

ATM (ataxia-telangiectasia mutated) kinase is a key factor in DNA damage responses, and mutations in ATM cause AT (ataxia-telangiectasia), a form of genomic instability syndrome.1 Various forms of genotoxic agents that generate DSB (double-stranded DNA breaks) activate ATM and induce DNA damage checkpoint pathways involving cell cycle arrest and repair of legions. ATM plays a central role in maintenance of genomic stability, and recent studies indicate that other environmental signals, including hypoxic conditions and oxidative stress, as well as alteration of chromatin structures, can activate ATM,2-4 which then transduces various cellular signaling pathways that help cells maintain their genetic integrity.5
Among various errors that could occur during the course of DNA replication, rereplication may be one of the most deleterious events that need to be avoided to minimize the genomic instability. Thus, cells are equipped with multiple systems that prevent rereplication.6 This regulation centers on the assembly of pre-RC (pre-replicative complex) that is essential for initiating DNA replication during S phase. Pre-RCs are generated on the chromosomes at the late M to early G1 and probably all through G1 phase through recruitment of Mcm (minichromosome maintenance) onto Orc (origin recognition complex), the core complex of which is constitutively bound to chromatin. This process is facilitated by Cdt1, which binds to Mcm and delivers it onto Orc.7 Once pre-RC is activated during S phase, and new DNA strands are synthesized, the pre-RC on the replicated DNA templates are inactivated. The expression of Cdt1 is strictly cell cycle-regulated, and the Cdt1 protein is present only during G1 when pre-RC formation is permitted. Aberrant expression of Cdt1 during S phase can generate pre-RC on the replicated chromosomes, which would be activated to cause rereplication. Rapid degradation of Cdt1 in S phase is ensured by 2 different ubiquitin ligases, namely SCFSkp2 dependent on Cdk-mediated phosphorylation and Cul4-DDB1Cdt2 ubiquitin ligase dependent on PCNA (proliferating cell nuclear antigen).
In the February 1, 2013 issue of Cell Cycle, the group led by Masatoshi Fujita reported a novel link between ATM and prevention of rereplication through regulation of Cdt1 stability.8 In ATM-depleted cells and cells from AT patients, expression of Cdt1 is deregulated during S phase, which renders these cells more prone to rereplicate their genomes. The authors have concluded that the ATM-Akt-SCFSkp2 pathway may play a role to ensure the complete degradation of Cdt1 during S phase (Fig. 1) on the basis of the following observations: (1) ATM inhibition at least partially downregulates the Skp2 protein level; (2) Skp2 depletion stabilizes Cdt1 during S phase; (3) ATM inhibition reduces Akt phosphorylation; (4) Akt depletion decreases the Skp2 protein levels; and (5) Akt inhibition partially stabilizes Cdt1 during S phase.
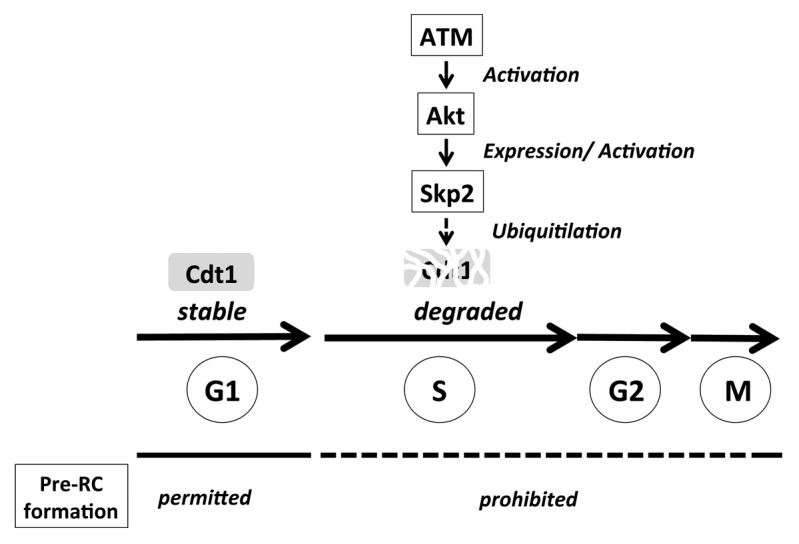
Figure 1. A novel role of ATM in prevention of rereplication during unperturbed cell cycle. ATM ensures full activation of Akt, which, in turn, facilitates the expression (or activation) of Skp2 that ubiquitinates Cdt1 for degradation. In the absence of ATM, Akt is not fully activated, resulting in reduction of Skp2 activity and stabilization of Cdt1 during S phase. This predisposes cells to rereplication, one of the major sources for genomic instability.
The report reveals yet another role of ATM in preventing genomic instability during unperturbed cell cycle progression. It is interesting that ATM depletion affects the level of Cdt1 (and p27 in some cells) but not that of p21 or CyclinD1, which is known to be regulated by Skp2. This may reflect the physiological importance of strict regulation of Cdt1 protein level to ensure “once and only once” replication, which is probably one of the most important rules of DNA replication for the maintenance of genome integrity. Among the remaining questions, how ATM regulates Akt activation would be one of the most critical issues to be clarified to understand the still elusive signaling cascade from ATM to cell growth and anti-apoptosis.
Iwahori S, et al. Cell Cycle. 2014;13:471–81. doi: 10.4161/cc.27274.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/28216
References
- 1.Kastan MB, et al. Nature. 2004;432:316–23. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- 2.Cam H, et al. Mol Cell. 2010;40:509–20. doi: 10.1016/j.molcel.2010.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guo Z, et al. Science. 2010;330:517–21. doi: 10.1126/science.1192912. [DOI] [PubMed] [Google Scholar]
- 4.Bakkenist CJ, et al. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 5.Hinz M, et al. Mol Cell. 2010;40:63–74. doi: 10.1016/j.molcel.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 6.Arias EE, et al. Genes Dev. 2007;21:497–518. doi: 10.1101/gad.1508907. [DOI] [PubMed] [Google Scholar]
- 7.Fujita M. Cell Div. 2006;1:22. doi: 10.1186/1747-1028-1-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iwahori S, et al. Cell Cycle. 2014;13:471–81. doi: 10.4161/cc.27274. [DOI] [PMC free article] [PubMed] [Google Scholar]