

The voltage-gated calcium channel (VGCC), CACNA1A, is associated with several neurological disorders, including familial hemiplegic migraine, episodic ataxia type 2, epilepsy, and spinocerebellar ataxia type 6 (SCA6).1 CACNA1A encodes the Cav2.1 (α1A) subunit, the main pore-forming subunit of the P/Q-type VGCC, responsible for the Ca2+ currents that mediate transmitter release in many CNS synapses. CACNA1A contains a CAG repeat in exon 47 that encodes the polyglutamine (polyQ) tract, which is expanded in SCA6. Until recently the prevailing hypothesis was that SCA6 was due to the presence of the expanded polyQ tract in the C terminus of a long splice variant of α1A, which perturbed channel function.2 However, studies attempting to implicate altered channel function in SCA6 have yielded conflicting findings.
Recently, we reported that a 75 kDa C-terminal “fragment” of α1A (α1ACT) containing the polyQ tract forms a stable polypeptide, enriched in nuclear fractions in cerebellum, that acts as a transcription factor for neuronally expressed genes. α1ACT enhances the expression of at least 3 genes, GRN, PMCA2, and BTG1, in pheochromocytoma cells (PC12) and cerebellar tissue, potentiates NGF-mediated neurite outgrowth in PC12 cells, and partially rescues the ataxic phenotype of CACNA1A knockout mice. However, α1ACT with expanded polyQ (α1ACTSCA6) lacks normal α1ACT function in gene expression and is pathogenic in vitro and in vivo. Nevertheless, it is still not clear whether the cell death induced by α1ACTSCA6 results from loss of normal α1ACT function or gain of abnormal function, such as aberrant gene activation or DNA damage.3
Because of the therapeutic implications, it has been important to establish the origin of α1ACT, in the hope that this might lead to rational and safe treatments. In eukaryotic cells, the majority of mRNA translation is cap-dependent, relying on the complex of proteins that assemble at the 7-methyl-guanosine cap at the 5′ end of an mRNA. Another form of translation, termed cap-independent, occurs when the 40S ribosome directly binds mRNA near the start codon at an internal ribosome entry site (IRES).4 IRESs have been suggested to be present in the 5′ untranslated region (UTR) of approximately 10–15% of cellular mRNAs, presumably to ensure translation of critical proteins during times of cell stress, cell cycle, or certain developmental stages when cap-dependent protein synthesis may be reduced.5 Using RNA folding simulation and expression studies, we have shown that a ~1-kb nucleotide sequence within the α1A mRNA forms a predicted complex hairpin structure and contains an IRES that directs α1ACT translation. We are currently screening a library of oligonucleotides and small molecules to identify drugs to suppress α1ACT expression without affecting α1A.
Most IRESs are thought to function through RNA binding proteins termed IRES trans-activating factors (ITAFs), some of which are tissue-specific.5 These ITAFs interact with RNA sequences or structures in the IRES to nucleate the formation of a translation initiation complex distal to the 5′ cap structure. Based on ITAF expression, the activity of some cellular IRESs varies between different cell types and tissues. To test for the differences in cellular factors that might influence IRES function, we tested an IRES-containing dual luciferase reporter in different cell lines. We found that the increase in IRES activity reflected by F-Luc/R-Luc ratios ranged from 14 to 45 in cultured cell lines, including Cos-7, N2a, NIH 3T3, HEK 293, SH-SY5Y, PC12 as well as primary rat granule neurons, with the highest activity of F-Luc seen in neuronal cell lines PC12 and SY5Y. These latter levels in neuronal cells were 1.8- to 2-fold greater, respectively, than in HEK 293 cells derived from human kidney cells. This observation suggests that certain neuron-specific cellular factors may influence the activity of the CACNA1A IRES. Taken together, these data further indicate that the CACNA1A gene contains a functional cellular IRES that mediates the expression of the second gene product, α1ACT, of the CACNA1A gene (Fig. 1).
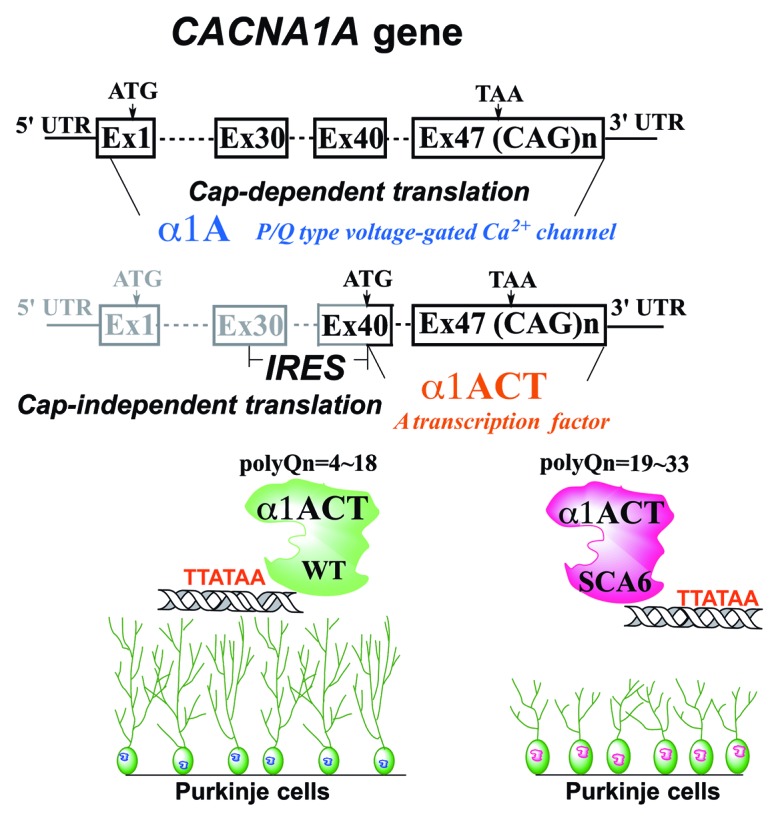
Figure 1. Schematic illustration of expression regulation and function of α1A and α1ACT. The CACNA1A gene, encoding the VGCC subunit α1A, is bicistronic. The second gene product, α1ACT, is generated from the α1A transcript by a cellular IRES located within the coding region of the α1A mRNA. α1ACT is a transcription factor that regulates expression of several neuronally-expressed genes, promoting neurite outgrowth. The α1ACTSCA6 reduces viability of cells and causes cerebellar cortical atrophy in an animal model.
There are currently only a few other genes known, namely PITSLRE/CDK11, USP18-sf, Cx43, Cx55.5, and Notch2, in which IRES elements found in the coding region, rather than in the 5′-UTR of the mRNA, initiate translation of smaller proteins at alternate start codons.6,7 We speculate that the presence of IRES elements in the coding region of cellular genes leading to the separate expression of C-terminal domains has important biological implications. Several proteins encoded by these “second cistrons” are novel isoforms or N-terminally truncated proteins, some of which enter the nucleus and possibly function as transcription factors. These observations suggest the presence of a previously unrecognized general strategy for coordinated genetic control. A systematic search for bicistronic genes might reveal the frequent usage of IRESs in coding regions that allows for the production of secondary proteins that perform distinct functions from the primary proteins.
The discovery of the role of RNA processing in neuronal function and survival is yielding a wealth of new therapeutic targets and new RNA-based tools for developing therapeutics.8 Additional studies supporting a Ca2+ channel-independent disease mechanism in SCA6 may pave the way for a search for therapeutic agents to selectively suppress IRES function and provide a better understanding of the mechanisms underlying regulation of gene expression and its perturbation in neurodegenerative disorders.
Du X, et al. Cell. 2013;154:118–33. doi: 10.1016/j.cell.2013.05.059.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/28199
References
- 1.Rajakulendran S, et al. Rev Neurol. 2012;8:86–96. doi: 10.1038/nrneurol.2011.228. [DOI] [PubMed] [Google Scholar]
- 2.Zhuchenko O, et al. Nat Genet. 1997;15:62–9. doi: 10.1038/ng0197-62. [DOI] [PubMed] [Google Scholar]
- 3.Du X, et al. Cell. 2013;154:118–33. doi: 10.1016/j.cell.2013.05.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Palmenberg AC, et al. Semin Virol. 1997;8:231–41. doi: 10.1006/smvy.1997.0126. [DOI] [Google Scholar]
- 5.Spriggs KA, et al. Biol Cell. 2008;100:27–38. doi: 10.1042/BC20070098. [DOI] [PubMed] [Google Scholar]
- 6.Cornelis S, et al. Mol Cell. 2000;5:597–605. doi: 10.1016/S1097-2765(00)80239-7. [DOI] [PubMed] [Google Scholar]
- 7.Ul-Hussain M, et al. Brain Res. 2012;1487:99–106. doi: 10.1016/j.brainres.2012.05.065. [DOI] [PubMed] [Google Scholar]
- 8.Blagden SP, et al. Clin Oncol. 2011;8:280–91. doi: 10.1038/nrclinonc.2011.16. [DOI] [PubMed] [Google Scholar]