

Abstract
Background: The U.S. Environmental Protection Agency (EPA) completed a toxicological review of tetrachloroethylene (perchloroethylene, PCE) in February 2012 in support of the Integrated Risk Information System (IRIS).
Objectives: We reviewed key findings and scientific issues regarding the human health effects of PCE described in the U.S. EPA’s Toxicological Review of Tetrachloroethylene (Perchloroethylene).
Methods: The updated assessment of PCE synthesized and characterized a substantial database of epidemiological, experimental animal, and mechanistic studies. Key scientific issues were addressed through modeling of PCE toxicokinetics, synthesis of evidence from neurological studies, and analyses of toxicokinetic, mechanistic, and other factors (tumor latency, severity, and background rate) in interpreting experimental animal cancer findings. Considerations in evaluating epidemiological studies included the quality (e.g., specificity) of the exposure assessment methods and other essential design features, and the potential for alternative explanations for observed associations (e.g., bias or confounding).
Discussion: Toxicokinetic modeling aided in characterizing the complex metabolism and multiple metabolites that contribute to PCE toxicity. The exposure assessment approach—a key evaluation factor for epidemiological studies of bladder cancer, non-Hodgkin lymphoma, and multiple myeloma—provided suggestive evidence of carcinogenicity. Bioassay data provided conclusive evidence of carcinogenicity in experimental animals. Neurotoxicity was identified as a sensitive noncancer health effect, occurring at low exposures: a conclusion supported by multiple studies. Evidence was integrated from human, experimental animal, and mechanistic data sets in assessing adverse health effects of PCE.
Conclusions: PCE is likely to be carcinogenic to humans. Neurotoxicity is a sensitive adverse health effect of PCE.
Citation: Guyton KZ, Hogan KA, Scott CS, Cooper GS, Bale AS, Kopylev L, Barone S Jr, Makris SL, Glenn B, Subramaniam RP, Gwinn MR, Dzubow RC, Chiu WA. 2014. Human health effects of tetrachloroethylene: key findings and scientific issues. Environ Health Perspect 122:325–334; http://dx.doi.org/10.1289/ehp.1307359
Introduction
Tetrachloroethylene (perchloroethylene, or PCE) is a widely used chlorinated solvent that is produced commercially for use in dry cleaning, textile processing, and metal-cleaning operations. PCE has been detected in drinking water, indoor environments, ambient air, groundwater, and soil. Many point sources of contamination exist in the United States [U.S. Environmental Protection Agency (EPA) 2013c], and PCE is also commonly found at Superfund hazardous waste sites (U.S. EPA 2013b). Regarding exposure to the general population, the Centers for Disease Control and Prevention (2013) reported that PCE levels assessed in the most recent biomonitoring survey [the 2003–2004 subsample of the National Health and Nutrition Examination Survey (NHANES)] appeared to be similar or slightly lower than levels reported in earlier NHANES surveys. The primary exposure routes are via inhalation, including as a result of vapor intrusion from contaminated soil and water (U.S. EPA 2013d), and ingestion of contaminated water.
The U.S. EPA identified PCE as a priority existing chemical for regulatory action review under the Toxic Substances Control Act (U.S. EPA 2012c) and as one of several volatile organic compounds to be regulated as a group in drinking water (U.S. EPA 2010). Supporting these and other agency actions, the U.S. EPA Integrated Risk Information System (IRIS) program released an updated human health assessment of PCE in February 2012 that included an extensive toxicological review (U.S. EPA 2012b), hereafter referred to as the Toxicological Review. The Toxicological Review was developed according to the general guidelines for risk assessment set forth by the National Research Council (NRC 1983, 1994) as well as relevant U.S. EPA Guidelines and Risk Assessment Forum technical panel reports (U.S. EPA 2013a). The literature search strategy was based on the Chemical Abstracts Service Registry Number (CASRN) and at least one common name. Primary peer-reviewed literature published during or before August 2011 was included. Public submissions to the U.S. EPA and peer-reviewed information (including health assessments developed by other organizations, review articles, and independent analyses of the health effects data) were also considered for inclusion. Toxicokinetic, mechanistic, and other data (e.g., tumor latency, severity, background rate) were considered in interpreting experimental animal cancer findings. Individual study evaluation considered essential design features (particularly, the study species and its population and the relevance of the exposure paradigm); other considerations are discussed in the following sections. The Toxicological Review (U.S. EPA 2012b) provides additional detail regarding the source literature databases, the relevant U.S. EPA guidance, and the study evaluation criteria.
The assessment development involved multiple internal and external peer review stages. Critical input was provided by a 2004 peer consultation workshop on PCE neurotoxicity (U.S. EPA 2004), a 2010 NRC panel report (NRC 2010), a 2011 peer review of the physiologically based pharmacokinetic (PBPK) model applications (U.S. EPA 2012b), and written and oral comments from scientists within the U.S. EPA, other federal agencies, and the Office of Management and Budget (U.S. EPA 2012a) as well as the public (Regulations.gov 2008). Herein we describe key findings and scientific issues addressed in the U.S. EPA’s 2012 Toxicological Review of PCE, covering the following topics:
The role of metabolism in toxicity, informed by the development and application of an updated PBPK model
The carcinogenicity of PCE, based on analyses of epidemiological studies, multiple laboratory animal bioassays, and mechanistic data
Noncancer toxicity, focusing on neurotoxicity as a sensitive outcome.
Role of Metabolism in PCE Toxicity
PBPK models can aid in integrating complex toxicokinetic information on the absorption, distribution, metabolism, and excretion of environmental chemicals and their metabolites. These models are constructed from physiologic information in addition to chemical- and metabolite-specific toxicokinetic data. Some models separate data sets for model calibration and evaluation, utilizing Bayesian methods to strengthen model predictions. PBPK models are used in human health assessments to predict the extent and nature of metabolism across species or exposure routes.
The metabolism of PCE yields multiple metabolites through two main irreversible pathways: a) oxidation via the microsomal mixed-function oxidase system [i.e., cytochrome P450s (CYPs)], and b) conjugation with glutathione (GSH) by glutathione S-transferases (GSTs) (Lash and Parker 2001). Oxidation (Figure 1, left) occurs predominantly in the liver to a ferric-oxide (Fe-O) intermediate, the primary fate of which is thought to be trichloroacetyl chloride (TCAC), which then hydrolyses to yield trichloroacetic acid (TCA). A secondary fate of oxidation is the epoxide (PCE-O), which decomposes to ethandioyl dichloride (EDD) and then to carbon monoxide (CO) and carbon dioxide (CO2) (Yoshioka et al. 2002). Oxalic acid (OXA) has been reported as both an in vivo and in vitro product of PCE oxidation (Pegg et al. 1979; Yoshioka et al. 2002) and may be derived either from the epoxide or directly from the Fe-O intermediate. PCE conjugation with GSH (Figure 1, right) in the liver or kidney forms trichlorovinyl glutathione (TCVG), which is further processed by γ-glutamyl transpeptidase (GGT) and cysteinylglycine dipeptidase (DP) in the kidney, forming the cysteine conjugate S-trichlorovinyl-l-cysteine (TCVC). TCVC may be bioactivated by β-lyase or by flavin-containing monooxygenases (FMO3s) or CYP3A to reactive species (Anders et al. 1988; Krause et al. 2003), or (reversibly) may undergo N-acetylation [by N-acetyltransferase (NAT)] to the mercapturate N-acetyl trichlorovinyl cysteine (NAcTCVC). NAcTCVC is then excreted in urine or sulfoxidated by CYP3A to reactive species (Werner et al. 1996). Dichloroacetic acid (DCA), excreted in urine, is thought to be an end product of β-lyase–mediated bioactivation (Lash and Parker 2001), although a small contribution from TCA dechlorination cannot be ruled out.
Figure 1.
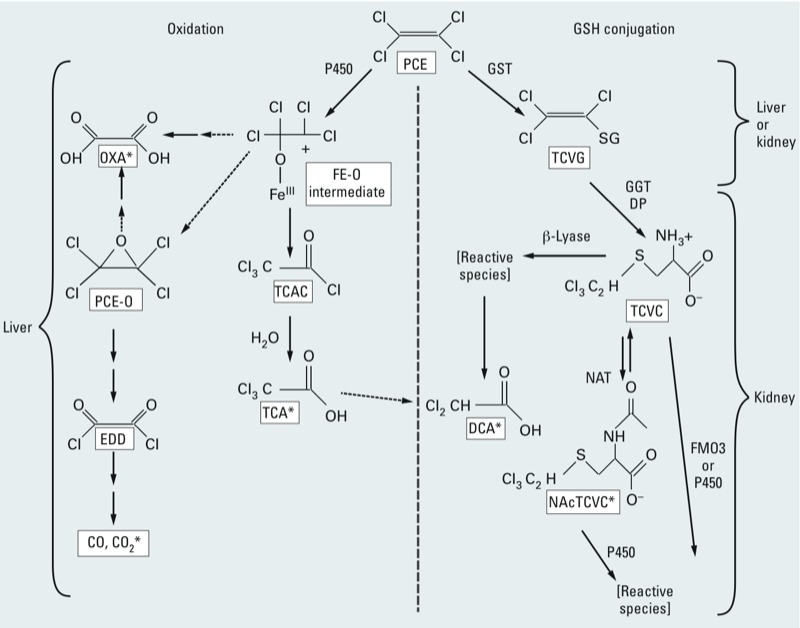
Simplified PCE metabolism scheme. PCE is metabolized in humans and experimental animal species by both oxidation (left) and GSH conjugation (right) metabolic pathways, yielding numerous toxicologically active compounds (Lash and Parker 2001). Tetrachlorethylene metabolism yields the oxidative metabolites TCAC, which hydrolyses to yield TCA, and the epoxide PCE-O, which decomposes in turn to EDD, CO, and CO2. OXA is also a product of PCE oxidation. GSH conjugation products include TCVG, the cysteine conjugate TCVC, and the mercapturate NAcTCVC and its sulfoxidation products. DCA is likely produced via β-lyase–mediated bioactivation, although TCA dechlorination may be an additional minor source. *Metabolites identified in blood, urine, or breath after in vivo PCE exposure (rodent or human).
PCE liver effects are thought to result from oxidative metabolites (Buben and O’Flaherty 1985), whereas metabolites resulting from GSH conjugation are hypothesized to cause kidney effects (Lash and Parker 2001). The identity of PCE metabolites involved in the induction of other PCE health effects is less clear, although PCE itself has been presumed to cause neurological effects (e.g., Boyes et al. 2009).
Many PBPK models for PCE have been developed to predict the relationship between external measures of exposure and internal dose [Bois et al. 1996 (updated by Chiu and Bois 2006); Chen and Blancato 1987; Chien 1997; Clewell et al. 2005; Covington et al. 2007 (updated by Qiu et al. 2010); Gearhart et al. 1993; Loizou 2001; Rao and Brown 1993; Reitz et al. 1996]. These PBPK models have led to a wide range of predictions for the amount of PCE metabolized in humans at low exposure levels, with estimates across the various models spanning an order of magnitude or more (Chiu and Ginsberg 2011).
In an attempt to reconcile these uncertainties, Chiu and Ginsberg (2011) developed a harmonized PBPK model that integrated the previous models and data. The harmonized model predicted oxidative metabolism with fairly high confidence. However, estimates of the extent of GSH conjugation in humans were substantially more uncertain, spanning more than three orders of magnitude. These predictions provide a plausible explanation for the apparently inconsistent findings among previously published models. In particular, previously conducted analyses that concluded low total metabolism (roughly 1% of PCE uptake) also assumed that TCA (derived from oxidative metabolism) represented 30–100% of total metabolism (e.g., Chen and Blancato 1989; Clewell et al. 2005; Covington et al. 2007; Qiu et al. 2010). These results are consistent with the Chiu and Ginsberg (2011) model predictions that oxidative metabolism is low in humans. On the other hand, previous analyses that concluded greater metabolism (> 20% of PCE uptake) either allowed for both oxidative metabolism and GSH conjugation, or made inferences based on the disappearance of the parent compound [e.g., Bois et al. 1990, 1996; Chiu and Bois 2006; Reitz et al. 1996; Ward et al. 1988]. These results are consistent with the Chiu and Ginsberg (2011) model predictions that the amount of GSH conjugation metabolism in humans is highly uncertain, and might be high, low, and/or highly variable.
Carcinogenicity
Evaluation of cancer epidemiology data. Much of the epidemiological research has been conducted in the dry-cleaning industry, in which PCE was widely used from 1960 onward in the United States and Europe. A recent comprehensive review of 109 occupational studies with exposure measures estimated a mean exposure of 59 ppm in dry-cleaning workers based on personal measurements (Gold et al. 2008). These measures varied considerably with the type of work, however, ranging from < 10 ppm for spotters, pressers, and counter clerks to > 100 ppm for machine operators. Exposures in metal and plastic degreasing industries were also high (mean of approximately 100 ppm).
A key consideration in the evaluation of these studies was the quality (e.g., the specificity) of the exposure assessment methods. The ability of a study to identify cancer hazards is strengthened by “higher quality or higher specificity” exposure-assessment approaches that allow for a delineation of exposure potential to individual study subjects. In particular, these exposure-assessment methodologies included a) biological monitoring data, b) cohort studies using a job-exposure matrix based on historical industrial monitoring data, c) case–control studies using a job-exposure matrix focusing on PCE based on information on job title and tasks or duties, d) additional sources of information such as union records or modules for specific jobs, and e) studies of residential PCE exposure using a statistical model of the water distribution system to estimate the delivered dose to a study subject’s home (Table 1). Because of the variability in exposure potential within dry-cleaning occupations, less specific exposure-assessment approaches (e.g., through broader job title groups or plant- or geographically based classifications) were given less weight but were not excluded from the hazard evaluation. Other study quality considerations included a lack of support for alternative explanations for observed associations (e.g., bias or confounding).
Table 1.
Results of epidemiological studies of PCE and bladder cancer, non-Hodgkin lymphoma, or multiple myeloma using higher quality exposure assessment methodology.
| Study type/reference, population, design | Exposure surrogate | Bladder cancer RR (95% CI) [n]a | non-Hodgkin lymphoma RR (95% CI) [n]a | Multiple myeloma RR (95% CI) [n]a |
|---|---|---|---|---|
| Cohort studies | ||||
| Antilla et al. 1995 (Finland): biologically monitored workers (SIR), blood PCE | Any PCE | Not reported | 3.76 (0.77, 11.0) [3] | (Expected = 0.38) [0] |
| Boice et al. 1999 (United States): aerospace workers (SMR, RR), job-exposure matrix | Any routine exposure to PCE | 0.70 (0.09, 2.53) [2] | 1.70 (0.73, 3.34) [8] | 0.40 (0.01, 2.25) [1] |
| Duration, among those with routine or intermittent exposure to PCE: | ||||
| No routine or intermittent exposure | Not reported | 1.0 (Referent) [32] | 1.0 (Referent) [24] | |
| < 1 year | 1.25 (0.43, 3.57) [4] | 0.46 (0.06, 3.48) [1] | ||
| 1–4 years | 1.11 (0.46, 2.70) [6] | 1.13 (0.38, 3.35) [4] | ||
| ≥ 5 years | 1.41 (0.67, 3.00) [10] | 0.24 (0.03, 1.84) [1] | ||
| Blair et al. 2003 (United States): laundry and dry-cleaning workers (SMR), union records | Little to no PCE exposure | 1.4 (0.4, 3.2) [5] | Not reported | Not reported |
| Medium-to-high PCE exposure | 1.5 (0.6, 3.1) [7] | |||
| Lynge et al. 2006 (Sweden, Denmark, Finland, Norway): nested case–control, census occupation codes and pension data/questionnaires | Dry-cleaner job title | 1.44 (1.07, 1.93) [93] | Not studied | Not studied |
| Employment duration (dry cleaner): | Not studied | Not studied | ||
| Never | 1.0 (Referent) [188] | |||
| < 1 year | 1.50 (0.57, 3.96) [6] | |||
| 2–4 years | 2.39 (1.09, 5.22) [10] | |||
| 5–9 years | 0.91 (0.52, 1.59) [17] | |||
| > 10 years | 1.57 (1.07, 2.29) [54] | |||
| Unknown duration | 1.97 (0.64, 6.05) [6] | |||
| Radican et al. 2008 (United States): aircraft maintenance workers (RR, internal referent), job-exposure matrix | Any PCE: | |||
| Males | Not reported | 2.32 (0.75, 7.15) [5] | 1.71 (0.42, 6.91) [3] | |
| Females | Not reported | 2.35 (0.52, 10.7) [2] | 7.84 (1.43, 43.1) [2] | |
| Seldén and Ahlborg 2011 (Sweden): dry-cleaning workers (SIR), census occupation codes, questionnaire, and company-provided data pertaining to solvent use | Any PCE: | |||
| Males | Not reported | 2.02 (1.13, 3.34) [15] | Not reported | |
| Females | Not reported | 1.14 (0.68, 1.81) [18] | Not reported | |
| Calvert et al. 2011 (United States): laundry and dry-cleaning workers (SMR), union employment records (PCE-only exposure based on history of solvent use by shops) | Any PCE | Not reported [0] | 2.46 (0.90, 5.36) [6] | Not reported |
| Case–control studies | ||||
| Aschengrau et al. 1993 [United States (Massachusetts)]: residential history, ordinal estimate of PCE-contaminated water from exposure model | Any PCE | 1.39 (0.67, 2.91) [13] | Not studied | Not studied |
| Any PCE > 90th percentile relative delivered dose | 4.03 (0.65, 25.10) [4] | Not studied | Not studied | |
| Pesch et al. 2000 (Germany): job- and task-exposure matrix | Any PCE (males): | Not studied | Not studied | |
| Medium exposure | 1.0 (0.7, 1.5) [37] | |||
| High exposure | 1.3 (0.8, 1.7) [47] | |||
| Substantial exposure | 1.8 (1.1, 3.1) [22] | |||
| Miligi et al. 2006b, Costantini et al. 2008b (Italy): job-exposure matrix | Any PCE: | Not studied | ||
| Very low/low intensity | 0.6 (0.3, 1.2) [18]c | Not reported [3] | ||
| Medium/high intensity | 1.2 (0.6, 2.5) [14]c | Not reported [2] | ||
| Seidler et al. 2007 (Germany): job-exposure matrix | PCE, cumulative exposure (ppm-years): | Not studied | ||
| 0 | 1.0 (Referent) [667] | 1.0 (Referent) [33] | ||
| > 0 to ≤ 9.1 | 1.1 (0.5, 2.3) [16]d | 1.8 (0.5, 6.7) [3] | ||
| > 9.1 to ≤ 78.8 | 1.0 (0.5, 2.2) [14]d | [0] | ||
| > 78.8 | 3.4 (0.7, 17.3) [6]d | [0] | ||
| Gold et al. 2010 (United States): all jobs held >12 months, job-exposure matrix | Any PCE | Not studied | Not studied | 1.5 (0.8, 2.9) [16] |
| Cumulative PCE exposure (ppm-weeks): | Not studied | Not studied | ||
| 0 | 1.0 [164] | |||
| 1–353 | 0.3 (0.04, 3.0) [1] | |||
| 354–1,430 | 0.5 (0.1, 4.4) [1] | |||
| 1,431–4,875 | 1.5 (0.4, 5.4) [4] | |||
| 4,876–13,500 | 3.3 (1.2, 9.5) [10] | |||
| Abbreviations: RR, relative risk; SIR, standardized incidence ratio; SMR, standardized mortality ratio. an refers to the number of exposed cases. bBoth Miligi et al. (2006) and Costantini et al. (2008) are based on the Italian Multicenter Case–control Study on Hematolymphopoietic Malignancies and Exposure to Solvents and Pesticides. Miligi et al. (2006) reported ORs for non-Hodgkin lymphoma and PCE; Costantini et al. (2008) reported ORs for multiple myeloma and PCE. cIncludes patients with non-Hodgkin lymphoma and chronic lymphocytic leukemia. dIncludes patients with non-Hodgkin and Hodgkin lymphoma. | ||||
The epidemiological evidence from cohort and case–control studies provides evidence of associations between PCE exposure and bladder cancer, non-Hodgkin lymphoma, and multiple myeloma in adults. Of these, bladder cancer and non-Hodgkin lymphoma were considered to have the strongest databases on the basis of the relative consistency of an observed association among studies with the higher quality exposure measurement and indication of increasing risk with increasing exposure among the studies using a cumulative exposure metric.
Table 1 summarizes cohort and case–control studies of bladder cancer, non-Hodgkin lymphoma, and multiple myeloma using a higher quality exposure assessment methodology; summaries of the other cancer sites can be found in the Toxicological Review (U.S. EPA 2012b). Studies of dry cleaners, launderers, and pressers used additional information to distinguish PCE-exposed workers from other workers (Blair et al. 2003; Calvert et al. 2011; Lynge et al. 2006; Pesch et al. 2000; Seldén and Ahlborg 2011). Specificity was also improved in studies in other work settings and in population-based case–control studies that used an individual-level exposure assignment (e.g., through a job-exposure matrix, PCE in blood as a biological marker, or a statistical model of water distribution to estimate a delivered PCE dose to a study subject’s home (Anttila et al. 1995; Aschengrau et al. 1993; Boice et al. 1999; Gold et al. 2010; Miligi et al. 2006; Radican et al. 2008; Seidler et al. 2007). Smoking history was considered a potential confounder only in the bladder cancer studies because it is not a known risk factor for non-Hodgkin lymphoma or multiple myeloma.
For bladder cancer, two moderate-sized (> 20 exposed cases) studies used a relatively specific exposure assessment method (Lynge et al. 2006; Pesch et al. 2000). Pesch et al. (2000) observed odds ratios (ORs) of 1.0, 1.2, and 1.8 for the medium, high, and substantial exposure categories, respectively; however, the pattern was more variable in the nested case–control study by Lynge et al. (2006), in which duration of dry-cleaning work was used as the exposure measure. Smoking is a risk factor for bladder cancer, and could also be related to TCE exposure. The case–control studies addressed this potential confounding by adjusting for smoking. The studies of dry-cleaning workers are also useful in that the study subjects are unlikely to have been exposed to other occupational bladder carcinogens. In the small studies (< 10 exposed cases) of non-Hodgkin lymphoma, an approximate doubling of risk compared with the referent population was seen (Anttila et al. 1995; Boice et al. 1999; Calvert et al. 2011; Radican et al. 2008; Seldén and Ahlborg 2011), and a relative risk of 3.4 (95% CI: 0.7, 17.3) was seen in the highest cumulative exposure group in Seidler et al. (2007). Multiple myeloma is a relatively rare type of cancer, and results for multiple myeloma are based on a smaller set of studies and fewer observed cases than those for non-Hodgkin lymphoma. The largest of these studies, with 16 exposed cases, reported an OR of 3.3 (95% CI: 1.2, 9.5) for the highest exposure group compared with the unexposed group (Gold et al. 2010). For each of the three cancer types above, the epidemiological data were considered to provide evidence suggestive of a causal association. For cancers of other sites, including esophageal, kidney, lung, liver, cervical, and breast cancer, results were more variable (data not shown). Studies of cancer in children exposed prenatally or postnatally to PCE were inadequate to allow for drawing firm conclusions (Brown Dzubow et al. 2010).
Evaluation of experimental evidence of carcinogenicity. There is clear evidence of PCE carcinogenicity in rodents: one oral gavage [National Cancer Institute (NCI) 1977] and two inhalation [Japan Industrial Safety and Health Association (JISA) 1993; National Toxicology Program (NTP) 1986] cancer bioassays, all in sexually mature animals. No data were available on cancer risks in experimental animals exposed to PCE during early life stages. As summarized in Table 2, a number of factors were considered in evaluating the rodent carcinogenicity findings. These included statistical analyses to adjust for survival differences and cause of death and mode-of-action analyses to inform judgments regarding human relevance of animal bioassay results and susceptible populations or life stages (U.S. EPA 2005b).
Table 2.
Summary of factors considered in evaluating carcinogenicity findings in experimental animals.
| Tumor type | Incidence (dose, sex, strain, route) | Tumor latency, severity, mortality, background rate | Toxicokinetic information | MOA information |
|---|---|---|---|---|
| Rat mononuclear cell leukemia | Significant increases in both sexes of F344/N (NTP 1986) and F344/DuCrj (JISA 1993) strains; dose-dependent increase in F344/DuCrj males (JISA 1993) | NTP 1986: decreased latency in females; increased severity in both sexes; background rate 56% in males, 36% in females JISA 1993: decreased latency in both sexes; background rate of ~ 20% in both sexes | No information available concerning active moiety(ies) | None hypothesized; studies demonstrating hemolysis and bone marrow toxicity in mice add some support to the biologic plausibility |
| Mouse hepatocellular tumors | Significant, dose-dependent increases in both sexes of B6C3F1 (NTP 1986) and Crj:BDF1 (JISA 1993) strains with inhalation exposures; no continued increase with increasing dose in gavage study of B6C3F1 strain (NCI 1977) | Decreased latency; increased mortality; increased metastases in inhalation studies; background rate of ~ 30% in males, ~ 8% in females | The metabolites TCA and DCA are mouse hepatocarcinogens, alone and in combination | Evidence is insufficient for the hypothesized MOAs evaluated: PPARα activation, mutagenicity, alterations in DNA methylation, oxidative stress secondary to cytotoxicity |
| Mouse hemangiomas, hemangiosarcomas | Significant, dose-dependent increases in males in one bioassay (Crj:BDF1 strain, JISA 1993) | Background rate of 2–4% in both sexes; decreased latency | No information available concerning active moiety(ies) | None hypothesized |
| Rat kidney tumors | Significant trend in males in one bioassay (F344/N strain, NTP 1986) | Low background rate (1/549 among historical controls for facility; ~ 0.2% in 1,968 untreated controls in the NTP program) | GSH conjugation metabolites are likely contributors to renal carcinogenicity | Evidence is insufficient for the hypothesized MOA evaluated: α2u-globulin nephropathy did not meet the U.S. EPA criteria for establishing this MOA; evidence of either a) cytotoxicity not associated with α2u-globulin accumulation, or b) peroxisome proliferation lacked specificity with regard to dose, sex and/or species; limited evidence of mutagenicity (positive Ames assays with GSH conjugation metabolites) |
| Abbreviations: MOA, mode of action; PPARα, peroxisome proliferator-activated receptor α. | ||||
Evaluation of rat tumors. The primary tumor finding in rats was a significant increase in the incidence of mononuclear cell leukemia in both sexes in independent inhalation bioassays using the F344/N (JISA 1993; NTP 1986) or F344/DuCrj (JISA 1993) strain (Figure 2). The NTP (1986) analyses of the PCE bioassay results revealed an increase in tumor incidence and severity in both sexes and a shortened time to death with mononuclear cell leukemia in female rats. These results were affirmed by statistical analyses performed recently by Thomas et al. (2007) who noted PCE to be 1 of only 5 of the 500 chemicals examined to produce “definitive” leukemia effects in both sexes of rats in NTP bioassays. JISA (1993) corroborated these results. There is a paucity of data in F344 rats on toxicokinetics or contributing metabolites or mechanisms to inform mode-of-action conclusions. Nonetheless, increases in hemolysis and bone marrow toxicity in NMRI mice after PCE exposure (Ebrahim et al. 2001; Marth 1987; Marth et al. 1985a, 1985b, 1989; Seidel et al. 1992)] add some support to the biologic plausibility of the observed leukemic effects (NRC 2010).
Figure 2.
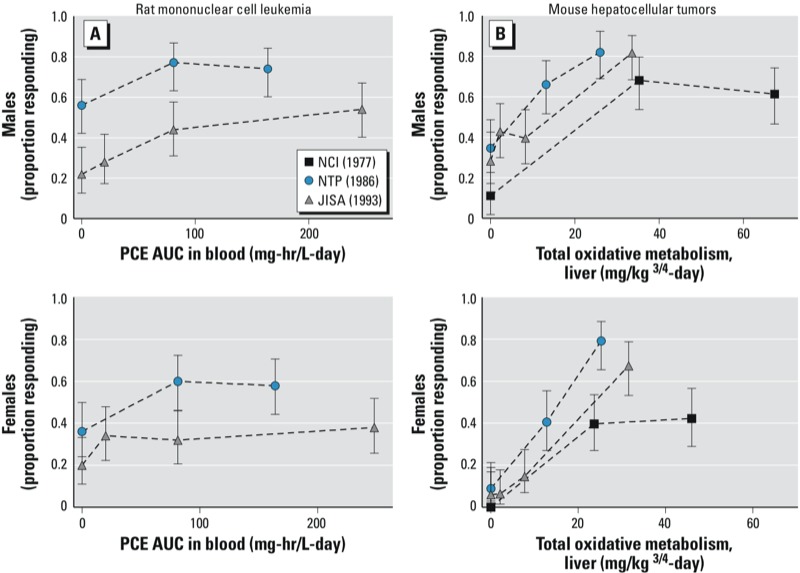
Dose–response relationships for rat mononuclear cell leukemias (A) and mouse hepatocellular tumors (B) in PCE bioassays. Three laboratories evaluated PCE in both mice and rats [oral gavage (NCI 1977); inhalation (JISA 1993; NTP 1986)]. The PBPK model of Chiu and Ginsberg (2011) was used to estimate internal dose for each site, allowing comparison of responses across routes of exposure. The best supported dose metric for mouse liver tumors was total oxidative metabolism in the liver (B), whereas that for rat mononuclear cell leukemia was PCE area under the curve (PCE AUC) in blood (A). The study in Osborne-Mendel rats (NCI 1977) was judged inconclusive because of high rates of respiratory disease and mortality with PCE and, thus, rat data from that study are not presented. Survival-adjusted responses are presented as proportion responding (incidence/number at risk).
Kidney tumors, rare in male rats, were increased in a single bioassay (NTP 1986) (see Supplemental Material, Table S1). For rat kidney carcinogenesis, mechanistic data informative for the evaluation of PCE’s carcinogenic mode of action were identified (summarized in Table 2). PCE metabolites from the GSH conjugation pathway are thought to mediate mechanistic events (other than α2u-globulin nephropathy), contributing to renal carcinogenesis (Lash and Parker 2001). The GSH conjugation metabolites—TCVG, the cysteine conjugate TCVC, and the mercapturate NAcTCVC—are mutagenic in Salmonella tests, consistent with the observation that PCE tested positive for mutagenicity in the few studies in which products of the GSH conjugation metabolic pathway would have been generated (Dekant et al. 1986; Dreessen et al. 2003; Vamvakas et al. 1987, 1989a, 1989b). However, in vivo evidence of genotoxicity in the kidney is limited to reports of modest effects after intraperitoneal exposure (Mazzullo et al. 1987; Walles 1986). Evidence for α2u-globulin nephropathy did not meet the U.S. EPA’s criteria for establishing that renal tumors resulted from this mode of action (U.S. EPA 1991). In addition, evidence of either cytotoxicity not associated with α2u-globulin accumulation, or of peroxisome proliferation, lacked specificity with regard to dose, sex, and/or species.
Another rare rat tumor, brain glioma, was also increased in both sexes in a single bioassay (NTP 1986) (see Supplemental Material, Table S1). This study also reported increases in the rate of testicular interstitial cell tumors, a tumor type of high incidence in unexposed male F344 rats. Mechanistic or other data to inform the interpretation of the increases observed in rat brain and testicular tumors in the NTP bioassay were not identified.
Evaluation of mouse tumors. All three bioassays reported an increase in liver tumors in mice exposed to PCE (JISA 1993; NCI 1977; NTP 1986) (Figure 2). Statistical analyses of the gavage study (NCI 1977) revealed that the incidence of hepatocellular carcinomas or adenomas was significantly increased and that tumor latency was significantly decreased. A significant association between increased mortality and PCE dose was seen, with liver tumors found in many mice that died before scheduled termination. Inhalation exposure to PCE induced significant, dose-related increases in the incidence of hepatocellular adenomas or carcinomas in both sexes of B6C3F1 (NTP 1986) and Crj:BDF1 (JISA 1993) mice. The incidence of hepatocellular carcinomas that metastasized to the lungs was also significantly increased in the inhalation studies.
The mouse liver tumor evaluation also considered data on the hepatocarcinogenicity of the PCE metabolites TCA and DCA. The primary urinary oxidative metabolite in rodents and humans, TCA, significantly increased the incidence of liver tumors in male and female B6C3F1 mice exposed via drinking water for 52–104 weeks (Bull et al. 1990, 2002; DeAngelo et al. 2008; Herren-Freund et al. 1987; Pereira 1996). Liver tumor incidence increased with increasing concentrations of TCA in drinking water (Bull et al. 1990, 2002; DeAngelo et al. 2008; Pereira 1996). Tumors in TCA-exposed animals developed rapidly, and significant increases were evident in less-than-lifetime studies of ≤ 82 weeks. In addition, TCA was hepatocarcinogenic in mice when coadministered in the drinking water for 52 weeks with the PCE metabolite DCA (Bull et al. 2002). DCA alone also causes liver cancer in mice (Bull et al. 1990; Daniel et al. 1992; DeAngelo et al. 1999; Herren-Freund et al. 1987).
The issue of whether TCA can solely account for the hepatocarcinogenicity of PCE was addressed using toxicokinetic analyses because the hepatocarcinogenic potencies of TCA and PCE have not been directly compared in a single rodent bioassay. The U.S. EPA’s analysis found that a wide range of possible contributions of TCA to PCE carcinogenicity, from as little as 12% to as much as 100%, is consistent with the available data (U.S. EPA 2012b). A more precise quantitative estimate of the relative contribution of TCA to PCE-induced liver tumors requires an appropriately designed study to better control for experimental variability in kinetics (e.g., dosing patterns in drinking water, metabolism, bioavailability) and dynamics (e.g., background tumor rates).
For hepatocarcinogenesis, a key issue was whether evidence for peroxisome proliferation is sufficient to establish the peroxisome proliferator-activated receptor α (PPARα)–activation mode of action. The relevance to humans of tumors induced by this mode of action has been debated (e.g., Corton 2008; Guyton et al. 2009; Klaunig et al. 2003; Rusyn and Corton 2012; Rusyn et al. 2006). For instance, a dissenting opinion provided in the NRC peer review (NRC 2010, Appendix B) stated that
the weight of evidence strongly favors a key role of PPARα activation in tetrachloroethylene-induced hepatocarcinogenesis in mice; furthermore, this mode of action lacks relevance for human hepatocarcinogenesis.
However, the NRC peer-review committee as a whole did not support these conclusions, stating in rebuttal that many gaps in knowledge remain with regard to the hepatocarcinogenic mechanisms of PCE. PCE-specific experiments have provided evidence that peroxisomal markers are increased, but at dose levels (i.e., 1,000 mg/kg/day) exceeding those causing liver toxicity, proliferation, or carcinogenicity (Odum et al. 1988; Philip et al. 2007). In particular, Philip et al. (2007) reported that CYP4A, a marker of PPARα-activation, was only increased in Swiss Webster mice at the highest PCE dose (1,000 mg/kg/day) and at the earliest (7 days), but not later, time points. In contrast, the study reported a robust dose-dependent proliferative response that persisted for 14–30 days postexposure at 150-, 500-, and 1,000-mg/kg/day levels of PCE. Liver toxicity and repair has been reported at lower doses in other studies in the B6C3F1 strain [e.g., at 100 mg/kg/day (Schumann et al. 1980)]. Moderate increases in peroxisome proliferation have been reported in rats (Odum et al. 1988), a species insensitive to PCE hepatocarcinogenicity. In total, these findings indicate that the modest peroxisome proliferative response to PCE may lack specificity with respect to species, tissue, and dose. The temporal sequence of events also remains to be established. Given these limitations, the database of PCE-specific studies was judged insufficient to demonstrate a causative role of this effect in hepatocarcinogenesis by PCE (U.S. EPA 2012b). PCE and/or its metabolites have been shown to induce a number of other mechanistic events that may also contribute to carcinogenicity, including mutagenicity, alterations in DNA methylation, and oxidative stress (U.S. EPA 2012b).
In addition to mouse liver tumors, hemangiosarcomas or hemangiomas of the liver, spleen, fat, and subcutaneous skin were reported in male mice in one inhalation study (JISA 1993) (see Supplemental Material, Table S2). This mouse tumor type was not reported in the NCI oral gavage bioassay (NCI 1977), and no increase was reported in the NTP inhalation bioassay (NTP 1986). Mechanistic or other data to inform the interpretation of this tumor type were not identified.
Conclusions on carcinogenic hazard. Supported by the analyses described above, and following the U.S. EPA’s Guidelines for Carcinogen Risk Assessment (U.S. EPA 2005a), PCE is characterized as “likely to be carcinogenic to humans” (U.S. EPA 2012b). This characterization is based on suggestive evidence of carcinogenicity in epidemiological studies and conclusive evidence that the administration of PCE, either by ingestion or by inhalation to sexually mature rats and mice, increases tumor incidence (JISA 1993; NCI 1977; NTP 1986). The specific carcinogenic active moiety(ies) and mode(s) of action are not fully characterized, and the hypothesis that mutagenicity contributes to the PCE carcinogenesis has not been ruled out, particularly for kidney carcinogenicity. No data were available on cancer risks in animals exposed to PCE during early life stages.
Noncancer Toxicity
The U.S. EPA’s analysis identified the central nervous system, kidney, liver, immune and hematologic systems, and development and reproduction, as target organs of PCE toxicity (U.S. EPA 2012c). Although sufficient for hazard identification, the supporting evidence for several end points was limited for characterizing the relationship of effect with dose at low exposures. Neurotoxicity was supported by a considerable database of human, animal, and mechanistic studies. In addition, neurological effects were generally observed at lower PCE concentrations compared with other noncancer health effects. Further, both the 2004 peer consultation workshop (U.S. EPA 2004) and the 2010 NRC peer review (NRC 2010) affirmed the conclusion that neurotoxicity is a sensitive end point because these effects were observed at lower concentrations and had substantial evidential support. The human and animal neurotoxicity findings supporting the U.S. EPA’s conclusions concerning PCE neurotoxicity are summarized below.
Neurotoxicity. Human studies regarding neurotoxicological hazard. The three primary neurological domains most consistently associated with subchronic or chronic PCE exposure in human studies were vision, visuospatial memory, and neuropsychological function (e.g., reaction time). Occupational and residential exposure studies support an association of visual deficits after chronic PCE exposure. Deficits in color vision, relative to unexposed study participants, were observed in an occupational study (Cavalleri et al. 1994) and in a residential study (Schreiber et al. 2002). In a longitudinal follow-up study to Cavalleri et al (1994), Gobba et al (1998) reported that there was a worsening in the color vision in workers (self-comparison) who were exposed to higher levels of PCE. In the dry-cleaning facilities, color vision deficits, reported as a color confusion index (CCI) (Iregren et al. 2002), were significantly greater in exposed workers than in unexposed controls, with mean CCI scores of 1.143 and 1.108, respectively (p = 0.03) (Cavalleri et al. 1994). An additional 6% (p < 0.01) deterioration in mean CCI score relative to their previous score (reported by Cavalleri et al. 1994) was seen in workers exposed to increasing concentrations of PCE (median, 1.67–4.35 ppm) 2 years later in a follow-up study of the same worker population (Gobba et al. 1998). Schreiber et al. (2002) reported lower CCI scores, in comparison with a nonexposed residential group, for adult and child residents living within a close proximity to dry cleaners, with mean CCI scores of 1.33 in exposed study participants and 1.20 in controls (p = 0.26). In the same study, no difference in CCI scores was observed in day-care workers working in a day-care center next to a dry cleaner in comparison with day-care workers in buildings with no PCE exposure. Two studies did not observe changes in color vision with PCE exposure, but they were limited by having no exposure characterization (Sharanjeet-Kaur et al. 2004) or by using less sensitive color vision testing (Nakatsuka et al. 1992). In addition, deficits in visual contrast sensitivity relative to unexposed study participants were reported in the two residential populations living or working in buildings co-located with dry cleaners (Schreiber et al. 2002; Storm et al. 2011). There was a decreasing trend (p < 0.05) for visual contrast sensitivity in the residential population achieving the maximum contrast sensitivity score at 6 cycles per degree, with 28.3% in the referent group versus 8.3% in the highest exposed (> 100 μg/m3) group (Storm et al. 2011).
Associations between exposure and visuospatial memory were also reported in each of the studies that examined this measure in humans. These associations (increased response times or cognition errors) were reported in occupational (Echeverria et al. 1994, 1995; Seeber 1989) and residential (Altmann et al. 1995) studies related to dry-cleaning PCE exposure. In the occupational studies (Echeverria et al. 1994, 1995; Seeber, 1989), cognition errors ranged from 4 to 30% at exposures of 12–23 ppm, depending on the subtest that was used. In the residential study (Altmann et al. 1995), visual memory and cognitive function scores were 15% lower in individuals with a mean exposure of 0.7 ppm PCE compared with unexposed individuals. No studies specifically examined visuospatial memory in children.
For neuropsychological function, two studies reported 10–20% increases in simple reaction time, in comparison with an unexposed group, in PCE-exposed occupational (Ferroni et al. 1992) and residential (Altmann et al. 1995) settings. However, another occupational study reported a 16% improvement in simple reaction time in comparison with unexposed individuals (Lauwerys et al. 1983).
Animal studies of neurotoxicological effects. Animal studies of subchronic PCE exposure also observed changes in visual function, cognitive function, and reaction time. In rats, acute inhalation exposure to PCE resulted in changes in visual-evoked potentials (Boyes et al. 2009; Mattsson et al. 1998). In one subchronic exposure study (Mattsson et al. 1998), a significant increase in amplitude and latency was observed in one peak of the visual-evoked potential responses from a flash stimulus at 5,424 mg/m3, but histopathological lesions were not observed in the examination of central and peripheral brain structures (e.g., visual cortex, optic nerve) of the visual system.
Significant decrements in the motor activity domain as measured by increased reaction time, increased number of false alarms, and decreased trial completions in a signal detection task (measures of decreased attention) were reported at ≥ 6,782 mg/m3 in an acute exposure study in rats (Oshiro et al. 2008). In addition, deficits in operant tasks that test cognitive performance were seen in rats and mice after acute oral (Warren et al. 1996) and intraperitoneal (Umezu et al. 1997) exposures to PCE. These findings support evidence of deficits in cognition and memory associated with PCE exposure in humans. However, no animal studies to date have evaluated the persistence of cognitive performance deficits from acute or chronic PCE exposure.
Observed changes in brain weight and levels of DNA, RNA, and neurotransmitters in experimental animals are consistent with evidence of neurobehavioral effects of PCE exposure in humans. Brain DNA, RNA and protein levels, as well as lipid composition, were altered after PCE inhalation. Changes were observed in the cerebellum, the hippocampus, and the frontal cortex in sexually mature animals (Rosengren et al. 1986; Savolainen et al. 1977a, 1977b; Wang et al. 1993) as well as after gestational exposure (Kyrklund and Haglid 1991; Nelson et al. 1979).
Conclusions regarding noncancer hazard. The U.S. EPA’s analysis identified the central nervous system, kidney, liver, immune and hematologic systems, and development and reproduction, as target organs of PCE toxicity (U.S. EPA 2012c). Neurotoxicity was identified as among the most sensitive outcomes, occurring at low exposures. The assessment of the neurotoxicity studies drew conclusions through an examination of affected domains (e.g., cognition, vision, motor activity). Human and experimental animal studies provided complementary evidence regarding the association of neurobehavioral deficits and PCE exposure. Studies of humans exposed by inhalation suggest that chronic PCE exposure can result in decrements in vision, visuospatial memory, and, possibly, other aspects of cognition and neuropsychological function, including reaction time. Animal studies provide substantial support for associations of PCE exposure with effects in these domains of neurotoxicity.
Summary
PCE is a widespread contaminant that is present in ambient air, indoor air, soil, drinking water, and groundwater. Once exposed, humans and laboratory animal species rapidly absorb PCE. PCE is then distributed to tissues via the systemic circulation, metabolized, and excreted primarily in breath as unchanged PCE or CO2, or in urine as metabolites. The role of metabolism in the toxicity of PCE was informed by the development and application of an updated PBPK model (Chiu and Ginsberg 2011). Low oxidative metabolism was predicted in humans, whereas GSH conjugation metabolism is more uncertain and may be high, low, and/or highly variable. These PBPK model predictions informed the extent and nature of metabolism in different target tissues, and the extrapolation across species and routes of exposure.
Following the U.S. EPA’s Guidelines for Carcinogen Risk Assessment (U.S. EPA 2005a), PCE was characterized as “likely to be carcinogenic to humans” by all routes of exposure. This characterization is based on suggestive evidence of carcinogenicity in epidemiological studies and conclusive evidence that the administration of PCE, either by ingestion or by inhalation to sexually mature rats and mice, increases tumor incidence (JISA 1993; NCI 1977; NTP 1986).
Neurotoxicity is identified as a sensitive outcome that follows either oral or inhalation exposure to PCE in humans and experimental animals. Associations between exposure and neurotoxic outcomes have been reported by human controlled exposure, occupational, and residential studies as well as experimental animal studies, providing evidence that PCE exposure results in visual changes, increased reaction time, and decrements in cognition.
The U.S. EPA’s analysis, approaches, and conclusions are consistent with multiple sets of peer-reviewer recommendations (U.S. EPA 2012a). In addition, the use of evidence tables, narrative syntheses, and other aspects of the assessment approach were in accord with later NRC recommendations for improving IRIS assessments (NRC 2011). The International Agency for Research on Cancer recently classified PCE as probably carcinogenic to humans (Group 2A) on the basis of sufficient evidence in animals and limited evidence in humans, consistent with the U.S. EPA’s conclusion (Guha et al. 2012). Finally, studies of the health effects of PCE published since the U.S. EPA’s assessment continue to report associations with neurological outcomes, including studies of illicit drug use (Aschengrau et al. 2011); mental illness (Aschengrau et al. 2012); visual effects (Getz et al. 2012); visuospatial functioning, learning and memory, motor, attention, and mood (Janulewicz et al. 2012); and Parkinson disease (Goldman et al. 2012). Similarly, recent analyses of PCE exposure and cancer continue to add support for human tumor sites identified in the Toxicological Review (Christensen et al. 2013; Lipworth et al. 2011; Ruder et al. 2013; Vizcaya et al. 2013; Vlaanderen et al. 2013; 2014).
Supplemental Material
Acknowledgments
We thank B. Sonawane, P. White, D. Bussard, V. Cogliano, R. Clark, P. Preuss, and P. Anastas [U.S. Environmental Protection Agency (EPA)] for providing management support for the assessment and this manuscript. This work has also benefitted from scientific peer reviewers, including members of the National Academies of Sciences National Research Council review panel.
Footnotes
The views expressed in this article are those of the authors and do not necessarily represent the views or policies of the U.S. EPA.
The authors declare they have no actual or potential competing financial interests.
References
- Altmann L, Neuhann HF, Krämer U, Witten J, Jermann E. Neurobehavioral and neurophysiological outcome of chronic low-level tetrachloroethene exposure measured in neighborhoods of dry cleaning shops. Environ Res. 1995;69:83–89. doi: 10.1006/enrs.1995.1028. [DOI] [PubMed] [Google Scholar]
- Anders MW, Lash L, Dekant W, Elfarra AA, Dohn DR, Reed DJ. Biosynthesis and biotransformation of glutathione S-conjugates to toxic metabolites. Crit Rev Toxicol. 1988;18:311–341. doi: 10.3109/10408448809037470. [DOI] [PubMed] [Google Scholar]
- Anttila A, Pukkala E, Sallmen M, Hernberg S, Hemminki K. Cancer incidence among Finnish workers exposed to halogenated hydrocarbons. J Occup Environ Med. 1995;37:797–806. doi: 10.1097/00043764-199507000-00008. [DOI] [PubMed] [Google Scholar]
- Aschengrau A, Ozonoff D, Paulu C, Coogan P, Vezina R, Heeren T, et al. Cancer risk and tetrachloroethylene-contaminated drinking water in Massachusetts. Arch Environ Health. 1993;48:284–292. doi: 10.1080/00039896.1993.9936715. [DOI] [PubMed] [Google Scholar]
- Aschengrau A, Weinberg JM, Janulewicz PA, Romano ME, Gallagher LG, Winter MR, et al. 2011Affinity for risky behaviors following prenatal and early childhood exposure to tetrachloroethylene (PCE)-contaminated drinking water: a retrospective cohort study. Environ Health 10102; 10.1186/1476-069X-10-102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschengrau A, Weinberg JM, Janulewicz PA, Romano ME, Gallagher LG, Winter MR, et al. 2012Occurrence of mental illness following prenatal and early childhood exposure to tetrachloroethylene (PCE)-contaminated drinking water: a retrospective cohort study. Environ Health 112; 10.1186/1476-069X-11-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair A, Petralia SA, Stewart PA. Extended mortality follow-up of a cohort of dry cleaners. Ann Epidemiol. 2003;13:50–56. doi: 10.1016/s1047-2797(02)00250-8. [DOI] [PubMed] [Google Scholar]
- Boice JD, Jr, Marano D, Fryzek J, Sadler C, McLaughlin JK. Mortality among aircraft manufacturing workers. Occup Environ Med. 1999;56:581–597. doi: 10.1136/oem.56.9.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bois FY, Gelman A, Jiang J, Maszle DR, Zeise L, Alexeef G. Population toxicokinetics of tetrachloroethylene. Arch Toxicol. 1996;70:347–355. doi: 10.1007/s002040050284. [DOI] [PubMed] [Google Scholar]
- Bois FY, Zeise L, Tozer TN. Precision and sensitivity of pharmacokinetic models for cancer risk assessment: tetrachloroethylene in mice, rats, and humans. Toxicol Appl Pharmacol. 1990;102:300–315. doi: 10.1016/0041-008x(90)90029-t. [DOI] [PubMed] [Google Scholar]
- Boyes WK, Bercegeay M, Oshiro WM, Krantz QT, Kenyon EM, Bushnell PJ, et al. Acute perchloroethylene exposure alters rat visual-evoked potentials in relation to brain concentrations. Toxicol Sci. 2009;108:159–172. doi: 10.1093/toxsci/kfn265. [DOI] [PubMed] [Google Scholar]
- Brown Dzubow R, Makris S, Siegel Scott C, Barone S., Jr Early lifestage exposure and potential developmental susceptibility to tetrachloroethylene. Birth Defects Res B Dev Reprod Toxicol. 2010;89:50–65. doi: 10.1002/bdrb.20222. [DOI] [PubMed] [Google Scholar]
- Buben JA, O’Flaherty EJ. Delineation of the role of metabolism in the hepatotoxicity of trichloroethylene and perchloroethylene: a dose-effect study. Toxicol Appl Pharmacol. 1985;78:105–122. doi: 10.1016/0041-008x(85)90310-2. [DOI] [PubMed] [Google Scholar]
- Bull RJ, Orner GA, Cheng RS, Stillwell L, Stauber AJ, Sasser LB, et al. Contribution of dichloroacetate and trichloroacetate to liver tumor induction in mice by trichloroethylene. Toxicol Appl Pharmacol. 2002;182:55–65. doi: 10.1006/taap.2002.9427. [DOI] [PubMed] [Google Scholar]
- Bull RJ, Sanchez IM, Nelson MA, Larson JL, Lansing AJ. Liver tumor induction in B6C3F1 mice by dichloroacetate and trichloroacetate. Toxicology. 1990;63:341–359. doi: 10.1016/0300-483x(90)90195-m. [DOI] [PubMed] [Google Scholar]
- Calvert GM, Ruder AM, Petersen MR. Mortality and end-stage renal disease incidence among dry cleaning workers. Occup Environ Med. 2011;68:709–716. doi: 10.1136/oem.2010.060665. [DOI] [PubMed] [Google Scholar]
- Cavalleri A, Gobba F, Paltrinieri M, Fantuzzi G, Righi E, Aggazzotti G. Perchloroethylene exposure can induce colour vision loss. Neurosci Lett. 1994;179:162–166. doi: 10.1016/0304-3940(94)90959-8. [DOI] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention. National Biomonitoring Program: Biomonitoring Summary, Halogenated Solvents. 2013. Available: http://www.cdc.gov/biomonitoring/HalogenatedSolvents_BiomonitoringSummary.html [accessed 26 September 2013]
- Chen C, Blancato J. Washington, DC: National Academies Press; 1987. Role of Pharmacokinetic Modeling in Risk Assessment: Perchloroethylene as an Example. [Google Scholar]
- Chen CW, Blancato JN. Incorporation of biological information in cancer risk assessment: example—vinyl chloride. Cell Biol Toxicol. 1989;5:417–444. doi: 10.1007/BF00118412. [DOI] [PubMed] [Google Scholar]
- Chien YC. The Influences of Exposure Pattern and Duration on Elimination Kinetics and Exposure Assessment of Tetrachloroethylene in Humans [PhD Dissertation]. New Brunswick, NJ:Rutgers University. 1997.
- Chiu WA, Bois FY. Revisiting the population toxicokinetics of tetrachloroethylene. Arch Toxicol. 2006;80:382–385. doi: 10.1007/s00204-006-0061-9. [DOI] [PubMed] [Google Scholar]
- Chiu WA, Ginsberg GL. Development and evaluation of a harmonized physiologically based pharmacokinetic (PBPK) model for perchloroethylene toxicokinetics in mice, rats, and humans. Toxicol Appl Pharmacol. 2011;253:203–234. doi: 10.1016/j.taap.2011.03.020. [DOI] [PubMed] [Google Scholar]
- Christensen KY, Vizcaya D, Richardson H, Lavoue J, Aronson K, Siemiatycki J. Risk of selected cancers due to occupational exposure to chlorinated solvents in a case–control study in Montreal. J Occup Environ Med. 2013;55:198–208. doi: 10.1097/JOM.0b013e3182728eab. [DOI] [PubMed] [Google Scholar]
- Clewell HJ, Gentry PR, Kester JE, Andersen ME. Evaluation of physiologically based pharmacokinetic models in risk assessment: an example with perchloroethylene. Crit Rev Toxicol. 2005;35:413–433. doi: 10.1080/10408440590931994. [DOI] [PubMed] [Google Scholar]
- Corton JC. Evaluation of the role of peroxisome proliferator-activated receptor α (PPARα) in mouse liver tumor induction by trichloroethylene and metabolites. Crit Rev Toxicol. 2008;38:857–875. doi: 10.1080/10408440802209796. [DOI] [PubMed] [Google Scholar]
- Costantini AS, Benvenuti A, Vineis P, Kriebel D, Tumino R, Ramazzotti V, et al. Risk of leukemia and multiple myeloma associated with exposure to benzene and other organic solvlents: evidence from the Italian multicenter Case–control Study. Am J Ind Med. 2008;51:803–811. doi: 10.1002/ajim.20592. [DOI] [PubMed] [Google Scholar]
- Covington TR, Gentry PR, Van Landingham CB, Andersen ME, Kester JE, Clewell HJ. The use of Markov chain Monte Carlo uncertainty analysis to support a public health goal for perchloroethylene. Regul Toxicol Pharmacol. 2007;47:1–18. doi: 10.1016/j.yrtph.2006.06.008. [DOI] [PubMed] [Google Scholar]
- Daniel FB, DeAngelo AB, Stober JA, Olson GR, Page NP. Hepatocarcinogenicity of chloral hydrate, 2-chloroacetaldehyde, and dichloroacetic acid in the male B6C3F1 mouse. Fundam Appl Toxicol. 1992;19:159–168. doi: 10.1016/0272-0590(92)90147-a. [DOI] [PubMed] [Google Scholar]
- DeAngelo AB, Daniel FB, Wong DM, George MH. The induction of hepatocellular neoplasia by trichloroacetic acid administered in the drinking water of the male B6C3F1 mouse. J Toxicol Environ Health A. 2008;71:1056–1068. doi: 10.1080/15287390802111952. [DOI] [PubMed] [Google Scholar]
- DeAngelo AB, George MH, House DE. Hepatocarcinogenicity in the male B6C3F1 mouse following a lifetime exposure to dichloroacetic acid in the drinking water: dose-response determination and modes of action. J Toxicol Environ Health A. 1999;58:485–507. doi: 10.1080/009841099157115. [DOI] [PubMed] [Google Scholar]
- Dekant W, Vamvakas S, Berthold K, Schmidt S, Wild D, Henschler D. Bacterial β-lyase mediated cleavage and mutagenicity of cysteine conjugates derived from the nephrocarcinogenic alkenes trichloroethylene, tetrachloroethylene and hexachlorobutadiene. Chem Biol Interact. 1986;60:31–45. doi: 10.1016/0009-2797(86)90015-3. [DOI] [PubMed] [Google Scholar]
- Dreessen B, Westphal G, Bünger J, Hallier E, Müller M. Mutagenicity of the glutathione and cysteine S-conjugates of the haloalkenes 1,1,2-trichloro-3,3,3-trifluoro-1-propene and trichlorofluoroethene in the Ames test in comparison with the tetrachloroethene-analogues. Mutat Res Genet Toxicol Environ Mutagen. 2003;539:157–166. doi: 10.1016/s1383-5718(03)00160-8. [DOI] [PubMed] [Google Scholar]
- Ebrahim AS, Babu E, Thirunavukkarasu C, Sakthisekaran D. Protective role of vitamin E, 2-deoxy-d-glucose, and taurine on perchloroethylene induced alterations in ATPases. Drug Chem Toxicol. 2001;24:429–437. doi: 10.1081/dct-100106267. [DOI] [PubMed] [Google Scholar]
- Echeverria D, Heyer N, Checkoway H, Brodkin CA, Bittner A Jr, Toutonghi G, et al. Seattle, WA: Battelle Centers for Public Health Research and Evaluation; 1994. A Behavioral Investigation of Occupational Exposures to Solvents: Perchloroethylene among Dry Cleaners, and Styrene among Reinforced Fiberglass Laminators. BSRC-100/94/040. [Google Scholar]
- Echeverria D, White RF, Sampaio C. A behavioral evaluation of PCE exposure in patients and dry cleaners: a possible relationship between clinical and preclinical effects. J Occup Environ Med. 1995;37:667–680. doi: 10.1097/00043764-199506000-00008. [DOI] [PubMed] [Google Scholar]
- Ferroni C, Selis L, Mutti A, Folli D, Bergamaschi E, Franchini I. Neurobehavioral and neuroendocrine effects of occupational exposure to perchloroethylene. Neurotoxicology. 1992;13:243–247. [PubMed] [Google Scholar]
- Gearhart JM, Mahle DA, Greene RJ, Seckel CS, Flemming CD, Fisher JW, et al. Variability of physiologically based pharmacokinetic (PBPK) model parameters and their effects on PBPK model predictions in a risk assessment for perchloroethylene (PCE). Toxicol Lett. 1993;68:131–144. doi: 10.1016/0378-4274(93)90126-i. [DOI] [PubMed] [Google Scholar]
- Getz KD, Janulewicz PA, Rowe S, Weinberg JM, Winter MR, Martin BR, et al. 2012Prenatal and early childhood exposure to tetrachloroethylene and adult vision. Environ Health Perspect 1201327–1332.; 10.1289/ehp.1103996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobba F, Righi E, Fantuzzi G, Predieri G, Cavazzuti L, Aggazzotti G. Two-year evolution of perchloroethylene-induced color-vision loss. Arch Environ Health. 1998;53:196–198. doi: 10.1080/00039899809605695. [DOI] [PubMed] [Google Scholar]
- Gold LS, De Roos AJ, Waters M, Stewart P. Systematic literature review of uses and levels of occupational exposure to tetrachloroethylene. JJ Occup Environ Hyg. 2008;5:807–839. doi: 10.1080/15459620802510866. [DOI] [PubMed] [Google Scholar]
- Gold LS, Stewart PA, Milliken K, Purdue M, Severson R, Seixas N, et al. The relationship between multiple myeloma and occupational exposure to six chlorinated solvents. Occup Environ Med. 2010;68:391–399. doi: 10.1136/oem.2009.054809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman SM, Quinlan PJ, Ross GW, Marras C, Meng C, Bhudhikanok GS, et al. Solvent exposures and Parkinson disease risk in twins. Ann Neurol. 2012;71:776–784. doi: 10.1002/ana.22629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guha N, Loomis D, Grosse Y, Lauby-Secretan B, El Ghissassi F, Bouvard V, et al. Carcinogenicity of trichloroethylene, tetrachloroethylene, some other chlorinated solvents, and their metabolites. Lancet Oncol. 2012;13:1192–1193. doi: 10.1016/s1470-2045(12)70485-0. [DOI] [PubMed] [Google Scholar]
- Guyton KZ, Chiu WA, Bateson TF, Jinot J, Scott CS, Brown RC, et al. 2009A reexamination of the PPAR-α activation mode of action as a basis for assessing human cancer risks of environmental contaminants. Environ Health Perspect 1171664–1672.; 10.1289/ehp.0900758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herren-Freund SL, Pereira MA, Khoury MD, Olson G. The carcinogenicity of trichloroethylene and its metabolites, trichloroacetic acid and dichloroacetic acid, in mouse liver. Toxicol Appl Pharmacol. 1987;90:183–189. doi: 10.1016/0041-008x(87)90325-5. [DOI] [PubMed] [Google Scholar]
- Iregren A, Andersson M, Nylen P. Color vision and occupational chemical exposures. II. Visual functions in non-exposed subjects. Neurotoxicology. 2002;23:735–745. doi: 10.1016/S0161-813X(02)00113-4. [DOI] [PubMed] [Google Scholar]
- Janulewicz PA, White RF, Martin BM, Winter MR, Weinberg JM, Vieira V, et al. Adult neuropsychological performance following prenatal and early postnatal exposure to tetrachloroethylene (PCE)-contaminated drinking water. Neurotoxicol Teratol. 2012;34:350–359. doi: 10.1016/j.ntt.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JISA (Japan Industrial Safety and Health Association). Carcinogenicity Study of Tetrachloroethylene by Inhalation in Rats and Mice. Hadano, Japan:JISA. 1993. Available: http://www.epa.gov/iris/supdocs/0106index.html [accessed 28 February 2014]
- Klaunig JE, Babich MA, Baetcke KP, Cook JC, Corton JC, David RM, et al. PPARα agonist-induced rodent tumors: modes of action and human relevance. Crit Rev Toxicol. 2003;33:655–780. doi: 10.1080/713608372. [DOI] [PubMed] [Google Scholar]
- Krause RJ, Lash LH, Elfarra AA. Human kidney flavin-containing monooxygenases and their potential roles in cysteine S-conjugate metabolism and nephrotoxicity. J Pharmacol Exp Ther. 2003;304:185–191. doi: 10.1124/jpet.102.042911. [DOI] [PubMed] [Google Scholar]
- Kyrklund T, Haglid K. Brain lipid composition in guinea pigs after intrauterine exposure to perchloroethylene. Pharmacol Toxicol. 1991;68:146–148. doi: 10.1111/j.1600-0773.1991.tb02054.x. [DOI] [PubMed] [Google Scholar]
- Lash LH, Parker JC. Hepatic and renal toxicities associated with perchloroethylene. Pharmacol Rev. 2001;53:177–208. [PubMed] [Google Scholar]
- Lauwerys R, Herbrand J, Buchet JP, Bernard A, Gaussin J. Health surveillance of workers exposed to tetrachloroethylene in dry-cleaning shops. Int Arch Occup Environ Health. 1983;52:69–77. doi: 10.1007/BF00380609. [DOI] [PubMed] [Google Scholar]
- Lipworth L, Sonderman JS, Mumma MT, Tarone RE, Marano DE, Boice JD, Jr, et al. Cancer mortality among aircraft manufacturing workers: an extended follow-up. J Occup Environ Med. 2011;53:992–1007. doi: 10.1097/JOM.0b013e31822e0940. [DOI] [PubMed] [Google Scholar]
- Loizou GD. The application of physiologically based pharmacokinetic modelling in the analysis of occupational exposure to perchloroethylene. Toxicol Lett. 2001;124:59–69. doi: 10.1016/s0378-4274(00)00283-6. [DOI] [PubMed] [Google Scholar]
- Lynge E, Andersen A, Rylander L, Tinnerberg H, Lindbohm ML, Pukkala E, et al. 2006Cancer in persons working in dry cleaning in the Nordic countries. Environ Health Perspect 114213–219.; 10.1289/ehp.8425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marth E. Metabolic changes following oral exposure to tetrachloroethylene in subtoxic concentrations. Arch Toxicol. 1987;60:293–299. doi: 10.1007/BF01234668. [DOI] [PubMed] [Google Scholar]
- Marth E, Stünzner D, Binder H, Möse JR. Tetrachloroethylene: effect of low concentrations of 1,1,2,2-tetrachloroethylene (perchloroethylene) on the mouse. II. Study of tetrachloroethylene in various organs and demonstration of histological changes in the examined organs [in German]. Zentralbl Bakteriol Mikrobiol Hyg. 1985a;181:541–547. [PubMed] [Google Scholar]
- Marth E, Stünzner D, Binder H, Möse JR. Tetrachloroethylene: effect of low concentrations of 1,1,2,2-tetrachloroethylene (perchloroethylene) on organisms in the mouse. I. Laboratory chemical research [in German]. Zentralbl Bakteriol Mikrobiol Hyg. 1985b;181:525–540. [PubMed] [Google Scholar]
- Marth E, Stünzner D, Köck M, Möse JR. Toxicokinetics of chlorinated hydrocarbons. J Hyg Epidemiol Microbiol Immunol. 1989;33:514–520. [PubMed] [Google Scholar]
- Mattsson JL, Albee RR, Yano BL, Bradley G, Spencer PJ. Neurotoxicologic examination of rats exposed to 1,1,2,2-tetrachloroethylene (perchloroethylene) vapor for 13 weeks. Neurotoxicol Teratol. 1998;20:83–98. doi: 10.1016/s0892-0362(97)00074-3. [DOI] [PubMed] [Google Scholar]
- Mazzullo M, Grilli S, Lattanzi G, Prodi G, Turina MP, Colacci A. Evidence of DNA binding activity of perchloroethylene. Res Comm Chem Pathol Pharmacol. 1987;58:215–235. [PubMed] [Google Scholar]
- Miligi L, Costantini AS, Benvenuti A, Kriebel D, Bolejack V, Tumino R, et al. Occupational exposure to solvents and the risk of lymphomas. Epidemiology. 2006;17:552–561. doi: 10.1097/01.ede.0000231279.30988.4d. [DOI] [PubMed] [Google Scholar]
- Nakatsuka H, Watanabe T, Takeuchi Y, Hisanaga N, Shibata E, Suzuki H, et al. Absence of blue-yellow color vision loss among workers exposed to toluene or tetrachloroethylene, mostly at levels below occupational exposure limits. Int Arch Occup Environ Health. 1992;64:113–117. doi: 10.1007/BF00381478. [DOI] [PubMed] [Google Scholar]
- NCI (National Cancer Institute). Bioassay of Tetrachloroethylene for Possible Carcinogenicity. CAS No. 127-18-4. NCI-CGTR-13; DHEW Publication No. (NIH) 77–813. Bethesda, Md:National Institutes of Health. 1977. Available: http://ntp.niehs.nih.gov/ntp/htdocs/LT_rpts/tr013.pdf [accessed 27 February 2014]
- Nelson BK, Taylor BJ, Setzer JV, Hornung RW. Behavioral teratology of perchloroethylene in rats. J Environ Pathol Toxicol Oncol. 1979;3:233–250. [PubMed] [Google Scholar]
- NRC (National Research Council). Risk Assessment in the Federal Government: Managing the Process. Washington, DC:National Academies Press. 1983. Available: http://www.nap.edu/openbook.php?record_id=366 [accessed 27 February 2014] [PubMed]
- NRC (National Research Council). Science and Judgment in Risk Assessment. Washington, DC:National Academies Press. 1994. Available: http://www.nap.edu/openbook.php?record_id=2125 [accessed 27 February 2014] [PubMed]
- NRC (National Research Council). Review of the Environmental Protection Agency’s Draft IRIS Assessment of Tetrachloroethylene. Washington, DC:National Academies Press. 2010. Available: http://www.nap.edu/openbook.php?record_id=12863 [accessed 27 February 2014] [PubMed]
- NRC (National Research Council). Review of the Environmental Protection Agency’s Draft IRIS Assessment of Formaldehyde. Washington, DC:National Academies Press. 2011. Available: http://www.nap.edu/openbook.php?record_id=13142 [accessed 27 February 2014] [PubMed]
- NTP (National Toxicology Program). Toxicology and Carcinogenesis Studies of Tetrachloroethylene (Perchloroethylene) (CAS no. 127-18-4) in F344/N Rats and B6C3F1 Mice (Inhalation Studies). TR 311. Research Triangle Park, NC:NTP. 1986. Available: http://ntp.niehs.nih.gov/ntp/htdocs/lt_rpts/tr311.pdf [accessed 27 February 2014] [PubMed]
- Odum J, Green T, Foster JR, Hext PM. The role of trichloroacetic acid and peroxisome proliferation in the differences in carcinogenicity of perchloroethylene in the mouse and rat. Toxicol Appl Pharmacol. 1988;92:103–112. doi: 10.1016/0041-008x(88)90232-3. [DOI] [PubMed] [Google Scholar]
- Oshiro WM, Krantz QT, Bushnell PJ. Characterization of the effects of inhaled perchloroethylene on sustained attention in rats performing a visual signal detection task. Neurotoxicol Teratol. 2008;30:167–174. doi: 10.1016/j.ntt.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Pegg DG, Zempel JA, Braun WH, Watanabe PG. Disposition of tetrachloro(14C)ethylene following oral and inhalation exposure in rats. Toxicol Appl Pharmacol. 1979;51:465–474. doi: 10.1016/0041-008x(79)90371-5. [DOI] [PubMed] [Google Scholar]
- Pereira MA. Carcinogenic activity of dichloroacetic acid and trichloroacetic acid in the liver of female B6C3F1 mice. Fundam Appl Toxicol. 1996;31:192–199. doi: 10.1006/faat.1996.0091. [DOI] [PubMed] [Google Scholar]
- Pesch B, Haerting J, Ranft U, Klimpel A, Oelschlägel B, Schill W. Occupational risk factors for urothelial carcinoma: agent-specific results from a case-control study in Germany. Int J Epidemiol. 2000;29:238–247. doi: 10.1093/ije/29.2.238. [DOI] [PubMed] [Google Scholar]
- Philip BK, Mumtaz MM, Latendresse JR, Mehendale HM. Impact of repeated exposure on toxicity of perchloroethylene in Swiss Webster mice. Toxicology. 2007;232:1–14. doi: 10.1016/j.tox.2006.12.018. [DOI] [PubMed] [Google Scholar]
- Qiu J, Chien YC, Bruckner JV, Fisher JW. Bayesian analysis of a physiologically based pharmacokinetic model for perchloroethylene in humans. J Toxicol Environ Health A. 2010;73:74–91. doi: 10.1080/15287390903249099. [DOI] [PubMed] [Google Scholar]
- Radican L, Blair A, Stewart P, Wartenberg D. Mortality of aircraft maintenance workers exposed to trichloroethylene and other hydrocarbons and chemicals: extended follow-up. J Occup Environ Med. 2008;50:1306–1319. doi: 10.1097/JOM.0b013e3181845f7f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao HV, Brown DR. A physiologically based pharmacokinetic assessment of tetrachloroethylene in groundwater for a bathing and showering determination. Risk Anal. 1993;13:37–49. doi: 10.1111/j.1539-6924.1993.tb00727.x. [DOI] [PubMed] [Google Scholar]
- Regulations.gov. Docket ID: EPA-HQ-ORD-2008-0461, Toxicological Review of Tetrachloroethylene (Perchloroethylene): In Support of the Summary Information in the Integrated Risk Information System (IRIS). 2008. Available: http://www.regulations.gov/#!docketDetail;D=EPA-HQ-ORD-2008-0461 [accessed 26 September 2013]
- Reitz RH, Gargas ML, Mendrala AL, Schumann AM. In vivo and in vitro studies of perchloroethylene metabolism for physiologically based pharmacokinetic modeling in rats, mice, and humans. Toxicol Appl Pharmacol. 1996;136:289–306. doi: 10.1006/taap.1996.0036. [DOI] [PubMed] [Google Scholar]
- Rosengren LE, Kjellstrand P, Haglid KG. Tetrachloroethylene: levels of DNA and S-100 in the gerbil CNS after chronic exposure. Neurobehav Toxicol Teratol. 1986;8:201–206. [PubMed] [Google Scholar]
- Ruder AM, Yiin JH, Waters MA, Carreón T, Hein MJ, Butler MA, et al. The Upper Midwest Health Study: gliomas and occupational exposure to chlorinated solvents. Occup Environ Med. 2013;70:73–80. doi: 10.1136/oemed-2011-100588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusyn I, Corton JC. Mechanistic considerations for human relevance of cancer hazard of di(2-ethylhexyl) phthalate. Mutat Res. 2012;750:141–158. doi: 10.1016/j.mrrev.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusyn I, Peters JM, Cunningham ML. Modes of action and species-specific effects of di-(2-ethylhexyl)phthalate in the liver. Crit Rev Toxicol. 2006;36:459–479. doi: 10.1080/10408440600779065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savolainen H, Pfäffli P, Tengén M, Vainio H. Biochemical and behavioural effects of inhalation exposure to tetrachlorethylene and dichlormethane. J Neuropathol Exp Neurol. 1977a;36:941–949. doi: 10.1097/00005072-197711000-00005. [DOI] [PubMed] [Google Scholar]
- Savolainen H, Pfäffli P, Tengén M, Vainio H. Trichloroethylene and 1,1,1-trichloroethane: effects on brain and liver after five days intermittent inhalation. Arch Toxicol. 1977b;38:229–237. doi: 10.1007/BF00293657. [DOI] [PubMed] [Google Scholar]
- Schreiber JS, Hudnell HK, Geller AM, House DE, Aldous KM, Force MS, et al. Apartment residents’ and day care workers’ exposures to tetrachloroethylene and deficits in visual contrast sensitivity. Environ Health Perspect. 2002;110:655–664. doi: 10.1289/ehp.02110655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann AM, Quast JF, Watanabe PG. The pharmacokinetics and macromolecular interactions of perchloroethylene in mice and rats as related to oncogenicity. Toxicol Appl Pharmacol. 1980;55:207–219. doi: 10.1016/0041-008x(80)90082-4. [DOI] [PubMed] [Google Scholar]
- Seeber A. Neurobehavioral toxicity of long-term exposure to tetrachloroethylene. Neurotoxicol Teratol. 1989;11:579–583. doi: 10.1016/0892-0362(89)90041-x. [DOI] [PubMed] [Google Scholar]
- Seidel HJ, Weber L, Barthel E. Hematological toxicity of tetrachloroethylene in mice. Arch Toxicol. 1992;66:228–230. doi: 10.1007/BF01974021. [DOI] [PubMed] [Google Scholar]
- Seidler A, Möhner M, Berger J, Mester B, Deeg E, Elsner G, et al. 2007Solvent exposure and malignant lymphoma: a population-based case-control study in Germany. J Occup Med Toxicol 22; 10.1186/1745-6673-2-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seldén AI, Ahlborg G. Cancer morbidity in Swedish dry-cleaners and laundry workers: historically prospective cohort study. Int Arch Occup Environ Health. 2011;84:435–443. doi: 10.1007/s00420-010-0582-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharanjeet-Kaur , Mursyid A, Kamaruddin A, Ariffin A. Effect of petroleum derivatives and solvents on colour perception. Clin Exp Optom. 2004;87:339–343. doi: 10.1111/j.1444-0938.2004.tb05064.x. [DOI] [PubMed] [Google Scholar]
- Storm JE, Mazor KA, Aldous KM, Blount BC, Brodie SE, Serle JB. Visual contrast sensitivity in children exposed to tetrachloroethylene. Arch Environ Occup Health. 2011;66:166–177. doi: 10.1080/19338244.2010.539638. [DOI] [PubMed] [Google Scholar]
- Thomas J, Haseman JK, Goodman JI, Ward JM, Loughran TP, Jr, Spencer PJ. A review of large granular lymphocytic leukemia in Fischer 344 rats as an initial step toward evaluating the implication of the endpoint to human cancer risk assessment. Toxicol Sci. 2007;99:3–19. doi: 10.1093/toxsci/kfm098. [DOI] [PubMed] [Google Scholar]
- Umezu T, Yonemoto J, Soma Y, Miura T. Behavioral effects of trichloroethylene and tetrachloroethylene in mice. Pharmacol Biochem Behav. 1997;58:665–671. doi: 10.1016/s0091-3057(97)00046-4. [DOI] [PubMed] [Google Scholar]
- U.S. EPA (U.S. Environmental Protection Agency). Alpha2u-globulin: Association with Chemically Induced Renal Toxicity and Neoplasia in the Male Rat. EPA/625/3-91/019f (ntis pb92143668). 1991. Available: http://www.epa.gov/raf/publications/alpha2u-globulin.htm [accessed 27 February 2014]
- U.S. EPA (U.S. Environmental Protection Agency). Summary Report of the Peer Review Workshop on the Neurotoxicity of Tetrachloroethylene (Perchloroethylene) Discussion Paper. 2004. Available: http://ofmpub.epa.gov/eims/eimscomm.getfile?p_download_id=434721 [accessed 26 September 2013]
- U.S. EPA (U.S. Environmental Protection Agency). Guidelines for Carcinogen Risk Assessment. EPA/630/p-03/001f. 2005a. Available: http://www.epa.gov/raf/publications/pdfs/CANCER_GUIDELINES_FINAL_3-25-05.PDF [accessed 27 February 2014]
- U.S. EPA (U.S. Environmental Protection Agency). Supplemental Guidance for Assessing Susceptibility from Early-Life Exposure to Carcinogens. EPA/630/r-03/003f. 2005b. Available: http://www.epa.gov/raf/publications/pdfs/childrens_supplement_final.pdf [accessed 27 February 2014]
- U.S. EPA (U.S. Environmental Protection Agency). A New Approach to Protecting Drinking Water and Public Health. EPA 815f10001. 2010. Available: http://water.epa.gov/lawsregs/rulesregs/sdwa/dwstrategy/upload/Drinking_Water_Strategyfs.pdf [accessed 26 September 2013]
- U.S. EPA (U.S. Environmental Protection Agency). IRIS Toxicological Review of Tetrachloroethylene (Perchloroethylene) (Interagency Science Discussion Draft). 2012a. Available: http://cfpub.epa.gov/ncea/iris_drafts/recordisplay.cfm?deid=238089 [accessed 26 September 2013]
- U.S. EPA (U.S. Environmental Protection Agency). Tetrachloroethylene (Perchloroethylene) (CASRN: 127-18-4). 2012b. Available: http://www.epa.gov/iris/toxreviews/0106tr.pdf [accessed 6 March 2014]
- U.S. EPA (U.S. Environmental Protection Agency). TSCA Work Plan Chemicals. 2012c. Available: http://www.epa.gov/oppt/existingchemicals/pubs/Work_Plan_Chemicals_Web_Final.pdf [accessed 26 September 2013]
- U.S. EPA (U.S. Environmental Protection Agency). Integrated Risk Information System (IRIS). IRIS Guidance Documents. 2013a. Available: http://www.epa.gov/iris/backgrd.html [accessed 26 September 2013]
- U.S. EPA (U.S. Environmental Protection Agency). National Priorities List (NPL). 2013b. Available: http://www.epa.gov/superfund/sites/npl/index.htm [accessed 26 September 2013]
- U.S. EPA (U.S. Environmental Protection Agency). Toxics Release Inventory (TRI) Program. 2013c. Available: http:/www.epa.gov/tri [accessed 26 September 2013]
- U.S. EPA (U.S. Environmental Protection Agency). Vapor Intrusion. 2013d. Available: http://www.epa.gov/oswer/vaporintrusion/ [accessed 26 September 2013]
- Vamvakas S, Dekant W, Berthold K, Schmidt S, Wild D, Henschler D. Enzymatic transformation of mercapturic acids derived from halogenated alkenes to reactive and mutagenic intermediates. Biochem Pharmacol. 1987;36:2741–2748. doi: 10.1016/0006-2952(87)90258-9. [DOI] [PubMed] [Google Scholar]
- Vamvakas S, Dekant W, Henschler D. Genotoxicity of haloalkene and haloalkane glutathione S-conjugates in porcine kidney cells. Toxicol In Vitro. 1989a;3:151–156. doi: 10.1016/0887-2333(89)90058-1. [DOI] [PubMed] [Google Scholar]
- Vamvakas S, Herkenhoff M, Dekant W, Henschler D. Mutagenicity of tetrachloroethene in the Ames test: metabolic activation by conjugation with glutathione. J Biochem Toxicol. 1989b;4:21–27. doi: 10.1002/jbt.2570040105. [DOI] [PubMed] [Google Scholar]
- Vizcaya D, Christensen KY, Lavoue J, Siemiatycki J. Risk of lung cancer associated with six types of chlorinated solvents: results from two case–control studies in Montreal, Canada. Occup Environ Med. 2013;70:81–85. doi: 10.1136/oemed-2012-101155. [DOI] [PubMed] [Google Scholar]
- Vlaanderen J, Straif K, Pukkala E, Kauppinen T, Kyyronen P, Martinsen JI, et al. Occupational exposure to trichloroethylene and perchloroethylene and the risk of lymphoma, liver, and kidney cancer in four Nordic countries. Occup Environ Med. 2013;70:393–401. doi: 10.1136/oemed-2012-101188. [DOI] [PubMed] [Google Scholar]
- Vlaanderen J, Straif K, Ruder A, Blair A, Hansen J, Lynge E, et al. Tetrachloroethylene exposure and bladder cancer risk: a meta-analysis of dry cleaning worker studies. Environ Health Perspect 2014 doi: 10.1289/ehp.1307055. , in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walles SAS. Induction of single-strand breaks in DNA of mice by trichloroethylene and tetrachloroethylene. Toxicol Lett. 1986;31:31–35. doi: 10.1016/0378-4274(86)90191-8. [DOI] [PubMed] [Google Scholar]
- Wang S, Karlsson JE, Kyrklund T, Haglid K. Perchloroethylene-induced reduction in glial and neuronal cell marker proteins in rat brain. Basic Clin Pharmacol Toxicol. 1993;72:273–278. doi: 10.1111/j.1600-0773.1993.tb01649.x. [DOI] [PubMed] [Google Scholar]
- Ward RC, Travis CC, Hetrick DM, Andersen ME, Gargas ML. Pharmacokinetics of tetrachloroethylene. Toxicol Appl Pharmacol. 1988;93:108–117. doi: 10.1016/0041-008x(88)90030-0. [DOI] [PubMed] [Google Scholar]
- Warren DA, Reigle TG, Muralidhara S, Dallas CE. Schedule-controlled operant behavior of rats following oral administration of perchloroethylene: time course and relationship to blood and brain solvent levels. J Toxicol Environ Health. 1996;47:345–362. doi: 10.1080/009841096161690. [DOI] [PubMed] [Google Scholar]
- Werner M, Birner G, Dekant W. Sulfoxidation of mercapturic acids derived from tri- and tetrachloroethene by cytochromes P450 3A: a bioactivation reaction in addition to deacetylation and cysteine conjugate β-lyase mediated cleavage. Chem Res Toxicol. 1996;9:41–49. doi: 10.1021/tx950075u. [DOI] [PubMed] [Google Scholar]
- Yoshioka T, Krauser JA, Guengerich FP. Tetrachloroethylene oxide: hydrolytic products and reactions with phosphate and lysine. Chem Res Toxicol. 2002;15:1096–1105. doi: 10.1021/tx020028j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.