe). To measure recombinant DAGLβ activity, proteomes were diluted to 0.3 mg/mL in assay buffer (50 mM HEPES, pH 7.2, 100 mM NaCl, 5 mM CaCl2, 0.1% v/v TX-100, 10% v/v DMSO) and subjected to ABPP analysis as described above.
Cycloaddition reactions with the clickable probes 32 and 33
Click chemistry was performed following previously described protocols.30 For in vitro experiments, membrane proteomes (1 mg/mL in PBS) were incubated with DMSO or varying concentrations of compound 32, or 33 (3 nM - 10 μM) at 37 °C for 30 min prior to incubation with 38 (1 μM) for 30 min. For in situ experiments, Neuro2A cells were incubated with DMSO, 32 or 33 (10 nM – 25 μM clickable probes) for 1 hr at 37 °C, lysed, and membrane fractions isolated for click-chemistry. The alkyne-labeled membrane proteomes (1 mg/mL in PBS) were then incubated with rhodamineazide (50 μM), followed by TCEP (1 mM), ligand (100 μM), and CuSO4 (1 mM). After 1 hr at 25 °C, reactions were analyzed by SDS-PAGE and in-gel fluorescence scanning.
Competitive ABPP-SILAC
Neuro2A cells were grown for 10 passages in either light or heavy SILAC DMEM medium supplemented with 10% (v/v) dialyzed FCS and 2 mM L-glutamine. The light medium was supplemented with 100 μg/mL L-arginine and 100 μg/mL L-lysine. Heavy medium was supplemented with 100 μg/mL [13C615N4]-L-Arginine and 100 μg/mL [13C615N2]-L-Lysine. The heavy cells (in 10 mL medium) were treated with test compound and light cells were treated with DMSO for 4 hr at 37 °C. Cells were washed with PBS (2×), harvested, and lysed by sonication in DPBS. Membrane and soluble proteomes were isolated as described above and adjusted to a final concentration of 2.0 mg/mL and labeled with 10 μM FP-biotin (500 μL total reaction volume) for 2 hr at 25 °C. After incubation, light and heavy proteomes were mixed in 1:1 ratio, and excess FP-biotin removed by CHCl3/MeOH extraction. To the light and heavy mixed proteomes (1 mL volume), we added 2 mL MeOH, 0.5 mL CHCl3, 1.5 mL H2O, vortexed, and centrifuged at 1,400 × g for 3 min. The top (aqueous) and bottom (organic) layers were removed and 600 μL of MeOH was added to the protein interface, which was subsequently transferred to a microfuge tube. Next, 150 μL of CHCl3 and 600 μL of H2O was added, vortexed, and centrifuged as described above. The top and bottom layers were removed, the protein interface sonicated in 600 μL of MeOH, and then centrifuged at 14,000 rpm for 5 min to pellet protein. The MeOH was removed and pellet resuspended in 6 M urea/25 mM ammonium bicarbonate. Samples were then reduced with 10 mM DTT for 15 min (65 °C) and alkylated with 10 mM iodoacetamide for 30 min at 25 °C in the dark. Afterwards, samples were treated with SDS (2% final, 5 min) and biotinylated proteins enriched with avidin beads (50 μL beads; conditions: 1 hr, 25 °C, 0.5% SDS in PBS). The beads were washed three times with 1% SDS in PBS followed by three washes with PBS. The urea concentration was reduced to 2 M with 2× volume DPBS. On-bead digestions were performed for 12 hr at 37 °C with sequence-grade modified trypsin (Promega; 2 μg) in the presence of 2 mM CaCl2. Peptide samples were acidified to a final concentration of 5% (v/v) formic acid and stored at -80 °C prior to analysis. LC-MS/MS analysis of ABPP-SILAC samples were performed as previously described.7, 8, 13
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health Grants DA017259 (B.F.C.) and DA033760 (B.F.C.), a Hewitt Foundation Postdoctoral Fellowship (K.L.H.), the Skaggs Institute for Chemical Biology, and Dainippon Sumitomo Pharma (K.T.)
Abbreviations Used
- SH
serine hydrolase
- Pip-1,2,3-TU
piperidyl-1,2,3-triazole urea
- ABPP
activity-based protein profiling
- CuAAC
copper-catalyzed azide alkyne cycloaddition
- SILAC
stable isotope labeling by amino acids in cell culture
- FP-Rh
fluorophosphonate-rhodamine
Footnotes
Notes: The authors declare the following competing financial interest(s): Dr. Cravatt is a founder and adviser to Abide Therapeutics a biotechnology company interested in developing serine hydrolase inhibitors as drugs to treat human disease.
Supporting Information. Supplemental methods, synthetic procedures and characterization of 1,2,3-triazole ureas, HRMS data, chiral separation of compound 2, and supplemental figures. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Simon GM, Cravatt BF. Activity-based proteomics of enzyme superfamilies: serine hydrolases as a case study. J Biol Chem. 2010;285:11051–11055. doi: 10.1074/jbc.R109.097600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Long JZ, Cravatt BF. The metabolic serine hydrolases and their functions in mammalian physiology and disease. Chem Rev. 2011;111:6022–6063. doi: 10.1021/cr200075y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Henness S, Perry CM. Orlistat: a review of its use in the management of obesity. Drugs. 2006;66:1625–1656. doi: 10.2165/00003495-200666120-00012. [DOI] [PubMed] [Google Scholar]
- 4.Thornberry NA, Weber AE. Discovery of JANUVIA (Sitagliptin), a selective dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes. Curr Top Med Chem. 2007;7:557–568. doi: 10.2174/156802607780091028. [DOI] [PubMed] [Google Scholar]
- 5.Racchi M, Mazzucchelli M, Porrello E, Lanni C, Govoni S. Acetylcholinesterase inhibitors: novel activities of old molecules. Pharmacol Res. 2004;50:441–451. doi: 10.1016/j.phrs.2003.12.027. [DOI] [PubMed] [Google Scholar]
- 6.Bachovchin DA, Cravatt BF. The pharmacological landscape and therapeutic potential of serine hydrolases. Nat Rev Drug Discov. 2012;11:52–68. doi: 10.1038/nrd3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bachovchin DA, Mohr JT, Speers AE, Wang C, Berlin JM, Spicer TP, Fernandez-Vega V, Chase P, Hodder PS, Schurer SC, Nomura DK, Rosen H, Fu GC, Cravatt BF. Academic cross-fertilization by public screening yields a remarkable class of protein phosphatase methylesterase-1 inhibitors. Proc Natl Acad Sci U S A. 2011;108:6811–6816. doi: 10.1073/pnas.1015248108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adibekian A, Martin BR, Wang C, Hsu KL, Bachovchin DA, Niessen S, Hoover H, Cravatt BF. Click-generated triazole ureas as ultrapotent in vivo–active serine hydrolase inhibitors. Nat Chem Biol. 2011;7:469–478. doi: 10.1038/nchembio.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Long JZ, Li W, Booker L, Burston JJ, Kinsey SG, Schlosburg JE, Pavon FJ, Serrano AM, Selley DE, Parsons LH, Lichtman AH, Cravatt BF. Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat Chem Biol. 2009;5:37–44. doi: 10.1038/nchembio.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang JW, Nomura DK, Cravatt BF. A potent and selective inhibitor of KIAA1363/AADACL1 that impairs prostate cancer pathogenesis. Chem Biol. 2011;18:476–484. doi: 10.1016/j.chembiol.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ahn K, Smith SE, Liimatta MB, Beidler D, Sadagopan N, Dudley DT, Young T, Wren P, Zhang Y, Swaney S, Van Becelaere K, Blankman JL, Nomura DK, Bhattachar SN, Stiff C, Nomanbhoy TK, Weerapana E, Johnson DS, Cravatt BF. Mechanistic and pharmacological characterization of PF-04457845: a highly potent and selective fatty acid amide hydrolase inhibitor that reduces inflammatory and noninflammatory pain. J Pharmacol Exp Ther. 2011;338:114–124. doi: 10.1124/jpet.111.180257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zuhl AM, Mohr JT, Bachovchin DA, Niessen S, Hsu KL, Berlin JM, Dochnahl M, Lopez-Alberca MP, Fu GC, Cravatt BF. Competitive activity-based protein profiling identifies aza-beta-lactams as a versatile chemotype for serine hydrolase inhibition. J Am Chem Soc. 2012;134:5068–5071. doi: 10.1021/ja300799t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hsu KL, Tsuboi K, Adibekian A, Pugh H, Masuda K, Cravatt BF. DAGLbeta inhibition perturbs a lipid network involved in macrophage inflammatory responses. Nat Chem Biol. 2012;8:999–1007. doi: 10.1038/nchembio.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hsu KL, Tsuboi K, Speers AE, Brown SJ, Spicer T, Fernandez-Vega V, Ferguson J, Cravatt BF, Hodder P, Rosen H. Probe Reports from the NIH Molecular Libraries Program [Internet] Bethesda (MD): National Center for Biotechnology Information (US); 2012. Optimization and characterization of a triazole urea inhibitor for diacylglycerol lipase beta (DAGL-β) updated Feb 25 2013. [PubMed] [Google Scholar]
- 15.Bisogno T, Howell F, Williams G, Minassi A, Cascio MG, Ligresti A, Matias I, Schiano-Moriello A, Paul P, Williams EJ, Gangadharan U, Hobbs C, Di Marzo V, Doherty P. Cloning of the first sn1-DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. J Cell Biol. 2003;163:463–468. doi: 10.1083/jcb.200305129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao Y, Vasilyev DV, Goncalves MB, Howell FV, Hobbs C, Reisenberg M, Shen R, Zhang MY, Strassle BW, Lu P, Mark L, Piesla MJ, Deng K, Kouranova EV, Ring RH, Whiteside GT, Bates B, Walsh FS, Williams G, Pangalos MN, Samad TA, Doherty P. Loss of Retrograde Endocannabinoid Signaling and Reduced Adult Neurogenesis in Diacylglycerol Lipase Knock-out Mice. J Neurosci. 2010;30:2017–2024. doi: 10.1523/JNEUROSCI.5693-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tanimura A, Yamazaki M, Hashimotodani Y, Uchigashima M, Kawata S, Abe M, Kita Y, Hashimoto K, Shimizu T, Watanabe M, Sakimura K, Kano M. The endocannabinoid 2-arachidonoylglycerol produced by diacylglycerol lipase alpha mediates retrograde suppression of synaptic transmission. Neuron. 2010;65:320–327. doi: 10.1016/j.neuron.2010.01.021. [DOI] [PubMed] [Google Scholar]
- 18.Hsu KL, Tsuboi K, Chang JW, Whitby LR, Speers AE, Pugh H, Cravatt BF. Discovery and optimization of piperidyl-1,2,3-triazole ureas as potent, selective, and in vivo-active inhibitors of alpha/beta-hydrolase domain containing 6 (ABHD6) J Med Chem. 2013 doi: 10.1021/jm400899c. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jessani N, Liu Y, Humphrey M, Cravatt BF. Enzyme activity profiles of the secreted and membrane proteome that depict cancer cell invasiveness. Proc Natl Acad Sci U S A. 2002;99:10335–10340. doi: 10.1073/pnas.162187599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kalisiak J, Sharpless KB, Fokin VV. Efficient Synthesis of 2-Substituted-1,2,3-triazoles. Org Lett. 2008;10:3171–3174. doi: 10.1021/ol8006748. [DOI] [PubMed] [Google Scholar]
- 21.Nagano JM, Hsu KL, Whitby LR, Niphakis MJ, Speers AE, Brown SJ, Spicer T, Fernandez-Vega V, Ferguson J, Hodder P, Srinivasan P, Gonzalez TD, Rosen H, Bahnson BJ, Cravatt BF. Selective inhibitors and tailored activity probes for lipoprotein-associated phospholipase A(2) Bioorg Med Chem Lett. 2013;23:839–843. doi: 10.1016/j.bmcl.2012.11.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew Chem Int Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 23.Speers AE, Cravatt BF. Profiling enzyme activities in vivo using click chemistry methods. Chem Biol. 2004;11:535–546. doi: 10.1016/j.chembiol.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 24.Schuurs-Hoeijmakers JH, Geraghty MT, Kamsteeg EJ, Ben-Salem S, de Bot ST, Nijhof B, van de V, II, van der Graaf M, Nobau AC, Otte-Holler I, Vermeer S, Smith AC, Humphreys P, Schwartzentruber J, Ali BR, Al-Yahyaee SA, Tariq S, Pramathan T, Bayoumi R, Kremer HP, van de Warrenburg BP, van den Akker WM, Gilissen C, Veltman JA, Janssen IM, Vulto-van Silfhout AT, van der Velde-Visser S, Lefeber DJ, Diekstra A, Erasmus CE, Willemsen MA, Vissers LE, Lammens M, van Bokhoven H, Brunner HG, Wevers RA, Schenck A, Al-Gazali L, de Vries BB, de Brouwer AP. Mutations in DDHD2, encoding an intracellular phospholipase A(1), cause a recessive form of complex hereditary spastic paraplegia. Am J Hum Genet. 2012;91:1073–1081. doi: 10.1016/j.ajhg.2012.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang ZQ, Liu G, Bollig-Fischer A, Giroux CN, Ethier SP. Transforming properties of 8p11-12 amplified genes in human breast cancer. Cancer Res. 2010;70:8487–8497. doi: 10.1158/0008-5472.CAN-10-1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu Y, Patricelli MP, Cravatt BF. Activity-based protein profiling: the serine hydrolases. Proc Natl Acad Sci U S A. 1999;96:14694–14699. doi: 10.1073/pnas.96.26.14694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kidd D, Liu Y, Cravatt BF. Profiling serine hydrolase activities in complex proteomes. Biochemistry. 2001;40:4005–4015. doi: 10.1021/bi002579j. [DOI] [PubMed] [Google Scholar]
- 28.Bachovchin DA, Ji T, Li W, Simon GM, Blankman JL, Adibekian A, Hoover H, Niessen S, Cravatt BF. Superfamily-wide portrait of serine hydrolase inhibition achieved by library-versus-library screening. Proc Natl Acad Sci U S A. 2010;107:20941–20946. doi: 10.1073/pnas.1011663107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jessani N, Niessen S, Wei BQ, Nicolau M, Humphrey M, Ji Y, Han W, Noh DY, Yates JR, 3rd, Jeffrey SS, Cravatt BF. A streamlined platform for high-content functional proteomics of primary human specimens. Nat Methods. 2005;2:691–697. doi: 10.1038/nmeth778. [DOI] [PubMed] [Google Scholar]
- 30.Speers AE, Adam GC, Cravatt BF. Activity-Based Protein Profiling in Vivo Using a Copper(I)-Catalyzed Azide-Alkyne [3 + 2] Cycloaddition. J Am Chem Soc. 2003;125:4686–4687. doi: 10.1021/ja034490h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.