

Abstract
NAD+ metabolism plays key roles not only in energy production but also in diverse cellular physiology. Aberrant NAD+ metabolism is considered a hallmark of cancer. Recently, the tumor suppressor p53, a major player in cancer signaling pathways, has been implicated as an important regulator of cellular metabolism. This notion led us to examine whether p53 can regulate NAD+ biosynthesis in the cell. Our search resulted in the identification of nicotinamide mononucleotide adenylyltransferase 2 (NMNAT-2), a NAD+ synthetase, as a novel downstream target gene of p53. We show that NMNAT-2 expression is induced upon DNA damage in a p53-dependent manner. Two putative p53 binding sites were identified within the human NMNAT-2 gene, and both were found to be functional in a p53-dependent manner. Furthermore, knockdown of NMNAT-2 significantly reduces cellular NAD+ levels and protects cells from p53-dependent cell death upon DNA damage, suggesting an important functional role of NMNAT-2 in p53-mediated signaling. Our demonstration that p53 modulates cellular NAD+ synthesis is congruent with p53’s emerging role as a key regulator of metabolism and related cell fate.
Keywords: p53, NMNAT-2, NAD+ biosynthesis, apoptosis
Introduction
Nicotinamide adenine dinucleotide (NAD+) and its derivatives, NADP+, NADH, and NADPH, have central roles in cellular metabolism and energy production as redox co-enzymes. Accumulating evidence reveals that NAD+ can also serve as a substrate for non-redox reactions, which are essential for diverse biological functions such as gene expression, Ca2+ mobilization, cell death, and aging.1 Sirtuins (SIRTs), homologs of yeast silent information regulator 2 (Sir2), are originally characterized as NAD+-dependent histone deacetylases (HDACs).2 SIRTs utilize NAD+ to remove the acetyl moiety from lysine on histones and other proteins, regulating mitochondrial metabolism and stress responses.3-6 NAD+ is also consumed by poly (ADP-ribose) polymerases (PARPs) and cyclic ADP (cADP)-ribose synthases. PARPs catalyze poly(ADP-ribosyl)ation, which plays diverse roles in cell survival, DNA repair, and cell death.7,8 Cyclic ADP-ribose synthases, a pair of ecto-enzymes known as the lymphocyte antigens CD38 and CD157, use NAD+ to produce the Ca2+-mobilizing second-messenger cADP-ribose.9,10
In view of the multifaceted roles NAD+ plays in mediating various aspects of cell physiology, it is not surprising that cancer cells generally display aberrant NAD+ metabolism. A first consideration in this regard is the Warburg effect, which relates to cancer cells’ dependence on cytoplasmic aerobic glycolysis rather than mitochondrial oxidation-phosphorylation for production of energy and biomass.11 The high rates of aerobic glycolysis perturbs NAD+ metabolism, altering the NADH/NAD+ redox ratio, thereby disrupting the cellular redox homeostasis and further promoting cancer progression.12,13
p53 plays important roles in cellular processes such as apoptosis, cell cycle progression, senescence, autophagy, and DNA repair.14-17 p53 also can serve as a key regulator of cellular metabolism, such as aerobic glycolysis.18,19 For example, p53 can induce hexokinase2 and glucose-6-phosphate dehydrogenase (G6PDH) genes to promote glycolysis.20-22 For proper functioning of p53, acetylation is imperative, which is regulated by NAD+-dependent SIRT1 deacetylase, the best known member of the SIRTs family. Acetylation of p53 augments its DNA binding ability, and loss of acetylation abrogates p53-dependent growth arrest and apoptosis.23 Functional properties of p53 can also be modified by poly(ADP-ribosyl)ation where PARP-1 physically participates in this process.24 Poly(ADP-ribosyl)ation modulates p53-mediated gene transcription (i.e., metastasis tumor antigens 1 [MTA1])25 and effectiveness of p53-mediated DNA damage response.26
Given p53’s role in the regulation of cellular metabolism, of which redox reactions are an essential part, we wanted to investigate whether p53 can directly regulate NAD+ biosynthesis. Furthermore, since SIRT1 requires NAD+ as a cofactor in regulating p53 activity, it would seem highly likely that a feedback loop exists whereby p53 can fine-tune this process by regulating NAD+ levels. Two major metabolic pathways are involved in NAD+ synthesis: the de novo pathway from tryptophan, and the salvage pathway that involves biosynthesis from other NAD+ precursors. NAD+ synthesis via the salvage pathway is conducted by 2 key enzymes: nicotinamide phosphoribosyltransferase (NAMPT) and nicotinamide mononucleotide adenylyltransferases (NMNATs) (Fig. 1A). Nicotinamide (NAM), the NAD+ precursor, is usually derived either from nutrients or released as by-product from enzymatic reactions.27 NAMPT catalyzed the first rate-limiting step to produce nicotinamide mononucleotide (NMN) from NAM, then NMN was further converted into NAD+ by NMNAT-1, NMNAT-2, and NMNAT-3.28 In this study, we determined whether NAD+ synthesizing enzymes can be modulated by p53 and demonstrate that p53 is a direct upstream regulator of NMNAT-2 gene expression.

Figure 1. p53 induces NMNAT-2 expression. (A) Schematic diagram of NAD+ biosynthesis via the salvage pathway. (B and C) Cellular mRNA levels of NAMPT, NMNAT-1, NMNAT-2, and NMNAT-3 in response to p53 induction were determined by semi- (B) or real-time qRT-PCR (C). Relative mRNA levels were normalized to those from controls cells (no doxycycline). p53 expression in H1299-p53-Tet On cells was induced by 1 μg/ml of doxycycline (C, bottom panel). Data represent mean values from 3 independent experiments, with error bars showing SEM.
Results
p53 induces the expression of NMNAT-2
To test the possible regulation of cellular NAD+ biosynthesis by p53, we first examined whether gene expression of NAD+ synthesizing enzymes in the NAD+ salvage pathways can be modulated by p53 (Fig. 1A). The salvage pathway was chosen for this study based on recent evidence linking this pathway to SIRT1 activity.29,30 Wild-type p53 was ectopically expressed in the absence of DNA damage, since DNA lesions can activate p53 through upstream elements such as ATM/ATR/DNA-PK that affect various cellular pathways, thereby complicating interpretation. The H1299 human non-small lung cancer cell line, which lacks expression of endogenous p53 due to homozygous partial deletion of the p53 gene, was used to construct a stable cell line expressing wild-type p53 under the control of doxycycline. mRNA levels of NAD+ synthetic enzymes, including NAMPT, NMNAT-1, NMNAT-2, and NMNAT-3, were compared in the presence or absence of p53 by semi-quantitative PCR. We found that only the NMNAT-2 mRNA level was significantly elevated by ectopic p53 expression (Fig. 1B). NMNAT-2 has 2 transcription variants (designated as tv1 and tv2) that differ only in their N-terminal sequences (28 aa in tv1 and 23 aa in tv2) (Fig. 2E). In initial experiments, primers amplifying a common region of NMNAT-2 mRNAs were used to detect both variants. The tight control of p53 expression by doxycycline was confirmed (Fig. 1B and C). Furthermore, we observed the induction of all 3 NMNAT genes by p53 over time by real-time qPCR (Fig. 1C). Although NMNAT-1 and NMNAT-3 were also induced, their expression levels were significantly lower than those of NMNAT-2. NMNAT-2 was therefore chosen for further study.

Figure 2. NMNAT-2 induction by DNA damage is p53-dependent. (A–C) NMNAT-2 expression was measured in U2OS and HCT116 cells treated with doxorubicin (Dox, 200 ng/ml) or etoposide (Etp, 4 μM). mRNA level of NMNAT-2 was measured using real-time qRT-PCR after 72 h (A and C). Cell-associated NMNAT-2 protein levels were compared by flow cytometry with intracellular staining after 48 h (B). (D) U2OS cells (control, p53-KD and NMNAT-2-KD) were treated with actinomycin D (Act. D, 20 nM) for 48 h. Protein levels of p53 and NMNAT-2 were compared. Actin was used as loading control. (E) Schematic illustration of the human NMNAT-2 gene and its encoded products. Transcription start sites and protein sequences of N-terminal of NMNAT-2 transcription variant 1 (tv1) and 2 (tv2) are shown. (F–I) U2OS cells (control or p53-KD) were treated with actinomycin D (20 nM), doxorubicin (200 ng/ml), or nutlin-3 (Nut, 10 μM) for the indicated times. Fold-increase of mRNA in U2OS p53-KD cells were compared with that of U2OS control cells. Data represent mean values from 3 independent experiments, with error bars showing SEM: *P < 0.05; **P < 0.01; ***P < 0.0001.
DNA damages induce NMNAT-2 in a p53-dependent manner
While ectopic expression of p53 induces the expression of NMNAT-2, we further examined whether activation of endogenous p53 by DNA damaging agents has the same effect on NMNAT-2. First, both doxorubicin (Dox) and etoposide (Etp) treatments caused increased expression of NMNAT-2 at mRNA and protein levels in U2OS cell (Fig. 2A and B). A similar observation was extended to another p53 wild-type cell line HCT116 (human colon cancer cell), where Etp induced NMNAT-2 transcription (Fig. 2C). This suggests that NMNAT-2 is a DNA damage-responsive gene. Second, expression of NMNAT-2 was examined in the context of p53 gene knockdown by retrovirus-mediated shRNA. In agreement with previous data shown in Figure 1, activation of p53 by actinomycin D treatment elevated the protein level of endogenous NMNAT-2 (Fig. 2D). Importantly, p53 knockdown abolished NMNAT-2 expression upon actinomycin D treatment (Fig. 2D). Specificity of the anti-NMNAT-2 antibody employed was also validated using NMNAT-2-knockdown cells, where actinomycin D treatment did not result in enhanced immunoreactivity (Fig. 2D). These results clearly demonstrate that NMNAT-2 expression is induced by DNA damage in a p53-dependent manner.
Due to the lack of transcript variant-specific antibodies, we sought to examine whether both NMNAT-2-tv1/tv2 can be induced by p53 activation with real-time qPCR using primer pairs specific to each NMNAT-2 transcript (tv1 and tv2). Expression of both variants was greatly enhanced by both DNA damage agents (actinomycin D and doxorubicin) and Nutlin-3a (a p53 stabilizer that disrupts interaction of p53 with its negative regulator MDM231) in a time-dependent manner in control but not in p53-KD U2OS cells (Fig. 2F-I). Therefore, p53 can induce both NMNAT-2 transcript variants. Interestingly, we observed that Nutlin-3a, which stabilizes p53 without causing DNA damage, is more potent than Dox for induction of NMNAT-2 tv1 (Fig. 2H), whereas induction of NMNAT-2 tv2 is more sensitive to Dox than to Nutlin-3a (Fig. 2I). This suggests that the expression of these 2 transcript variants is likely differentially regulated depending on the nature of events upstream of p53. More studies are needed to address this issue.
p53 directly binds to 2 putative responsive elements within human NMNAT-2 gene
Our results strongly suggest that NMNAT-2 is a downstream transcriptional target of p53. To confirm this possibility, we set out to identify p53 binding elements in the human NMNAT-2 gene. It has been well established that p53 consensus binding sites consist of 2 copies of the 10-base pair motif 5′-RRRCWWGYYY -3′ separated by 0–13 base pairs (R for purine, Y for pyrimidine, W is an A or T).32 Analysis of the human NMNAT-2 gene, located on chromosome1–1q25, revealed the existence of a ~114-kilobase intronic region that separates the transcription initiation sites of tv1 and tv2 (Fig. 2E). Candidates for p53-response elements (REs) were predicted with the p53MH algorithm, using the first 150 kilobases of the gene as input.33 Direct binding between p53 and predicted biding sites were assessed by chromatin immunoprecipitation (ChIP) analysis before and after DNA damage. Chromatin fragments (~500 base pairs) associated with p53 protein were amplified with specific primers to each candidate p53RE. Two well-known p53 target genes (p21/WAF1 and MDM2) were used as positive controls. Using this approach, 2 putative p53 REs (designated as BS#1 and BS#2) that associated with p53 protein were identified (Fig. 3A and data not shown). Interestingly, BS#1 and BS#2 are both located in the first intronic region of NMNAT-2 gene (Fig. 3B). This is not surprising, as functional p53 REs in intronic regions have also been found in various p53 target genes such as Tigar,17,34 Bax,35 and mdm2.36

Figure 3. NMNAT-2 is a direct target of p53. (A) p53 binds to 2 putative binding sites. U2OS cells treated with actinomycin D (20 nM) were subjected to chromatin immunoprecipitation (ChIP) analysis. Chromatin fragments co-immunoprecipitated with p53 protein were amplified using specific primers spanning individual p53REs. p53REs in the p21WAF1/CIP1 and mdm2 gene were used as positive controls. Mouse monoclonal anti-p53 antibody was used for p53 pull-down and mouse normal IgG for negative control. Results are representative of 2 independent experiments. Arrowheads indicate the amplified REs. (B) Location of p53 binding sites (BS) upstream of NMNAT-2 gene was depicted. Sequences of wild-type and mutant p53 BS#1 and BS#2 are shown. Consensus sequences of p53-response elements (REs) are designated in bold lettering with nucleotide substitutions indicated by asterisks. Wild-type nucleotides of p53BS#1 and #2 highlighted in red were substituted with indicated nucleotides in green as depicted in mutant-type. (C and D) p53REs identified in human NMNAT-2 gene are transcriptionally active. Firefly luciferase reporter plasmids carrying individual p53BS#1 and #2 (wild-type or mutant) were co-transfected with Renilla luciferase (for normalization) into U2OS cells (control or p53-KD). Cells were treated with actinomycin D (10 nM) or nutlin-3 (10 nM) for 48 h. Luciferase activities of each reporter were normalized to those from non-treated U2OS control cells. Data are presented as mean values with error bars showing SEM from 3 independent experiments. *P < 0.05; **P < 0.01; ***P < 0.0001.
To definitively demonstrate that p53 BS#1 and BS#2 are functional bona fide p53 REs, luciferase reporter assays were performed. p53 REs contain 2 consensus sites of RRRCWWGYYY, in which the fourth cytosine “C” and seventh guanine “G” are conserved nucleotides and essential for p53-binding. These 2 pyrimidine nucleotides (C/G) were substituted with purine (A/T) to generate mutant BS#1 and mutant BS#2 as negative controls (Fig. 3B). BS#1, BS#2, or their mutant counterparts were placed upstream of firefly luciferase reporter gene and co-transfected with Renilla luciferase reporter as internal control into either control or p53-KD U2OS cells. Both wild-type BS#1 and BS#2 were capable of driving the expression of firefly luciferase reporter gene in control, but not p53-KD U2OS cells, upon actinomycin D or Nutlin-3 treatments (Fig. 3C and D). Importantly, mutant BS#1 and BS#2 failed to express the reporter gene under the same conditions, strongly suggesting that BS#1 and BS#2 are bona fide p53-response elements. Taken together, our data clearly demonstrate that NMNAT-2 is a direct transcriptional target of p53.
p53 and NMNAT-2 regulates cellular NAD+ level upon DNA damage
We then examined whether the induction of NMNAT-2 by p53 is indeed relevant to cellular NAD+ metabolism. Cellular NAD+ levels were monitored upon DNA damage in control, p53-KD, and NMNAT-2-KD U2OS cells. We observed that in control cells, NAD+ level increased by 50% after DNA damage when compared with non-treated cells, indicating the elevated cellular demands for NAD+ under stressed conditions (Fig. 4). Strikingly, knocking down both NMNAT-2 isoforms caused a drastic decrease of NAD+ level in comparison to non-treated cells, suggesting the major roles of NMNAT-2 proteins in replenishing the cellular NAD+ pool upon DNA damage. In p53-KD cells, increase of NAD+ level is comparable to that of control cells at earlier stage (36 h). However, these cells failed to maintain their NAD+ pool under prolonged exposure to DNA damage compared with control cells. Our data demonstrates that p53 facilitates cellular NAD+ biosynthesis through induction of NMNAT-2 to maintain a higher cellular NAD+ level under prolonged DNA damage condition.
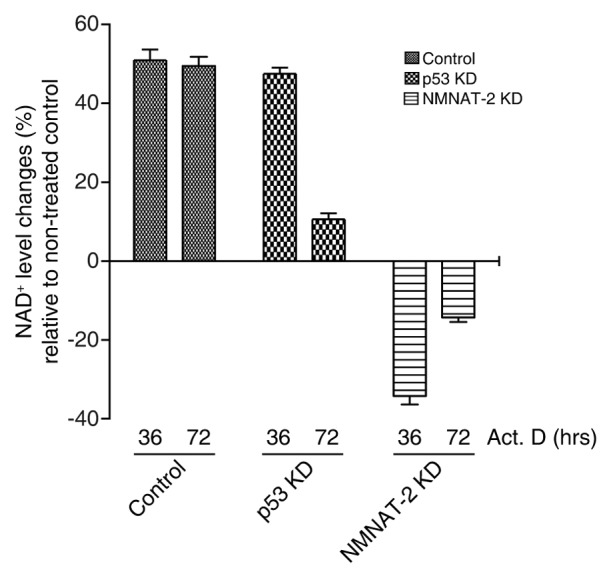
Figure 4. NMNAT-2 regulates cellular NAD+ level. U2OS cells (control, p53-KD, and NMNAT-2-KD) were treated with actinomycin D (20 nM) for the indicated times, and cellular NAD+ level was then measured. Data presented are mean values from 3 independent experiments with error bars showing SEM.
NMNAT-2 is involved in p53-mediated cell death
p53 is a major pro-apoptotic regulator of DNA damage-induced cell death. To determine whether NMNAT-2 is involved in this process, DNA damage-induced cell death was compared in control, p53-KD, and NMNAT-2-KD cells after treatment with DNA damaging reagents (actinomycin D or camptothecin). While actinomycin D caused significant cell death (33%) in control cells, p53-KD cells were relatively tolerant, showing 12% of cell death (Fig. 5A). Importantly, the level of DNA damage-induced cell death in NMNAT-2-KD cells was comparable to that in p53-KD cells. Knocking down NMNAT-2 also reduced camptothecin-induced cell death (Fig. 5B), showing that the protective effect of NMNAT-2 is not limited to actinomycin D treatment. Therefore, our results demonstrate that NMNAT-2 plays an important role in p53-mediated cell death upon DNA damage. Further studies should reveal whether this is due to a direct effect of NMNAT-2 or an indirect effect from changes in NAD+ levels via p53-mediated NMNAT-2 induction.
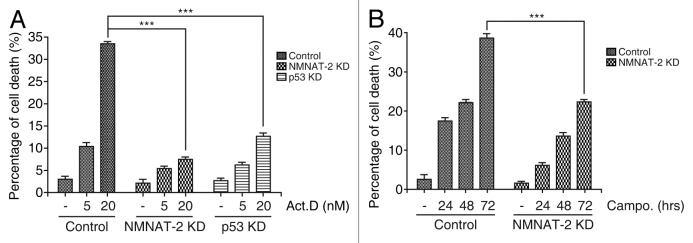
Figure 5. NMNAT-2 is required for p53-dependent cell death. Knocking down NMNAT-2 protects U2OS cells from DNA damage-induced cell death. (A) U2OS cells (control, NMNAT-2-KD and p53-KD) were treated with actinomycin D (5 or 20 nM) for 48 h. The percentage of dead cells was determined by sub-G1 DNA content analysis using flow cytometry. (B) Cells were treated with camptothecin (0.5 μM) for 24 h. Data presented as mean values from 3 independent experiments with error bars showing SEM. Cell death in NMNAT-2-KD or p53-KD cells was compared with that of U2OS control cells. *P < 0.05; **P < 0.01; ***P < 0.0001.
Discussion
Dysregulation of cellular energy metabolism are relevant to various types of disorders, including aging-related diseases, diabetes, and cancers.37-39 As a key metabolic regulator, NAD+ is the essential hub coenzyme involved in not only cellular redox reactions, but also the activities of crucial regulators including Sirtuins and PARPs, whose substrates play important roles in almost all aspects of cell life.40,41 Under stress conditions (e.g., oxidative stress, DNA damage, etc.), cells require higher levels of cellular NAD+ to drive a myriad of cellular processes, hence the need to promote NAD+ biosynthesis to meet the demand. Given that the tumor suppressor p53 can also be activated by stress signals, thereby invoking a vast array of responses leading to cell cycle arrest (quiescence), cell death, DNA repair, and reprogramming of cellular metabolism,16,17 it would seem intuitive that NAD+ and p53 are intimately linked. A most pertinent and important observation in this regard was the demonstration, in 2001, that SIRT1, whose activity is NAD+-dependent, is an important p53 deacetylase,6 placing NAD+ as an upstream modulator of p53 stability and activation. It would therefore seem logical to speculate that a negative feedback loop exists whereby p53 can fine-tune its own activity by back regulating NAD+ levels. Such a mechanism would be akin to the p53–Mdm2 negative feedback system, whereby p53 activates Mdm2 transcription, and Mdm2, in turn, loops back and targets p53 for degradation.42 Our present demonstration that p53 is an upstream regulator of a major NAD+ synthesizing enzyme, NMNAT-2, suggests a feedback loop likely exists between p53 and NAD+, as represented by p53 → NMNAT-2 → NAD+ → SIRT1 → p53.
It is noteworthy that in terms of biological outcome, research on the SIRT1-p53 axis has focused primarily on the aging aspect.43-45 Our present finding suggests that NMNAT-2 is likely a major regulator in this process. In this regard, it is noteworthy that recently, Gomes AP et al. demonstrate that during aging, declining nuclear NAD+ levels induce a pseudohypoxia state, disrupting nuclear–mitochondrial communication and resulting in decline in mitochondrial function.46 Specifically, decreasing nuclear NAD+ levels in cells of aging mice significantly compromises the SIRT1-HIF1α-cMyc-TFAM cascade, leading to loss of TFAM and mitochondrial encoded oxidative phosphorylation (OXPHOS) components. As expected, gene knockdown experiments show that nuclear NAD+ levels are regulated by the nuclear-localized NMNAT-1, rather than the Golgi-localized NMNAT-2 and the mitochondria-localized NMNAT-3. Interestingly, our data show that p53 also slightly induces the expression of NMNAT-1 and NMNAT-3, albeit to a much lesser extent than that of NMNAT-2 (Fig. 1C). Therefore, although the present study focuses on the p53 regulation of NMNAT-2, we do not exclude the existence of possible links between p53 and other NMNATs. Indeed, it is not inconceivable that all 3 NMNAT isoforms are somehow involved in the NAD+ negative feedback loop system mentioned above. Thus, follow-up investigation of p53-NMNAT1/3 linkages will need to be performed, particularly when one considers the possible connection between p53-mediated aging regulation and mitochondrial oxidative phosphorylation (OXPHOS) and biogenesis.
Our present study also provides a new perspective on the established link between p53 and the mammalian target of rapamycin (mTOR) that is also associated with cellular metabolism and senescence.47,48 While p53 has long been considered to be an inducer of senescence, recent evidence suggests that under conditions that strongly enhance p53 stability (e.g., using Nutlin-3a), p53 inhibits mTOR activities and promotes quiescence while suppressing senescence.49-51 Considering the fact that mTOR is sensitive to cellular energy status changes, it is tempting to speculate that p53 modulates mTOR activities, at least in part, through NMNAT-2/NAD+-mediated events, including modification of p53 itself (for example, by SIRT1/2). It is also noteworthy that studies by Leontieva et al. show that p53 induction by Nutlin-3a and the DNA damaging agent doxorubicin (Dox) (at low concentrations) produces different outcomes (quiescence and senescence, respectively).50 Interestingly, our present data shows that expression of the NMNAT-2 tv1 transcript variant is more sensitive to Nutlin-3a treatment than to Dox, whereas Dox is more potent for the induction of tv2, raising the possibility that differential inductions of the 2 NMNAT-2 isoforms under stress conditions dictate the outcome of cellular fate. A detailed study of the mechanisms regulating the expression and/or stability of the 2 variants coupled with their functional analysis should be a top priority for further pursuance.
It is also of great interest to note that NMNAT-2 is found mainly in the brain, where SIRT1 is known to modulate a variety of neuronal functions.52-54 Both NMNAT-2 and SIRT1 have been shown to maintain healthy axons and suppress various neurological disorders, including tauopathy, which is associated with Alzheimer disease.52,55-58 The present study adds an important piece (i.e., p53) to this complex puzzle, as it provides clues as to how NMNAT-2 and SIRT1 are functionally linked within the brain. Therefore p53-controlled expression of NNMAT-2 under stress conditions may play an important role in tauopathy and other types of neuronal diseases.
Overall, our findings shed light on p53 as an important player in the generation of NAD+, a hub molecule involved in numerous metabolic processes, which is, again, congruent with the relatively new role of p53 as a major metabolism regulator.
Materials and Methods
For cell cultures, reagents, plasmid construction, flow cytometry analysis, and qRT-PCR, see Supplementary Materials.
NAD+/NADH quantification assay
To measure cellular NAD+, cells were lysed and processed with the NAD+/NADH quantification kit (Biovision) according to the manufacturer’s instructions.
Chromatin immunoprecipitation
U2OS cells were treated with actinomycin D (10 nM) or Nutlin-3 (10 μM) for 48 h then subjected to chromatin immunoprecipitation (ChIP) analysis. ChIP assays were performed with slight modification, as previously described.59,60 Briefly, cells were fixed with 1% formaldehyde, and then whole-cell lysates were prepared. Protein lysate was used for ChIP with the indicated antibodies, followed by DNA purification using Quickspin PCR purification kit. Recovered DNA fragments were analyzed by PCR with the indicated primer sets listed in Table S1.
Dual luciferase reporter assay
To examine the functionality of p53-responsive elements (REs) identified in human NMNAT-2 gene, both wild-type and mutant p53-binding sites (BS#1 and BS#2) were cloned upstream of the firefly luciferase gene. The constructed firefly luciferase reporter plasmids containing NMNAT-2 p53BS were transfected into cells together with Renilla luciferase reporter plasmid (TK-Renilla luciferase, Promega) as an internal control. Twenty-four hours post-transfection, cells were treated with actinomycin D (10 nM) or Nutlin-3 (10 μM). Forty-eight hours later, luciferase activities were measured using dual luciferase reporter assay kit (Promega) and GloMax luminometer (Promega). Firefly luciferase activity was normalized to Renilla luciferase activity. These values were further normalized to protein concentration of each sample and presented as relative amount change to non-treated cells.
Cell death analysis
Cells treated with DNA damaging reagents were collected by trypsinization and washed twice with cold PBS without Ca2+ or Mg2+. Cells were resuspended in 1 ml cold PBS, and 3 ml 95% cold ethanol was added slowly (drop-wise) to the cell suspension with gentle vortexing. Cells were fixed at 4 °C for 2 h, centrifuged at 500 × g for 10 min, and the cell pellet was washed twice with 10 ml of cold PBS then resuspended in 500 μl PI/Triton X-100 staining solution [0.1% (v/v) Triton X-100 in PBS with 20 μg/ml RNase A (DNase free) and 50 μg/ml propidium iodide]. Single-cell suspension was made by gentle pipetting then incubated at room temperature for 30 min or stored at 4 °C overnight (protected from light). Samples were analyzed by flow cytometry within 24 h to avoid cell breakdown. Flow cytometer was set to FL2-A and FL2-W as linear. Data was analyzed with FCS express software.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by an operating grant from the Canadian Institute of Health Research (CIHR) to P.W.K.L., Cancer Research Training Program postdoctoral fellowships through the Beatrice Hunter Cancer Research Institute (D.A.), a Government of Canada Post-Doctoral Research Fellowship (D.A.), Canadian Institutes of Health Research (S.G.), and Nova Scotia Health Research Foundation (D.C.).
Glossary
Abbreviations:
- NAD
nicotinamide adenine dinucleotide
- NMN
nicotinamide mononucleotide
- NAM
nicotinamide
- NMNAT
nicotinamide mononucleotide adenylyltransferase
- SIRT
sirtuins
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/28128
References
- 1.Di Stefano G, Manerba M, Vettraino M. NAD metabolism and functions: a common therapeutic target for neoplastic, metabolic and neurodegenerative diseases. Curr Top Med Chem. 2013;13:2918–29. doi: 10.2174/15680266113136660207. [DOI] [PubMed] [Google Scholar]
- 2.Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 3.Ahn BH, Kim HS, Song S, Lee IH, Liu J, Vassilopoulos A, Deng CX, Finkel T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci U S A. 2008;105:14447–52. doi: 10.1073/pnas.0803790105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, Grueter CA, Harris C, Biddinger S, Ilkayeva OR, et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. 2010;464:121–5. doi: 10.1038/nature08778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van der Horst A, Tertoolen LG, de Vries-Smits LM, Frye RA, Medema RH, Burgering BM, van der HA FOXO4 is acetylated upon peroxide stress and deacetylated by the longevity protein hSir2(SIRT1) J Biol Chem. 2004;279:28873–9. doi: 10.1074/jbc.M401138200. [DOI] [PubMed] [Google Scholar]
- 6.Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–59. doi: 10.1016/S0092-8674(01)00527-X. [DOI] [PubMed] [Google Scholar]
- 7.Wang ZQ, Stingl L, Morrison C, Jantsch M, Los M, Schulze-Osthoff K, Wagner EF. PARP is important for genomic stability but dispensable in apoptosis. Genes Dev. 1997;11:2347–58. doi: 10.1101/gad.11.18.2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Murcia JM, Niedergang C, Trucco C, Ricoul M, Dutrillaux B, Mark M, Oliver FJ, Masson M, Dierich A, LeMeur M, et al. Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc Natl Acad Sci U S A. 1997;94:7303–7. doi: 10.1073/pnas.94.14.7303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim H, Jacobson EL, Jacobson MK. Synthesis and degradation of cyclic ADP-ribose by NAD glycohydrolases. Science. 1993;261:1330–3. doi: 10.1126/science.8395705. [DOI] [PubMed] [Google Scholar]
- 10.Howard M, Grimaldi JC, Bazan JF, Lund FE, Santos-Argumedo L, Parkhouse RM, Walseth TF, Lee HC. Formation and hydrolysis of cyclic ADP-ribose catalyzed by lymphocyte antigen CD38. Science. 1993;262:1056–9. doi: 10.1126/science.8235624. [DOI] [PubMed] [Google Scholar]
- 11.Warburg O. On respiratory impairment in cancer cells. Science. 1956;124:269–70. [PubMed] [Google Scholar]
- 12.Heikal AA. Intracellular coenzymes as natural biomarkers for metabolic activities and mitochondrial anomalies. Biomark Med. 2010;4:241–63. doi: 10.2217/bmm.10.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7:11–20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 14.Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol. 2008;9:402–12. doi: 10.1038/nrm2395. [DOI] [PubMed] [Google Scholar]
- 15.Brady CA, Attardi LD. p53 at a glance. J Cell Sci. 2010;123:2527–32. doi: 10.1242/jcs.064501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009;137:413–31. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 17.Menendez D, Inga A, Resnick MA. The expanding universe of p53 targets. Nat Rev Cancer. 2009;9:724–37. doi: 10.1038/nrc2730. [DOI] [PubMed] [Google Scholar]
- 18.Berkers CR, Maddocks OD, Cheung EC, Mor I, Vousden KH. Metabolic regulation by p53 family members. Cell Metab. 2013;18:617–33. doi: 10.1016/j.cmet.2013.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lago CU, Sung HJ, Ma W, Wang PY, Hwang PM. p53, aerobic metabolism, and cancer. Antioxid Redox Signal. 2011;15:1739–48. doi: 10.1089/ars.2010.3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mathupala SP, Heese C, Pedersen PL. Glucose catabolism in cancer cells. The type II hexokinase promoter contains functionally active response elements for the tumor suppressor p53. J Biol Chem. 1997;272:22776–80. doi: 10.1074/jbc.272.36.22776. [DOI] [PubMed] [Google Scholar]
- 21.Cheung EC, Vousden KH. The role of p53 in glucose metabolism. Curr Opin Cell Biol. 2010;22:186–91. doi: 10.1016/j.ceb.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 22.Vousden KH, Ryan KM. p53 and metabolism. Nat Rev Cancer. 2009;9:691–700. doi: 10.1038/nrc2715. [DOI] [PubMed] [Google Scholar]
- 23.Luo J, Li M, Tang Y, Laszkowska M, Roeder RG, Gu W. Acetylation of p53 augments its site-specific DNA binding both in vitro and in vivo. Proc Natl Acad Sci U S A. 2004;101:2259–64. doi: 10.1073/pnas.0308762101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Simbulan-Rosenthal CM, Rosenthal DS, Luo RB, Samara R, Jung M, Dritschilo A, Spoonde A, Smulson ME. Poly(ADP-ribosyl)ation of p53 in vitro and in vivo modulates binding to its DNA consensus sequence. Neoplasia. 2001;3:179–88. doi: 10.1038/sj.neo.7900155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee MH, Na H, Kim EJ, Lee HW, Lee MO. Poly(ADP-ribosyl)ation of p53 induces gene-specific transcriptional repression of MTA1. Oncogene. 2012;31:5099–107. doi: 10.1038/onc.2012.2. [DOI] [PubMed] [Google Scholar]
- 26.Valenzuela MT, Guerrero R, Núñez MI, Ruiz De Almodóvar JM, Sarker M, de Murcia G, Oliver FJ. PARP-1 modifies the effectiveness of p53-mediated DNA damage response. Oncogene. 2002;21:1108–16. doi: 10.1038/sj.onc.1205169. [DOI] [PubMed] [Google Scholar]
- 27.Sauve AA. NAD+ and vitamin B3: from metabolism to therapies. J Pharmacol Exp Ther. 2008;324:883–93. doi: 10.1124/jpet.107.120758. [DOI] [PubMed] [Google Scholar]
- 28.Berger F, Lau C, Dahlmann M, Ziegler M. Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J Biol Chem. 2005;280:36334–41. doi: 10.1074/jbc.M508660200. [DOI] [PubMed] [Google Scholar]
- 29.Nakahata Y, Sahar S, Astarita G, Kaluzova M, Sassone-Corsi P. Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science. 2009;324:654–7. doi: 10.1126/science.1170803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang T, Berrocal JG, Frizzell KM, Gamble MJ, DuMond ME, Krishnakumar R, Yang T, Sauve AA, Kraus WL. Enzymes in the NAD+ salvage pathway regulate SIRT1 activity at target gene promoters. J Biol Chem. 2009;284:20408–17. doi: 10.1074/jbc.M109.016469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–8. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 32.el-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B. Definition of a consensus binding site for p53. Nat Genet. 1992;1:45–9. doi: 10.1038/ng0492-45. [DOI] [PubMed] [Google Scholar]
- 33.Hoh J, Jin S, Parrado T, Edington J, Levine AJ, Ott J. The p53MH algorithm and its application in detecting p53-responsive genes. Proc Natl Acad Sci U S A. 2002;99:8467–72. doi: 10.1073/pnas.132268899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, Gottlieb E, Vousden KH. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126:107–20. doi: 10.1016/j.cell.2006.05.036. [DOI] [PubMed] [Google Scholar]
- 35.Thornborrow EC, Patel S, Mastropietro AE, Schwartzfarb EM, Manfredi JJ. A conserved intronic response element mediates direct p53-dependent transcriptional activation of both the human and murine bax genes. Oncogene. 2002;21:990–9. doi: 10.1038/sj.onc.1205069. [DOI] [PubMed] [Google Scholar]
- 36.Zauberman A, Flusberg D, Haupt Y, Barak Y, Oren M. A functional p53-responsive intronic promoter is contained within the human mdm2 gene. Nucleic Acids Res. 1995;23:2584–92. doi: 10.1093/nar/23.14.2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dang CV. Links between metabolism and cancer. Genes Dev. 2012;26:877–90. doi: 10.1101/gad.189365.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11:85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- 39.Curtis R, Geesaman BJ, DiStefano PS. Ageing and metabolism: drug discovery opportunities. Nat Rev Drug Discov. 2005;4:569–80. doi: 10.1038/nrd1777. [DOI] [PubMed] [Google Scholar]
- 40.Schreiber V, Dantzer F, Ame JC, de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol. 2006;7:517–28. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- 41.Verdin E. The Many Faces of Sirtuins: Coupling of NAD metabolism, sirtuins and lifespan. Nat Med. 2014;20:25–7. doi: 10.1038/nm.3447. [DOI] [PubMed] [Google Scholar]
- 42.Wade M, Li YC, Wahl GM. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer. 2013;13:83–96. doi: 10.1038/nrc3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Herskovits AZ, Guarente L. Sirtuin deacetylases in neurodegenerative diseases of aging. Cell Res. 2013;23:746–58. doi: 10.1038/cr.2013.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rufini A, Tucci P, Celardo I, Melino G. Senescence and aging: the critical roles of p53. Oncogene. 2013;32:5129–43. doi: 10.1038/onc.2012.640. [DOI] [PubMed] [Google Scholar]
- 45.Longo VD, Kennedy BK. Sirtuins in aging and age-related disease. Cell. 2006;126:257–68. doi: 10.1016/j.cell.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 46.Gomes AP, Price NL, Ling AJ, Moslehi JJ, Montgomery MK, Rajman L, White JP, Teodoro JS, Wrann CD, Hubbard BP, et al. de CR, Rolo AP, Turner N, Bell EL, and Sinclair DA. Declining NAD(+) Induces a Pseudohypoxic State Disrupting Nuclear-Mitochondrial Communication during. Aging Cell. 2013;155:1624–38. doi: 10.1016/j.cell.2013.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Akeno N, Miller AL, Ma X, Wikenheiser-Brokamp KA. p53 suppresses carcinoma progression by inhibiting mTOR pathway activation. Oncogene. 2014 doi: 10.1038/onc.2013.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008;134:451–60. doi: 10.1016/j.cell.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Serrano M. Shifting senescence into quiescence by turning up p53. Cell Cycle. 2010;9:4256–7. doi: 10.4161/cc.9.21.13785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leontieva OV, Gudkov AV, Blagosklonny MV. Weak p53 permits senescence during cell cycle arrest. Cell Cycle. 2010;9:4323–7. doi: 10.4161/cc.9.21.13584. [DOI] [PubMed] [Google Scholar]
- 51.Demidenko ZN, Korotchkina LG, Gudkov AV, Blagosklonny MV. Paradoxical suppression of cellular senescence by p53. Proc Natl Acad Sci U S A. 2010;107:9660–4. doi: 10.1073/pnas.1002298107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gilley J, Coleman MP. Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS Biol. 2010;8:e1000300. doi: 10.1371/journal.pbio.1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Min SW, Cho SH, Zhou Y, Schroeder S, Haroutunian V, Seeley WW, Huang EJ, Shen Y, Masliah E, Mukherjee C, et al. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron. 2010;67:953–66. doi: 10.1016/j.neuron.2010.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yan T, Feng Y, Zheng J, Ge X, Zhang Y, Wu D, Zhao J, Zhai Q. Nmnat2 delays axon degeneration in superior cervical ganglia dependent on its NAD synthesis activity. Neurochem Int. 2010;56:101–6. doi: 10.1016/j.neuint.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 55.Gan L, Mucke L. Paths of convergence: sirtuins in aging and neurodegeneration. Neuron. 2008;58:10–4. doi: 10.1016/j.neuron.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ali YO, Ruan K, Zhai RG. NMNAT suppresses tau-induced neurodegeneration by promoting clearance of hyperphosphorylated tau oligomers in a Drosophila model of tauopathy. Hum Mol Genet. 2012;21:237–50. doi: 10.1093/hmg/ddr449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hicks AN, Lorenzetti D, Gilley J, Lu B, Andersson KE, Miligan C, Overbeek PA, Oppenheim R, Bishop CE. Nicotinamide mononucleotide adenylyltransferase 2 (Nmnat2) regulates axon integrity in the mouse embryo. PLoS One. 2012;7:e47869. doi: 10.1371/journal.pone.0047869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ljungberg MC, Ali YO, Zhu J, Wu CS, Oka K, Zhai RG, Lu HC. CREB-activity and nmnat2 transcription are down-regulated prior to neurodegeneration, while NMNAT-2 over-expression is neuroprotective, in a mouse model of human tauopathy. Hum Mol Genet. 2012;21:251–67. doi: 10.1093/hmg/ddr492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mattia M, Gottifredi V, McKinney K, Prives C. p53-Dependent p21 mRNA elongation is impaired when DNA replication is stalled. Mol Cell Biol. 2007;27:1309–20. doi: 10.1128/MCB.01520-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kaeser MD, Iggo RD. Chromatin immunoprecipitation analysis fails to support the latency model for regulation of p53 DNA binding activity in vivo. Proc Natl Acad Sci U S A. 2002;99:95–100. doi: 10.1073/pnas.012283399. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.