

Abstract
Recently, we described the aminothiazole lead (4-biphenyl-4-yl-thiazol-2-yl)-(6-methyl-pyridin-2-yl)-amine (1), which exhibits many desirable properties, including excellent stability in liver microsomes, oral bioavailability of ∼40% and high exposure in the brains of mice. Despite its good pharmacokinetic properties, compound 1 exhibited only modest potency in mouse neuroblastoma cells overexpressing the disease-causing prion protein PrPSc. Accordingly, we sought to identify analogs of 1 with improved antiprion potency in ScN2a-cl3 cells while retaining comparable or superior properties. We now report the discovery of improved lead compounds such as (6-methyl-pyridin-2-yl)-[4-(4-pyridin-3-yl-phenyl)-thiazol-2-yl]-amine (15) and cyclopropanecarboxylic acid (4-biphenyl-thiazol-2-yl)-amide (34), which exhibited brain exposure/EC50 ratios at least ten-fold greater than that of 1.
Keywords: 2-Aminothiazoles, neurological agents, pharmacokinetic optimization, prion disease, structure-activity relationships
Introduction
A rapidly expanding body of evidence indicates that prions cause many different neurodegenerative disorders including Alzheimer's, Parkinson's and Creutzfeldt–Jakob (CJD) diseases as well as the frontotemporal dementias and some forms of amyotrophic lateral sclerosis (ALS).[1, 2] The human prion diseases caused by the aberrantly folded prion protein (PrP) include kuru, CJD, fatal insomnia and Gerstmann–Sträussler–Scheinker disease.[2, 3] Bovine spongiform encephalopathy, or “mad cow” disease, is the most well-known among the animal PrP prion diseases that also encompass scrapie of sheep and goats, feline spongiform encephalopathy, and chronic wasting disease of deer and elk. The unique characteristic common to all of these disorders, whether sporadic, dominantly inherited, or acquired by infection, is the aberrant refolding of cellular PrP (PrPC) into a disease-causing isoform (PrPSc).[4-6] The exact mechanism by which α-helix–rich PrPC is transformed to β-sheet–rich PrPSc is unknown, but auxiliary proteins such as chaperones seem likely to participate in the replication process, as is the case for yeast prions.[7, 8] Oligomers of PrPSc are thought to be cytotoxic and responsible for neuronal dysfunction [9-12]. Prion diseases are invariably fatal, and no effective treatment for these devastating disorders is currently available.
Attempting to reduce levels of PrPSc by inhibiting its production and/or increasing its clearance would seem to be among the viable therapeutic approaches for prion diseases. Moreover, lowering levels of glycosyl phosphatidylinositol–anchored PrPC on the surface of neurons may represent a useful complementary therapy.[13] Several groups have screened for antiprion compounds; typically, scrapie-infected murine neuroblastoma cell (ScN2a) have been used. Both biologics (antibodies) and small organic molecules with antiprion activity have been identified using assays based on PrPSc levels in ScN2a cells. Among known antiprion small molecules are Congo red,[14] acridines,[15] diphenyloxazoles and diphenylthiazoles,[16] pyrazolones,[17] 2-aminopyridines,[18] statins, and indole-3-glyoxylamides.[19]
Previously, we reported on the identification of several classes of antiprion compounds including 2-aminothiazoles (AMT).[20] Subsequent structure-activity relationship (SAR) studies of the AMT series led to the discovery of several blood-brain barrier (BBB)–penetrant AMT lead compounds.[21] From lead optimization studies in which ∼30 early leads were profiled in PK studies, the AMT lead 1 (Figure 1) and a related analog were selected as possessing optimal pharmacokinetic (PK) properties for evaluation in animal models of prion disease. For example, compound 1 was found to achieve sustained brain exposure in mice on oral dosing (%F ∼ 40), with brain area under the curve (AUC) values for both bound and unbound drug well in excess of the EC50 values for the compound.[22]
Figure 1.
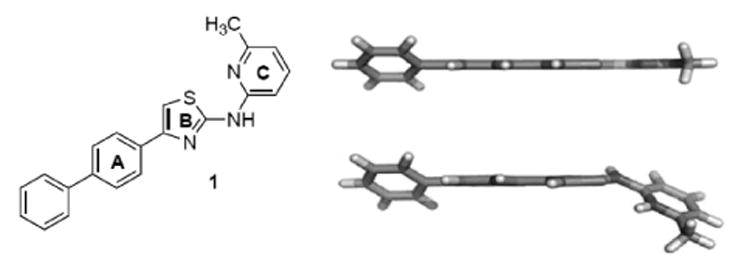
Structure of lead compound 1 and conformational analysis of neutral (top) and protonated (bottom) forms of 1 by density functional theory (DFT).
Despite its promising PK properties, 1 exhibited only moderate potency (EC50 = 1.29 μM) in ScN2a-cl3 cells and was challenging to formulate at the relatively high doses that were predicted to be required to reach therapeutic brain levels. Accordingly, we sought to identify close analogs of 1 that might exhibit substantially improved potency in ScN2a-cl3 cells while retaining the favorable PK and metabolic properties of 1. With reduced dosing requirements, we expected to mitigate the formulation issues encountered with 1, while also reducing the potential for drug-associated toxicities during the necessarily chronic dosing regimens required in animal models of prion disease. Here, we report the discovery of new 2-AMT analogs that exhibit substantially (up to ∼20-fold) improved antiprion potency, while retaining or improving upon the PK properties of 1.
Results and Discussion
Chemistry
Compound 1 contains four aromatic and heteroaromatic rings (Figure 1) joined directly by sp2-hybridized carbon or nitrogen atoms, producing a highly conjugated structure. Density functional theory (DFT)-based quantum mechanical calculations suggest that this molecule adopts a largely coplanar conformation at neutral pH, with only the terminal ring of the biaryl system slightly out of plane in the lowest energy conformation. At more acidic pH, the aminopyridine C-ring is protonated, and as a result, 1 adopts a conformation in which the B- and C-rings are no longer coplanar (Figure 1). Earlier SAR studies provided compelling evidence that a coplanar conformation of the A-B ring system is required for antiprion effects in cells.[21] The DFT analysis of 1, however, suggested that the terminal biaryl A-ring system may have less rigid requirements with regard to coplanarity. Accordingly, we sought to explore diverse heteroaryl and heterocyclic substitutions at this terminal ring, including the introduction of ortho substituents that would intentionally favor a nonplanar conformation of this system. In parallel, we sought to explore substituents that would alter the pKa of the pyridine C-ring, favoring nonplanar B-C ring conformers via steric or protonation effects.
The synthesis of the desired AMTs was accomplished using Hantzsch-type condensation reactions of bromomethyl ketones with thioureas (Schemes 1–3). The key aminothiazole intermediate 2 was formed via Hantzsch reaction of 2-bromo-1-(4-bromophenyl)ethanone with thiourea 5, which is derived from 2-amino-6-methylpyridine 4.[21] Bromide 2 could be converted to pinacol borate 3 via palladium-catalyzed Suzuki-Miyaura reaction (Scheme 1). Both aryl bromide 2 and pinacol borate ester 3 proved to be versatile intermediates for subsequent Suzuki-Miyaura or Buchwald-Hartwig coupling reactions, yielding the desired biaryl A-ring analogs 12–21 (Table 1) and 25–33 (Table 2).
Scheme 1.

Synthesis of key synthetic intermediates 2 and 3 used in subsequent coupling reactions to prepare 12–21 and 25–33. Reaction conditions: (a) BzSCN, acetone, reflux; (b) NaOH, MeOH, reflux; (c) bromoacetophenone, EtOH, reflux; (d) bis(pinacolato)diboron, Pd (dppf)2Cl2, AcOK, dioxane, 80 °C. (e) For compounds 12-21 from 2: ArB(OH)2, 1,4-dioxane, K3PO4, Pd2(dba)3, tricyclohexylphosphine, microwave (150 °C, 30 min); (f) For compounds 25-33 from 3: ArBr, Pd(dppf)2Cl2, K2CO3, 1,4-dioxane/water (5:1), 80 °C, overnight.
Scheme 3.

Synthesis of intermediates 10 and 11 used in subsequent coupling reactions to afford analogs 39–47. Reaction conditions: (a) thiourea, EtOH; (b) cyclopropanecarbonyl chloride, (iPr)2EtN, CH2Cl2; (c) Bis (pinacolato)diboron, Pd (dppf)2Cl2, KOAc, dioxane, 80 °C; (d) For compounds 39-41 from 10: ArB(OH)2, 1,4-dioxane, K3PO4, Pd2(dba)3, tricyclohexylphosphine, microwave (150 °C, 30 min); (e) For compounds 42-47 from 11: ArBr, Pd(dppf)2Cl2, K2CO3, 1,4-dioxane/water (5:1), 80 °C, overnight.
Table 1.
Antiprion potency for AMT analogs.
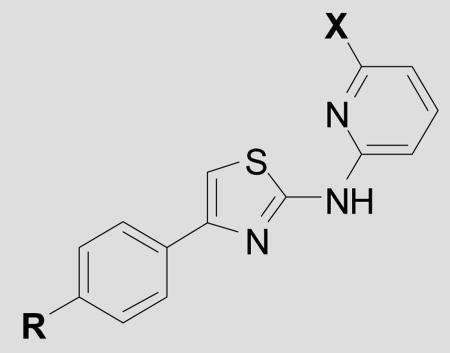
| ||||
|---|---|---|---|---|
| Cmpd | R = | X = | EC50 ± SEM(nM)[a] | n[B] |
| 1 | Phenyl | CH3 | 1290 ± 122 | 85 |
| 12 | pyridin-4-yl | CH3 | 87 ± 43 | 3 |
| 13 | pyridin-4-yl N-oxide | CH3 | 559 ± 120 | 3 |
| 14 | 2-Me-pyridin-4-yl | CH3 | 400 ± 61 | 3 |
| 15 | pyridin-3-yl | CH3 | 68 ± 13 | 5 |
| 16 | pyridin-3-yl N-oxide | CH3 | 203 ± 10 | 3 |
| 17 | 2-Me-pyridin-3-yl | CH3 | 618 ± 54 | 3 |
| 18 |
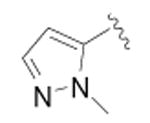
|
CH3 | 89 ± 35 | 3 |
| 19 |
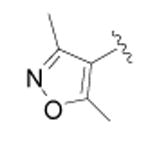
|
CH3 | 2133 ± 226 | 3 |
| 20 | N-Me-piperazinyl | CH3 | 840 ± 132 | 3 |
| 21 | morpholino | CH3 | 51 ± 7 | 3 |
| 22 | phenyl | MeOCH2CH2O- | >10,000 | 3 |
| 23 | phenyl | Me2N- | 699 ± 87 | 3 |
| 24 | phenyl | N-Me-piperazinyl | 1183 ± 206 | 5 |
antiprion potency in ScN2a-cl3 cells by ELISA; SEM is calculated as standard deviation divided by the square root of n.
number of repetitions.
Table 2.
Antiprion potency for AMT analogs with terminal 4-pyridyl or 3-pyridyl rings.
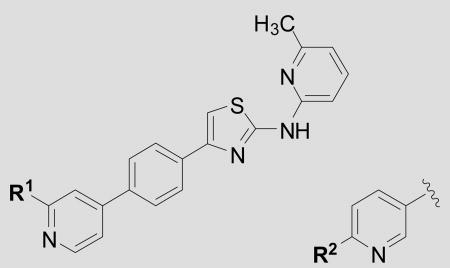
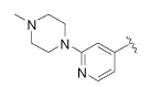
| ||||
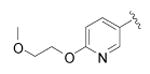
|---|---|---|---|---|
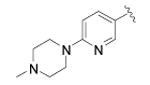
| Cmpd | R1 = | R2 = | EC50 ± SEM(nM)[a] | n[B] |
| 12 | H | --- | 87 ± 43 | 3 |
| 15 | --- | H | 68 ± 13 | 5 |
| 25 | --- | MeO | 140 ± 23 | 3 |
| 26 | MeOCH2CH2O- | --- | 153 ± 36 | 3 |
| 27 | --- | MeOCH2CH2O- | 137 ± 13 | 3 |
| 28 | i-PrO- | --- | 2840 ± 22 | 3 |
| 29 | --- | i-PrO- | 5860 ± 327 | 3 |
| 30 | Me2N- | --- | 123 ± 35 | 3 |
| 31 | --- | Me2N- | 69 ± 21 | 3 |
| 32 | N-Me-piperazinyl | --- | 759 ± 374 | 3 |
| 33 | --- | N-Me-piperazinyl | 96 ± 31 | 4 |
antiprion potency in ScN2a-cl3 cells by ELISA; SEM is calculated as standard deviation divided by the square root of n.
number of repetitions.
To alter B-C ring diaxial conformational preferences, we sought to introduce amine substituents neighboring the nitrogen atom on the pyridine C-ring of AMT analogs. The commercially available aminopyridine 6 was converted in two steps to the thiourea 7 using the standard procedure. The intermediate 7 was next employed in Hantzsch reaction to afford the key intermediate 8. As expected, fluoropyridine 8 readily participated in SNAr reaction with various amines or alkoxides to afford the desired analogs 22–24 (Table 1, Scheme 2).
Scheme 2.

Synthesis of key synthetic intermediate 8 used in subsequent SNAr reactions to prepare analogs 22–24. Reaction conditions: (a) BzNCS, acetone, reflux; (b) NaOH, MeOH, reflux; (c) 1-([1,1′-biphenyl]-4-yl)-2-bromoethanone, NaHCO3, CH3CN, reflux; (d) ROH and Na or R1R2NH, 120-130 °C, 4-12 hr.
More dramatic alteration of the C-ring moiety was explored with commercial and synthetic analogs of 1. Although the majority of such analogs were devoid of useful antiprion activity, we did find that small alkyl amides (acetyl, cyclopropylcarbonyl) could serve as viable surrogates for the pyridine C-ring of 1. Among the amide C-groups examined, cyclopropylamides were deemed of greatest interest due to their good potency and the expectation of superior hydrolytic/proteolytic stability as compared to acetamides, which were also active. We therefore sought means to access cyclopropylamide C-group analogs of 1 with diverse A-ring substitutions. Hence, Hantzsch reaction of 9 with thiourea afforded an AMT intermediate that was then treated with cyclopropylcarbonyl chloride to afford the aryl bromide 10 (Scheme 3). We found that bromide 10 and the corresponding pinacol borate ester 11 readily participated in Buchwald-Hartwig or Suzuki-Miyaura reactions, affording analogs 39–47 (Table 3).
Table 3.
Antiprion potency for AMT analogs with amide C-groups.
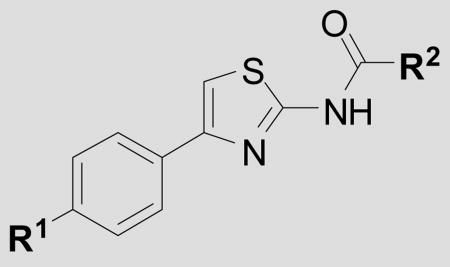
| ||||
|---|---|---|---|---|
| Cmpd | R1 = | R2 = | EC50 ± SEM(nM)[a] | n[B] |
| 34 | phenyl | cyclopropyl | 248 ± 67 | 4 |
| 35 | phenyl | CH3 | 119 ± 10 | 3 |
| 36 | phenyl | MeOCH2- | 1588 ± 295 | 3 |
| 37 | phenyl | i-Pr | >10,000 | 3 |
| 38 | phenyl | phenyl | >10,000 | 3 |
| 39 | pyridin-4-yl | cyclopropyl | 173 ± 16 | 3 |
| 40 | 2-Me-pyridin-3-yl | cyclopropyl | >10,000 | 3 |
| 41 | 4-Me2N-pyridin-3-yl | cyclopropyl | 109 ± 12 | 3 |
| 42 |
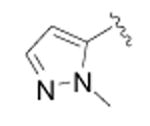
|
cyclopropyl | >10,000 | 3 |
| 43 |
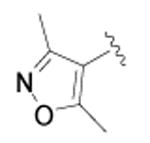
|
cyclopropyl | >10,000 | 3 |
| 44 |

|
cyclopropyl | 1315 ± 233 | 4 |
| 45 |
|
cyclopropyl | 682 ± 100 | 3 |
| 46 |
|
cyclopropyl | 118 ± 11 | 3 |
| 47 |
|
cyclopropyl | 70 ± 23 | 4 |
antiprion potency in ScN2a-cl3 cells by ELISA; SEM is calculated as standard deviation divided by the square root of n.
number of repetitions.
Structure-activity Relationships
All EC50 values were determined in ScN2a-cl3 cells as described previously,[21] and mean EC50 values from three or more separate determinations are reported in the tables, along with standard error of the mean (SEM) values. To confirm that lowering of PrPSc levels was due to drug-like action and not from the compound affecting the viability of ScN2a-cl3 cells, each compound was evaluated for its effects on cell viability, using the fluorescent probe calcein-AM.[21] As with the AMT analogs described in our earlier communications, the new AMTs reported herein do not affect the viability of ScN2a-cl3 cells at relevant concentrations (LC50 > 10 μM for all analogs reported herein).
Our SAR studies began with the premise that the pyridyl C-ring and distal ring of the biaryl A-ring system would be most tolerant of modification and offered the best opportunity for identifying more potent analogs. This conjecture was informed by our best understanding of SAR for the AMT class and the results of the DFT-based conformational analysis, as described above. In fact, seemingly minor structural changes such as replacement of the terminal phenyl ring with pyridyl was not only tolerated but produced analogs (12 and 15) with >10-fold greater potency than 1 in ScN2a-cl3 cells (Table 1). The corresponding pyridine N-oxides 13 and 16 were less potent than the reduced forms, but still more potent than 1.
Perturbation of the pyridine-phenyl dihedral angle with ortho substituents (in 14 and 17) was also tolerated, in contrast to the detrimental effects we observed for similar perturbation of the A-B ring dihedral.[21] Heterocycles other than pyridine could also be introduced at the terminal position, including pyrazole (18), piperazine (20), and morpholine (21). The isoxazole analog 19 was the only heteroaromatic analog inferior to 1, although this effect may be less related to the isoxazole ring than to the presence of two methyl groups in positions that must severely limit the number of energetically reasonable conformations available to this analog.
In our earlier SAR studies of the C-ring we had noted that distal ring substitutions, including fusion to a second ring system (e.g., quinoline for pyridine), produced more potent analogs.[21] However, optimization of brain exposure for the AMT series led us to analogs such as 1, which bears a methyl group proximal to the pyridine ring nitrogen. Possibly, the excellent metabolic stability and prolonged half-life of 1 derives in part from the presence of flanking substituents on either side of the pyridine C-ring nitrogen.[22] In the current study, we sought to retain flanking substituents while exploring amines or ethers in place of the methyl substituent in 1. New analogs like 22–24 alter the basicity of the pyridine ring while introducing greater steric bulk. Evaluation of 22–24 in ScN2a-cl3 cells revealed that pKa appears relatively unimportant with respect to antiprion activity. Hence, weak base 1 (pKa ∼5.6) is nearly equipotent to the increasingly basic analog 23 and 24 (Table 1). Furthermore, the observation that analog 24 retains activity suggests that reasonable steric bulk can be accommodated at the position proximal to the pyridine nitrogen atom (i.e., substituent X, Table 1). The methoxyethoxy ether analog 22, in contrast, was found to be inactive, suggesting an unfavourable entropic and/or steric effect arising from the larger cone angle of the acyclic ether substituent in 22 as compared to the cyclic piperazine ring in 24.
With compounds 12 and 15 representing improved leads, we next explored the introduction of ether or amine substituents on the terminal pyridine ring (Table 2). A smallish para-methoxy substituent as in 25 had little effect on potency, so more sterically demanding side chains were explored. In fact, a methoxyethoxy ether was well tolerated at either the meta or para position (analogs 26 and 27). Branched side chains were less well tolerated, however, with isopropyl ethers 28 and 29 somewhat less potent than 1 and much less potent than the parent pyridine analogs 12 and 15. As with the C-ring modifications discussed above, modifying basicity via A-ring substitutions had a negligible effect on potency: The dimethylaminopyridines 30 and 31 were essentially equipotent to their parent pyridine analogs. Introduction of an N-methylpiperazine side chain was better tolerated at the para position (33) than in the meta position (32). In summary, the introduction of amine or ether side chains in the terminal pyridyl A-ring produced several new analogs that retained the good potency of 12 and 15. With altered basicities and polar surface areas, the in vivo behaviour of these new AMT analogs was of considerable interest.
A parallel SAR effort was directed at exploring more diverse substituents in place of the pyridyl C-ring in 1. Various aliphatic and aromatic substituents were examined with little success. However, small aliphatic amides proved to be perfectly viable C-groups, with the acetamide 35 and cyclopropylamide 34 proving to be ten- and five-fold more potent than 1, respectively. Steric requirements are strict in this series however, as the marginally larger methoxyacetamide 36 was notably less potent than 34 or 35, while isopropylamide 37 and benzamide 38 were without measurable antiprion activity (Table 3).
The SAR of amide-type C-groups was distinct from that of the original AMT series, where aromatic C-rings as large as quinoline were not only tolerated but exhibited enhanced potency.[21] We soon found that other SAR trends also failed to translate between the original pyridyl C-ring series and the new amide C-group series. Hence, a reinvestigation of “optimal” biaryl A-rings was undertaken in the context of a cyclopropyl C-group (Table 3). While a terminal pyridine ring was well tolerated in both contexts (compare 12 and 39), an ortho methyl substituent was not permitted in the cyclopropylamide series (analogs 40, 42, 43). Moving to the meta and para positions, we found that para substitution (46 and 47) was better tolerated than meta substitution (44 and 45). Overall, SAR in the cyclopropylamide C-group series suggests more stringent steric requirements about the biaryl A-ring. Another interpretation of this SAR is that cyclopropylamide analogs require a more nearly coplanar biaryl A-ring system for effective binding. Overall, it appears that AMT analogs bind slightly differently depending on the nature of the C-ring/group.
In vivo Pharmacokinetics
In vivo evaluation of the new AMT analogs was conducted throughout the course of the optimization described above. Here, we summarize our findings and evaluate specific physiochemical properties that appear to impact the brain exposure of AMT analogs in mice. Following an oral dose of 10 mg/kg, compound 1 achieved a brain AUC value 9.78 ± 2.07 μM *h, a brain/plasma AUC ratio > 1, and a brain AUC/EC50 ratio of 7.6 (Table 4). These values represented a benchmark at the outset of the current study. Specifically, we sought to improve brain AUC/EC50 ratio in new analogs, with the long-term aim of achieving animal efficacy at the lowest possible daily dose. Gratifyingly, the highly potent pyridin-3-yl analog 15 was found to achieve a brain AUC value comparable to 1, resulting in a dramatically improved brain AUC/EC50 ratio of 160 (as compared to 7.6 for 1). The N-oxide 16 (the oxidized form of 15) achieved overall exposure comparable to 15, but was predominantly distributed in plasma rather than brain. The same was true of 27, the pyridin-3-yl analog bearing a methoxyethoxy side chain. Despite having poorer brain penetration than 1, analogs 16 and 27 nevertheless exhibited improved brain AUC/EC50 ratios as compared to 1 (Table 4).
Table 4.
In vivo PK parameters and microsome stability for selected AMT analogs following a single 10 mg/kg oral dose in mice.
| Cmpd | Cmax (μM) Brain | AUC (μM *h) | T1/2[a] (min) | Brain AUC/EC50 [b] | |
|---|---|---|---|---|---|
| Brain | Plasma | ||||
| 1 | 2.45 ± 0.74 | 9.78 ± 2.07 | 6.99 ± 0.73 | > 60 | 7.6 |
| 12 | 0.50 ± 0.05 | 1.68 ± 0.10 | ND[c] | >60 | 19 |
| 15 | 2.46 ± 0.85 | 10.90 ± 1.75 | 15.60 ± 0.06 | >60 | 160 |
| 26 | 0.33 ± 0.16 | 1.04 ± 0.13 | ND[c] | ND[c] | 6.8 |
| 32 | 1.17 ± 0.19 | 5.22 ± 0.10 | 1.57 ± 0.15 | 6.8 | 6.9 |
| 27 | 0.97 ± 0.22 | 3.27 ± 0.44 | 6.75 ± 0.45 | >60 | 24 |
| 16 | 0.90 ± 0.24 | 3.85 ± 0.52 | 11.50 ± 0.71 | >60 | 19 |
| 34 | 4.34 ± 0.53 | 18.60 ± 3.75 | 12.70 ± 1.11 | >60 | 75 |
| 45 | 0.25 ± 0.11 | 1.29 ± 0.02 | 0.76 ± 0.01 | 4.4 | 1.9 |
| 46 | 0.15 ± 0.01 | 0.32 ± 0.00 | 0.26 ± 0.01 | 55 | 2.7 |
| 47 | 0.06 ± 0.03 | 0.19 ± 0.08 | 0.38 ± 0.19 | 2.5 | 2.7 |
stability to mouse liver microsomes.
Ratio of brain AUC (μM *h) to EC50 (μM).
not determined.
Given the exceptional in vivo profile of pyridin-3-yl analog 15, it was surprising to find that regioisomeric analog 12 exhibited much inferior brain exposure and an only slightly improved AUC/EC50 ratio compared to 1. Related pyridin-4-yl analogs like 26 and 32 were similarly disappointing in vivo, with AUC/EC50 ratios only comparable to 1. The N-methylpiperazine analog 32 exhibited the best brain/plasma AUC ratio (>3) of the AMT analogs studied but its modest potency produced a brain AUC/EC50 ratio slightly inferior to 1. Of the pyridine C-ring analogs examined in mice, pyridin-3-yl analog 15 emerged as the clear winner. Hence, an increase in brain AUC/EC50 ratio from 7.6 to 160 was realized by the introduction of a single nitrogen atom (compare 1 and 15).
We also examined the in vivo PK properties of AMT analogs bearing cyclopropylamide C-groups, beginning with the progenitor of this series, compound 34. This analog exhibited the highest brain exposure of all the AMT analogs examined, producing an excellent brain AUC/EC50 ratio of 75. Disappointingly, however, none of the other cyclopropylamide analogs evaluated demonstrated anywhere near the brain exposure achieved by 34. Indeed, cyclopropylamide analogs 45, 46 and 47 exhibited very poor exposure in both brain and plasma, resulting in brain AUC/EC50 ratios that were inferior to 1. Possibly the poor metabolic stability of the N-methylpiperazine analogs 45 and 47 is partly to blame for their poor performance. It is of interest to compare the congeneric pairs 34/1 and 32/45, which differ only in the nature of the C-group/ring. With regard to brain AUC levels, cyclopropylamide in 34 is superior to the pyridine in 1, while in the case of 32 and 45, pyridine is favored over cyclopropylamide. Such findings highlight the necessarily empirical nature of lead optimization and the difficulty of predicting in vivo exposure levels, even among closely related structural analogs.
In an attempt to understand better which physiochemical properties most influence the brain exposure of AMT analogs, we correlated experimental PK parameters for the compounds in Table 4 with readily calculated properties like molecular weight, clogP, and total polar surface area (TPSA). These properties have been proposed[23] as potential predictors of permeability across the BBB. We excluded from this analysis 32, 45 and 47, as these analogs appear to have a metabolic liability associated with the N-methylpiperazine ring and are therefore not representative of the AMT class in general (Table 4). Correlation coefficients (r2 values) were determined using the software package Vortex (Dotmatics, Ltd.) and are presented above (Table 5). With regard to measured brain AUC values, clogP was very poorly correlated (r2 = 0.13) while molecular weight (r2 = 0.54) and TPSA (r2 = 0.60) were better predictors. Of course, brain AUC values are clearly dependent on other PK parameters such as oral bioavailability, metabolism, clearance, etc., which will vary unpredictably between analogs. Given this, the brain/plasma AUC ratio might be regarded as a more direct measure of BBB penetration. Unfortunately, none of the calculated properties showed a compelling correlation with brain/plasma AUC ratio, although TPSA was again best (r2 = 0.00, 0.21 and 0.32, for clogP, MW, and TPSA, respectively). Possibly this poor correlation reflects the inability of crude measures like TPSA and clogP to account for active efflux by the drug transporter P-glycoprotein, which plays an important role in small molecule permeability across the BBB.[24]
Table 5.
Correlation of selected calculated properties with experimentally determined PK parameters, expressed as correlation coefficients (r2 values).
| MW | clogP[a] | TPSA[a] | |
|---|---|---|---|
| brain AUC | 0.54 | 0.13 | 0.60 |
| plasma AUC | 0.43 | 0.10 | 0.27 |
| total AUC | 0.70 | 0.10 | 0.60 |
| brain/plasma AUC ratio | 0.21 | 0.00 | 0.32 |
calculated in Vortex using “XlogP” and “TPSA_NOPS” predictors.
Conclusion
In this study we sought to improve the potency of new AMT leads while maintaining the favorable in vivo PK profile of our previous best lead compound 1. The majority of the new analogs described in herein showed improved potency as compared to 1, and several exhibited EC50 values below 100 nM in ScN2a-cl3 cells (Tables 1–3). Of ten new AMT leads evaluated in single-dose mouse PK studies, half (12, 15, 16, 27, and 34) demonstrated brain AUC/EC50 ratios that were substantially improved over 1. Compounds 15 and 34 appear to be particularly promising leads, with brain AUC/EC50 ratios of 160 and 75 following a single oral dose of 10 mg/kg. These analogs would thus be predicted to achieve therapeutic brain concentrations at oral doses much lower than might be required with 1, thus satisfying the original objectives of our study. The evaluation of 15 and 34 in animal models of prion disease will be of significant interest.
Experimental Section
EC50 and LC50 Assays
The methods employed to evaluate the effects of compounds on PrPSc levels and cell viability were analogous to previously published protocols.[21]
Microsome Stability and in vivo PK Studies
Procedures and analysis for the microsome stability and PK studies were performed as previously reported.[22] All animal studies were approved by the Institutional Animal Care and Use Committee at the University of California San Francisco.
Computational Methods
Compound 1 structure was prepared and minimized in the Ligprep module of Schrödinger Software Suite 2010.[25] Nitrogen pKa values were predicted using Epik and two protonation states were generated: neutral and with +1 charge. Each of the states, initially in planar configuration, was then subjected to geometry optimization in Jaguar[25] using DFT[26, 27] with B3LYP functional and 6-31G** basis set. The DIIS convergence scheme and default convergence criteria were used: maximum iteration number of 48 and energy change of 5e-05 hartrees. The calculated properties provided in Table 5 were calculated with Vortex software (Dotmatics, Limited).
Compound Characterization and Purity
Reagents and solvents were purchased from Aldrich Chemical, Acros Organics, Alfa Aesar, AK Scientific, or TCI America and used as received unless otherwise indicated. Compounds 34–38 were obtained from commercial sources. Air- and/or moisture-sensitive reactions were carried out under an argon atmosphere in oven-dried glassware using anhydrous solvents from commercial suppliers. Air- and/or moisture-sensitive reagents were transferred via syringe or cannula and introduced into reaction vessels through rubber septa. Solvent removal was accomplished with a rotary evaporator at ∼10–50 Torr. Microwave irradiation was carried out with a CEM Intellivent Explorer system. Automated silica gel column chromatography was carried out using a Biotage SP1 system and silica gel cartridges from Biotage. Analytical TLC plates from EM Science (Silica Gel 60 F254) were employed for TLC analyses. 1H NMR spectra were recorded on a Varian INOVA-400 400 MHz spectrometer for compounds 1, 12–21, 25, and 39–43 or Bruker Avance III plus 400 MHz for compounds 22-24, 26–33, and 44–47. Chemical shifts are reported in δ units (ppm) relative to trimethylsilyl (TMS) as an internal standard. Coupling constants (J) are reported in hertz (Hz). Characterization data are reported as follows: chemical shift, multiplicity (s=singlet, d=doublet, t=triplet, q=quartet, br=broad, m=multiplet), coupling constants, number of protons, mass-to-charge ratio.
All analogs submitted for testing were judged to be of 95% or higher purity based on analytical LC/MS analysis. For compounds 1, 12–21, 25, and 39–43, LC/MS analyses were performed on a Waters Micromass ZQ/Waters 2795 Separation Module/Waters 2996 Photodiode Array Detector system controlled by MassLynx 4.0 software. Separations were carried out on an XTerra MS C18 5-μm, 4.6 × 50 mm column at ambient temperature using a mobile phase of water-acetonitrile containing 0.05% trifluoroacetic acid. Gradient elution was employed wherein the acetonitrile:water ratio was increased linearly from 5 to 95% acetonitrile over 2.5 min, maintained at 95% acetonitrile for 1.5 min, then decreased to 5% acetonitrile over 0.5 min, and maintained at 5% acetonitrile for 0.5 min. Compound purity was determined by integrating peak areas of the liquid chromatogram, monitored at 254 nm.
LC/MS analyses for compounds 22-24, 26–33, and 44–47 were performed on an Agilent LC/MSD 1200 Series, a quadruple mass spectrometer equipped with a Welchrom XB-C18 column (50 × 4.6 mm, 5 μm) at ambient temperature using a mobile phase of water-acetonitrile containing 0.05% trifluoroacetic acid with a flow rate of 1.5 mL/min. Gradient elution was employed wherein the acetonitrile:water ratio was increased linearly from 5 to 95% acetonitrile over 2.5 minutes, maintained at 95% acetonitrile for 1.5 min, then decreased to 5% acetonitrile over 0.5 min, and maintained at 5% acetonitrile for 0.5 min. Compound purity was determined by integrating peak areas of the liquid chromatogram, monitored at 254 nm.
General Procedure A for preparing aminothiazole analogs from thioureas
An ethanolic solution (∼10 mL) of the desired thiourea (1 mmol) and the requisite bromoacetophenone (1 mmol) was heated at reflux for 3–5 h, or until the reaction was judged complete (LC/MS). The reaction mixture was then poured into water-ice (20 mL) and stirred for another 30 min. A solution of aqueous 1 N Na2CO3 was then added to produce a solution of pH ∼8. The aminothiazole product typically precipitated from this solution and was collected by filtration and washed with water. Crude aminothiazoles were purified by column chromatography on silica gel (∼40–80% ethyl acetate-hexanes) or using reverse-phase HPLC. Relevant fractions were collected and concentrated to afford the desired product.
(4-Biphenyl-4-yl-thiazol-2-yl)-(6-methyl-pyridin-2-yl)-amine (1)
(6-Methyl-pyridin-2-yl)-thiourea[21] was reacted with commercially available 1-biphenyl-4-yl-2-bromo-ethanone, according to General Procedure A to afford the title compound in 85% yield; mp 218–222 °C. 1H NMR (400 MHz, DMSO-d6) δ 2.47 (s, 3H), 6.79 (d, J = 7.14 Hz, 1H), 6.90 (d, J = 8.24 Hz, 1H), 7.32–7.40 (m, 1H), 7.42–7.51 (m, 3H), 7.55–7.63 (m, 1H), 7.67–7.76 (m, 4H), 7.96–8.02 (m, 2H), 11.35 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 23.4, 106.2, 107.5, 114.9, 126.1, 126.4, 126.8, 127.0, 127.4, 128.9, 128.9, 129.1, 134.0, 138.1, 138.9, 139.7, 148.1, 151.1, 155.1, 159.7; MS (ESI) m/z 344 (MH+).
[4-(4-Bromo-phenyl)-thiazol-2-yl]-(6-methyl-pyridin-2-yl)-amine (2)
An ethanolic solution of (6-methyl-pyridin-2-yl)-thiourea (5, 1 mmol)[21] and commercially available 2-bromo-1-(4-bromo-phenyl)-ethanone (1 mmol, 1:1 equiv) was reacted according to general procedure A. The crude product was purified by column chromatography on silica gel (40 to 100% ethyl acetate-hexane) and relevant fractions were collected and concentrated to afford the aryl bromide 2 in 80% yield. 1H NMR (400 MHz, DMSO-d6) δ 2.47 (s, 3H), 6.79 (d, J = 7.33Hz, 1H), 6.88 (d, J = 8.24 Hz, 1H), 7.56–7.64 (m, 3H), 7.82–7.89 (m, 2H), 7.49 (s, 1H), 11.35 (s, 1H),; MS (ESI) m/z 347 (MH+).
N-(6-methylpyridin-2-yl)-4-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)thiazol-2-amine (3)
A modification of the Miyaura borylation procedure[28] was employed. A solution of 2 (5.0 g, 14.5 mmol), bis(pinacolato)diboron (4.8 g, 18.8 mmol), Pd(dppf)2Cl2 (1.2 mg, 1.5 mmol) and KOAc (4.3 g, 43.3 mmol) in dioxane (100 mL) was heated to 80 °C under N2 overnight. The reaction mixture was loaded directly on a rotary evaporator to remove most of the solvents under vacuum. Water (200 mL) was added to the resulting residue, which was then extracted with dichloromethane (3 × 60 mL). The combined organic layers were dried over Na2SO4, filtered, and evaporated to give the crude product as a brown residue. This material was purified by silica gel column chromatography using ethyl acetate/hexane to afford compound 3 as a yellow solid (2.7 g, 64%). 1H NMR (400 MHz, CDCl3) δ 1.35 (12H, s), 2.54 (3H, s), 6.33–6.35 (1H, m), 6.66–6.68 (1H, m), 7.11 (1H, s), 7.29–7.33 (1H, m), 7.81–7.83 (2H, m), 7.89–7.91 (2H, m), 9.72 (1H, s).
1-(6-Fluoropyridin-2-yl)thiourea (7)
To a solution of benzoyl isothiocyanate (3.2 g, 19.6 mmol) in acetone (30 mL) was added 6-fluoropyridin-2-amine (6, 2.0 g, 17.9 mmol) in acetone (15 ml) dropwise, and then the reaction mixture was heated to reflux for 3 h. The reaction mixture was poured on to crushed ice, filtered, and washed with water to afford N-((6-fluoropyridin-2-yl)carbamothioyl)benzamide as a yellow solid (4.9 g, 91% crude yield). LCMS (ESI) m/z 276 (MH+).
To a solution of N-((6-fluoropyridin-2-yl)carbamothioyl)benzamide as a yellow solid (4.9 g, 17.9 mmol) in THF (72 ml) was added 2 N NaOH (18 mL), the reaction mixture was then heated at reflux for 3 h. The reaction mixture was then diluted with water (60 mL) and extracted with dichloromethane (3 × 60 mL). The combined organic layers were washed with brine and water, dried over Na2SO4, and concentrated to intermediate 7 as a yellow solid (1.7 g, 56% yield). 1H NMR (400 MHz, CDCl3) δ 6.62 (1H, dd, J=2.0, 8.0 Hz), 6.76 (1H, dd, J= 1.2, 8.0 Hz), 7.73–7.76 (1H, m), 10.21(1H, br s); MS (ESI) m/z 172 (MH+).
4-([1,1′-Biphenyl]-4-yl)-N-(6-fluoropyridin-2-yl)thiazol-2-amine (8)
A mixture of intermediate 7 (1.7 g, 10.0 mmol), 1-([1,1′-biphenyl]-4-yl)-2-bromoethanone (2.78 g, 10.0 mmol) and NaHCO3 (1.0 g, 12 mmol) in acetonitrile (25 mL) was stirred at 80 °C for 6 h. The reaction mixture was concentrated to give a brown residue, which was suspended in dichloromethane (50 mL). The suspension was filtered and the filtrate was concentrated to afford intermediate 8 as a yellow solid (1.4 g, 40% yield). 1H NMR (400 MHz, CDCl3) δ 6.43 (1H, dd, J=2.4, 8.0 Hz), 6.50 (1H, dd, J= 1.2, 8.0 Hz), 7.12 (1H, s), 7.35 (1H, t, J= 7.2 Hz), 7.45 (2H, t, J= 8.0 Hz), 7.51 (1H, q, J=8.0 Hz), 7.60–7.64 (4H, m), 7.94 (2H, d, J= 8.0 Hz), 9.79 (1H, br s); MS (ESI) m/z 348 (MH+).
Cyclopropanecarboxylic acid [4-(4-bromo-phenyl)-thiazol-2-yl]-amide (10)
A mixture of commercially available 2-bromo-1-(4-bromo-phenyl)-ethanone (9, 1 mmol) and 1.0 equiv of thiourea (1 mmol) in EtOH (∼10 mL) was stirred overnight, or until the reaction was judged completed (LC/MS). The reaction mixture was evaporated and dissolved in ethyl acetate, the organic layer was washed with an aqueous solution of sodium bicarbonate. The organic layer was separated, dried over MgSO4, filtered, and concentrated to afford 4-(4-bromo-phenyl)-thiazol-2-ylamine in 95% yield. This material was used without further purification.1H NMR (400 MHz, DMSO-d6) δ 7.08 (s, 3H), 7.51–7.57 (m, 2H), 7.70–7.77 (m, 2H); MS (ESI) m/z 256 (MH+).
4-(4-Bromo-phenyl)-thiazol-2-ylamine (2.61 mmol) was reacted with 2 equiv of cyclopropanecarbonyl chloride (5.33 mmol) and 4 equiv of DIPEA (10.4 mmol) in dichloromethane (40 mL). The reaction mixture was stirred at room temperature overnight or until the reaction was judged completed (LC/MS). The reaction mixture was washed with 1 N HCl (40 mL) and the organic layer was separated, washed with an aqueous solution of sodium bicarbonate and brine. The organic layer was separated, dried over MgSO4, filtered, and the solvent evaporated. The resulting solid was further washed with ether and dried to afford 10 in 35% yield. 1H NMR (400 MHz, DMSO-d6) δ 0.87–0.96 (m, 4H), 1.93–2.01 (m, 1H), 7.59–7.68 (m, 3H), 7.81–7.88 (m, 2H); LCMS (ESI) m/z 324 (MH+).
N-(4-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)thiazol -2-yl)cyclopropanecarboxamide (11)
A modification of the Miyaura borylation procedure[28] was employed. A solution of 10 (500 mg, 1.55 mmol), bis(pinacolato)diboron (510 mg, 2.01 mmol), Pd(dppf)2Cl2 (120 mg, 0.15 mmol) and KOAc (460 mg, 4.65 mmol) in dioxane (8.0 ml) was heated to 80 °C under N2 overnight. The reaction mixture was placed on a rotary evaporator to remove most of the solvents under vacuum. Water (20 mL) was added to the resulting residue, which was extracted with dichloromethane (3 × 30 mL). The combined organic layers were dried over Na2SO4, filtered, and evaporated to give the crude product as a brown residue, which was purified by silica gel column chromatography using ethyl acetate/petroleum ether (1:20) to afford 11 as an off-white solid (460 mg, 80% yield). 1H NMR (400 MHz, CDCl3) δ 0.72–0.77 (2H, m), 1.06–1.11 (2H, m), 1.25 (1H, m), 1.36 (12H, s), 7.18 (1H, s), 7.80–7.86 (4H, m), 10.30 (1H, br, s).
General Procedure B for preparing aminothiazole analogs from intermediate 2
Coupling conditions described by Fu[29] were employed. A 10-mL microwave reaction vial was charged with 2 (0.42 mmol), the appropriate boronic acid (2.0 equiv), tricyclohexylphosphine (0.04 mmol), and tris (dibenzylideneacetone)dipalladium (0.016 mmol) dissolved in degassed 1,4 dioxane (0.8 mL). A degassed 1.26-M solution of K3PO4 (0.92 mmol) was then added along with a stir bar. The reaction tube was capped and the contents were further degassed for 1 min by bubbling argon through the solution. The reaction was then subjected to microwave irradiation at 150 °C for 30 min. After cooling, the crude reaction mixture was subjected to one of three workup procedures. In workup A, the precipitated product was collected; washed with water, methanol, and ether; and then subjected to preparative HPLC purification. In workup B (for products that did not precipitate), the reaction mixture was partitioned between ethyl acetate and aqueous sodium bicarbonate and the organic layer separated, dried, and solvent removed under vacuum to afford crude product, which was purified by automated silica gel column chromatography using ethyl acetate/hexane. In workup C, the precipitated product was filtered, redissolved in ∼2 mL DMSO, filtered through a 0.02-μm Whatman Anatop filter, and gently swirled on an orbital stirrer with 70 mg of Quadrapure MPA resin overnight to remove palladium. The resin was then removed by filtration and the product was precipitated from ice water to afford the product.
General Procedure C for preparing aminothiazole analogs from intermediate 3
A solution of pinacol boronate 3 (300 mg, 0.76 mmol), the appropriate aryl bromide (1.3 equiv), Pd (dppf)2Cl2 (62 mg, 0.08 mmol, 10 mol%) and K2CO3 (316 mg, 2.29 mmol) in dioxane (25 mL) and water (5 mL) was heated to 80 °C under N2 overnight. The reaction mixture was loaded directly on a rotary evaporator to remove most of the solvents under vacuum. Water (10 mL) was added to the resulting residue, which was then extracted with dichloromethane (3 × 20 mL). The combined organic layers were dried over Na2SO4, filtered, and evaporated to give the crude product, which was then purified by preparative HPLC to afford the product.
General Procedure D for preparing aminothiazole analogs from intermediate 10
A 10-mL microwave reaction vial was charged with 10 (0.23 mmol), the appropriate boronic acid (0.46 mmol, 2.0 equiv), tris (dibenzylideneacetone)dipalladium (0.008 mmol) and tricyclohexylphosphine (0.02 mmol) dissolved in degassed 1,4 dioxane (0.8 mL). A degassed 1.26 M solution of K3PO4 (0.518 mmol) was then added along with a stir bar. The reaction tube was capped and the contents were further degassed for 1 min by bubbling argon through the solution. The reaction was then subjected to microwave irradiation at 150 °C for 30 min. After cooling, reaction products were isolated by one of the following workups. In workup A, the precipitated product was washed with water, MeOH, and ether, then subjected to preparative HPLC purification. In workup B (for products that did not precipitate), the reaction mixture was partitioned between ethyl acetate and aqueous sodium bicarbonate. The organic layer was separated and dried, and the solvent was removed under vacuum to afford crude product that was purified on silica gel column chromatography using ethyl acetate/hexane.
General Procedure E for preparing aminothiazole analogs from intermediate 11
A solution of pinacol boronate 11 (200 mg, 0.54 mmol), the appropriate aryl bromide (1.2 equiv), Pd (dppf)2Cl2 (44 mg, 0.054 mmol, 10 mol%), and K2CO3 (224 mg, 1.62 mmol) in dioxane (25 mL) and water (5 mL) was heated to 80 °C under N2 overnight. The reaction mixture was loaded directly on a rotary evaporator to remove most of the solvents under vacuum. Water (10 mL) was added to the resulting residue, which was extracted with dichloromethane (3 × 20 mL). The combined organic layers were dried over Na2SO4, filtered, and evaporated to give the crude product, which was then purified by preparative HPLC to afford the final product.
(6-Methyl-pyridin-2-yl)-[4-(4-pyridin-4-yl-phenyl)-thiazol-2-yl]-amine (12)
Intermediate 2 was reacted with pyridin-4-ylboranediol according to General Procedure B and workup C to afford the title compound (13 mg) in 9% yield. mp 238–242 °C; 1H NMR (400 MHz, DMSO-d6) δ 2.48 (s, 3H), 6.80 (d, J = 7.51 Hz, 1H), 6.90 (d, J = 8.06 Hz, 1H), 7.59 (t, J = 8.0 Hz, 1H), 7.57 (s, 1H), 7.76 (d, J = 8.0 Hz, 2H), 7.87–7.92 (m, J = 8.24 Hz, 2H), 8.03–8.09 (m, J = 8.42 Hz, 2H), 8.64 (d, J = 5.86 Hz, 2H), 11.38 (s, 1H); MS (ESI) m/z 345 (MH+).
4-(4-(2-((6-Methylpyridin-2-yl)amino)thiazol-4-yl)phenyl)pyridine 1-oxide (13)
Pinacoboronate intermediate 3 was reacted with 4-bromopyridine 1-oxide according to General Procedure C to afford the title compound as a yellow solid (100 mg) in 36% yield. 1H NMR (400 MHz, DMSO-d6) δ 2.42 (3H, s), 6.79–6.80 (1H, m), 6.88–6.90 (1H, m), 7.56–7.62 (2H, m), 7.83–7.88 (4H, m), 8.02–8.04 (2H, m), 8.29–8.28 (2H, m), 11.38 (1H, s); LCMS (ESI) m/z 361.1 (MH+).
(6-Methyl-pyridin-2-yl)-{4-[4-(3-methyl-pyridin-4-yl)-phenyl]-thiazol-2-yl}-amine trifluoro acetic acid salt (14)
Intermediate 2 was reacted with commercially available 3-picoline-4-boronic acid according to General Procedure B and workup A to afford the title compound (20 mg) in 29% yield. 1H NMR (400 MHz, DMSO-d6) δ 2.44 (s, 3H), 2.48 (s, 3H), 6.81 (d, J = 7.33 Hz, 1H), 6.92 (d, J = 8.06 Hz, 1H), 7.57–7.65 (m, 4H), 7.81 (d, J = 5.68 Hz, 1H), 8.06–8.12 (m, 2H), 8.74 (d, J = 5.86 Hz, 1H), 8.81 (s, 1H); MS (ESI) m/z 359 (MH+).
(6-Methyl-pyridin-2-yl)-[4-(4-pyridin-3-yl-phenyl)-thiazol-2-yl]-amine (15)
Intermediate 2 was reacted with pyridin-3-ylboranediol according to General Procedure B and was further treated according to workup C to afford the title compound (52 mg) in 37% yield; mp 176–117 °C; 1H NMR (400 MHz, DMSO-d6) δ 2.48 (s, 3 H) 6.81 (d, J=7.14 Hz, 1 H) 6.91 (d, J=8.24 Hz, 1 H) 7.52–7.67 (m, 2 H) 7.79 (dd, J=8.15, 5.22 Hz, 1 H) 7.89 (d, J=8.61 Hz, 2 H) 8.08 (d, J=8.42 Hz, 2 H) 8.50 (d, J=7.69 Hz, 1 H) 8.72 (d, J=4.94 Hz, 1 H) 9.12 (br. s., 1 H) 1.37 (br. s., 1 H); 13C NMR (100 MHz, DMSO-d6) δ 23.4, 106.7, 107.5, 115.0, 123.9, 126.3, 127.1, 133.9, 134.6, 135.1, 135.8, 138.2, 147.4, 147.9 148.4, 151.1, 155.2, 159.7; MS (ESI) m/z 345 (MH+).
3-(4-(2-((6-Methylpyridin-2-yl)amino)thiazol-4-yl)phenyl)pyridine 1-oxide (16)
Pinacoboronate intermediate 3 was reacted with 3-bromopyridine 1-oxide according to General Procedure C to afford the title compound as a yellow solid (42 mg) in 23% yield. 1H NMR (400 MHz, DMSO-d6) δ 2.50 (3H, s), 6.80–6.81 (1H, m), 6.89–6.91 (1H, m), 7.49–7.61 (3H, m), 7.63 (1H, s), 7.71–7.73 (2H, m), 8.03–8.05 (2H, m), 8.21–8.23 (1H, m), 8.64 (1H, s), 11.38 (1H, s); MS (ESI) m/z 361.1 (MH+).
(6-Methyl-pyridin-2-yl)-{4-[4-(2-methyl-pyridin-3-yl)-phenyl]-thiazol-2-yl}-amine trifluoro acetic acid salt (17)
Intermediate 2 was reacted with commercially available 2-methyl-3-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-pyridine according to General Procedure B and workup B. Automated silica gel chromatography followed by preparative HPLC purification afforded the title compound (44 mg) in 64% yield; mp 174–176 °C; 1H NMR (400 MHz, DMSO-d6) δ 2.45 (s, 3H), 2.68 (s, 3H), 6.81 (d, J = 7.3 Hz, 1H), 6.92 (d, J = 8.2 Hz, 1H), 7.49–7.72 (m, 4H), 7.89 (d, J =5.7 Hz, 1H), 8.08 (d, J = 7.3 Hz, 2H), 8.36 (d, J = 7.9 Hz, 1H), 8.79 (d, J = 5.5 Hz, 1H), 11.37 (br. s., 1H); MS (ESI) m/z 359 (MH+).
{4-[4-(2-Methyl-2H-pyrazol-3-yl)-phenyl]-thiazol-2-yl}-(6-methyl-pyridin-2-yl)-amine (18)
Intermediate 2 (50 mg, 0.145 mmol) was reacted with commercially available (1-methyl-1H-pyrazol-5-yl)boranediol (36.5 mg, 0.290 mmol) according to General Procedure B except that catalyst loading was increased (0.21 mmol tricyclohexyl phosphine, 0.087 mmol of palladium catalyst) and with microwave irradiation at 150 °C for 1 h. Workup B afforded in the title compound (32 mg) in 62% yield. 1H NMR (400 MHz, CDCl3) δ 2.54 (s, 3H), 3.87 (s, 3H), 6.31 (d, J = 1.83 Hz, 1H), 6.35 (d, J = 8.24 Hz, 1H), 6.68 (d, J = 7.33 Hz, 1H), 7.10 (s, 1H), 7.30 (t, J = 7.78 Hz, 1H), 7.42 (d, J = 8.42 Hz, 2H), 7.51 (d, J = 1.83 Hz, 1H), 7.96 (d, J = 8.24 Hz, 2H), 9.79 (s, 1H); MS (ESI) m/z 348 (MH+).
{4-[4-(3,5-Dimethyl-isoxazol-4-yl)-phenyl]-thiazol-2-yl}-(6-methyl-pyridin-2-yl)-amine (19)
Intermediate 2 (50 mg, 0.145 mmol) was reacted with commercially available (3, 5-dimethyl-1,2-oxazol-4-yl)boranediol (29.7 mg, 0.211 mmol) according to General Procedure B and workup B. The material was further purified by preparative HPLC purification, which afforded the title compound as a trifluoroacetate salt (43 mg) in 62% yield. 1H NMR (400 MHz, CDCl3) δ 2.32 (s, 3H), 2.46 (s, 3H), 2.62 (s, 3H), 6.96 (t, J = 3.66 Hz, 2H), 7.31 (d, J = 7.69 Hz, 1H), 7.38–7.43 (m, 2H), 7.70 (t, J = 7.87 Hz, 1H), 7.84–7.90 (m, 2H); MS (ESI) m/z 363 (MH+).
{4-[4-(4-Methyl-piperazin-1-yl)-phenyl]-thiazol-2-yl}-(6-methyl-pyridin-2-yl)-amine (20)
Intermediate 2 (500 mg, 1.45 mmol), N-methyl piperazine (311 mg, 0.345 ml, 3.11 mmol), sodium tert-butoxide (350 mg, 3.60 mmol), tris (dibenzylideneacetone)dipalladium (63 mg, 0.068 mmol) and (-)BINAP (63 mg, 0.1 mmol) were dissolved in dry dimethylformamide that had been degassed by bubbling with argon for 2 min. The tube was further degassed for 1 min and then subjected to microwave irradiation (140 °C for 1 h). The crude residue was diluted with ethyl acetate and extracted twice with saturated aqueous sodium bicarbonate. The combined organic layers were dried with brine and MgSO4, filtered, and evaporated. The crude residue was purified using automated silica gel chromatography (methylene chloride/methanol, 9:1). The product thus obtained was taken up in 8 ml of 1:1 methanol:dichloromethane, and 1 M HCl in ether was added until a precipitate formed. The hydrochloride salt was filtered to afford the title compound (34 mg) in 5% yield. 1H NMR (400 MHz, D2O) δ 2.46 (s, 3H), 2.98 (s, 3H), 3.05 (m, 2H), 3.18–3.29 (m, 2H), 3.65 (d, J = 12.27 Hz, 2H), 3.80 (d, J = 13.37 Hz, 2H), 6.82 (d, J = 8.42 Hz, 2H), 6.90 (d, J = 8.79 Hz, 1H), 7.04–7.13 (m, 2H), 7.33 (d, J = 8.24 Hz, 2H), 8.01 (t, J = 8.15 Hz, 1H); MS (ESI) m/z 366 (MH+).
(6-Methyl-pyridin-2-yl)-[4-(4-morpholin-4-yl-phenyl)-thiazol-2-yl]-amine (21)
Intermediate 2 (200 mg, 0.580 mmol) and morpholine (126 mg, 0.126 ml, 1.45 mmol), were reacted according to the procedure described above for 6-methyl-N-{4-[4-(4-methylpiperazin-1-yl)phenyl]-1,3-thiazol-2-yl}pyridin-2-amine (20). The crude reaction residue was purified by automated silica gel chromotagraphy (1:1 hexane/ethyl acetate). Half of this material was further purified by preparative HPLC to afford the trifluoroacetate salt (2.9 mg) in 2.1% yield. 1H NMR (400 MHz, CDCl3) δ 2.60 (s, 3H) 3.22–3.29 (m, 4H), 3.85–3.91 (m, 4H), 6.70 (s, 1H), 6.93 (d, J = 7.51 Hz, 1H), 6.96–7.02 (m, 2H), 7.20 (br. s., 1H), 7.63–7.68 (m, 1H), 7.68–7.72 (m, 2H); MS (ESI) m/z 353 (MH+).
4-([1,1′-Biphenyl]-4-yl)-N-(6-(2-methoxyethoxy)pyridin-2-yl)thiazol-2-amine (22)
To a reaction vial loaded with 2-methoxyethan-1-ol (10 mL), sodium metal (106 mg, 4.6 mmol) was added; the resulting mixture was stirred at room temperature until a clear solution was obtained. 2-Fluropyridine intermediate 8 (200 mg, 0.58 mmol) was added to the reaction mixture, and the reaction was heated to 125 °C for 14 h. After cooling to room temperature, the reaction mixture was diluted with water (30 mL) and extracted with dichloromethane (3 × 50 mL). The combined organic layers were washed with brine, then dried over Na2SO4, and concentrated to give a brown residue, which was purified by preparative HPLC to afford the product 22 as a yellow solid (49 mg, 21% yield). 1H NMR (400 MHz, CDCl3) δ 3.48 (3H, s), 3.83–3.85 (2H, m), 4.69–4.71 (2H, m), 6.23 (1H, d, J = 8.0 Hz), 6.38 (1H, d, J = 8.0 Hz), 7.05 (1H, s), 7.26–7.35 (1H, m), 7.37–7.47 (3H, m), 7.62–7.65 (4H, m), 7.92–7.94 (2H, m), 8.77 (1H, s); MS (ESI) m/z 404 (MH+).
N2-(4-([1,1′-biphenyl]-4-yl)thiazol-2-yl)-N6,N6-dimethylpyridine-2,6-diamine (23)
To a 200 mL reaction vessel, intermediate 24 (450 mg, 1.3 mmol) and dimethylamine aqueous solution (100 mL, 33%) were added. The vessel was then sealed and heated to 130 °C overnight. After cooling to room temperature, the reaction mixture was extracted with ethyl acetate (3 × 100 mL). The combined organic layers were washed with brine, then dried over Na2SO4, and concentrated to give a brown residue, which was purified by preparative HPLC to afford the product 23 as a gray solid (54 mg, 11% yield). 1H NMR (400 MHz, CDCl3) δ 3.20 (6H, s), 5.93 (1H, d, J = 8.0 Hz), 6.02 (1H, d, J = 8.0 Hz), 7.02 (1H, s), 7.30–7.36 (2H, m), 7.43–7.47 (2H, m), 7.63–7.65 (4H, m), 7.92–7.95 (2H, m), 8.52 (1H, s); MS (ESI) m/z 373 (MH+).
4-([1,1′-Biphenyl]-4-yl)-N-(6-(4-methylpiperazin-1-yl)pyridin-2-yl)thiazol-2-amine (24)
A vial was charged with intermediate 8 (130 mg, 0.4 mmol) and N-methylpiperazine (5 mL), then sealed and heated to 120 °C for 4 h. After cooling to room temperature, the reaction mixture was poured slowly to ice water (30 mL); the resulting light brown precipitates were collected by filtration. Further purification by preparative HPLC gave 24 as a white solid (69 mg, 37% yield). 1H NMR (400 MHz, CDCl3) δ 2.37 (3H, s), 2.56–2.58 (4H, m), 3.69–3.72 (4H, m), 6.07–6.09 (1H, m), 6.19–6.21 (1H, m), 7.02 (1H, s), 7.33–7.46 (4H, m), 7.63–7.65 (4H, m), 7.91–7.93 (2H, m), 8.26–8.32 (1H, s); MS (ESI) m/z 428.1 (MH+).
{4-[4-(6-Methoxy-pyridin-3-yl)-phenyl]-thiazol-2-yl}-(6-methyl-pyridin-2-yl)-amine (25)
4-(4-Bromophenyl)-N-(6-methylpyridin-2-yl)-1,3-thiazol-2-amine 2 (50 mg, 0.145 mmol) was reacted with (6-methoxypyridin-3-yl)boranediol (33.2 mg, 0.217 mmol) according to General Procedure B and workup C to afford the title compound (16 mg) in 29% yield; mp 217–222 °C; 1H NMR (400 MHz, DMSO-d6) δ 2.48 (s, 3H), 3.91 (s, 3H), 6.79 (dd, J = 0.55, 7.33 Hz, 1H), 6.87–6.95 (m, 2H), 7.48 (s, 1H), 7.57–7.63 (m, 1H), 7.70–7.76 (m, J = 8.06 Hz, 2H), 7.96–8.02 (m, J = 7.87 Hz, 2H), 8.07 (ddd, J = 0.82, 2.61, 8.65 Hz, 1H), 8.48–8.60 (m, 1H), 11.35 (s, 1H); MS (ESI) m/z 375 (MH+).
4-(4-(2-(2-methoxyethoxy)pyridin-4-yl)phenyl)-N-(6-methylpyridin-2-yl)thiazol-2-amine (26)
Intermediate 3 was reacted with 4-bromo-2-(2-methoxyethoxy)pyridine according to General Procedure C to afford the title compound as a yellow solid (74 mg) in 35% yield. 1H NMR (400 MHz, DMSO-d6) δ 2.56 (3H, s), 3.47 (3H, s), 3.77–3.80 (2H, m), 4.52–4.54 (2H, m), 6.52–6.54 (1H, m), 6.72–6.73 (1H, m), 6.07–6.10 (2H, m), 7.10–7.13 (1H, m), 7.40–7.44 (1H, m), 7.64–7.66 (2H, m), 7.96–7.98 (2H, m), 8.17–8.18 (1H, m), 8.19 (1H, s); MS (ESI) m/z 419.1 (MH+).
4-(4-(6-(2-methoxyethoxy)pyridin-3-yl)phenyl)-N-(6-methylpyridin-2-yl)thiazol-2-amine (27)
Intermediate 3 was reacted with 5-bromo-2-(2-methoxyethoxy)pyridine according to General Procedure C to afford the title compound as a yellow solid (123 mg) in 58% yield. 1H NMR (400 MHz, DMSO-d6) δ 2.48 (3H, s), 3.34 (3H, s), 3.67–3.70 (2H,m), 4.42–4.44 (2H, m), 6.79–6.80 (1H, m), 6.89–6.95 (2H, m), 7.49 (1H,s), 7.59–7.62 (1H,m), 7.72–7.74 (2H,m), 7.99–8.00 (1H,m), 8.06–8.09 (1H, m), 8.08–8.09 (1H, m), 8.53 (1H, m), 11.37 (1H, s); MS (ESI) m/z 419.1 (MH+).
4-(4-(2-isopropoxypyridin-4-yl)phenyl)-N-(6-methylpyridin-2-yl)thiazol-2-amine (28)
Pinacoboronate intermediate 3 was reacted with 4-bromo-2-isopropoxypyridine according to General Procedure C to afford the title compound as a yellow solid (57 mg) in 28% yield. 1H NMR (400 MHz, DMSO-d6) δ 1.35–1.39 (6H, m), 2.55 (3H, s), 5.32–5.39 (1H, m), 6.37–6.39 (1H, m), 6.68–6.70 (1H, m), 6.93 (1H, s), 7.07–7.12 (2H, m), 7.30–7.34 (1H, m), 7.63–7.65 (2H, m), 7.97–7.99 (2H, m), 8.18–8.19 (1H, m), 9.72 (1H, s); MS (ESI) m/z 403.1 (MH+).
4-(4-(6-Isopropoxypyridin-3-yl)phenyl)-N-(6-methylpyridin-2-yl)thiazol-2-amine (29)
Intermediate 3 was reacted with 5-bromo-2-isopropoxypyridine according to General Procedure C to afford the title compound as a yellow solid (42 mg) in 21% yield. 1H NMR (400 MHz, DMSO-d6) δ 1.35–1.39 (6H, m), 2.55 (3H, m), 5.32–5.38 (1H,m), 6.60(x002C6)=6.62 (1H, m), 6.74–6.77 (2H, m), 7.06 (1H, s), 7.46–7.50 (1H,m), 7.56–7.58 (2H, m), 7.80–7.81 (1H, m), 7.93–7.95 (2H, m), 8.37–8.42 (2H, m); MS (ESI) m/z 403.1 (MH+).
4-(4-(2-(dimethylamino)pyridin-4-yl)phenyl)-N-(6-methylpyridin-2-yl)thiazol-2-amine (30)
Intermediate 3 was reacted with 4-bromo-N,N-dimethylpyridin-2-amine according to General Procedure C to afford the title compound as a yellow solid (33 mg) in 17% yield. 1H NMR (400 MHz, DMSO-d6) δ 2.57 (3H, s), 3.16 (6H, s), 6.50–6.52 (1H, m), 6.72–6.74 (2H, m), 6.80 (1H, m), 7.40–7.42 (1H, m), 7.42–7.44 (2H, m), 7.96–7.98 (2H, m), 8.22–8.24 (1H, m), 9.01 (1H, s); MS (ESI) m/z 388.1 (MH+).
4-(4-(6-(Dimethylamino)pyridin-3-yl)phenyl)-N-(6-methylpyridin-2-yl)thiazol-2-amine (31)
Intermediate 3 was reacted with 5-bromo-N,N-dimethylpyridin-2-amine according to General Procedure C to afford the title compound as a yellow solid (127 mg) in 43% yield. 1H NMR (400 MHz, DMSO-d6) δ 2.47 (3H, s), 3.08 (6H, s), 6.73–6.75 (2H, m), 6.85–6.87 (1H, m), 7.39 (1H, s), 7.55–7.59 (1H, m), 7.65–7.68 (2H, m), 7.88–7.90 (1H, m), 7.92–7.96 (2H, m), 8.49 (1H, s); MS (ESI) m/z 388.1 (MH+).
4-(4-(2-(4-methylpiperazin-1-yl)pyridin-4-yl)phenyl)-N-(6-methylpyridin-2-yl)thiazol-2-amine (32)
Intermediate 3 was reacted with 1-(4-bromopyridin-2-yl)-4-methylpiperazine according to General Procedure C to afford the title compound as a yellow solid (110 mg) in 49% yield. 1H NMR (400 MHz, DMSO-d6) δ 2.23 (3H, s), 2.42–2.47 (4H, m), 2.50 (3H, s), 3.58 (4H, m), 6.75–6.76 (1H, m), 6.84–6.86 (1H, m), 6.99–7.01 (1H, m), 7.10 (1H, s), 7.49 (1H, s), 7.55–7.59 (1H, m), 7.82–7.84 (2H, m), 8.00–8.02 (2H, m), 8.16–8.18 (1H, m), 11.42 (1H, s); MS (ESI) m/z 443.2 (MH+).
4-(4-(6-(4-Methylpiperazin-1-yl)pyridin-3-yl)phenyl)-N-(6-methylpyridin-2-yl)thiazol-2-amine (33)
Intermediate 3 was reacted with 1-(5-bromopyridin-2-yl)-4-methylpiperazine according to General Procedure C to afford the title compound as a yellow solid (128 mg) in 57% yield. 1H NMR (400 MHz, DMSO-d6) δ 2.23 (3H, s), 2.31–2.33 (4H, m), 2.40 (3H,s), 3.52–3.57 (4H, m), 6.76–6.78 (1H, m), 6.86–6.94 (2H, m), 7.42 (1H,s), 7.56–7.59 (1H, m), 7.67–7.69 (2H, m), 7.89–7.97 (3H, m), 8.50–8.52 (1H, m), 11.37 (1H, s); MS (ESI) m/z 443.2 (MH+).
Cyclopropanecarboxylic acid [4-(4-pyridin-4-yl-phenyl)-thiazol-2-yl]-amide trifluoroacetic acid salt (39)
Intermediate 10 was reacted with commercially available 4-pyridine-boronic acid according to General Procedure D. The crude material was further purified via workup A to afford the title compound in 12% yield; mp 233–239 °C; 1H NMR (400 MHz, DMSO-d6) δ 0.88–0.98 (m, 4H), 1.95–2.04 (m, 1H), 7.82–7.86 (s, 1H), 8.05–8.15 (m, 4H), 8.25 (d, J = 5.49 Hz, 2H), 8.87 (d, J = 5.49 Hz, 2H); MS (ESI) m/z 322 (MH+).
Cyclopropanecarboxylic acid {4-[4-(2-methyl-pyridin-3-yl)-phenyl]-thiazol-2-yl}-amide (40)
Intermediate 10 was reacted with commercially available 2-methyl-3-(4,4,5,5-tetramethyl-[1,3, 2]dioxaborolan-2-yl)-pyridine according to General Procedure D. The crude product was further purified via workup B to afford the title compound in 50% yield; mp 264–266 °C; 1H NMR (400 MHz, CDCl3) δ 0.55–0.66 (m, 2H), 1.00–1.09 (m, 2H), 1.22–1.36 (m, 1H), 2.57 (s, 3H), 7.15–7.25 (m, 2H),11.66 (br. s., 1H), 7.34–7.46 (m, 2H), 7.54 (dd, J = 1.65, 7.69 Hz, 1H), 7.88–7.96 (m, 2H), 8.54 (dd, J = 1.74, 4.85 Hz, 1H); MS (ESI) m/z 336 (MH+).
Cyclopropanecarboxylic acid {4-[4-(6-dimethylamino-pyridin-3-yl)-phenyl]-thiazol-2-yl}-amide (41)
Intermediate 10 was reacted with commercially available 6-(dimethylamino)pyridine-3-boronic acid according to General Procedure D except that the workup was modified. The crude product was filtered on paper, redissolved in ∼2 mL DMSO and filtered through a 0.02-μm Whatman Anatop filter. The filtrate was swirled gently on an orbital stirrer with 70 mg of Quadrapure MPA resin overnight. The resin was then removed by filtration and the product precipitated from ice water to afford the title compound in 58% yield. 1H NMR (400 MHz, DMSO-d6) δ 0.89–0.96 (m, 4H), 1.94–2.04 (m, 1H), 3.07 (s, 6H), 6.73 (d, J = 8.97 Hz, 1H), 7.60 (s, 1H), 7.68 (d, J = 8.24 Hz, 2H), 7.88 (dd, J = 2.56, 8.97 Hz, 1H), 7.93 (d, J = 8.24 Hz, 2H), 8.49 (d, J = 2.56 Hz, 1H), 12.53 (s, 1H); MS (ESI) m/z 365 (MH+).
Cyclopropanecarboxylic acid {4-[4-(2-methyl-2H-pyrazol-3-yl)-phenyl]-thiazol-2-yl}-amide (42)
Intermediate 10 was reacted with commercially available 1-methyl-1H-pyrazole-5-boronic acid according to General Procedure D to afford the title compound in 95% yield; mp 215–217 °C; 1H NMR (400 MHz, CDCl3) δ 0.68 (m, 2H), 1.08 (m, 2H), 1.28–1.38 (m, 1H), 3.95 (s, 3H), 6.35 (s, 1H), 7.21 (s, 1H), 7.46–7.53 (d, J = 7.33, 2H), 7.55 (s, 1H), 7.88–7.98 (d, J = 7.51, 2H), 11.02 (br. s., 1H); MS (ESI) m/z 325 (MH+).
Cyclopropanecarboxylic acid {4-[4-(3,5-dimethyl-isoxazol-4-yl)-phenyl]-thiazol-2-yl}-amide (43)
Intermediate 10 was reacted with commercially available 3,5-dimethylisoxazole-4-boronic acid according to General Procedure D with purification on silica to afford the title compound in 54% yield; mp 222–223 °C; 1H NMR (400 MHz, CDCl3) δ 0.59 (m, 2H), 0.98–1.08 (m, 2H), 1.22 –1.32 (m, 1H), 2.32 (s, 3H), 2.45 (s, 3H), 7.19 (s, 1H), 7.29–7.37 (m, 2H), 7.88–7.95 (m, 2H), 11.51 (s, 1H); MS (ESI) m/z 340 (MH+).
N-(4-(4-(2-(2-methoxyethoxy)pyridin-4-yl)phenyl)thiazol-2-yl)cyclopropanecarboxamide (44)
Pinacoboronate intermediate 11 was reacted with 4-bromo-2-(2-methoxyethoxy)pyridine according to General Procedure E to afford the title compound as a white solid (106 mg) in 43% yield. 1H NMR (400 MHz, DMSO-d6) δ 0.93 (4H, t), 1.99 (1H, m), 2.82 (3H, s), 3.68 (2H, t), 4.43 (2H, t), 7.18 (1H, s), 7.37 (1H, d), 7.73 (1H, s), 7. 87 (2H, d), 8.02 (2H, d), 8.21 (1H, d), 12.55 (1H, s); MS (ESI) m/z 396 (MH+).
N-(4-(4-(2-(4-methylpiperazin-1-yl)pyridin-4-yl)phenyl)thiazol-2-yl)cyclopropanecarboxamide (45)
Pinacoboronate intermediate 11 was reacted with 1-(4-bromopyridin-2-yl)-4-methylpiperazine according to General Procedure E to afford the title compound as a white solid (27 mg) in 9% yield. 1H NMR (400 MHz, CDCl3) δ 0.86 (2H, m), 1.14 (2H, m), 1.50 (1H, m), 2.67 (4H, s), 3.72 (4H, s), 6.90 (2H, m), 7.18 (1H, s), 7.65 (2H, d), 7. 90 (2H, d), 8.25 (1H, d), 10.21 (1H, s); MS (ESI) m/z 420 (MH+).
N-(4-(4-(6-(2-methoxyethoxy)pyridin-3-yl)phenyl)thiazol-2-yl)cyclopropanecarboxamide (46)
Pinacoboronate intermediate 11 was reacted with 5-bromo-2-(2-methoxyethoxy)pyridine according to General Procedure E to afford the title compound as a white solid (28 mg) in 13% yield. 1H NMR (400 MHz, DMSO-d6) δ 0.92 (4H, t), 1.99 (1H, m), 2.82 (3H, s), 3.68 (2H, t), 4.43 (2H, t), 6.93 (1H, d), 7.74 (2H, d), 7.98 (2H, d), 8.52 (1H, d), 12.54 (1H, s); MS (ESI) m/z 396 (MH+).
N-(4-(4-(6-(4-methylpiperazin-1-yl)pyridin-3-yl)pheyl)thiazol-2-yl)cyclopropanecarboxamide (47)
Pinacoboronate intermediate 11 was reacted with 1-(5-bromopyridin-2-yl)-4-methylpiperazine according to General Procedure E to afford the title compound as a white solid (17 mg) in 6% yield. 1H NMR (400 MHz, DMSO-d6) δ 0.93 (4H, t), 1.99 (1H, m), 2.24 (3H, s), 2.45 (4H, m), 3.56 (4H, m), 6.93 (1H, d), 7.61 (1H, s), 7.69 (2H, d), 7.92 (3H, m), 8.50 (1H, d), 12.54 (1H, s); MS (ESI) m/z 420 (MH+).
Acknowledgments
This work was funded by grants from the National Institutes of Health (AG021601, AG031220, AG002132, AG10470) as well as by gifts from the Sherman Fairchild Foundation, Larry L. Hillblom Foundation, Lincy Foundation and Robert Galvin. The authors thank Mr. Phillip Benner for the preparation, animal dosing, and sample collection in PK studies; and Dr. Kurt Giles and the staff of the Hunter's Point animal facility for expert animal studies.
References
- 1.Jucker M, Walker LC. Ann Neurol. 2011;70:532–540. doi: 10.1002/ana.22615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prusiner SB. Science. 2012;336:1511–1513. doi: 10.1126/science.1222951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aguzzi A, Sigurdson C, Heikenwaelder M. Annu Rev Pathol. 2008;3:11–40. doi: 10.1146/annurev.pathmechdis.3.121806.154326. [DOI] [PubMed] [Google Scholar]
- 4.Pan KM, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, Mehlhorn I, Huang Z, Fletterick RJ, Cohen FE, Prusiner SB. Proc Natl Acad Sci USA. 1993;90:10962–10966. doi: 10.1073/pnas.90.23.10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chien P, Weissman JS, DePace AH. Annu Rev Biochem. 2004;73:617–656. doi: 10.1146/annurev.biochem.72.121801.161837. [DOI] [PubMed] [Google Scholar]
- 6.Soto C, Castilla J. Nat Med. 2004;10:S63–S67. doi: 10.1038/nm1069. [DOI] [PubMed] [Google Scholar]
- 7.Jansen K, Schäfer O, Birkmann E, Post K, Serban H, Prusiner SB, Riesner D. Biol Chem. 2001;382:683–691. doi: 10.1515/BC.2001.081. [DOI] [PubMed] [Google Scholar]
- 8.Shorter J, Lindquist S. EMBO J. 2008;27:2712–2724. doi: 10.1038/emboj.2008.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prusiner SB, Scott M, Foster D, Pan KM, Groth D, Mirenda C, Torchia M, Yang SL, Serban D, Carlson GA, Hoppe PC, Westaway D, DeArmond SJ. Cell. 1990;63:673–686. doi: 10.1016/0092-8674(90)90134-z. [DOI] [PubMed] [Google Scholar]
- 10.Masel J, Genoud N, Aguzzi A. J Mol Biol. 2005;345:1243–1251. doi: 10.1016/j.jmb.2004.10.088. [DOI] [PubMed] [Google Scholar]
- 11.Caughey B, Baron GS, Chesebro B, Jeffrey M. Annu Rev Biochem. 2009;78:177–204. doi: 10.1146/annurev.biochem.78.082907.145410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Novitskaya V, Bocharova OV, Bronstein I, Baskakov IV. J Biol Chem. 2006;281:13828–13836. doi: 10.1074/jbc.M511174200. [DOI] [PubMed] [Google Scholar]
- 13.Prusiner SB, May BCH, Cohen FE. In: Prion Biology and Diseases. Prusiner SB, editor. Cold Spring Harbor Laboratory Press; Cold Spring Harbor: 2004. pp. 961–1014. [Google Scholar]
- 14.Caspi S, Sasson SB, Taraboulos A, Gabizon R. J Biol Chem. 1998;273(6):3484–3489. doi: 10.1074/jbc.273.6.3484. [DOI] [PubMed] [Google Scholar]
- 15.Dollinger S, Lober S, Klingenstein R, Korth C, Gmeiner P. J Med Chem. 2006;49:6591–6595. doi: 10.1021/jm060773j. [DOI] [PubMed] [Google Scholar]
- 16.Heal W, Thompson MJ, Mutter R, Cope H, Louth JC, Chen B. J Med Chem. 2007;50:1347–1353. doi: 10.1021/jm0612719. [DOI] [PubMed] [Google Scholar]
- 17.Kimata A, Nakagawa H, Ohyama R, Fukuuchi T, Ohta S, Doh-ura K, Suzuki T, Miyata N. J Med Chem. 2007;50:5053–5056. doi: 10.1021/jm070688r. [DOI] [PubMed] [Google Scholar]
- 18.May BCH, Zorn JA, Witkop J, Sherrill J, Wallace AC, Legname G, Prusiner SB, Cohen FE. J Med Chem. 2007;50:65–73. doi: 10.1021/jm061045z. [DOI] [PubMed] [Google Scholar]
- 19.Thompson MJ, Louth JC, Ferrara S, Jackson MP, Sorrell FJ, Cochrane EJ, Gever J, Baxendale S, Silber BM, Roehl HH, Chen B. Eur J Med Chem. 2011;46:4125–4132. doi: 10.1016/j.ejmech.2011.06.013. [DOI] [PubMed] [Google Scholar]
- 20.Ghaemmaghami S, May BCH, Renslo AR, Prusiner SB. J Virol. 2010;84:3408–3412. doi: 10.1128/JVI.02145-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gallardo-Godoy A, Gever J, Fife KL, Silber BM, Prusiner SB, Renslo AR. J Med Chem. 2011;54:1010–1021. doi: 10.1021/jm101250y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Silber BM, Rao S, Fife KL, Gallardo-Godoy A, Renslo AR, Dalvie DK, Giles K, Freyman Y, Elepano M, Gever JR, Lam B, Jacobson MP, Huang Y, Benet LZ, Prusiner SB. Pharm Res. doi: 10.1007/s11095-012-0912-4. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hitchcock SA, Pennington LD. J Med Chem. 2006;49:7559–7583. doi: 10.1021/jm060642i. [DOI] [PubMed] [Google Scholar]
- 24.Dolghih E, Bryant C, Renslo AR, Jacobson MP. PLoS Comput Biol. 2011;7:e1002083. doi: 10.1371/journal.pcbi.1002083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jaguar. version 7.8. Schrödinger, L.L.C.; New York: 2011. [Google Scholar]
- 26.Hohenberg P, Kohn W. Phys Rev. 1964;136:B864–B871. [Google Scholar]
- 27.Kohn W, Sham LJ. Phys Rev. 1965;140(4A):A1133–A1138. [Google Scholar]
- 28.Ishiyama T, Murata M, Miyaura N. J Org Chem. 1995;60:7508–7510. [Google Scholar]
- 29.Kudo N, Perseghini M, Fu GC. Angew Chem Int Ed. 2006;45:1282–1284. doi: 10.1002/anie.200503479. [DOI] [PubMed] [Google Scholar]