

Abstract
The recently identified C9ORF72 gene accounts for a large proportion of amyotrophic lateral sclerosis and frontotemporal lobar degenerations. Since several forms of these disorders are associated with parkinsonism, we hypothesized that some patients with Parkinson’s disease or other forms of parkinsonism might carry pathogenic C9OFR72 expansions. Therefore, we looked for C9ORF72 repeat expansions in 1,446 parkinsonian unrelated patients consisted of 1,225 clinically diagnosed with Parkinson’s disease, 123 with progressive supranuclear palsy, 21 with corticobasal degeneration syndrome, 43 with Lewy body dementia and 25 with multiple system atrophy-parkinsonism. Of the 1,446 parkinsonian patients, five carried C9ORF72 expansions: three patients with typical Parkinson’s disease, one with corticobasal degeneration syndrome and another with progressive supranuclear palsy. This study shows that: i) although rare, C9ORF72 repeat expansions may be associated with clinically typical Parkinson’s disease, but also with other parkinsonism; ii) in several patients, parkinsonism was dopa-responsive and remained pure, without associated dementia, for more than 10 years; iii) interestingly, all C9ORF72 repeat expansion carriers had positive family histories of parkinsonism, degenerative dementias or amyotrophic lateral sclerosis. This study also provides the tools for identifying parkinsonian patients with C9ORF72 expansions, with important consequences for genetic counseling.
Keywords: Adolescent; Adult; Aged; Aged, 80 and over; Female; Humans; Male; Middle Aged; Open Reading Frames; genetics; Parkinson Disease; diagnosis; genetics; Pedigree; Proteins; genetics; Trinucleotide Repeat Expansion; genetics; Young Adult
Introduction
Parkinson’s disease (PD) is common in the aging population, ranking second in prevalence after Alzheimer’s disease. The prevalence of this condition is age-dependent, affecting more than 1% of persons over the age of 65 years, but this rate is increased to 4–5 % by the age of 85 years (De Lau and Breteler, 2006). PD is a clinical motor syndrome with variable combinations of akinesia, rigidity, rest tremor, and postural instability, linked to a characteristic pattern of neurodegeneration of dopaminergic neurons, particularly in the substantia nigra pars compacta and the presence of intraneuronal cytoplasmic inclusions, known as Lewy bodies, in surviving dopaminergic neurons. In addition, cognitive dysfunction is frequent in PD (Barton et al., 2012) and also in atypical forms of parkinsonism such as Lewy body dementia (LBD), progressive supranuclear palsy (PSP), and corticobasal degeneration syndrome (CBDS). A GGGGCC repeat expansion in C9ORF72 was recently reported to be the most common genetic cause of amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degenerations (FTLD) (DeJesus et al., 2011; Renton et al., 2011). Since the clinical and neuropathological features of ALS, neurodegenerative dementias and parkinsonism overlap, we looked for C9ORF72 repeat expansions in 1,446 parkinsonian unrelated patients.
Subjects and Methods
Subjects
The study included a total of 1,446 unrelated patients: 1,225 clinically diagnosed with PD, 123 with PSP, 21 with CBDS, 43 with LBD and 25 with multiple system atrophy-parkinsonism (MSA-P) (Table 1).
Table 1.
Demographic and clinical characteristics of patients with parkinsonism and C9ORF72 expansion carriers
| Parkinsonian disorders | PD | LBD | PSP | CBDS | MSA-P |
|---|---|---|---|---|---|
| Patients, n | 1235a | 43 | 123 | 21 | 25 |
| Sex, M/F | 734/501 | 31/12 | 65/58 | 12/9 | 16/9 |
| Age at disease onset, years (range) | 48.3±12.5 (6–86) | 63.6±11.9 (34–83) | 64.1±8.2 (40–79) | 64.2±8.8 (50–80) | 56.3±13.2 (35–81) |
| Age at examination, years (range) | 56.8±13.1 (14–87) | 71.1±10.0 (43–85) | 67.8±9.8 (44–83) | 67.8±8.5 (52–82) | 63.7±14.7 (36–87) |
| Dementia (%) | 10 | 100 | 100 | 100 | 54 |
| Patients with family histories of other neurodegenerative disordersb, n (%) | 47 (4) | 11 (26) | 15 (12) | 7 (33) | 17 (68) |
| C9ORF72 expansion carriers, n | 3 | 0 | 1 | 1 | 0 |
| Expansion carriers with family histories of other neurodegenerative disordersb (%) | 6 | 0 | 7 | 14 | 0 |
40% of the patients had family histories of typical PD.
Other neurodegenerative disorders include parkinsonism, degenerative dementias or ALS.
CBDS = corticobasal degeneration syndrome, LBD = Lewy body dementia, MSA-P = multiple system atrophy-parkinsonism, PD = Parkinson’s disease, PSP = progressive supranuclear palsy.
Patients with PD were recruited through the French network for the study of Parkinson’s disease Genetics (PDG). PD was diagnosed according to the UK Parkinson’s Disease Society Brain Bank (PDSBB) clinical diagnostic criteria (Hughes et al., 1992). Most (>80%) PD patients fulfilled the criteria for definite PD: at least 2 of 3 cardinal signs (akinesia and/or rigidity and/or tremor) and a positive and sustained response to levodopa therapy. PSP (Litvan et al., 1996), CBDS (Boeve et al., 2003), LBD (McKeith et al., 2005), and MSA (Gilman et al., 2008) were diagnosed according to the international consensus diagnosis criteria. Patients underwent standard neurological examinations, including the Unified Parkinson’s Disease Rating scale (UPDRS) (Goetz et al., 2008), the Hoehn and Yahr scale (Hoehn and Yahr, 2001), to evaluate patients’ motor disability, response to levodopa, and the Mini Mental State Examination (MMSE) (Folstein et al., 1975), Frontal assessment battery (FAB) (Dubois et al., 2000), Wisconsin Card Sorting Test (WCST) (Heaton, 1981) and Mattis Dementia Rating Scale (MDRS) (Mattis, 1976) to evaluate cognitive function.
Patients with a positive family history were defined as individuals with at least one first degree relative with a confirmed diagnosis of typical PD or other neurodegenerative disorders including parkinsonism, degenerative dementias or ALS. Of note, 40% of the 1,225 PD patients included in the study reported family histories of classical PD; this is because they were selected in an effort to enrich the cohort in individuals who might have more genetic susceptibility factors. Four percent had family histories of other neurodegenerative disorders (Table 1). All the 1,225 unrelated PD patients were excluded for the common Leucine-rich repeat kinase 2 (LRRK2) G2019S mutation. All index cases compatible with autosomal dominant PD were excluded for SNCA multiplications (n = 340); patients compatible with autosomal recessive PD (n = 60) as well as sporadic cases (n = 420), with an age at onset ≤50 years were excluded for PARK2, PARK6 and PARK7.
Control subjects, 445 Caucasians, mostly of French descent (174 males, 271 females, mainly spouses) without family histories of parkinsonism and excluded for the LRRK2 G2019S mutation, were examined at age 64.9±8.5 years (range 51–89).
Local ethics committees approved the study and written informed consent was obtained from all participants.
Methods
Genomic DNA from all participants was obtained from venous blood using standard protocols. The GGGGCC repeat expansions in the first intron of C9ORF72 were searched for, using the repeat primed PCR reaction adapted from Renton et al. (2011). In short, 80 ng of genomic DNA were mixed with 14 μl FastStart PCR Master- Mix (Roche Diagnostics), 0.18 mM 7-deaza-dGTP (Roche Diagnostics), Q solution (Qiagen Inc.), 7% dimethyl sulfoxide (DMSO), 0.9 mM Mgcl2, 0.7 μM reverse primer consisting of four GGGGCC repeats and an anchor tail (TACGCATCCCAGTTTGAGACGGGGGCCGGGGCCGGGGCCGGGG), 1.4 μM 6FAM-fluorescent forward primer located 280 base pairs 3′ prime to the repeat sequence (AGTCGCTAGAGGCGAAAGC) and 1.4 μM anchor primer corresponding to the anchor tail of the reverse primer (TACGCATCCCAGTTTGAGACG). A touchdown PCR cycling protocol was used in which the initial annealing temperature was lowered from 70°C to 56°C in 2°C increments with a 3 minute-extension in each cycle (Renton et al., 2011). Fragment length analysis was performed on an ABI 3730 genetic analyzer (Applied Biosystems), and data analyzed using Peak Scanner software version v1.0 (Applied Biosystems).
To determine the accurate sizing of the non-expanded allele and score all alleles in heterozygous or homozygous state, we performed a classical FAM-fluorescent PCR assay, using previously described primers (Renton et al., 2011). To determine the mean number of C9ORF72 hexanucleotide repeats in non-mutation carriers, we summed the number of repeats on both non-expanded alleles.
To confirm the size of the largest expanded alleles, we performed a Southern blot analysis of DNA from the putative C9ORF72 expansion carriers and also from patients with an intermediate length hexanucleotide repeat-containing allele for whom DNA was available. Briefly, ten μg of genomic DNA extracted from blood were digested with MboI restriction endonuclease (New England Biolabs) and the digested samples were separated on a 0.8% agarose gel in 1x TBE buffer at 1V/cm overnight. DNA was alkali-transferred onto a GeneScreen Plus filter (NEN Life Sciences Products Inc) according to the manufacturer’s protocol. A double labeled probe (NeBlot kit New England Biolabs, [a-32P]dATP and [a-32P]dCTP 3000ci/mmol, PerkinElmer) corresponding to chromosome 9 position 27,573,698-27,573,938 in the hg19 assembly was hybridized to blots in modified Church and Gilbert hybridization buffer (0.5 M NaH2PO4/Na2HPO4, pH 6.8, 7% SDS, 1 mM EDTA, 1% BSA) at 65°C overnight (Church and Gilbert, 1984). Blot membranes were then washed two times at room temperature and twice at 65°C in 40 mM NaH2PO4/Na2HPO4, pH 6.8, 1% SDS. Films were exposed at −80°C against an intensifying screen for two or more days.
Results
A total of 1,446 patients with parkinsonism, including 1,225 clinically diagnosed with PD and 464 healthy controls were successfully genotyped with both assays to study the C9ORF72 repeat in all patients: a repeat-primed PCR to identify expansion carriers and a classical fluorescent PCR to allow the accurate sizing of the non-expanded allele.
Five of the 1,446 patients with parkinsonism showed the typical sawtooth tail pattern detected by repeat-primed PCR analysis (Renton et al., 2011), characteristic of C9ORF72 repeat expansions in the pathogenic size (≥ 60 repeat units) (Fig. 1A) and a single peak and potentially unamplifiable repeat expansions detected by fluorescent fragment length PCR analysis (Fig. 1B) (Table 2). The presence of these large expanded alleles was further confirmed on Southern blots (Fig. 1C), which showed additional expanded alleles >12 kilobases in size in the five patients with parkinsonism. Although we confirmed the presence of repeat expansions by Southern blot, significant repeat size heterogeneity resulting in a smear on the blot complicated accurate sizing of the repeats in the five patients and the positive control (FTLD patient with a C9ORF72 expansion).
Figure 1.
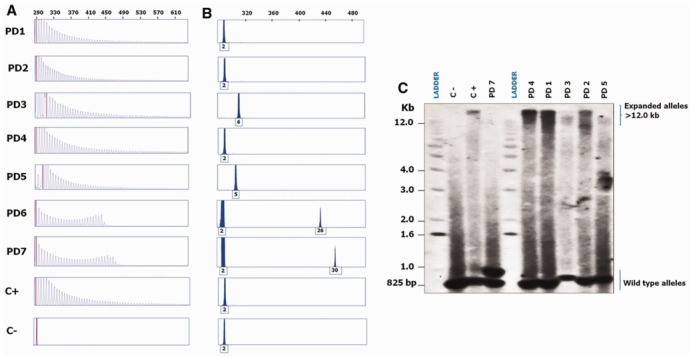
Molecular genetic analyses of hexanucleotide repeat expansions in the seven C9ORF72 patients with pathogenically expanded or intermediate alleles. (A) Repeat-primed PCR products separated on an ABI 3730 genetic analyzer (Applied Biosystems); data were analyzed using Peak Scanner software version v1.0 (Applied Biosystems). Similar to the positive control (FTLD patient with a C9ORF72 expansion), stutter amplifications characteristic of C9ORF72 repeat expansions were seen in patients PD1, PD2, PD3, PD4 and PD5. The electrophoregrams of patients PD6 and PD7with intermediate length alleles and the negative control with a non-expanded allele are also shown. (B) Fluorescent fragment length analysis of PCR products containing GGGGCC hexanucleotide repeats, the numbers of which are indicated under the peaks. Patients PD1, PD2, PD3, PD4 and PD5 as well as the FTLD patient had a single peak and potentially unamplifiable repeat expansions. Patients PD6 and PD7 showed heterozygous peaks corresponding to 2/26 and 2/30 hexanucleotide repeats, respectively. (C) Southern blot profiles of a control with non-expanded alleles (C−) at the expected size (~825 base pairs), a FTLD patient with a known C9ORF72 expansion (C+), PD7 with 2 and 30 hexanucleotide repeats, and patients PD1, PD2, PD3, PD4 and PD5 with additional expanded alleles >12 kilobases in size.
Table 2.
Clinical characteristics of C9ORF72 expansion carriers
| Patients | Diagnosis | Age at onset | DD at examination | DD at last visit | Symptoms at onset | Parkinsonism | UPDRS Part IIIa on/off (/108) | Hoehn & Yahr stage (/5) | Cognitive deficit (DD before cognitive deficit occurred) | MMSE (/30)/FAB (/18) at examination | Gazepalsy | Dystoniab | Other symptoms | Family history of neuropsychi atric disorders |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PD1 | PD | 48 | 6 | 11 | P | R,A,T (Bilateral, LL>UL) | 7/20 | 4 | + (8) | 25/na | - | - | Sleep disorders | Dementia, PD |
| PD2 | PD | 64 | 5 | 6 | P and dystonia | R,A (Left, LL>UL) | 13/na | 3 | + (5) | 26/15 | - | LL | Depression, sleep apnoea | Depression, P |
| PD3 | PD | 29 | 6 | 12 | P and dystonia | R,A,T (Left, UL>LL) | 9/37 | 2 | - | 27/18 | - | LL | Enhanced reflexes | Depression, dementias, P, ALS |
| PD4 | PSP | na | na | na | P | + | na | na | + (at onset) | na | + | na | na | Dementia, PSP, P |
| PD5 | CBDS | 50 | 2 | 2 | P, anarthria, orofacial apraxia, dystonia | R,A (Left, UL>LL) | na | na | + (at onset) | na | - | UL | Enhanced reflexes | Dementia |
A = akinesia, ALS = amyotrophic lateral sclerosis, CBDS = corticobasal degeneration syndrome, DD = disease duration, FAB = Frontal assessment battery, FTLD = frontotemporal lobar degenerations, LL = Lower limb, MMSE = Mini mental status examination, na = not available, P = Parkinsonism, PD = Parkinson’s disease, PSP = progressive supra-nuclear palsy, R = rigidity, T = rest tremor, UL = upper limb.
UPDRS Part III motor “on/off” score = Unified Parkinson’s Disease Rating Scale rated under/off treatment with levodopa at evaluation time.
In the subgroup of 1,225 PD patients, there were three carriers of C9ORF72 repeat expansions (0.2%, 95% confidence intervals (CI) 0.09–0.7) who had isolated parkinsonian symptoms at onset. Ages at onset ranged from 29 to 64 years. In the first patient (PD1), parkinsonism began at age 48 with bilateral rest tremor and akinesia. At age 54, she developed axial rigidity and a severe gait disorder. She showed cognitive deficits (MMSE score 25/30) at age 56 and died at 59. The second patient (PD2) developed a levodopa-responsive akinetic rigid syndrome and asymmetric lower limb dystonia at age 64. She developed dyskinesias, visual hallucinations under dopamine agonists and a mild cognitive deficit at age 69 that had no effect on her autonomy in daily life (MMSE 26/30; FAB 15/18). The third patient (PD3) developed left hemi-parkinsonism at age 29 with upper limb rest tremor, akinesia, rigidity and dystonia (UPDRS III: 17/108; Hoehn and Yahr: 1.5/5). Her parkinsonian symptoms worsened progressively; dopamine agonists were partially effective. At age 34, she had motor fluctuations and dyskinesias (UPDRS III off medication: 37/108; Hoehn and Yahr: 2/5). Before deep brain stimulation, at age 37, her FAB score was 18/18, her MMSE 27/30, her WCST 18/20 and her MDRS 138/144. At 40, she had no dementia, nor psychiatric signs. She is still working as a secretary.
Although these three patients were clinically diagnosed with typical PD, they all had family histories of atypical PD, degenerative dementias or ALS (Fig. 2). No expansions were identified in patients with family histories of typical PD.
Figure 2.
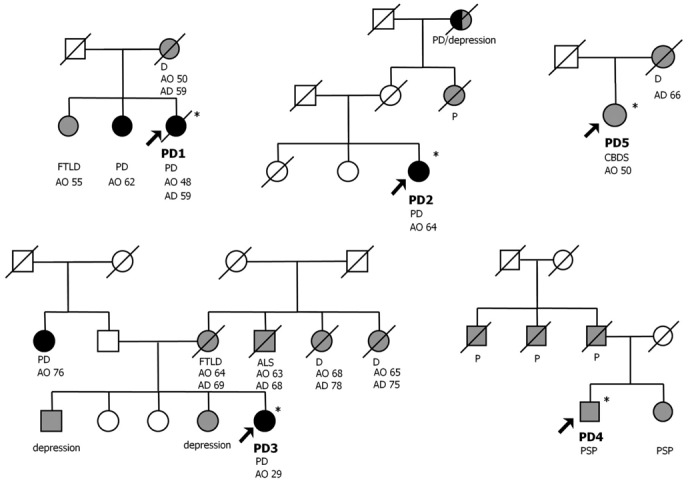
Pedigree structures of families with repeat expansions that had family histories of parkinsonism, degenerative dementias or ALS are shown. Patients with PD are represented by black symbols, those with parkinsonism, including PSP, CBDS, and degenerative dementias or ALS by grey symbols, unaffected individuals by open symbols. The arrows point to the probands. *DNA available. AD = age at death, ALS = amyotrophic lateral sclerosis, AO = age at onset, CBDS = corticobasal degeneration syndrome, D = dementia, FTLD = frontotemporal lobar degenerations, P = parkinsonism, PD = Parkinson’s disease, PSP = progressive supranuclear palsy.
We also found a C9ORF72 repeat expansion in a PSP patient (PD4) with parkinsonism, gaze-palsy and dementia (1/123, 0.8%, 95% CI 0.2–4.4), and in a CBDS patient (PD5) with left akinetic rigid parkinsonism, upper limb dystonia, orofacial apraxia, anarthric speech and frontal lobe syndrome (1/21, 5%, 95% CI 1.1–22.8) (Table 2). In addition, two patients with PSP, aged 59 (PD6) and 74 at onset (PD7), had intermediate repeat lengths of 26 and 30, respectively (Fig. 1). Both patients presented typical features of isolated PSP, including parkinsonism, supranuclear gaze palsy, postural instability early in the disease course, dysarthria and mild frontal dementia.
No expansions were found in patients with MSA-P or with LBD or in controls. The average C9ORF72 repeat number was 4.5±3.3 (range 2–22) in the 445 control subjects.
Discussion
In a recent study, 35% of patients with C9ORF72 expansions developed parkinsonism within the first two years after onset of FTD and ALS (Boeve et al., 2012). Using a stepwise protocol according to previously described methods, combining fluorescent fragment length, repeat-primed PCR and Southern blot analyses (DeJesus et al., 2011), we have now identified disease-causing repeat expansions in a subset of patients from a large cohort with different forms of parkinsonism, extending the clinical phenotype associated with these expansions from degenerative dementias including Alzheimer’s disease (Majounie et al., 2012a) and ALS to parkinsonism. Two patients had typical PD for many years (5 to 8) before cognitive deficits or dementias developed; one patient had no cognitive deficits, 12 years after PD onset. This study shows that: i) although rare, C9ORF72 repeat expansions may be associated with clinically typical PD, but also with PSP and CBDS; ii) the parkinsonism could be levodopa-responsive and, as in PD, remains pure, without associated cognitive deficits or dementias for more than 10 years; iii) interestingly, all C9ORF72 mutation carriers had positive family histories of parkinsonism, degenerative dementias or ALS. Among the patients with positive family histories, the proportion of patients with expansions in C9ORF72 reached 6% (3/47, 95% CI 2.3–17.2) in PD, 7% (1/15, 95% CI 1.6–30.2) in PSP and 14% (1/7, 95% CI 3.2–52.7) in CBDS.
We also identified two PSP patients with intermediate numbers of GGGGCC repeats, ranging from 26 to 30. Notably, a 29-hexanucleotide repeat allele was found in a patient from a Dutch c9FTLD/ALS family (Simón-Sánchez et al., 2012) suggesting that intermediate alleles might be pathogenic in patients with PSP. Of note, the maximum size of the repeat in most controls was reported not to exceed 24 units (DeJesus et al., 2011; Renton et al., 2011; Gijselinck et al., 2012; Rutherford et al., 2012; Sabatelli et al., 2012). In our study and that of Millecamps et al. (2012), repeat lengths ranged from 2 to 23 in the control group of French origin (n>600). Although, the repeat size in our patients was clearly larger than in ethnically matched control subjects and their ages at onset were in the range observed in typical patients with C9ORF72 repeat expansions, we cannot completely exclude that the observed repeat expansions and the presence of typical PD are unrelated (i.e. the patients have two different neurological diseases). A recent study in a large cohort of 781 Caucasian PD cases, one third of which reported family histories of PD, failed to identify any pathogenic hexanucleotide expansions in C9ORF72, although a patient with typical PD harbored a marginally 38-GGGGCC hexanucleotide repeat allele (Majounie et al., 2012b). Large multi-centre studies worldwide are needed to determine the frequency of C9ORF72 mutations in idiopathic PD, particularly when positive family histories of parkinsonism, degenerative dementias or ALS are present.
The present study, therefore, suggests that patients with PD or other parkinsonian syndromes, particularly when a family history of other neurodegenerative disorders is present, should be screened for C9ORF72 expansions. This provides the tools for identifying potential carriers with important consequences for genetic counseling.
Acknowledgments
We are grateful to the patients and their families. We thank Merle Ruberg for critical reading of the manuscript, the DNA and Cell Bank of the CRICM for sample preparation, Agnès Camuzat and Léna Guillot-Noel for technical assistance.
Funding
This work was supported by PSP-France Association, France-Parkinson Association. A.D. and A.B. were supported by the French Agency for Research (ANR) program grants.
Abbreviations
- ALS
amyotrophic lateral sclerosis
- CBDS
corticobasal degeneration syndrome
- CI
confidence intervals
- FAB
Frontal assessment battery
- FTLD
frontotemporal lobar degenerations
- LRRK2
Leucine-rich repeat kinase 2
- LBD
Lewy body dementia
- MDRS
Mattis Dementia Rating Scale
- MMSE
Mini Mental State Examination
- MSA-P
multiple system atrophy-parkinsonism
- PD
Parkinson’s disease
- PSP
progressive supranuclear palsy
- UPDRS
Unified Parkinson’s Disease Rating scale
References
- Barton B, Grabli D, Bernard B, Czernecki V, Goldman JG, Stebbins G, et al. Clinical validation of movement disorder society-recommended diagnostic criteria for Parkinson’s disease with dementia. Mov Disord. 2012;27:248–53. doi: 10.1002/mds.24059. [DOI] [PubMed] [Google Scholar]
- Boeve BF, Lang AE, Litvan I. Corticobasal degeneration and its relationship to progressive supranuclear palsy and frontotemporal dementia. Ann Neurol. 2003;54 (Suppl 5):S15–9. doi: 10.1002/ana.10570. [DOI] [PubMed] [Google Scholar]
- Boeve BF, Boylan KB, Graff-Radford NR, DeJesus-Hernandez M, Knopman DS, Pedraza O, et al. Characterization of frontotemporal dementia and/or amyotrophic lateral sclerosis associated with the GGGGCC repeat expansion in C9ORF72. Brain. 2012;135:765–83. doi: 10.1093/brain/aws004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Church GM, Gilbert W. Genomic sequencing. Proc Natl Acad Sci U S A. 1984;81:1991–5. doi: 10.1073/pnas.81.7.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lau LM, Breteler MM. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006;5:525–35. doi: 10.1016/S1474-4422(06)70471-9. [DOI] [PubMed] [Google Scholar]
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–56. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois B, Slachevsky A, Litvan I, Pillon B. The FAB: a frontal Assessment Battery at bedside. Neurology. 2000;55:1621–6. doi: 10.1212/wnl.55.11.1621. [DOI] [PubMed] [Google Scholar]
- Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–98. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- Gijselinck I, Van Langenhove T, van der Zee J, Sleegers K, Philtjens S, Kleinberger G, et al. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol. 2012;11:54–65. doi: 10.1016/S1474-4422(11)70261-7. [DOI] [PubMed] [Google Scholar]
- Gilman S, Wenning GK, Low PA, Brooks DJ, Mathias CJ, Trojanowski JQ, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology. 2008;71:670–6. doi: 10.1212/01.wnl.0000324625.00404.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz CG, Tilley BC, Shaftman SR, Stebbins GT, Fahn S, Martinez-Martin P, et al. Movement Disorder Society-sponsored revision of the Unified Parkinson’s Disease Rating Scale (MDS-UPDRS): scale presentation and clinimetric testing results. Mov Disord. 2008;23:2129–70. doi: 10.1002/mds.22340. [DOI] [PubMed] [Google Scholar]
- Heaton RK. A manual for the Wisconsin Card Sorting Test. Odessa, FL: Psychological Assessment Resources; 1981. [Google Scholar]
- Hoehn MM, Yahr MD. Parkinsonism: onset, progression, and mortality. 1967. Neurology. 2001;57:S11–26. [PubMed] [Google Scholar]
- Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry. 1992;55:181–4. doi: 10.1136/jnnp.55.3.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litvan I, Agid Y, Calne D, Campbell G, Dubois B, Duvoisin RC, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology. 1996;47:1–9. doi: 10.1212/wnl.47.1.1. [DOI] [PubMed] [Google Scholar]
- Mattis S. Mental status examination for organic mental syndrome in the elderly patient. In: Bellack L, Karusu TB, editors. Geriatric psychiatry. New York: Grune & Stratton; 1976. [Google Scholar]
- Majounie E, Abramzon Y, Renton AE, Perry R, Bassett SS, Pletnikova O, et al. Repeat expansion in C9ORF72 in Alzheimer’s disease. N Engl J Med. 2012a;366:283–4. doi: 10.1056/NEJMc1113592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majounie E, Abramzon Y, Renton AE, Keller MF, Traynor BJ, Singleton AB. Large C9orf72 repeat expansions are not a common cause of Parkinson’s disease. Neurobiol Aging. 2012b;33:2527, e1–2. doi: 10.1016/j.neurobiolaging.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeith IG, Dickson DW, Lowe J, Emre M, O’Brien JT, Feldman H, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65:1863–72. doi: 10.1212/01.wnl.0000187889.17253.b1. [DOI] [PubMed] [Google Scholar]
- Millecamps S, Boillée S, Le Ber I, Seilhean D, Teyssou E, Giraudeau M, et al. Phenotype difference between ALS patients with expanded repeats in C9ORF72 and patients with mutations in other ALS-related genes. J Med Genet. 2012;49:258–63. doi: 10.1136/jmedgenet-2011-100699. [DOI] [PubMed] [Google Scholar]
- Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–68. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford NJ, Heckman MG, Dejesus-Hernandez M, Baker MC, Soto-Ortolaza AI, Rayaprolu S, et al. R. Length of normal alleles of C9ORF72 GGGGCC repeat do not influence disease phenotype. Neurobiol Aging. 2012 Jul 26; doi: 10.1016/j.neurobiolaging.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatelli M, Conforti FL, Zollino M, Mora G, Monsurrò MR, Volanti P, et al. C9ORF72 hexanucleotide repeat expansions in the Italian sporadic ALS population. Neurobiol Aging. 2012;33:1848.e15–20. doi: 10.1016/j.neurobiolaging.2012.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simón-Sánchez J, Dopper EG, Cohn-Hokke PE, Hukema RK, Nicolaou N, Seelaar H, et al. The clinical and pathological phenotype of C9ORF72 hexanucleotide repeat expansions. Brain. 2012;135:723–35. doi: 10.1093/brain/awr353. [DOI] [PubMed] [Google Scholar]