

Abstract
Pancreatic carcinoma is the major clinical entity where the nucleoside analog gemcitabine is used for first-line therapy. Overcoming cellular resistance toward gemcitabine remains a major challenge in this context. This raises the need to identify factors that determine gemcitabine sensitivity in pancreatic carcinoma cells. We previously found the MAPK-activated protein kinase 2 (MK2), part of the p38/MK2 stress response pathway, to be required for DNA replication fork stalling when osteosarcoma-derived cells were treated with gemcitabine. As a consequence, inhibition or depletion of MK2 protects these cells from gemcitabine-induced death (Köpper, et al. Proc Natl Acad Sci USA 2013; 110:16856–61). Here, we addressed whether MK2 also determines the sensitivity of pancreatic cancer cells toward gemcitabine. We found that MK2 inhibition reduced the intensity of the DNA damage response and enhanced survival of the pancreatic cancer cell lines BxPC-3, MIA PaCa-2, and Panc-1, which display a moderate to strong sensitivity to gemcitabine. In contrast, MK2 inhibition only weakly attenuated the DNA damage response intensity and did not enhance long-term survival in the gemcitabine-resistant cell line PaTu 8902. Importantly, in BxPC-3 and MIA PaCa-2 cells, inhibition of MK2 also rescued increased H2AX phosphorylation caused by inhibition of the checkpoint kinase Chk1 in the presence of gemcitabine. These results indicate that MK2 mediates gemcitabine efficacy in pancreatic cancer cells that respond to the drug, suggesting that the p38/MK2 pathway represents a determinant of the efficacy by that gemcitabine counteracts pancreatic cancer.
Keywords: Chk1, DNA damage, MAPKAPK2, MK2, chemotherapy, gemcitabine, pancreatic carcinoma
Introduction
Among the common cancers, pancreatic cancer is the most deadly; patients diagnosed with pancreatic carcinoma have an average 5-year survival rate below 5%.1 For the first-line treatment of pancreatic adenocarcinomas, the nucleoside analog gemcitabine is commonly used as a monotherapy.2,3 Although gemcitabine significantly improves disease-free survival when used in adjuvant therapy4 and extends survival in palliative therapy,5 the clinical benefit is modest. Moreover, tumors frequently prove resistant against gemcitabine, while cellular resistance mechanisms are poorly understood.6 Thus, it is of great interest to identify factors that determine the gemcitabine sensitivity of pancreatic tumors. Such factors might then be targeted pharmacologically to increase treatment efficacy. This is in accordance with a general strategy in cancer treatment, i.e., to combine signaling inhibitors with drugs that target broadly active cellular machineries, e.g., for DNA replication.7 For instance the checkpoint kinase Chk1 governs the cellular response to gemcitabine, and its knockdown or inhibition strongly sensitizes pancreatic cancer cells toward the drug.8-10
We recently characterized the MAP kinase-activated protein kinase 2 (MK2) as a major mediator of the response to gemcitabine in the osteosarcoma-derived cell line U2OS.11 MK2 was originally identified in stress response signaling downstream of the MAP kinase p38 and recently reported to regulate G2 arrest, as well.12-14 We found that the depletion or inhibition of MK2 decreased the gemcitabine-induced phosphorylation of the histone variant 2AX (the phosphorylated form is called γH2AX), a hallmark of the DNA damage response (DDR). MK2 inhibition also promoted cell proliferation in the presence of gemcitabine. Furthermore, the impairment of DNA replication by gemcitabine was rescued by MK2 inhibition, and this rescue was dependent on translesion synthesis (TLS) polymerases. Importantly, depletion or inhibition of MK2 also counteracted the cytotoxic effects of Chk1 inhibition or depletion.11
Here, we investigated whether MK2 also determines the cellular response to gemcitabine in cells derived from pancreatic adenocarcinomas. Indeed, we found that inhibition of MK2 reduced gemcitabine-induced H2AX phosphorylation and improved proliferation in the presence of gemcitabine in pancreatic cancer-derived cell lines that respond to the drug. MK2 inhibition also rescued the increased H2AX phosphorylation that resulted from Chk1 inhibition in these cells. These findings demonstrate that MK2 is a determinant of gemcitabine sensitivity and replicative stress in pancreatic cancer cells.
Results
To investigate whether MK2 also modulates the gemcitabine-induced DDR in pancreatic cancer cells, we selected 4 cell lines derived from pancreatic adenocarcinomas with reportedly different sensitivities toward gemcitabine.15,16 These were treated with gemcitabine in the presence or absence of a pharmacological MK2 inhibitor III17 (subsequently referred to as “MK2 inhibitor”), and H2AX phosphorylation as a read-out for the DDR intensity was detected by immunoblot analysis (Fig. 1A‒D). The final inhibitor concentration was chosen according to previous studies to efficiently block MK2 target phosphorylation.17 In BxPC-3, MIA PaCa-2, and Panc-1 cells, H2AX phosphorylation was induced by gemcitabine, whereas inhibition of MK2 reduced this effect. In contrast, γH2AX levels in gemcitabine-resistant PaTu 8902 cells16 only slightly increased upon gemcitabine treatment (from a relatively high baseline level), and little if any impact of MK2 inhibition was evident. Panc-1 cells depleted of MK2 by siRNA-mediated knockdown also displayed lower γH2AX levels after gemcitabine treatment than control-transfected cells (Fig. 1E), as revealed by quantitative immunofluorescence analysis, and in line with the effect of MK2 inhibitor. Corresponding representative original images are shown in Figure S1. Thus, MK2 activity is required for gemcitabine-induced H2AX phosphorylation in the gemcitabine-responsive pancreatic cancer cell lines tested, whereas it does not change H2AX phosphorylation in the gemcitabine-insensitive PaTu 8902 cells.

Figure 1. Effect of MK2 inhibition and depletion on gemcitabine-induced H2AX phosphorylation in pancreatic cancer cell lines. BxPC-3 (A), MIA PaCa-2 (B), Panc-1 (C), and PaTu 8902 cells (D) were treated with 100 nM gemcitabine and MK2 inhibitor or DMSO for 24 h. H2AX phosphorylation was analyzed by immunoblot. (E) Panc-1 cells were depleted of MK2 by siRNA-mediated knockdown. Forty-eight hours later, the cells were treated with 300 nM gemcitabine for 22 h or left untreated. The cells were fixed and stained for immunofluorescence analysis, and γH2AX fluorescence intensity was quantified. Mean ± SD from 3 technical replicates. (**P = 0.009).
Next, we addressed the question whether MK2 mediates the impact of gemcitabine on cell viability, as it does in the osteosarcoma-derived cell line U2OS.11 Indeed, we found that, while treatment with gemcitabine alone strongly reduced the proliferation of BxPC-3, MIA PaCa-2, and Panc-1 cells, simultaneous inhibition of MK2 completely reversed this effect (Fig. 2A‒C). Proliferation of PaTu 8902 cells was hardly affected by gemcitabine, in line with the reported insensitivity of the cells toward the drug (Fig. 2D). Interestingly, MK2 inhibition slightly increased proliferation regardless of gemcitabine treatment in these cells, perhaps reflecting a reduction in their constitutive replicative stress. Thus, inhibition of MK2 protects gemcitabine-sensitive pancreatic cancer cells from the attenuation of proliferation induced by the drug. This is not the case for PaTu 8902 cells, in accordance with our observation that γH2AX levels remain unchanged by MK2 inhibitor or gemcitabine in these cells as well (Fig. 1D).
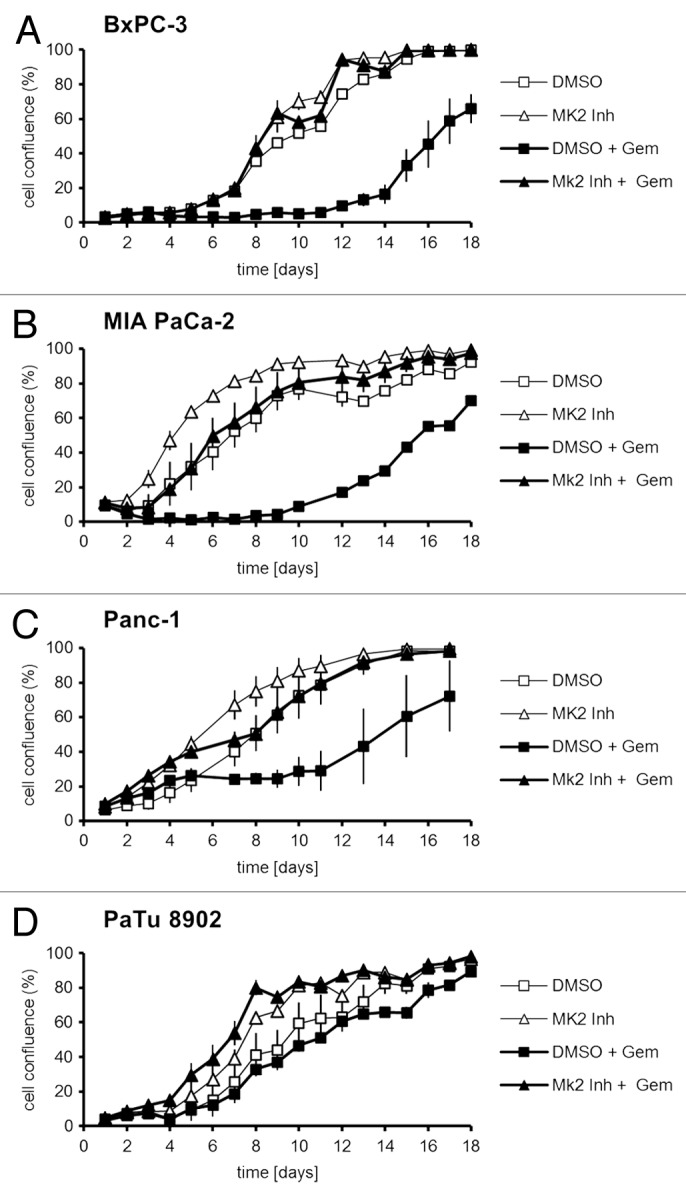
Figure 2. Proliferation of pancreatic cancer cell lines upon treatment with gemcitabine and/or MK2 inhibitor. BxPC-3 (A), MIA PaCa-2 (B), Panc-1 (C), and PaTu 8902 (D) cells were treated with 100 nM gemcitabine and MK2 inhibitor or DMSO for 24 h on day 1. Then the drugs were washed out, and cell confluence was quantified by light microscopy and digital image analysis until day 18.
We previously reported that, in U2OS cells, MK2 is not only essential for the DDR following gemcitabine treatment, but also for the increased γH2AX accumulation resulting from simultaneous gemcitabine treatment and inhibition of Chk1.11 Chk1 is a master regulator of the DDR.18 One of its major tasks is the coordination of DNA replication,19,20 and, thereby, Chk1 attenuates replicative stress.21 Accordingly, inhibition of Chk1 has the potential to overcome drug resistance in cancer cells in general18 and in pancreatic cancer cells in particular,8 and different Chk1 inhibitors are currently being tested in clinical trials.22,23 Most importantly in the context of this report, inhibition of Chk1 sensitizes pancreatic cancer cells toward gemcitabine.9,10 Therefore, we tested whether the response of pancreatic cancer cells toward gemcitabine, together with Chk1 inhibition, also depends on MK2. To this end, we combined gemcitabine treatment with inhibition of MK2, Chk1, or both kinases in the cell lines BxPC-3, MIA PaCa-2, and PaTu 8902. In BxPC-3 and MIA PaCa-2 cells, inhibition of Chk1 with the pharmacological inhibitor SB21807824 (subsequently called “Chk1 inhibitor”) strongly increased H2AX phosphorylation, but simultaneous inhibition of MK2 impaired this effect (Fig. 3A and B). Chk1 inhibitor concentration was based on previous studies to ensure efficient block of target phosphorylation.24 In PaTu 8902 cells, on the other hand, neither Chk1 inhibition alone nor combined treatment with MK2 inhibitor affected γH2AX levels in the presence of gemcitabine (Fig. 3C). We conclude that Chk1 inhibition only increases the response to gemcitabine in cell lines generally responsive to the drug, but not in gemcitabine-insensitive PaTu 8902 cells. Importantly, MK2 activity is required for the sensitizing effect of Chk1 inhibition, further supporting the notion of MK2 as a determinant of gemcitabine sensitivity in pancreatic cancer cells.
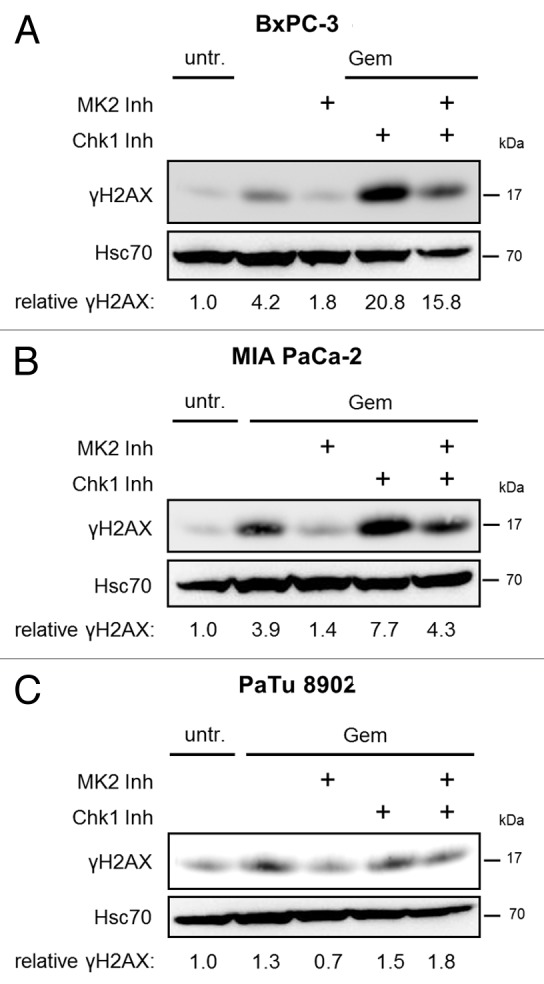
Figure 3. Gemcitabine-induced H2AX phosphorylation in dependence of MK2 and Chk1 inhibition in pancreatic cancer cell lines. BxPC-3 (A), MIA PaCa-2 (B), and PaTu 8902 (C) cells were treated with 100 nM gemcitabine and MK2 inhibitor, Chk1 inhibitor or both for 24 h. Then, H2AX phosphorylation was analyzed by immunoblot. “Relative γH2AX” indicates relative γH2AX intensities normalized to Hsc70 intensities. See Table S1 for raw data.
Discussion
The results presented here identify MK2 as a determinant of gemcitabine sensitivity in pancreatic cancer cells. This finding expands the known cellular functions of MK2 by an aspect with potential clinical relevance.
Our results suggest that MK2 represents a mediator of gemcitabine toxicity in pancreatic tumor cells, as was found previously in the osteosarcoma cell line U2OS. At first glance, this seems in contrast with a recent report that describes MK2 deficiency as synthetic lethal with p53 deficiency in non-small cell lung cancer upon treatment with cisplatin.25 However, these results need not necessarily contradict each other. Cisplatin acts by forming direct adducts with the DNA, leading to intra- and interstrand crosslinks.26 This type of DNA damage is fundamentally different from the misincorporation of nucleoside analogs. We therefore propose that MK2 may act as a mediator of chemoresponse in the case of nucleoside analogs, but may act in a protective manner when cells are exposed to different chemotherapeutics, e.g., platinum compounds.
Interestingly, in recent years a number of studies reported a correlation between expression of the heat shock protein of 27 kDa (Hsp27) and gemcitabine responsiveness in pancreatic cancer cells.27,28 Hsp27 is an important substrate of MK2 and extensively phosphorylated by the kinase. This phosphorylation modulates Hsp27 activity.29 The conclusions of these studies, however, are contradictory: some find Hsp27 overexpression to correlate with gemcitabine sensitivity,28,30 while others report that cells overexpressing Hsp27 are more resistant to the drug.27 This raises the need for rigorous clinical assessment of whether MK2 levels and activity correlate with the response of tumors to nucleoside analogs.
The phosphorylation status of Hsp27 has been investigated in the context of gemcitabine sensitivity, raising the possibility of indirectly assessing MK2 activity. Unfortunately, however, the reported results are as contradictory as for Hsp27 protein phosphorylation levels: while Taba and colleagues report that Hsp27 phosphorylation correlates with gemcitabine resistance in pancreatic cancer cells,31 Nakashima and colleagues find the opposite to be true.32 As Hsp27 phosphorylation depends on MK2 activity, the results presented in this report are in line with the latter study. Thus, while the predictive value of Hsp27 levels and phosphorylation for gemcitabine sensitivity remains to be validated in the clinics, the fact that correlations were repeatedly reported supports the notion of MK2—the kinase directly upstream of Hsp27—as a determinant of gemcitabine sensitivity.
We previously reported that, in the context of the DDR in U2OS cells, MK2 activity antagonizes that of Chk1.11 The results presented here show that this is also the case in gemcitabine-responsive pancreatic cancer cell lines (Fig. 3A and B). This finding is of interest with respect to the application of Chk1 inhibitors as chemosensitizers: first, it raises the possibility that the efficacy of Chk1 inhibitors as sensitizers toward gemcitabine might depend on the cellular levels of MK2. Our results suggest that low MK2 activity might dampen the effect of Chk1 inhibition. Second, MK2 and Chk1 display highly similar substrate specificity.33 It is therefore not surprising that the Chk1 inhibitor UCN-01 has been found to also block MK2 activity.14 Hence, the poor antitumor activity of UCN-01 in clinical trials22 might in part also be attributed to the simultaneous inhibition of MK2 that counteracts the effect of Chk1 inhibition. A similar counterproductive off-target effect might arise with other Chk1 inhibitors. Taken together, our study supports the notion that, while MK2 inhibitiors are expected to counteract gemcitabine efficacy, selective Chk1 inhibition might well help to sensitize tumor cells toward gemcitabine.
Along the same line, the protective effect of MK2 depletion or inhibition against certain chemotherapeutics might also be of importance for the clinical application of multikinase inhibitors designed to sensitize cells for chemotherapeutic treatment. Sorafenib (Bay43–9006; Nexavar, Bayer Healthcare Pharmaceuticals) is such an inhibitor that—among other pathways—also blocks MAPK14/p38 signaling.34,35 It has been shown to exhibit high anticancer efficacy in acute myeloid leukemia and other neoplasms.36 However, our results suggest that, due to its inhibitory effect on p38/MK2 signaling, sorafenib might also contribute to drug resistance in some cancers when combined with antimetabolites such as gemcitabine or other nucleoside analogs, compromising its efficacy.
Finally, our finding that MK2 is a determinant of gemcitabine sensitivity in pancreatic cancer cells raises the possibility of directly exploiting the p38/MK2 pathway as a pharmacological target in cancer therapy in general and in the treatment of pancreatic carcinoma in particular. As inhibition of MK2 dampens the DDR and promotes cell survival, the targeted activation of the p38/MK2 pathway, e.g., in the context of inflammation, may constitute a promising approach to sensitize tumor cells toward gemcitabine treatment.
Material and Methods
Cell culture, transfections, and treatments
MIA PaCa-2 and PaTu 8902 cells were cultured in DMEM (Invitrogen) supplemented with 10% FCS and antibiotics. Panc-1 and BxPC-3 cells were cultured in RPMI (Invitrogen) supplemented with 10% FCS and antibiotics. Specific knockdown of target genes was done using pre-designed Silencer Select siRNAs or control siRNAs (Ambion/Invitrogen) with a final concentration of 5 nM and Lipofectamine 2000 (Invitrogen), applied in a forward transfection protocol. Gemcitabine (Gemzar, Eli Lilly) was dissolved in H2O and applied in the concentrations indicated. MK2 and Chk1 kinases were inhibited by using 10 μM MK2 Inhibitor III or 2.5 μM SB218078 (both Calbiochem/Merck), dissolved in DMSO as a stock, respectively. Controls were treated with DMSO.
Immunoblot analysis and antibodies
Cell lysates were separated by SDS PAGE and transferred to nitrocellulose membranes. For detection of specific proteins, the membranes were incubated with antibodies diluted in 5% BSA in Tris-buffered saline solution containing 0.1% Tween-20. Mouse anti-γH2AX antibody (JBW301, Millipore/Merck) and anti-Hsc70 antibody were then detected with peroxidase-coupled secondary antibodies (Jackson). Quantification of band intensities was done with the ImageJ software.
High-content immunofluorescence microscopy
Quantitative detection of γH2AX by high-content immunofluorescence was done as described,11 using anti-γH2AX (JBW301, Millipore/Merck) and Alexa Fluor-546 anti-mouse IgG (Invitrogen). Nuclei were stained with Hoechst 33342 (5 μg/mL). Automated microscopy was performed with a Pathway HT Cell Imaging System using the AttoVision image acquisition software (Becton Dickinson) and single-cell-based image analysis. The Hoechst signal was used to identify cell nuclei. For quantification of signal intensities, the average fluorescence resulting from the respective staining was determined per nucleus. Immunofluorescence intensity is shown as mean ± SD. Unpaired Student t test was used for the calculation of P values.
Proliferation assay
For cell proliferation analysis, cells were seeded in triplicate in 96-well plates and, after 24 h, treated with inhibitors and drugs for 24 h, and then further incubated without drugs. Cell confluence was measured on consecutive days by bright-field microscopy using a Celigo Adherent Cell Cytometer (Brooks).
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by the DFG, the Deutsche Krebshilfe, the German José Carreras Leukemia foundation, and the Wilhelm Sander foundation. F.K. was a fellow of the International Max Planck Research School, Master PhD program in Molecular Biology, during this work, and he was supported by the Göttingen Graduate School for Neurosciences, Biophysics and Molecular Biosciences. A.M.B. was supported by the Jacob-Henle-Program of the University Medical Center Göttingen, and by the Deutsche Krebshilfe. F.K. and A.M.B. were also supported by the Studienstiftung des Deutschen Volkes.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/28292
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Freitas D, Fernandes GdosS, Hoff PM, Cunha JE. Medical management of pancreatic adenocarcinoma. Pancreatology. 2009;9:223–32. doi: 10.1159/000199433. [DOI] [PubMed] [Google Scholar]
- 3.Stathis A, Moore MJ. Advanced pancreatic carcinoma: current treatment and future challenges. Nat Rev Clin Oncol. 2010;7:163–72. doi: 10.1038/nrclinonc.2009.236. [DOI] [PubMed] [Google Scholar]
- 4.Oettle H, Post S, Neuhaus P, Gellert K, Langrehr J, Ridwelski K, Schramm H, Fahlke J, Zuelke C, Burkart C, et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. JAMA. 2007;297:267–77. doi: 10.1001/jama.297.3.267. [DOI] [PubMed] [Google Scholar]
- 5.Burris HA, 3rd, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo AM, Tarassoff P, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15:2403–13. doi: 10.1200/JCO.1997.15.6.2403. [DOI] [PubMed] [Google Scholar]
- 6.Long J, Zhang Y, Yu X, Yang J, LeBrun DG, Chen C, Yao Q, Li M. Overcoming drug resistance in pancreatic cancer. Expert Opin Ther Targets. 2011;15:817–28. doi: 10.1517/14728222.2011.566216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dobbelstein M, Moll UM. Targeting Tumor-Supportive Cellular Machineries in Anticancer Drug Development. Nat Rev Drug Discov. doi: 10.1038/nrd4201. In press. [DOI] [PubMed] [Google Scholar]
- 8.Azorsa DO, Gonzales IM, Basu GD, Choudhary A, Arora S, Bisanz KM, Kiefer JA, Henderson MC, Trent JM, Von Hoff DD, et al. Synthetic lethal RNAi screening identifies sensitizing targets for gemcitabine therapy in pancreatic cancer. J Transl Med. 2009;7:43. doi: 10.1186/1479-5876-7-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parsels LA, Morgan MA, Tanska DM, Parsels JD, Palmer BD, Booth RJ, Denny WA, Canman CE, Kraker AJ, Lawrence TS, et al. Gemcitabine sensitization by checkpoint kinase 1 inhibition correlates with inhibition of a Rad51 DNA damage response in pancreatic cancer cells. Mol Cancer Ther. 2009;8:45–54. doi: 10.1158/1535-7163.MCT-08-0662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parsels LA, Qian Y, Tanska DM, Gross M, Zhao L, Hassan MC, Arumugarajah S, Parsels JD, Hylander-Gans L, Simeone DM, et al. Assessment of chk1 phosphorylation as a pharmacodynamic biomarker of chk1 inhibition. Clin Cancer Res. 2011;17:3706–15. doi: 10.1158/1078-0432.CCR-10-3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Köpper F, Bierwirth C, Schön M, Kunze M, Elvers I, Kranz D, Saini P, Menon MB, Walter D, Sørensen CS, et al. Damage-induced DNA replication stalling relies on MAPK-activated protein kinase 2 activity. Proc Natl Acad Sci U S A. 2013;110:16856–61. doi: 10.1073/pnas.1304355110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reinhardt HC, Hasskamp P, Schmedding I, Morandell S, van Vugt MA, Wang X, Linding R, Ong SE, Weaver D, Carr SA, et al. DNA damage activates a spatially distinct late cytoplasmic cell-cycle checkpoint network controlled by MK2-mediated RNA stabilization. Mol Cell. 2010;40:34–49. doi: 10.1016/j.molcel.2010.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reinhardt HC, Yaffe MB. Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr Opin Cell Biol. 2009;21:245–55. doi: 10.1016/j.ceb.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reinhardt HC, Aslanian AS, Lees JA, Yaffe MB. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell. 2007;11:175–89. doi: 10.1016/j.ccr.2006.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fryer RA, Barlett B, Galustian C, Dalgleish AG. Mechanisms underlying gemcitabine resistance in pancreatic cancer and sensitisation by the iMiD™ lenalidomide. Anticancer Res. 2011;31:3747–56. [PubMed] [Google Scholar]
- 16.Akada M, Crnogorac-Jurcevic T, Lattimore S, Mahon P, Lopes R, Sunamura M, Matsuno S, Lemoine NR. Intrinsic chemoresistance to gemcitabine is associated with decreased expression of BNIP3 in pancreatic cancer. Clin Cancer Res. 2005;11:3094–101. doi: 10.1158/1078-0432.CCR-04-1785. [DOI] [PubMed] [Google Scholar]
- 17.Anderson DR, Meyers MJ, Vernier WF, Mahoney MW, Kurumbail RG, Caspers N, Poda GI, Schindler JF, Reitz DB, Mourey RJ. Pyrrolopyridine inhibitors of mitogen-activated protein kinase-activated protein kinase 2 (MK-2) J Med Chem. 2007;50:2647–54. doi: 10.1021/jm0611004. [DOI] [PubMed] [Google Scholar]
- 18.Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003;3:421–9. doi: 10.1016/S1535-6108(03)00110-7. [DOI] [PubMed] [Google Scholar]
- 19.Feijoo C, Hall-Jackson C, Wu R, Jenkins D, Leitch J, Gilbert DM, Smythe C. Activation of mammalian Chk1 during DNA replication arrest: a role for Chk1 in the intra-S phase checkpoint monitoring replication origin firing. J Cell Biol. 2001;154:913–23. doi: 10.1083/jcb.200104099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petermann E, Maya-Mendoza A, Zachos G, Gillespie DA, Jackson DA, Caldecott KW. Chk1 requirement for high global rates of replication fork progression during normal vertebrate S phase. Mol Cell Biol. 2006;26:3319–26. doi: 10.1128/MCB.26.8.3319-3326.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiao Z, Xue J, Sowin TJ, Rosenberg SH, Zhang H. A novel mechanism of checkpoint abrogation conferred by Chk1 downregulation. Oncogene. 2005;24:1403–11. doi: 10.1038/sj.onc.1208309. [DOI] [PubMed] [Google Scholar]
- 22.Ma CX, Janetka JW, Piwnica-Worms H. Death by releasing the breaks: CHK1 inhibitors as cancer therapeutics. Trends Mol Med. 2011;17:88–96. doi: 10.1016/j.molmed.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carrassa L, Damia G. Unleashing Chk1 in cancer therapy. Cell Cycle. 2011;10:2121–8. doi: 10.4161/cc.10.13.16398. [DOI] [PubMed] [Google Scholar]
- 24.Jackson JR, Gilmartin A, Imburgia C, Winkler JD, Marshall LA, Roshak A. An indolocarbazole inhibitor of human checkpoint kinase (Chk1) abrogates cell cycle arrest caused by DNA damage. Cancer Res. 2000;60:566–72. [PubMed] [Google Scholar]
- 25.Morandell S, Reinhardt HC, Cannell IG, Kim JS, Ruf DM, Mitra T, Couvillon AD, Jacks T, Yaffe MB. A reversible gene-targeting strategy identifies synthetic lethal interactions between MK2 and p53 in the DNA damage response in vivo. Cell Rep. 2013;5:868–77. doi: 10.1016/j.celrep.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov. 2005;4:307–20. doi: 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]
- 27.Baylot V, Andrieu C, Katsogiannou M, Taieb D, Garcia S, Giusiano S, Acunzo J, Iovanna J, Gleave M, Garrido C, et al. OGX-427 inhibits tumor progression and enhances gemcitabine chemotherapy in pancreatic cancer. Cell Death Dis. 2011;2:e221. doi: 10.1038/cddis.2011.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu QH, Zhao CY, Zhang J, Chen Y, Gao L, Ni CY, Zhu MH. Role of heat shock protein 27 in gemcitabine-resistant human pancreatic cancer: comparative proteomic analyses. Mol Med Rep. 2012;6:767–73. doi: 10.3892/mmr.2012.1013. [DOI] [PubMed] [Google Scholar]
- 29.Vidyasagar A, Wilson NA, Djamali A. Heat shock protein 27 (HSP27): biomarker of disease and therapeutic target. Fibrogenesis Tissue Repair. 2012;5:7. doi: 10.1186/1755-1536-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schäfer C, Seeliger H, Bader DC, Assmann G, Buchner D, Guo Y, Ziesch A, Palagyi A, Ochs S, Laubender RP, et al. Heat shock protein 27 as a prognostic and predictive biomarker in pancreatic ductal adenocarcinoma. J Cell Mol Med. 2012;16:1776–91. doi: 10.1111/j.1582-4934.2011.01473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taba K, Kuramitsu Y, Ryozawa S, Yoshida K, Tanaka T, Maehara S, Maehara Y, Sakaida I, Nakamura K. Heat-shock protein 27 is phosphorylated in gemcitabine-resistant pancreatic cancer cells. Anticancer Res. 2010;30:2539–43. [PubMed] [Google Scholar]
- 32.Nakashima M, Adachi S, Yasuda I, Yamauchi T, Kawaguchi J, Itani M, Yoshioka T, Matsushima-Nishiwaki R, Hirose Y, Kozawa O, et al. Phosphorylation status of heat shock protein 27 plays a key role in gemcitabine-induced apoptosis of pancreatic cancer cells. Cancer Lett. 2011;313:218–25. doi: 10.1016/j.canlet.2011.09.008. [DOI] [PubMed] [Google Scholar]
- 33.Manke IA, Nguyen A, Lim D, Stewart MQ, Elia AE, Yaffe MB. MAPKAP kinase-2 is a cell cycle checkpoint kinase that regulates the G2/M transition and S phase progression in response to UV irradiation. Mol Cell. 2005;17:37–48. doi: 10.1016/j.molcel.2004.11.021. [DOI] [PubMed] [Google Scholar]
- 34.Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099–109. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- 35.Chapuy B, Schuelper N, Panse M, Dohm A, Hand E, Schroers R, Truemper L, Wulf GG. Multikinase inhibitor sorafenib exerts cytocidal efficacy against Non-Hodgkin lymphomas associated with inhibition of MAPK14 and AKT phosphorylation. Br J Haematol. 2011;152:401–12. doi: 10.1111/j.1365-2141.2010.08526.x. [DOI] [PubMed] [Google Scholar]
- 36.Ibrahim N, Yu Y, Walsh WR, Yang JL. Molecular targeted therapies for cancer: sorafenib mono-therapy and its combination with other therapies (review) Oncol Rep. 2012;27:1303–11. doi: 10.3892/or.2012.1675. [review] [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.