

Abstract
Senescence-accelerated OXYS rats are an experimental model of accelerated aging that was established from Wistar stock via selection for susceptibility to cataractogenic effects of a galactose-rich diet and via subsequent inbreeding of highly susceptible rats. Currently, we have the 102nd generation of OXYS rats with spontaneously developing cataract and accelerated senescence syndrome, which means early development of a phenotype similar to human geriatric disorders, including accelerated brain aging. In recent years, our group found strong evidence that OXYS rats are a promising model for studies of the mechanisms of brain aging and neurodegenerative processes similar to those seen in Alzheimer disease (AD). The manifestation of behavioral alterations and learning and memory deficits develop since the fourth week of age, i.e., simultaneously with first signs of neurodegeneration detectable on magnetic resonance imaging and under a light microscope. In addition, impaired long-term potentiation has been demonstrated in OXYS rats by the age of 3 months. With age, neurodegenerative changes in the brain of OXYS rats become amplified. We have shown that this deterioration happens against the background of overproduction of amyloid precursor protein (AβPP), accumulation of β-amyloid (Aβ), and hyperphosphorylation of the tau protein in the hippocampus and cortex. The development of AMD-like retinopathy in OXYS rats is also accompanied by increased accumulation of Aβ in the retina. These published data suggest that the OXYS strain may serve as a spontaneous rat model of AD-like pathology and could help to decipher the pathogenesis of AD.
Keywords: Alzheimer disease, brain aging, senescence-accelerated OXYS rats
Introduction
Alzheimer disease (AD) is the most common neurodegenerative disorder that causes dementia as a result of atrophic changes in the brain, leading to disruption of attention, memory, and executive function, and ultimately to death within 3–9 y of the diagnosis. The major risk factor of AD is age. Moreover, the incidence of AD doubles every 5 y beyond 65 y of age.1 More than 35 million people worldwide have AD, and it is estimated that the number of these patients will exceed 115 million by 2050.2,3
The conventional view of AD, according to the “amyloid hypothesis”, is that β-amyloid (Aβ), the product of proteolytic cleavage of the large transmembrane Aβ precursor protein (AβPP), occupies a central place in the pathology of disease.4,5 AD is characterized by increased load of Aβ in the brain, especially the peptides 42 amino acids in length, leading to accumulation of Aβ, amyloid plaques, neurofibrillary tangles, synaptic failure, neuronal cell death, excitotoxicity, inflammation, mitochondrial dysfunction, and oxidative stress.3 In contrast, there is growing evidence that mitochondrial damage and oxidative stress lead to activation of the Aβ cascade.3,6,7 In regard to inflammatory cells, microglia can form a protective network for the brain to control neurotrophic factors, limit oxidative stress, and foster neuronal regeneration. Microglia also may limit the deposition and toxicity of Aβ and promote neuronal survival.8 In addition, Streit and colleagues9-13 have shown convincingly in a series of recent studies that neurodegeneration in AD is not the result of an overactive immune response, but rather of a decline of immune functions. Several factors, including lifestyle, diet, environmental exposure, apolipoprotein allele E4 (ApoE), and several other genetic variants have been reported to be involved in late-onset AD.14 It is noteworthy that AD frequently develops against a background of complex manifestations of aging and along with other age-related diseases, including age-related macular degeneration (AMD). AMD is a progressive, complex disease and is the most common cause of blindness in the aging population. At present, it is widely debated whether AMD shares some pathological hallmarks with AD, because a number of similarities between AD and AMD have been described.15-18 Like AD, AMD is characterized by Aβ deposition and is associated with changes in the complement system and ApoE.19,20
To date, there is no perfect rodent model able to fully mimic human neurodegenerative diseases, AD in particular.21,22 Transgenic/mutant strains of mice and rats with overproduction of AβPP, presenilin, tau protein, and ApoE cannot provide an all-encompassing picture of the pathophysiology of AD.23,24 Moreover, all these models are characterized by an increased expression of genes and mutations that are typical in early onset AD, which accounts for only ~5% of all cases. On the other hand, the remaining cases (~95%) of AD, which generally occur after the age of 65, are sporadic forms of AD.4 This observation calls for creation of new models, which will allow for studies of the pathogenesis of this particular form of AD.25 Only one widely accepted spontaneous model of sporadic form of AD exists: senescence-accelerated SAMP mice.3,22,26
This review is focused on our recent results, which offer promising evidence that the mechanisms of brain aging and neurodegenerative processes in senescence-accelerated OXYS rats are similar to those in AD. The OXYS rat strain was developed at the Institute of Cytology and Genetics, Russian Academy of Sciences (Novosibirsk), from Wistar stock via selection for susceptibility to cataractogenic effect of a galactose-rich diet and via inbreeding of highly susceptible rats.27 After 5 cycles of inbreeding, feeding of galactose-rich diet and selection, the subsequent generations of rats developed cataracts spontaneously, without the galactose-rich diet. These rats were registered in the Rat Genome Database as the OXYS rat strain (http://rgd.mcw.edu/). At present, we have the 102nd generation of OXYS rats with spontaneously developing cataract and accelerated senescence syndrome, which is characterized by early development of a phenotype similar to human geriatric disorders. This pathological phenotype primarily includes accelerated thymus involution,28-30 retinopathy similar to human AMD,31-34 high blood pressure,35 and senile osteoporosis.36-38 Accelerated brain aging manifests itself in OXYS rats as early development of behavioral and cognitive alterations against the background of neurodegenerative changes driven by an AD-like metabolic pathway.39-43
Behavioral Impairments and Learning Deficits in OXYS Rats
Aging in humans and in experimental animals is associated with slow deterioration of cognitive function, particularly of learning and memory, and with an increased risk of neurodegenerative disorders.44-46 Both aging and age-associated neurodegenerative changes are linked to the development of behavioral impairments; consequently, increased anxiety, worsened neuromuscular coordination, and decreased performances in exploratory tests are considered markers of neurological aging.
We showed that behavior of young OXYS rats is similar to the behavior of old Wistar rats.40,41,43 At the age of 3 mo, OXYS rats exhibit significantly reduced locomotor and exploratory activities in the open field test and increased anxiety in the elevated plus maze test. These impairments progressively increased with age in OXYS rats, compared with Wistar rats.
In OXYS rats, the development of cataract and retinopathy coincides with behavioral aberrations; thus, the behavioral changes might be influenced by impairment of sensory organs. In 3-mo-old OXYS rats, however, only early or mild stages of cataracts and retinopathy are present; therefore, vision cannot be significantly impaired. The behavior of 3-mo-old OXYS rats does not correlate with the initial sings of cataract, but at old age, when severity of cataract and retinopathy considerably increases, their negative effects on performance in behavioral tests are apparent in animals with pronounced changes of retina and lenses.
The early period of postnatal development is thought to be a critical period for the future development of healthy brain function. Therefore, for elucidation of causative factors underlying the development of behavioral deficits and for identification of their connection with changes at the molecular level, it is necessary to conduct studies of the brain function and behavior in the early postnatal period. Bearing this in mind, we examined the behavior of OXYS rats at the age of 4 wk. We did not find significant behavioral differences between OXYS and Wistar rats either in the open field or in the elevated plus maze tests.43
OXYS rats develop early learning and memory deficits, but the studies using a variety of learning techniques (passive avoidance, object recognition, 8-arm radial maze, and Morris water maze) have demonstrated the existence of test-dependent differences in learning ability. Investigation of learning ability in a single-trial passive avoidance test showed that at 3 mo of age, OXYS rats have significant associative learning deficits compared with Wistar rats.47 During familiarization with the experimental chamber, OXYS rats showed a higher rate of transition to the dark compartment compared with Wistar rats. This difference can also be attributed to a weak response to novelty, which can impair learning. During conditioning, OXYS rats showed increased responsiveness to pain stimuli and a delayed escape from the dangerous compartment. All these factors can interfere with acquisition of new information and prevent the formation of memory traces. These deficits progressively worsened with age.48
In the Morris water maze, the old rats show reduced spatial memory compared with young rats, while young and middle-aged animals do not differ from each other.49 We have shown that young (age 3 mo) and middle-aged (age 12–16 mo) Wistar rats display the same level of spatial learning, whereas learning and memory deficits in OXYS rats became progressively worse between ages 3 and 16 mo, pointing to gradual deterioration of cognitive function.40-43 In contrast, OXYS rats demonstrate learning and reference memory deficits in the 8-arm radial maze already at 3 mo of age (data in preparation).
Neuronal synaptic dysfunction is a significant factor contributing to the memory loss in AD.50 Age-associated cognitive decline in OXYS rats, particularly impairment of spatial learning, is associated with changes in the hippocampal synaptic plasticity including a deficit in long-term potentiation (LTP) starting at 3 mo of age.51 The well-known fact that LTP is a substrate for memory has helped to focus the research on a specific and easily studied form of plasticity. No neurobiological candidate other than LTP satisfies these requirements for an encoding mechanism. Plasticity, the process through which synapses modulate their strength and form new connections with other neurons, serves a particularly important role in the response to injury and disease, including AD.52
Signs of Neurodegeneration in OXYS Rats
The development of behavioral alterations in OXYS rats coincides with the first signs of neurodegeneration identifiable by means of light microscopy and magnetic resonance imaging (MRI). AD is associated with dysfunction and eventually death of brain neurons. Critical for learning and memory, the hippocampus is one of the first brain regions to be affected by AD.53 The analysis of the neuronal populations CA1 and CA3 (Fig. 1) and dentate gyrus regions of the hippocampus of OXYS rats demonstrated a progressive age-dependent loss of neuronal cells. The neuronal populations in all regions of the hippocampus of OXYS rats are increased at 5 mo of age compared with age-matched Wistar rats. We hypothesized that enhanced neurogenesis at this age could serve as an endogenous compensatory mechanism of repair. This phenomenon may also be explained by the increased production of immature neurons in the hippocampus of OXYS rats. In any case, the dysfunctional neurogenesis in the brain leads subsequently to subtle early manifestations of the disease, which could, in turn, render neurons more vulnerable to AD and contribute to memory impairment. Neuronal loss is usually prominent in the hippocampus, especially the CA1 region, and manifests itself further throughout the cerebral cortex, increasing with disease progression.54,55 In the CA1, CA3, and dentate gyrus regions of the hippocampus in OXYS rats, the significant structural changes of neurons develop by the age of 3–5 mo and increase with age (Maksimova et al., in press). The most pronounced neurodegenerative changes occur in the CA1 and CA3 regions (Fig. 1) of the hippocampus of OXYS rats: the percentage of dead or damaged neurons is significantly larger than that in Wistar rats at 5 and 15 mo of age. In addition, 5-mo-old OXYS rats have a significantly lower average body size and smaller nuclei of pyramidal neurons in the CA1 region of the hippocampus compared with age-matched Wistar rats. With age, these changes progress, and 15-mo-old OXYS rats exhibit a significant decrease in the amount of bodies and nuclei in all regions of the hippocampus that we studied. Thus, the increased neuronal populations in the hippocampus of 5-mo-old OXYS rats may represent a mechanism directed toward the replacement of dead or damaged neurons. Both the increase and decrease in neurogenesis have been observed in the brains of transgenic rodents that partly mimic AD pathology.56,57 It has been shown that there are several possible reasons for the limited repair capacity of neurogenesis in AD.58-60 First, the rate or extent of cell loss may be too great for a quantitatively significant replacement to be achieved. Second, the neurons that are produced may be ineffective, because they do not develop into fully mature functional neurons, because they do not develop into the right type of neurons, or because they are incapable of integrating into the surviving brain circuitry. Third, the microenvironment of the AD brain may be toxic to new neurons.
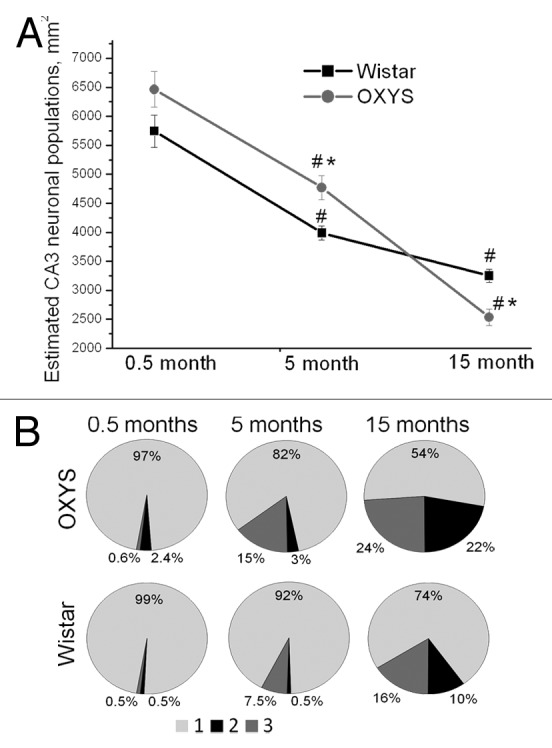
Figure 1. Quantification of neuronal populations in the hippocampal CA3 region of OXYS rats. (A) No cell loss in the CA3 region was detected at 0.5 mo; furthermore, 5-mo-old OXYS rats showed increased neuronal populations (P < 0.05); however, 15-mo-old OXYS rats showed a cell loss (P < 0.05) when compared with age-matched Wistar rats (control). *A statistically significant difference between the strains of the same age; #significant age-related differences compared with the previous age within the strain. (B) The neurodegenerative changes occur in the CA3 region of the hippocampus of OXYS rats: the percentage of dead or damaged neurons is significantly larger than that in Wistar rats at 5 and 15 mo of age. 1 (light gray) corresponds intact neurons, 2 (black) to dead neurons, and 3 (gray) to damaged neurons. Adapted from (Maksimova et al., in press).
The early development of neurodegenerative processes in OXYS rats is perhaps due to the fact that the brain development is accompanied by hypoxia caused by a delay in the formation of microvasculature.43 This notion is supported by a decrease the mitotic activity of vascular endothelial cells in the pia mater of the brain of OXYS rats compared with Wistar rats in the early postnatal development, when a development gap was identified in the formation of both arterial and venous microvessels. The same state of the microcirculatory system was found in the mesentery, suggesting that the observed deficits most likely represent a manifestation of systemic alterations rather than isolated local events.43 By the end of the first month of postnatal development, the differences in proliferation rate between OXYS and Wistar rats disappeared; however, alterations in energy metabolism were found in OXYS rats during the first month of life, particularly in phosphocreatine metabolism and phospholipid turnover, which are suggestive of adaptation to hypoxia in the OXYS rat brain.61
With age, the adaptive resources of OXYS rats decline. Examination of cerebral vessels and parameters of cerebral blood flow on MRI (in 12-mo-old OXYS rats) revealed structural and functional changes, including reduced reactivity of vessels typical for chronic ischemia,62,63 a condition that inevitably contributes to the progression of neurodegenerative changes. Chronic ischemia induced by diffuse insufficiency of blood supply to the brain leads to deterioration of brain function and to the development of cognitive and behavioral deficits in elderly people. The signs of tissue hypoxia and ischemia were also identified in the retina of OXYS rats. According to fundoscopy findings, the incidence of chorioretinal degeneration sharply increased in OXYS rats by the age of 4.5 mo, when retinopathy is observed in all animals. Morphological analysis showed that OXYS rats exhibit rapid expansion of the choriocapillaris, with concomitant evidence of disturbances of blood flow. The specific area of vessels with signs of partial occlusion of retinal vessels is significantly greater in young OXYS rats compared with Wistar rats.32,33 Overall, the changes in the chorioretinal complex of OXYS rats64,65 reflected a typical reaction to chronic hypoxia, one of the leading factors in the pathogenesis of AMD.
To sum up, the brain development in OXYS rats occurs under conditions of hypoxia, which are detectable in the early postnatal period, and this phenomenon might strongly affect the future development of brain function and behavioral impairments in these animals. The accumulation of different vasculotoxic and neurotoxic macromolecules in the brain as a result of hypoxia and the reduced cerebral blood flow can initiate neuronal dysfunction and neurodegenerative changes regardless of or prior to Aβ deposition.66 Neurovascular dysfunction as an integral part of AD may influence the onset and progression of cognitive decline and the establishment of a chronic neurodegenerative process.
Using MRI, we detected demyelinating lesions in the brain of 3-mo-old OXYS rats; these lesions progress with age (Fig. 2). Such lesions were not found in young Wistar rats.41,63,67 Demyelination occurs both during healthy brain aging and in AD, but the magnitude of changes is significantly different.68 With age, demyelination is detectable in Wistar rats, but in middle-aged and aged animals the number of demyelinating foci is smaller than that in age-matched OXYS rats (Fig. 2B).

Figure 2. T2-weighed MRI images of demyelinating foci in 3-, 12-, and 24-mo-old Wistar and OXYS rats. (A) The presence of foci of demyelization in 3-, 12-, and 24-mo-old Wistar and OXYS rats (% animals). (B) OXYS rats have a greater number of demyelinating foci compared with Wistar rats. (C) Axial slices of the brain of 3-, 12-, and 24-mo-old Wistar and OXYS rats. The arrows point to foci of demyelization. The data are shown as mean ± SEM. Legend: *P < 0.05 for differences between the strains. Adapted from reference 34.
In addition, according to MRI, the cross-section area of lateral ventricles, which is proportional to their volume, is larger even in 3-mo-old OXYS rats and still larger in middle-aged and aged animals compared with aged-matched Wistar rats (Fig. 3). Alterations in cerebrospinal fluid dynamics are associated with normal aging and AD.15 Enlarged cerebral lateral ventricular volumes are the evidence of early neurodegenerative changes that caused an imbalance between production and turnover of the cerebrospinal fluid, a problem that is also inherent in AD. As a result, with age, cerebrospinal fluid turnover becomes inversely related to accumulation of Aβ in the brain.
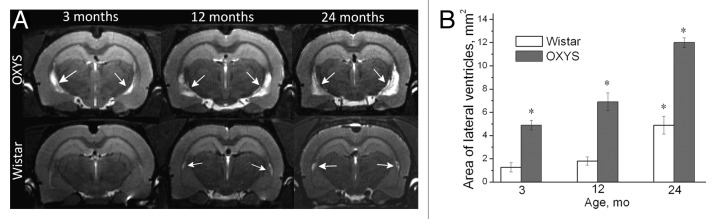
Figure 3. T2-weighed MRI images of lateral ventricles in 3-, 12-, and 24-mo-old Wistar and OXYS rats. (A) Axial slices of the brain of 3-, 12-, and 24-mo-old Wistar and OXYS rats. Increases in the size of lateral ventricles in OXYS rats are visible (the arrows). (B) In 3-, 12-, and 24-mo-old OXYS rats there was an enlargement of the lateral ventricles compared with Wistar rats. The data are shown as mean ± SEM. Legend: *P < 0.05 for differences between the strains. Adapted from reference 34.
Significant cognitive deficits and progressive neurodegenerative changes in OXYS rats develop along with a 4% reduction in the brain volume by the age of 12 mo compared with 3-mo-old animals of the same strain. The volume of the Wistar rats’ brains during this period is increased by 4%. It is known that the reduction of the brain volume may be indicative of the development of neurodegenerative disease.69-71 The body weight of OXYS rats at 12 mo of age reaches its maximum and does not yet begin typical age-related decline. Consequently, the decrease of brain volume of OXYS rats by age 12 mo probably points to progressive neurodegenerative changes. Thus, our data demonstrate that progressive neurodegenerative changes play a major role in the accelerated brain aging of OXYS rats.
Oxidative Stress and Mitochondrial Dysfunction in OXYS Rats
Mitochondrial dysfunction and oxidative stress are 2 interdependent and mutually reinforcing damage mechanisms that figure prominently both in the physiological aging process and in neurodegeneration.72-75 It is generally believed that oxidative stress—defined as a disturbed equilibrium between the pro- and antioxidant systems, leading to accumulation of oxidatively damaged macromolecules—is involved in age-related changes in cognitive function and in pathogenesis of neurodegenerative diseases such as AD.76
Previously, the accelerated senescence in OXYS rats had been linked to the “congenital overproduction of free radicals”, which was a key feature in the registration in the Rat Genome Database (http://rgd.mcw.edu/). On the other hand, our research into oxidative stress markers in OXYS rats has demonstrated that accumulation as well as imbalance of these markers in redox regulation appears later than the main manifestations of accelerated senescence.48,77,78 Higher superoxide dismutase (SOD) activity and a decreased level of reduced glutathione in the brain of young OXYS rats indicate effective functioning of the antioxidant system: probably the result of increased production of free radicals. SOD activity is affected by reactive oxygen species and can increase in response to oxidative stress; therefore, one could say that the observed increase in SOD activity in the brain of young OXYS rats reflects either the adaptation to more rapid oxidative metabolism or increased generation of reactive oxygen species (ROS) in the brain of OXYS rats.
In the course of the neurodegenerative process, several regions of the brain become particularly vulnerable to damage by oxidative stress. In AD, the activation of free radical formation initiated by neurodegeneration and neuronal death occurs in brain regions involved in memory and learning, specifically the hippocampus, cortex, and subcortical nucleus. The emotional and cognitive dysfunction in OXYS rats manifests earlier than does age-related accumulation of oxidized proteins and lipids in the brain.78 In all regions of OXYS rats’ brains, there is no generalized activation of lipid peroxidation (LPO) processes, which reflect the oxidative status of tissues. In young OXYS rats, however, 4 brain structures—i.e., midbrain, hippocampus, nuclei accumbens, and striatum—are characterized by an increased level of LPO products. The above regions are involved in memory and learning.
The level of LPO has been commonly accepted as an oxidative stress marker, whose increase is associated with oxidative damage and insufficiency of the antioxidant system. It is now evident, however, that the concept has been oversimplified, and that the intensity of ROS generation in various brain regions strongly depends on energy metabolism and electrophysiological activity of the brain.79,80 Furthermore, species-specific traits of animals’ behavior are linked to peculiarities of brain oxidative metabolism. These peculiarities are determined by the ratio of oxygen supply and consumption to activities of oxidative enzymes. The alterations in the energy metabolism in OXYS rats during the first month of life, the most important period of brain development, together with enhanced membrane lipid catabolism might be a possible reason for the delayed formation of the capillary system of the brain.
Although we did not assess considerable differences between OXYS and Wistar rats in oxidative stress markers under normal conditions, these differences became apparent under the conditions of emotional stress. Insufficiency of the antioxidant system together with the initially activated adrenal–cortical system in OXYS rats resulted in a decrease in their stress resistance compared with Wistar rats. According to other authors, stress resistance in mammals is detectable even early in the life of long-lived mutants and is believed to be the cause, rather than an effect, of delayed aging processes.81 Many genes that affect life span have been shown recently to influence stress resistance. Besides, there is a complicated interaction between stress resistance, mitochondrial function, and life span that has not yet been fully studied.
Mitochondria are responsible for supplying energy, but they are also the main generator of ROS. The latter come from oxidative phosphorylation and are neutralized by several antioxidant enzymes, such as SOD, catalase, and cytochrome oxidase. Mitochondrial aberrations increasing with age were found in OXYS rats.82,83 Functional disturbances include a low phosphorylation rate during oxidation of glutamate, malate, and succinate. Biochemical changes manifested themselves as a decrease in cytochrome content and F1F0-ATP-synthetase activity and an increase in cytochrome b5 concentration. Ultrastructural changes include a decrease in mitochondrial volume and surface density and appearance of mitochondria with destroyed cristae and lysed matrix. Besides, our results indicate that the rate of O2 and H2O2 production in the electron transport chain of liver mitochondria is lower in OXYS rats. This state of mitochondria in OXYS rats may be described as mild uncoupling. There is strong evidence that mild uncoupling attenuates mitochondrial ROS production and protects against cellular damage. Thus, mild uncoupling is viewed by many researchers as a protective mechanism that minimizes ROS production in adult tissues and preserves mitochondrial function with age. We also concluded43 that mitochondrial mild uncoupling found in OXYS rats most likely represents an adaptive response to some pathological conditions. A slight decrease in the transmembrane electrical potential (Ψ) and intensification of nonphosphorylative respiration maintain the low level of the concentrations of O2 and single-electron reducing agents (e.g., ubisemiquinone radical QH), thereby reducing the risk of O2 formation. Normal aging is accompanied by similar changes. Moreover, the increase in proton conductance of the inner mitochondrial membrane is considered uncoupling for saving.84,85 Despite insignificant production of ROS, the inhibition of oxidative phosphorylation in mitochondria of OXYS rats and age-related energy deficiency shift the prooxidant–antioxidant balance toward prooxidants, which is followed by oxidative stress. OXYS rats have high content of cytochrome b5 on the outer mitochondrial membrane. It should be emphasized that ubiquinone possesses both prooxidant (reduction of O2 to O2–) and antioxidant activities (scavenging of OH– and lipid and tocopherol radicals). The decrease in ubiquinone content suppresses O2 production, but intensifies oxidation of lipids by free radicals.
Our data from an RNA-Seq study (RNA sequencing, also known as whole-transcriptome shotgun sequencing) showed changes of mitochondrial gene expression in the retina of OXYS rats.86 Significantly decreased expression was found for Cox8b (cytochrome c oxidase, subunit VIIIb), an enzyme of the mitochondrial respiratory chain that is also involved in the AD pathway.87 Furthermore, at the age of 3 mo, mRNA levels of many mitochondrial genes in the OXYS rats’ retinas are the same as those of 18-mo-old Wistar rats.
Thus, our data demonstrate that progressive mitochondrial dysfunction plays a major part in orchestrating accelerated aging of the brain and in the development of oxidative stress-associated disorders in OXYS rats, suggesting that mild mitochondrial uncoupling is highly effective at supporting in vivo antioxidant mechanisms.
The AD-Like Metabolic Pathway in OXYS Rats
Aβ is produced during normal cellular metabolism and is secreted to the extracellular milieu of the human brain, which is in contact with cerebrospinal fluid, where Aβ is also detectable.52 In other words, a physiological role of Aβ in the central nervous system has been documented.88 Aβ is involved in ion channel modulation, kinase activation, regulation of cholesterol transport, protection against metal-induced oxidative damage, synaptic plasticity, neuronal survival, and transcriptional regulation of AD-associated genes. The pathological accumulation of Aβ, the canonical dysfunction of AD, is associated with an imbalance between its production and clearance as a result of disease-causing changes in the processing of AβPP in the brain.89 AβPP is a transmembrane protein expressed in the adult brain, where it participates in synaptogenesis and synaptic plasticity.26 OXYS rats overproduce AβPP protein by 13 mo of age, and the protein levels remain high subsequently at 23 mo of age.41 Thus, histopathologically, Aβ accumulates in the hippocampus and cortex of OXYS rats with age (Fig. 4A–C). Total Aβ content increases by 333% between the ages 3 and 23 mo. It is noteworthy that in 3-mo-old OXYS rats, when the behavioral alterations become manifested and first signs of neurodegeneration become detectable by MRI and light microscopy, there are no differences between OXYS and Wistar rats in the Aβ1–42 and AβPP levels in the cortex and hippocampus. In general, in the rodent models of AD, Aβ accumulation occurs later than do anomalous behavioral patterns49,90,91 and significant loss of neurons.55

Figure 4. Time course of progression of AD-like pathology, as a percentage of either 0.5- or 3-mo-old OXYS rats in a group. (A) Rats exhibited a 47% loss of neurons in the CA1 region of the hippocampus between 0.5 and 12 mo of age. In addition, a 102% increase in the phospho-tau T181 level occurred between 3 and 23 mo of age. Total Aβ expression increased by 333% between 3 and 23 mo of age. (B) Aβ1–42 level is increased in the hippocampus of 12- and 23-mo-old but not 3-mo-old OXYS rats compared with disease-free (control) Wistar rats. Staining for Aβ1–42 (red). DAPI staining (blue) corresponds to cellular nuclei. Scale bars: 20 μm. (C) Photomicrographs demonstrate the Aβ deposits in the brain of OXYS rats detected by MOAB-2, clone 6C3. Scale bars: 50 μm. (D) Immunostaining for Tau (green) and phospho-tau T181 (red) in the cortex of 18-mo-old OXYS and Wistar rats. DAPI staining (blue) corresponds to cellular nuclei. The arrow shows colocalization of Tau and phospho-tau T181 in OXYS and Wistar rats, whereas the arrowhead shows phospho-tau T181 in OXYS rats. Scale bars: 20 μm. (E) The percentages of dead or damaged neurons in the CA1 region of the hippocampus in 15-mo-old OXYS rats are significantly greater compared with Wistar rats. Neurons were stained with cresyl violet. Adapted from reference 34 and Maksimova et al.
It should also be noted that the abnormal excessive accumulation of Aβ in the brain of OXYS rats by 13 mo of age happens after the appearance of mitochondrial dysfunction, which take place at 3 mo of age.82,83 It has been shown that the decreased expression of mitochondrial respiratory chain complexes (I and III) and the impairment of mitochondrial respiration are detected before accumulation of Aβ and formation of plaques,92,93 suggesting that mitochondrial dysfunction and oxidative stress may play a role at an early stage of AD.94,95 In addition, a stronger reduction in mitochondrial membrane potential and in ATP levels was found in double transgenic APP/PS1 (APPS/L/PS1 M141L) mice.96 Overall, these data imply that Aβ-dependent mitochondrial dysfunction starts early and accelerates substantially with age.97
At present, it is debated whether the development of AD and AMD involves common metabolic pathways.15-18 AMD is characterized by accumulation of extracellular deposits called drusen, in which Aβ is a key constituent. These deposits result in dysfunction of the transepithelial barrier, RPE atrophy, and subsequent degeneration of the neural retina. Aβ causes RPE alterations and pathological changes leading to retinal degeneration. It also serves as an activator of the complement cascade and is associated with activated macrophages. The presence of these cells may be indicative of an attempt to clear Aβ, but it has also been suggested that they may be accomplices in the development of AMD.98 Recently, we showed that Aβ1–42 accumulates with age in the OXYS retina, but its level is not elevated at the early stages of retinopathy (age 3 mo, Fig. 5). The Aβ1–42 level increased in retina of middle-aged and old OXYS rats, when these rats present with severe stages of the disease with pronounced neurodegenerative changes and a progressive loss of photoreceptors, which could possibly be a result of apoptosis.99 Several components of signaling pathways known to inhibit apoptosis (Birc3, Cdc2, Bcl2l10, Aven, and others) are downregulated in young OXYS rats.
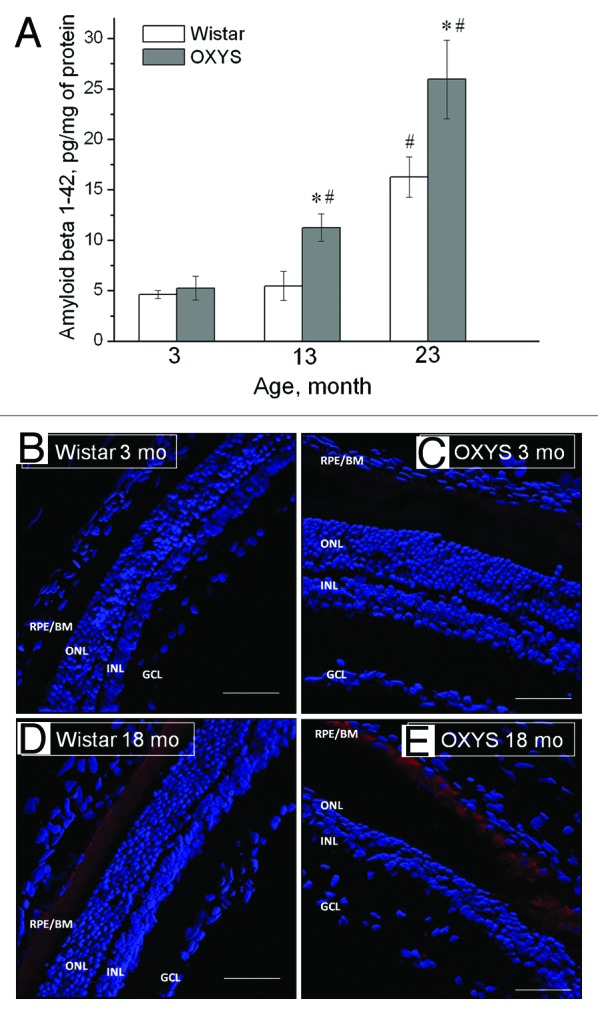
Figure 5. Aβ1–42 level is increased in the retina of OXYS rats compared with disease-free (control) Wistar rats. (A) Levels of Aβ1–42 of OXYS and Wistar rats at the age of 3, 13, and 23 mo (ELISA). *A statistically significant difference between the strains of the same age; #significant age-related differences compared with the previous age within the strain. (B–E) Confocal immunofluorescent images depict a weak Aβ (red) signal detected in a 3-mo-old Wistar rat (B), a stronger signal in a 3-mo-old OXYS rat (C), and strong staining in 18-mo-old rats of both strains (D and E). Cell nuclei are stained with DAPI (blue). The scale bar is 50 μm. RPE/BrM, retinal pigment epithelium/Bruch membrane; ONL, outer nuclear layer; INL, inner nuclear layer; GCL, ganglion cell layer. Adapted from reference 86.
In addition, the region of quantitative trait loci responsible for the manifestations of accelerated senescence in OXYS rats34 is enriched in genes participating in neurodegeneration. By means of the DNA arrays, in the OXYS retina, we found alterations in expression of several genes involved in the AD metabolic pathway: neurodegenerative changes and transcriptional changes of Picalm and Apba.99 PICALM (phosphatidylinositol clathrin assembly lymphoid–myeloid leukemia) appears to serve a critical function in clathrin-mediated endocytosis. Thus, both overexpression and underexpression of PICALM disrupt the processes of endocytosis, which plays an important role in the nervous system.100 Recent genome-wide association studies (GWAS) have found that PICALM mutations are significantly associated with AD, suggesting that PICALM has a hand in AβPP endocytosis and Aβ generation.101,102
APBA2 is a neuronal adaptor protein that interacts directly with AβPP and stabilizes it, thereby inhibiting the production of proteolytic fragments (Saito et al. 2008). There are no aberrations in the expression of APP in the OXYS retina. As noted above, the vast majority of AD cases are sporadic. A number of genetic mutations have been identified that result in familial forms of AD.103,104 The APP-processing pathway occupies the center stage in identification and characterization of the mutations that produce familial forms of AD, but only ~10% of these involve modifications in the APP gene itself.105 Although in familial forms of AD, APP mutations result in an increased Aβ production or aggregation, in sporadic AD, failure of the clearance mechanisms might be more important.106
There is a growing body of convincing evidence107 that any disturbances in immune defenses can accompany AD disease, not only upregulation, but also downregulation, which can be explained within the framework of the immunosenescence theory. Our data indicate that AD-like pathology and accelerated senescence in OXYS rats in general may derive from an imbalance of immune response, including alterations in inflammation.86 Pathway analysis of RNA-Seq data revealed significant downregulation of immune system genes in OXYS rats’ retinas that include many regulators of immunity, such as leukocyte markers, chemokines, cytokines, complement components, interferon-inducible proteins, and MHC (major histocompatibility complex) genes. The possible causes of the immune imbalance can be the accelerated thymus involution, a reduced level of delayed hypersensitivity reaction, and a decline of T cell-mediated immunity in OXYS rats.28-30 Progressive involution of the thymus, a hallmark of aging, leads to a decline of those immune functions that are related to T cell-dependent immunity.108,109 Because immunosenescence is progressive and generalized deterioration of immune functions that affects all cells and organs of the innate and adaptive immunity, it can be assumed that the immune imbalance (most likely provoked by accelerated thymic involution) can create a specific metabolic background for development and progression of the many manifestations of the accelerated senescence in OXYS rats. Also such altered cellular immune responses in OXYS rats may be the result of dysregulation of inter- and intracellular signal transduction. The questions still linger as to whether the observed decline in immune response is a cause or a consequence of the cross-talk among various impaired systems in OXYS rats, which we will study in the near future.
Another important characteristic of AD-like pathology observed in OXYS rats is hyperphosphorylation of tau.41 The pathophysiological relevance of tau phosphorylation in AD is still a subject of debate.104 Accumulating evidence indicates that the initiator of the disease process is alterations in the Aβ peptide.110 Aβ stimulates phosphorylation of the tau protein through glycogen synthase kinase, leading to neurofibrillary tangles: an effect directly causing synaptic damage and memory impairment. According to other authors, the aberrant accumulation of Aβ leads to mitochondrial dysfunction resulting in production of ROS, which, in turn, stimulates phosphorylation of tau.3,75 We uncovered hyperphosphorylation of tau in OXYS rats, noticeable before accumulation of Aβ (Fig. 4A). The significant increase in the level of the tau protein and its phosphorylation in the cortex and hippocampus were found already in 3-mo-old OXYS rats compared with age-matched Wistar rats; both of these parameters were elevated further at 13 and 23 mo of age.41 As shown in Figure 4A, the phospho-tau T181 level increases by 102% between 3 and 23 mo of age.
The time point of development of tau pathology in OXYS rats coincides with the manifestation of mitochondrial dysfunction, an imbalance in immune response, behavioral abnormalities, and first signs of neurodegenerative changes. The hyperphosphorylation of tau reduces its affinity for microtubules that can cause destabilization of the microtubules and an increase in the amount of unbound tau. These processes disrupt cellular transport systems and cause a synapse loss and, ultimately, many neurons die, thus leading to dysfunctional neural circuits and cognitive decline.110 It is likely that the synapse loss and neuronal dysfunction in young OXYS rats is due to impaired cellular transport and may contribute to early cognitive deficit and, later in the course of the disease, to an extensive neuronal loss.
What becomes a starting point of AD-like pathology in OXYS rats for the time being remains unclear. The significant tau and Aβ pathology associated with neurodegenerative diseases develop in OXYS rats with age. In this regard, rapidly developing rotenone-induced Parkinson-like pathology in OXYS rats is worth mentioning.67
Conclusion
Undoubtedly AD is a multifactorial disease and involves several etiopathogenic mechanisms. In the last decade, research efforts on aging and age-related deficits rapidly expanded and greatly advanced our understanding of the molecular and cellular mechanisms that result in neuronal dysfunction and death and of the genetic and environmental risk factors of AD. Nevertheless, many questions remain to be answered about how changes in the late-onset sporadic form of AD, which accounts for over 95% of all cases, lead to neurodegeneration and how to prevent or reverse these changes. To date, there has been only limited success in the development of an experimental animal model of sporadic AD.25
Our studies of OXYS rats demonstrated that this strain constitutes an interesting rodent model of sporadic form of AD. OXYS rats are characterized by the progressive cognitive and behavioral dysfunction associated with a loss of synapses, neuronal cell death, hyperphosphorylation of tau, and increased cerebral AβPP and Aβ levels. At the same time, the accumulation of Aβ occurs later than the above manifestations of accelerated brain aging in OXYS rats. This is consistent with the idea that Aβ may not be playing a direct or indirect role in cell death, but, in theory, that cell death has an impact on Aβ accumulation.
Although it has long been accepted that accumulation of oxidative damage to brain macromolecules is a major factor in the development of age-related deficits, there are still many paradoxical and inconsistent findings. When investigating tissue oxidative damage in OXYS rats, we did not find a cause–effect relationship between the manifestations of accelerated senescence, such as impairment of brain function, and the ubiquitous accumulation of oxidatively damaged macromolecules, which precedes the appearance of behavioral and cognitive deficits. Only indirect involvement of oxidative stress in the development of these deficits has been demonstrated, which can be described as a build-up of tension in the antioxidant system, e.g., increased SOD activity and decreased glutathione level, as well as activation of LPO processes in the relevant brain regions responsible for learning and memory.
A deficiency in mitochondrial function may underlie the abnormal oxidative and tissue stress responses observed in OXYS rats. It is now known that oxidative stress can be either the cause or the consequence of cerebral functional deficits. In the case of OXYS rats, it is more likely to be the consequence of other functional problems, and although antioxidants can improve some behavioral deficits in OXYS rats, their positive effect is most likely a result of the ability to improve mitochondrial function. The mitochondria-targeted antioxidant SkQ1 is the most effective antioxidant in reducing the severity of the manifestations of accelerated brain aging; for example, it slows down pathological accumulation of AβPP and Aβ and attenuates hyperphosphorylation of the tau protein in OXYS rats.40,41
Based on all of our results discussed above, we can conclude that mitochondrial dysfunction and the imbalance in immune response are key for the development of AD-like pathology in OXYS rats. We believe that further experiments with the OXYS rat strain will yield new insights into the complex mechanisms underlying AD and may lead to new therapeutic strategies to combat this disease.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by grants from the Russian Foundation for Basic Research (project #12-04-00091) and by grants from the presidium of the Russian Academy of Science (No. 14) and partially by Grants of the Government of the Russian Federation #2012-220-03-435 and #14.B25.31.0033.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/28255
References
- 1.Feng Y, Wang X. Antioxidant therapies for Alzheimer’s disease. Oxid Med Cell Longev. 2012;2012:472932. doi: 10.1155/2012/472932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med. 2010;362:329–44. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- 3.Morley JE, Armbrecht HJ, Farr SA, Kumar VB. The senescence accelerated mouse (SAMP8) as a model for oxidative stress and Alzheimer’s disease. Biochim Biophys Acta. 2012;1822:650–6. doi: 10.1016/j.bbadis.2011.11.015. [DOI] [PubMed] [Google Scholar]
- 4.Teich AF, Arancio O. Is the amyloid hypothesis of Alzheimer’s disease therapeutically relevant? Biochem J. 2012;446:165–77. doi: 10.1042/BJ20120653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liao FF, Wang R, Park EA. Repression of Alzheimer’s beta-secretase. Aging (Albany NY) 2013;5:789–90. doi: 10.18632/aging.100612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Swerdlow RH, Burns JM, Khan SM. The Alzheimer’s disease mitochondrial cascade hypothesis. J Alzheimers Dis. 2010;20(Suppl 2):S265–79. doi: 10.3233/JAD-2010-100339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Swerdlow RH, Khan SMA. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med Hypotheses. 2004;63:8–20. doi: 10.1016/j.mehy.2003.12.045. [DOI] [PubMed] [Google Scholar]
- 8.Shang YC, Chong ZZ, Wang S, Maiese K. Prevention of β-amyloid degeneration of microglia by erythropoietin depends on Wnt1, the PI 3-K/mTOR pathway, Bad, and Bcl-xL. Aging (Albany NY) 2012;4:187–201. doi: 10.18632/aging.100440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Streit WJ, Braak H, Xue QS, Bechmann I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol. 2009;118:475–85. doi: 10.1007/s00401-009-0556-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Streit WJ, Xue QS. Life and death of microglia. J Neuroimmune Pharmacol. 2009;4:371–9. doi: 10.1007/s11481-009-9163-5. [DOI] [PubMed] [Google Scholar]
- 11.Streit WJ, Xue QS. The Brain’s Aging Immune System. Aging Dis. 2010;1:254–61. [PMC free article] [PubMed] [Google Scholar]
- 12.Streit WJ, Xue QS. Alzheimer’s disease, neuroprotection, and CNS immunosenescence. Front Pharmacol. 2012;3:138. doi: 10.3389/fphar.2012.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Streit WJ, Xue QS. Microglial senescence. CNS Neurol Disord Drug Targets. 2013;12:763–7. doi: 10.2174/18715273113126660176. [DOI] [PubMed] [Google Scholar]
- 14.Reddy PH, Tripathi R, Troung Q, Tirumala K, Reddy TP, Anekonda V, Shirendeb UP, Calkins MJ, Reddy AP, Mao P, et al. Abnormal mitochondrial dynamics and synaptic degeneration as early events in Alzheimer’s disease: implications to mitochondria-targeted antioxidant therapeutics. Biochim Biophys Acta. 2012;1822:639–49. doi: 10.1016/j.bbadis.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chiu C, Miller MC, Caralopoulos IN, Worden MS, Brinker T, Gordon ZN, Johanson CE, Silverberg GD. Temporal course of cerebrospinal fluid dynamics and amyloid accumulation in the aging rat brain from three to thirty months. Fluids Barriers CNS. 2012;9:3. doi: 10.1186/2045-8118-9-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dasari B, Prasanthi JR, Marwarha G, Singh BB, Ghribi O. Cholesterol-enriched diet causes age-related macular degeneration-like pathology in rabbit retina. BMC Ophthalmol. 2011;11:22. doi: 10.1186/1471-2415-11-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaarniranta K, Salminen A, Haapasalo A, Soininen H, Hiltunen M. Age-related macular degeneration (AMD): Alzheimer’s disease in the eye? J Alzheimers Dis. 2011;24:615–31. doi: 10.3233/JAD-2011-101908. [DOI] [PubMed] [Google Scholar]
- 18.Ohno-Matsui K. Parallel findings in age-related macular degeneration and Alzheimer’s disease. Prog Retin Eye Res. 2011;30:217–38. doi: 10.1016/j.preteyeres.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 19.Laws SM, Hone E, Gandy S, Martins RN. Expanding the association between the APOE gene and the risk of Alzheimer’s disease: possible roles for APOE promoter polymorphisms and alterations in APOE transcription. J Neurochem. 2003;84:1215–36. doi: 10.1046/j.1471-4159.2003.01615.x. [DOI] [PubMed] [Google Scholar]
- 20.Loeffler DA, Camp DM, Bennett DA. Plaque complement activation and cognitive loss in Alzheimer’s disease. J Neuroinflammation. 2008;5:9. doi: 10.1186/1742-2094-5-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Braidy N, Muñoz P, Palacios AG, Castellano-Gonzalez G, Inestrosa NC, Chung RS, Sachdev P, Guillemin GJ. Recent rodent models for Alzheimer’s disease: clinical implications and basic research. J Neural Transm. 2012;119:173–95. doi: 10.1007/s00702-011-0731-5. [DOI] [PubMed] [Google Scholar]
- 22.Sabbagh JJ, Kinney JW, Cummings JL. Animal systems in the development of treatments for Alzheimer’s disease: challenges, methods, and implications. Neurobiol Aging. 2013;34:169–83. doi: 10.1016/j.neurobiolaging.2012.02.027. [DOI] [PubMed] [Google Scholar]
- 23.Savonenko AV, Melnikova T, Hiatt A, Li T, Worley PF, Troncoso JC, Wong PC, Price DL. Alzheimer’s therapeutics: translation of preclinical science to clinical drug development. Neuropsychopharmacology. 2012;37:261–77. doi: 10.1038/npp.2011.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tang J, Ghosh A. Treating transgenic Alzheimer mice with a β-secretase inhibitor, what have we learned? Aging (Albany NY) 2011;3:14–6. doi: 10.18632/aging.100267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iqbal K, Bolognin S, Wang X, Basurto-Islas G, Blanchard J, Tung YC. Animal models of the sporadic form of Alzheimer’s disease: focus on the disease and not just the lesions. J Alzheimers Dis. 2013;37:469–74. doi: 10.3233/JAD-130827. [DOI] [PubMed] [Google Scholar]
- 26.Del Valle J, Duran-Vilaregut J, Manich G, Casadesús G, Smith MA, Camins A, Pallàs M, Pelegrí C, Vilaplana J. Early amyloid accumulation in the hippocampus of SAMP8 mice. J Alzheimers Dis. 2010;19:1303–15. doi: 10.3233/JAD-2010-1321. [DOI] [PubMed] [Google Scholar]
- 27.Solov’eva NA, Morozkova TS, Salganik RI. [Development of a rat subline with symptoms of hereditary galactosemia and study of its biochemical characteristics] Genetika. 1975;11:63–71. [PubMed] [Google Scholar]
- 28.Markova EV, Obukhova LA, Kolosova NG. Activity of cell immune response and open field behavior in Wistar and OXYS rats. Bull Exp Biol Med. 2003;136:377–9. doi: 10.1023/B:BEBM.0000010957.87077.ae. [DOI] [PubMed] [Google Scholar]
- 29.Obukhova LA, Skulachev VP, Kolosova NG. Mitochondria-targeted antioxidant SkQ1 inhibits age-dependent involution of the thymus in normal and senescence-prone rats. Aging (Albany NY) 2009;1:389–401. doi: 10.18632/aging.100043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Obukhova LA, Vais VB, Bakeeva LE, Sergeeva SV, Kolosova NG. Structural and functional basis for accelerated thymic involution in OXYS rats. Adv Gerontol. 2013;26:229–35. [PubMed] [Google Scholar]
- 31.Markovets AM, Fursova AZ, Kolosova NG. Therapeutic action of the mitochondria-targeted antioxidant SkQ1 on retinopathy in OXYS rats linked with improvement of VEGF and PEDF gene expression. PLoS One. 2011;6:e21682. doi: 10.1371/journal.pone.0021682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Markovets AM, Saprunova VB, Zhdankina AA, Fursova AZh, Bakeeva LE, Kolosova NG. Alterations of retinal pigment epithelium cause AMD-like retinopathy in senescence-accelerated OXYS rats. Aging (Albany NY) 2011;3:44–54. doi: 10.18632/aging.100243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kolosova NG, Muraleva NA, Zhdankina AA, Stefanova NA, Fursova AZ, Blagosklonny MV. Prevention of age-related macular degeneration-like retinopathy by rapamycin in rats. Am J Pathol. 2012;181:472–7. doi: 10.1016/j.ajpath.2012.04.018. [DOI] [PubMed] [Google Scholar]
- 34.Korbolina EE, Kozhevnikova OS, Stefanova NA, Kolosova NG. Quantitative trait loci on chromosome 1 for cataract and AMD-like retinopathy in senescence-accelerated OXYS rats. Aging (Albany NY) 2012;4:49–59. doi: 10.18632/aging.100427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bobko AA, Sergeeva SV, Bagryanskaya EG, Markel AL, Khramtsov VV, Reznikov VA, Kolosova NG. 19F NMR measurements of NO production in hypertensive ISIAH and OXYS rats. Biochem Biophys Res Commun. 2005;330:367–70. doi: 10.1016/j.bbrc.2005.02.166. [DOI] [PubMed] [Google Scholar]
- 36.Muraleva NA, Ofitserov EN, Tikhonov VP, Kolosova NG. Efficacy of glucosamine alendronate alone & in combination with dihydroquercetin for treatment of osteoporosis in animal model. Indian J Med Res. 2012;135:221–7. [PMC free article] [PubMed] [Google Scholar]
- 37.Muraleva NA, Sadovoĭ MA, Kolosova NG. [Development of osteoporosis in prematurely aging OXYS rats] Adv Gerontol. 2010;23:233–42. [PubMed] [Google Scholar]
- 38.Muraleva NA, Sadovoĭ MA, Kolosova NG. [Effect of alendronate on bone tissue status of senescence-accelerated OXYS rats] Adv Gerontol. 2011;24:143–6. [PubMed] [Google Scholar]
- 39.Rykova VI, Leberfarb EY, Stefanova NA, Shevelev OB, Dymshits GM, Kolosova NG. Brain proteoglycans in postnatal development and during behavior decline in senescence-accelerated OXYS rats. Adv Gerontol. 2011;24:234–43. [PubMed] [Google Scholar]
- 40.Stefanova NA, Fursova AZh, Kolosova NG. Behavioral effects induced by mitochondria-targeted antioxidant SkQ1 in Wistar and senescence-accelerated OXYS rats. J Alzheimers Dis. 2010;21:479–91. doi: 10.3233/JAD-2010-091675. [DOI] [PubMed] [Google Scholar]
- 41.Stefanova NA, Muraleva NA, Skulachev VP, Kolosova NG. Alzheimer’s disease-like pathology in senescence-accelerated OXYS rats can be partially retarded with mitochondria-targeted antioxidant SkQ1. J Alzheimers Dis. 2014;38:681–94. doi: 10.3233/JAD-131034. [DOI] [PubMed] [Google Scholar]
- 42.Stefanova NA, Fursova AZh, Sarsenbaev KN, Kolosova NG. Effects of Cistanche deserticola on behavior and signs of cataract and retinopathy in senescence-accelerated OXYS rats. J Ethnopharmacol. 2011;138:624–32. doi: 10.1016/j.jep.2011.10.017. [DOI] [PubMed] [Google Scholar]
- 43.Kolosova NG, Stefanova NA, Sergeeva SV. OXYS rats: a prospective model for evaluation of antioxidant availability in prevention and therapy of accelerated aging and age-related cognitive decline. In: R. GQaM, ed. Handbook of Cognitive Aging: Causes, Proceses. NY: Nova Science Publishers, 2009:47-82. [Google Scholar]
- 44.Boguszewski P, Zagrodzka J. Emotional changes related to age in rats--a behavioral analysis. Behav Brain Res. 2002;133:323–32. doi: 10.1016/S0166-4328(02)00018-9. [DOI] [PubMed] [Google Scholar]
- 45.Tanisawa K, Mikami E, Fuku N, Honda Y, Honda S, Ohsawa I, Ito M, Endo S, Ihara K, Ohno K, et al. Exome sequencing of senescence-accelerated mice (SAM) reveals deleterious mutations in degenerative disease-causing genes. BMC Genomics. 2013;14:248. doi: 10.1186/1471-2164-14-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jaszberenyi M, Rick FG, Szalontay L, Block NL, Zarandi M, Cai RZ, Schally AV. Beneficial effects of novel antagonists of GHRH in different models of Alzheimer’s disease. Aging (Albany NY) 2012;4:755–67. doi: 10.18632/aging.100504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Loskutova LV, Kolosova NG. Emotional state and one-trial learning in OXYS rats with hereditarily elevated production of oxygen radicals. Bull Exp Biol Med. 2000;130:746–8. doi: 10.1007/BF02766083. [DOI] [PubMed] [Google Scholar]
- 48.Kolosova NG, Shcheglova TV, Sergeeva SV, Loskutova LV. Long-term antioxidant supplementation attenuates oxidative stress markers and cognitive deficits in senescent-accelerated OXYS rats. Neurobiol Aging. 2006;27:1289–97. doi: 10.1016/j.neurobiolaging.2005.07.022. [DOI] [PubMed] [Google Scholar]
- 49.Van Dam D, D’Hooge R, Staufenbiel M, Van Ginneken C, Van Meir F, De Deyn PP. Age-dependent cognitive decline in the APP23 model precedes amyloid deposition. Eur J Neurosci. 2003;17:388–96. doi: 10.1046/j.1460-9568.2003.02444.x. [DOI] [PubMed] [Google Scholar]
- 50.Fujimura RK, Reiner T, Ma F, Phillips V, de las Pozas A, Dickson DW, Roos BA, Howard GA, Perez-Stable C. Changes in the expression of genes associated with intraneuronal amyloid-beta and tau in Alzheimer’s disease. J Alzheimers Dis. 2010;19:97–109. doi: 10.3233/JAD-2010-1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Beregovoy NA, Sorokina NS, Starostina MV, Kolosova NG. Age-specific peculiarities of formation of long-term posttetanic potentiation in OXYS rats. Bull Exp Biol Med. 2011;151:71–3. doi: 10.1007/s10517-011-1262-7. [DOI] [PubMed] [Google Scholar]
- 52.Parihar MS, Brewer GJ. Amyloid-β as a modulator of synaptic plasticity. J Alzheimers Dis. 2010;22:741–63. doi: 10.3233/JAD-2010-101020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mu Y, Gage FH. Adult hippocampal neurogenesis and its role in Alzheimer’s disease. Mol Neurodegener. 2011;6:85. doi: 10.1186/1750-1326-6-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brun A, Englund E. Regional pattern of degeneration in Alzheimer’s disease: neuronal loss and histopathological grading. Histopathology. 1981;5:549–64. doi: 10.1111/j.1365-2559.1981.tb01818.x. [DOI] [PubMed] [Google Scholar]
- 55.Wright AL, Zinn R, Hohensinn B, Konen LM, Beynon SB, Tan RP, Clark IA, Abdipranoto A, Vissel B. Neuroinflammation and Neuronal Loss Precede Ab Plaque Deposition in the hAPP-J20 Mouse Model of Alzheimer's Disease. PLoS ONE. 2013;•••:8. doi: 10.1371/journal.pone.0059586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu Y, He J, Zhang Y, Luo H, Zhu S, Yang Y, Zhao T, Wu J, Huang Y, Kong J, et al. Increased hippocampal neurogenesis in the progressive stage of Alzheimer’s disease phenotype in an APP/PS1 double transgenic mouse model. Hippocampus. 2009;19:1247–53. doi: 10.1002/hipo.20587. [DOI] [PubMed] [Google Scholar]
- 57.Perry EK, Johnson M, Ekonomou A, Perry RH, Ballard C, Attems J. Neurogenic abnormalities in Alzheimer’s disease differ between stages of neurogenesis and are partly related to cholinergic pathology. Neurobiol Dis. 2012;47:155–62. doi: 10.1016/j.nbd.2012.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A. Tau is essential to beta -amyloid-induced neurotoxicity. Proc Natl Acad Sci U S A. 2002;99:6364–9. doi: 10.1073/pnas.092136199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jin K, Peel AL, Mao XO, Xie L, Cottrell BA, Henshall DC, Greenberg DA. Increased hippocampal neurogenesis in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2004;101:343–7. doi: 10.1073/pnas.2634794100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Song B, Davis K, Liu XS, Lee HG, Smith M, Liu X. Inhibition of Polo-like kinase 1 reduces beta-amyloid-induced neuronal cell death in Alzheimer’s disease. Aging (Albany NY) 2011;3:846–51. doi: 10.18632/aging.100382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sergeeva S, Bagryanskaya E, Korbolina E, Kolosova N. Development of behavioural dysfunctions in accelerated-senescence OXYS rats is associated with early postnatal alterations in brain phosphate metabolism. Exp Gerontol. 2006;41:141–50. doi: 10.1016/j.exger.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 62.Agafonova IG, Kolosova NG, Mishchenko NP, Chaikina EL, Stonik VA. Effect of histochrome on brain vessels and research and exploratory activity of senescence-accelerated OXYS rats. Bull Exp Biol Med. 2007;143:467–71. doi: 10.1007/s10517-007-0158-z. [DOI] [PubMed] [Google Scholar]
- 63.Agafonova IG, Kotel’nikov VN, Mischenko NP, Kolosova NG. Evaluation of effects of histochrome and mexidol on structural and functional characteristics of the brain in senescence-accelerated OXYS rats by magnetic resonance imaging. Bull Exp Biol Med. 2011;150:739–43. doi: 10.1007/s10517-011-1238-7. [DOI] [PubMed] [Google Scholar]
- 64.Zhdankina AA, Fursova AZh, Logvinov SV, Kolosova NG. Clinical and morphological characteristics of chorioretinal degeneration in early aging OXYS rats. Bull Exp Biol Med. 2008;146:455–8. doi: 10.1007/s10517-009-0298-4. [DOI] [PubMed] [Google Scholar]
- 65.Stefanova NA, Zhdankina AA, Fursova AZh, Kolosova NG. [Potential of melatonin for prevention of age-related macular degeneration: experimental study] Adv Gerontol. 2013;26:122–9. [PubMed] [Google Scholar]
- 66.Sagare AP, Bell RD, Zlokovic BV. Neurovascular dysfunction and faulty amyloid β-peptide clearance in Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2 doi: 10.1101/cshperspect.a011452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kolosova NG, Akulov AE, Stefanova NA, Moshkin MP, Savelov AA, Koptyug IV, Panov AV, Vavilin VA. Effect of malate on the development of rotenone-induced brain changes in Wistar and OXYS rats: An MRI study. Doklady biological sciences: Proceedings of the Academy of Sciences of the USSR, Biological sciences sections / translated from Russian. 2011;437:72–5. doi: 10.1134/S0012496611020049. [DOI] [PubMed] [Google Scholar]
- 68.Andrews-Hanna JR, Snyder AZ, Vincent JL, Lustig C, Head D, Raichle ME, Buckner RL. Disruption of large-scale brain systems in advanced aging. Neuron. 2007;56:924–35. doi: 10.1016/j.neuron.2007.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.de la Torre JC. Cerebral hypoperfusion, capillary degeneration, and development of Alzheimer disease. Alzheimer Dis Assoc Disord. 2000;14(Suppl 1):S72–81. doi: 10.1097/00002093-200000001-00012. [DOI] [PubMed] [Google Scholar]
- 70.Gökcay A, Kitis O, Ekmekci O, Karasoy H, Sener RN. Proton MR spectroscopy in Rett syndrome. Comput Med Imaging Graph. 2002;26:271–5. doi: 10.1016/S0895-6111(02)00016-2. [DOI] [PubMed] [Google Scholar]
- 71.Díez-Vives C, Gay M, García-Matas S, Comellas F, Carrascal M, Abian J, Ortega-Aznar A, Cristòfol R, Sanfeliu C. Proteomic study of neuron and astrocyte cultures from senescence-accelerated mouse SAMP8 reveals degenerative changes. J Neurochem. 2009;111:945–55. doi: 10.1111/j.1471-4159.2009.06374.x. [DOI] [PubMed] [Google Scholar]
- 72.Navarro A, Boveris A. Brain mitochondrial dysfunction in aging, neurodegeneration, and Parkinson’s disease. Front Aging Neurosci. 2010;2 doi: 10.3389/fnagi.2010.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McManus MJ, Murphy MP, Franklin JL. The mitochondria-targeted antioxidant MitoQ prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer’s disease. J Neurosci. 2011;31:15703–15. doi: 10.1523/JNEUROSCI.0552-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chakrabarti S, Munshi S, Banerjee K, Thakurta IG, Sinha M, Bagh MB. Mitochondrial Dysfunction during Brain Aging: Role of Oxidative Stress and Modulation by Antioxidant Supplementation. Aging Dis. 2011;2:242–56. [PMC free article] [PubMed] [Google Scholar]
- 75.Skulachev VP. Mitochondria-targeted antioxidants as promising drugs for treatment of age-related brain diseases. J Alzheimers Dis. 2012;28:283–9. doi: 10.3233/JAD-2011-111391. [DOI] [PubMed] [Google Scholar]
- 76.Massaad CA, Pautler RG, Klann E. Mitochondrial superoxide: a key player in Alzheimer’s disease. Aging (Albany NY) 2009;1:758–61. doi: 10.18632/aging.100088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kolosova NG, Grishanova AIu, Krysanova ZhS, Zueva TV, Sidorova IuA, Sinitsyna OI. [Age-related changes in protein and lipid oxidation in the liver of prematurely aging rats OXYS] Biomed Khim. 2004;50:73–8. [PubMed] [Google Scholar]
- 78.Kolosova NG, Shcheglova TV, Amstislavskaya TG, Loskutova LV. Comparative analysis of LPO products in brain structures of Wistar and OXYS rats of different age. Bull Exp Biol Med. 2003;135:593–6. doi: 10.1023/A:1025445822566. [DOI] [PubMed] [Google Scholar]
- 79.Kobayashi M, Takeda M, Sato T, Yamazaki Y, Kaneko K, Ito K, Kato H, Inaba H. In vivo imaging of spontaneous ultraweak photon emission from a rat’s brain correlated with cerebral energy metabolism and oxidative stress. Neurosci Res. 1999;34:103–13. doi: 10.1016/S0168-0102(99)00040-1. [DOI] [PubMed] [Google Scholar]
- 80.Federico A, Cardaioli E, Da Pozzo P, Formichi P, Gallus GN, Radi E. Mitochondria, oxidative stress and neurodegeneration. J Neurol Sci. 2012;322:254–62. doi: 10.1016/j.jns.2012.05.030. [DOI] [PubMed] [Google Scholar]
- 81.Lithgrow GJ, Miller RA. Genetic modulation of life span: a role for stress resistance: Determination of aging rate by coordinated resistance to multiple forms of stress. In: Guarente LPP, L.; Wallace, D.C., ed. Molecular biology of aging. NY: Cold Spring Harbor Laboratory Press, 2008:427-81. [Google Scholar]
- 82.Shabalina IG, Kolosova NG, Grishanova AIu, Solov’ev VN, Salganik RI, Solov’eva NA. [Oxidative phosphorylation activity, F0F1-ATPase and level of liver mitochondrial cytochromes in rats with congenitally increased ability for free radical formation] Biokhimiia. 1995;60:2045–52. [PubMed] [Google Scholar]
- 83.Kolosova NG, Aidagulova SV, Nepomnyashchikh GI, Shabalina IG, Shalbueva NI. Dynamics of structural and functional changes in hepatocyte mitochondria of senescence-accelerated OXYS rats. Bull Exp Biol Med. 2001;132:814–9. doi: 10.1023/A:1013014919721. [DOI] [PubMed] [Google Scholar]
- 84.Amara CE, Shankland EG, Jubrias SA, Marcinek DJ, Kushmerick MJ, Conley KE. Mild mitochondrial uncoupling impacts cellular aging in human muscles in vivo. Proc Natl Acad Sci U S A. 2007;104:1057–62. doi: 10.1073/pnas.0610131104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Caldeira da Silva CC, Cerqueira FM, Barbosa LF, Medeiros MH, Kowaltowski AJ. Mild mitochondrial uncoupling in mice affects energy metabolism, redox balance and longevity. Aging Cell. 2008;7:552–60. doi: 10.1111/j.1474-9726.2008.00407.x. [DOI] [PubMed] [Google Scholar]
- 86.Kozhevnikova OS, Korbolina EE, Ershov NI, Kolosova NG. Rat retinal transcriptome: effects of aging and AMD-like retinopathy. Cell Cycle. 2013;12:1745–61. doi: 10.4161/cc.24825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Maurer I, Zierz S, Möller HJ. A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients. Neurobiol Aging. 2000;21:455–62. doi: 10.1016/S0197-4580(00)00112-3. [DOI] [PubMed] [Google Scholar]
- 88.Stanga S, Lanni C, Govoni S, Uberti D, D’Orazi G, Racchi M. Unfolded p53 in the pathogenesis of Alzheimer’s disease: is HIPK2 the link? Aging (Albany NY) 2010;2:545–54. doi: 10.18632/aging.100205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science. 2010;330:1774. doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Capetillo-Zarate E, Staufenbiel M, Abramowski D, Haass C, Escher A, Stadelmann C, Yamaguchi H, Wiestler OD, Thal DR. Selective vulnerability of different types of commissural neurons for amyloid beta-protein-induced neurodegeneration in APP23 mice correlates with dendritic tree morphology. Brain. 2006;129:2992–3005. doi: 10.1093/brain/awl176. [DOI] [PubMed] [Google Scholar]
- 91.Rijal Upadhaya A, Capetillo-Zarate E, Kosterin I, Abramowski D, Kumar S, Yamaguchi H, Walter J, Fändrich M, Staufenbiel M, Thal DR. Dispersible amyloid β-protein oligomers, protofibrils, and fibrils represent diffusible but not soluble aggregates: their role in neurodegeneration in amyloid precursor protein (APP) transgenic mice. Neurobiol Aging. 2012;33:2641–60. doi: 10.1016/j.neurobiolaging.2011.12.032. [DOI] [PubMed] [Google Scholar]
- 92.Chen G, Chen KS, Knox J, Inglis J, Bernard A, Martin SJ, Justice A, McConlogue L, Games D, Freedman SB, et al. A learning deficit related to age and beta-amyloid plaques in a mouse model of Alzheimer’s disease. Nature. 2000;408:975–9. doi: 10.1038/35046031. [DOI] [PubMed] [Google Scholar]
- 93.Reddy PH, McWeeney S, Park BS, Manczak M, Gutala RV, Partovi D, Jung Y, Yau V, Searles R, Mori M, et al. Gene expression profiles of transcripts in amyloid precursor protein transgenic mice: up-regulation of mitochondrial metabolism and apoptotic genes is an early cellular change in Alzheimer’s disease. Hum Mol Genet. 2004;13:1225–40. doi: 10.1093/hmg/ddh140. [DOI] [PubMed] [Google Scholar]
- 94.Caspersen C, Wang N, Yao J, Sosunov A, Chen X, Lustbader JW, Xu HW, Stern D, McKhann G, Yan SD. Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 2005;19:2040–1. doi: 10.1096/fj.05-3735fje. [DOI] [PubMed] [Google Scholar]
- 95.Keil U, Bonert A, Marques CA, Scherping I, Weyermann J, Strosznajder JB, Müller-Spahn F, Haass C, Czech C, Pradier L, et al. Amyloid beta-induced changes in nitric oxide production and mitochondrial activity lead to apoptosis. J Biol Chem. 2004;279:50310–20. doi: 10.1074/jbc.M405600200. [DOI] [PubMed] [Google Scholar]
- 96.Eckert A, Hauptmann S, Scherping I, Rhein V, Müller-Spahn F, Götz J, Müller WE. Soluble beta-amyloid leads to mitochondrial defects in amyloid precursor protein and tau transgenic mice. Neurodegener Dis. 2008;5:157–9. doi: 10.1159/000113689. [DOI] [PubMed] [Google Scholar]
- 97.Quintanilla RA, Orellana JA, von Bernhardi R. Understanding risk factors for Alzheimer’s disease: interplay of neuroinflammation, connexin-based communication and oxidative stress. Arch Med Res. 2012;43:632–44. doi: 10.1016/j.arcmed.2012.10.016. [DOI] [PubMed] [Google Scholar]
- 98.Hoh Kam J, Lenassi E, Jeffery G. Viewing ageing eyes: diverse sites of amyloid Beta accumulation in the ageing mouse retina and the up-regulation of macrophages. PLoS One. 2010;5 doi: 10.1371/journal.pone.0013127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kozhevnikova OS, Korbolina EE, Stefanova NA, Muraleva NA, Orlov YL, Kolosova NG. Association of AMD-like retinopathy development with an Alzheimer’s disease metabolic pathway in OXYS rats. Biogerontology. 2013;14:753–62. doi: 10.1007/s10522-013-9439-2. [DOI] [PubMed] [Google Scholar]
- 100.Baig S, Joseph SA, Tayler H, Abraham R, Owen MJ, Williams J, Kehoe PG, Love S. Distribution and expression of picalm in Alzheimer disease. J Neuropathol Exp Neurol. 2010;69:1071–7. doi: 10.1097/NEN.0b013e3181f52e01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Xiao Q, Gil SC, Yan P, Wang Y, Han S, Gonzales E, Perez R, Cirrito JR, Lee JM. Role of phosphatidylinositol clathrin assembly lymphoid-myeloid leukemia (PICALM) in intracellular amyloid precursor protein (APP) processing and amyloid plaque pathogenesis. J Biol Chem. 2012;287:21279–89. doi: 10.1074/jbc.M111.338376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Williams A, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009;41:1088–93. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tanzi RE, Bertram L. Twenty years of the Alzheimer’s disease amyloid hypothesis: a genetic perspective. Cell. 2005;120:545–55. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 104.Kumar S, Walter J. Phosphorylation of amyloid beta (Aβ) peptides - a trigger for formation of toxic aggregates in Alzheimer’s disease. Aging (Albany NY) 2011;3:803–12. doi: 10.18632/aging.100362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Randall AD, Witton J, Booth C, Hynes-Allen A, Brown JT. The functional neurophysiology of the amyloid precursor protein (APP) processing pathway. Neuropharmacology. 2010;59:243–67. doi: 10.1016/j.neuropharm.2010.02.011. [DOI] [PubMed] [Google Scholar]
- 106.Marcello E, Epis R, Saraceno C, Di Luca M. Synaptic dysfunction in Alzheimer’s disease. Adv Exp Med Biol. 2012;970:573–601. doi: 10.1007/978-3-7091-0932-8_25. [DOI] [PubMed] [Google Scholar]
- 107.Stern JH. Mini-Chaperones for Early AMD. Invest Ophthalmol Vis Sci. 2013;54:2799. doi: 10.1167/iovs.13-12101. [DOI] [PubMed] [Google Scholar]
- 108.Gruver AL, Hudson LL, Sempowski GD. Immunosenescence of ageing. J Pathol. 2007;211:144–56. doi: 10.1002/path.2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Aw D, Silva AB, Palmer DB. Is thymocyte development functional in the aged? Aging (Albany NY) 2009;1:146–53. doi: 10.18632/aging.100027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Spires-Jones TL, Stoothoff WH, de Calignon A, Jones PB, Hyman BT. Tau pathophysiology in neurodegeneration: a tangled issue. Trends Neurosci. 2009;32:150–9. doi: 10.1016/j.tins.2008.11.007. [DOI] [PubMed] [Google Scholar]