

Abstract
Oncogenic mutation or misregulation of small GTPases in the Ras and Rho families can promote unregulated cell cycle progression in cancer. Post-translational modification by prenylation of these GTPases allows them to signal at the cell membrane. Splice variants of SmgGDS, named SmgGDS-607 and SmgGDS-558, promote the prenylation and membrane trafficking of multiple Ras and Rho family members, which makes SmgGDS a potentially important regulator of the cell cycle. Surprisingly little is known about how SmgGDS-607 and SmgGDS-558 affect cell cycle-regulatory proteins in cancer, even though SmgGDS is overexpressed in multiple types of cancer. To examine the roles of SmgGDS splice variants in the cell cycle, we compared the effects of the RNAi-mediated depletion of SmgGDS-558 vs. SmgGDS-607 on cell cycle progression and the expression of cyclin D1, p27, and p21 in pancreatic, lung, and breast cancer cell lines. We show for the first time that SmgGDS promotes proliferation of pancreatic cancer cells, and we demonstrate that SmgGDS-558 plays a greater role than SmgGDS-607 in cell cycle progression as well as promoting cyclin D1 and suppressing p27 expression in multiple types of cancer. Silencing both splice variants of SmgGDS in the cancer cell lines produces an alternative signaling profile compared with silencing SmgGDS-558 alone. We also show that loss of both SmgGDS-607 and SmgGDS-558 simultaneously decreases tumorigenesis of NCI-H1703 non-small cell lung carcinoma (NSCLC) xenografts in mice. These findings indicate that SmgGDS promotes cell cycle progression in multiple types of cancer, making SmgGDS a valuable target for cancer therapeutics.
Keywords: SmgGDS, Rap1GDS1, pancreatic cancer, breast cancer, lung cancer, GTPase, cell cycle, mouse tumorigenesis, proliferation, RNAi
Introduction
Cancer is the second leading cause of death in the United States, resulting in over 580 000 expected deaths this year.1 Over 41% of these deaths will be caused by pancreatic, lung, and breast cancers combined.1 Mutations and/or misregulation of multiple members of the Ras and Rho families of small GTPases have been reported to be involved in the malignancy of these types of cancers.2-7 One promising area of research in cancer treatment is inhibiting the signaling networks controlled by these GTPases, either through inhibition of the GTPase itself8,9 or by inhibiting their effectors and activators.10 GTPases are described as molecular switches that bind GTP in an active state and hydrolyze GTP to GDP, resulting in an inactive state.11 Mutation of GTPases such as K-Ras can result in a constitutively active (CA) mutant that does not hydrolyze GTP effectively and therefore continuously generates proliferative and anti-apoptosis signaling cascades that contribute to cancer development and progression.12,13 These CA mutations of a GTPase often occur as a result of a single nucleotide mutation, which makes it difficult to target in cancer therapeutics.14 Furthermore, targeting downstream effectors of a GTPase for therapy is difficult due to the high level of redundant cross-talk between signaling pathways.9
An important post-translational modification for the active signaling of a GTPase is its prenylation, which adds a hydrophobic farnesyl or geranylgeranyl moiety to the C-terminal cysteine of the GTPase, thus allowing it to anchor at the plasma membrane, where it can interact with effector proteins.15,16 GTPases such as K-Ras, Rac1, RhoA, and Rap1 have a string of basic amino acids just upstream of the site of prenylation known as the polybasic region (PBR), and this PBR provides a second signal for localization to the negatively charged plasma membrane.15 K-Ras is farnesylated by interacting with a farnesyltransferase (FTase), whereas Rap1, Rac1, and RhoA are geranylgeranylated by interacting with a geranylgeranyltransferase (GGTase).17 Farnesyltransferase inhibitors (FTIs) and geranylgeranyltransferase inhibitors (GGTIs) have promising effects in cells, which can inhibit even a mutated CA GTPase from signaling at cell membranes.18 However, in clinical trials FTIs and GGTIs have proven to be less effective, due to alternate prenylation of the GTPases and toxicity of the inhibitors.18,19
Since prenyltransferase inhibitors are clinically less effective than originally anticipated, other therapeutic targets that regulate the prenylation and trafficking of GTPases are needed. One such target is SmgGDS, which is expressed as 2 splice variants that promote the prenylation and trafficking of multiple Ras and Rho family members that have a PBR.20,21 The long form of SmgGDS, named SmgGDS-607, associates only with non-prenylated GTPases and controls their entry into the prenylation pathway, whereas the shorter form of SmgGDS, named SmgGDS-558, associates only with prenylated GTPases and regulates their trafficking to the plasma membrane.20 SmgGDS is overexpressed in multiple types of cancer, including non-small cell lung carcinoma (NSCLC),22 prostate cancer,23 and breast cancer.24 Previous studies found that depleting both SmgGDS splice variants simultaneously diminishes cell proliferation in NSCLC and prostate cancer cell lines.22,23 Only 2 previous studies have addressed the role of each splice variant separately in cancer malignancy.20,24 One of these studies showed that NSCLC colony formation depends more on SmgGDS-558 than SmgGDS-607,20 whereas the other study showed that SmgGDS-558 plays a greater role than SmgGDS-607 in promoting the proliferation, NFκB activity, and tumorigenesis of breast cancer cells.24
The link between the SmgGDS splice variants and their regulation of GTPases needs more exploration, as there are a multitude of signaling pathways that are downstream of these GTPases. One important cellular function that is regulated by GTPases is the cell cycle.25,26 Rho, Ras, Rap1, and Rac1 have all been linked to the regulation of the cell cycle, either directly or indirectly.25-30 One way that the cell cycle is regulated is by the synthesis and degradation of proteins involved in this pathway.31-37 For example, Ras and Rho are known regulators of the synthesis and degradation of Cyclin D1 and p27kip1 (p27), both important mediators of the G1 phase.34-36 Furthermore, p21Cip1/Waf1 (p21) is another protein that is downstream of Ras and Rho and regulates progression of the G1 and G2 phases of the cell cycle.37
The different roles identified for SmgGDS-607 and SmgGDS-558 in the regulation and trafficking of GTPases demonstrates the need to identify how the SmgGDS splice variants play a role in multiple types of cancer, and to rigorously identify their role in signaling pathways and on the expression of proteins involved in these pathways. We utilized 2 cell lines each from NSCLC, breast cancer, and pancreatic cancer to compare the roles of the 2 SmgGDS splice variants in these cancers. We compared the effects of the RNAi-mediated depletion of SmgGDS-607 or SmgGDS-558 individually, or both splice variants simultaneously, on the proliferation, cell cycle progression, and cell cycle protein expression in these cancer cell lines. Our results demonstrate that SmgGDS-558, but not SmgGDS-607, is needed for cell cycle progression in all 3 types of cancer. We found that in NCI-H1703 NSCLC cells, the loss of SmgGDS-558 alone is not enough to affect tumor growth, but the loss of both SmgGDS splice variants together significantly decreases tumor growth. This is the first study to identify the importance of SmgGDS in pancreatic cancer, as well as the first study that assesses the role of each SmgGDS splice variant in controlling cell cycle progression and cell cycle protein expression. Our results indicate that SmgGDS-558 may be an effective target for cancer therapeutics in multiple cancers and cancer phenotypes.
Results
SmgGDS splice variants are expressed in pancreatic cancer, non-small cell lung cancer (NSCLC), and breast cancer cell lines
SmgGDS consists of 13 armadillo (ARM) domains designated A–M (Fig. 1A). SmgGDS-558 is a splice variant of SmgGDS-607 and lacks the C ARM domain. In order to assess the effect of SmgGDS-607 and SmgGDS-558 in multiple cancers, we used 6 cell lines, representing 3 different cancers. The pancreatic cancer cell lines MiaPaCa-2 (MiaPaCa) and PANC-1 (Panc1), NSCLC cell lines NCI-H23 (H23) and NCI-H1703 (H1703), and breast cancer cell lines MCF-7 and MDA-MB-231 were utilized throughout this study (Fig. 1B). The SmgGDS splice variants were detected in all 6 cancer cell lines and are represented by a slower migrating form (SmgGDS-607) and a more rapidly migrating form (SmgGDS-558) on immunoblots (Fig. 1B, all cell lines, lane 1). We generated siRNA to target specific ARM domains of the SmgGDS splice variants as well as a control siRNA-labeled Scramble #3 (Sc #3) (Fig. 1). The siRNA C2 targets the C ARM domain that is present only in SmgGDS-607; the siRNA BD targets the junction between the B and D ARM domains, which is present only in SmgGDS-558, and the siRNA I1 targets the I ARM domain that is present in both splice variants (Fig. 1A). The depletion of SmgGDS-607 with siRNA C2 results in the loss of the slow migrating form of SmgGDS (Fig. 1B, all cell lines, lane 2), and the depletion of SmgGDS-558 with siRNA BD results in the loss of rapidly migrating form of SmgGDS (Fig. 1B, all cell lines, lane 3). Depletion of both SmgGDS-607 and SmgGDS-558 simultaneously with siRNA I1 results in the loss of both forms of SmgGDS (Fig. 1B, all cell lines, lane 4).
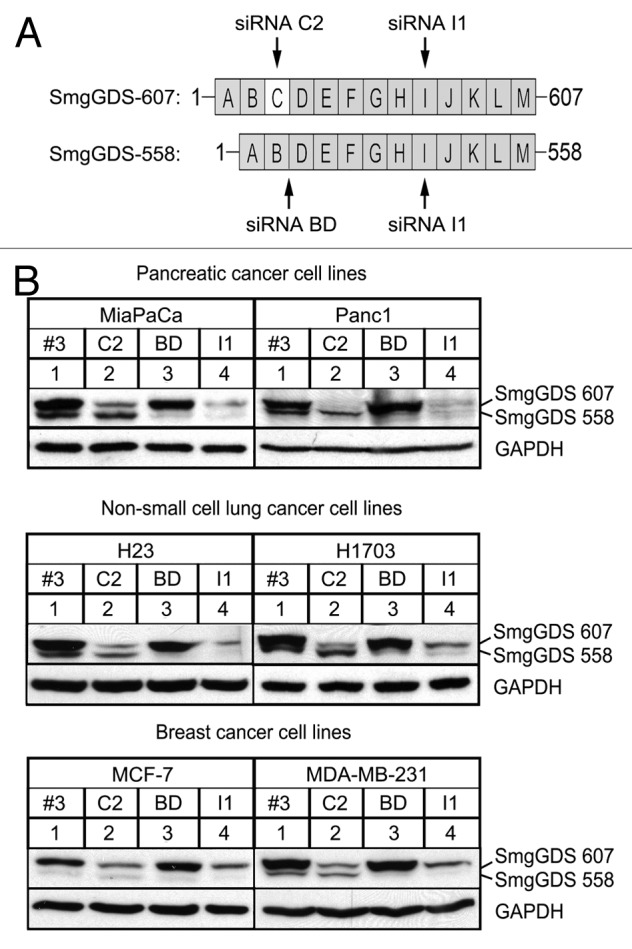
Figure 1. SmgGDS-607 and SmgGDS-558 are expressed in pancreatic cancer, NSCLC, and breast cancer, and can be silenced utilizing SmgGDS siRNA. (A) SmgGDS splice variants 607 and 558 are comprised of 13 or 12 armadillo repeats (ARMs), respectively. SmgGDS-607 has an ARM C, which is spliced out of SmgGDS-558. The following siRNAs were utilized in this study: the siRNA C2 targets only SmgGDS-607, whereas siRNA BD targets only SmgGDS-558. The siRNA I1 targets both splice variants and the non-targeting control siRNA is labeled as Scramble #3. (B) Pancreatic cancer cells lines MiaPaCa and Panc1 (top), non-small cell lung cancer cell lines H23 and H1703 (middle), and breast cancer cell lines MCF-7 and MDA-MB-231 (bottom) were transfected with 25 nM of the indicated siRNAs, lysed after 72 h (h), and subjected to ECL-western blotting using antibodies to SmgGDS and GAPDH. Results are representative of 3 independent experiments.
Loss of SmgGDS-558 alone or both SmgGDS-607 and SmgGDS-558 simultaneously decreases cell proliferation in cancer
To assess the effects of SmgGDS on cell proliferation in the cancer cell lines, we measured [3H]-thymidine uptake (Fig. 2A) and relative cell count (Fig. 2B) after silencing the SmgGDS splice variants’ expression. Previous studies show that depleting SmgGDS-558 alone or both splice variants of SmgGDS simultaneously significantly decreased [3H]-thymidine uptake in the MCF-7 and MDA-MB-231 breast cancer cell lines.24 We found that the loss SmgGDS-558 alone or both splice variants simultaneously also significantly decreases cell proliferation in the NSCLC and pancreatic cancer cell lines (Fig. 2A and B). Silencing SmgGDS-607 had no effect on cell proliferation in most of the cell lines (Fig. 2A and B). However, silencing SmgGDS-607 diminished [3H]-thymidine uptake in Panc-1 cells (Fig. 2A) and decreased both [3H]-thymidine uptake and cell count in H1703 cells (Fig. 2). Taken together, these data support the hypothesis that SmgGDS-558 is a more important promoter of cell proliferation than SmgGDS-607 in multiple types of cancer.

Figure 2. Cell proliferation is diminished more by silencing SmgGDS-558 than by silencing SmgGDS-607, and silencing both splice variants together enhances this effect. The indicated cell lines were assayed for cell proliferation utilizing [3H]-thymidine uptake (A) or relative cell count (B) 72 h after transfecting the cells with 25 nM of the indicated siRNAs. The values are normalized to cells transfected with non-targeting scramble #3 siRNA. Results are the mean ± SE from 3 or more independent experiments conducted with either quadruplicate or sextuplicate (A) samples in each experiment. (*P < 0.01 by one-way ANOVA with Dunnett post-hoc multiple comparisons test.)
SmgGDS is a mediator of the cell cycle
The effect of SmgGDS on cell proliferation in multiple cancer cell lines led us to ascertain the effects of the loss of SmgGDS on the passage of cells through the cell cycle. This assay was a 2-fold test designed to measure growth arrest and apoptosis/cell death of the cells after depletion of SmgGDS. An initial study that examined silencing of both SmgGDS-607 and SmgGDS-558 simultaneously using siRNA I1 in NSCLC cell lines detected a G1/G0 and G2/M cell cycle arrest in the H1703 cell line.22 Surprisingly, the H1703 NSCLC cell line was the outlier of all of the cell lines we tested.
In every cell line, we found that depleting SmgGDS-607 with siRNA C2 had no significant effect on the percent of cells in each phase of the cell cycle (Fig. 3). Depleting only SmgGDS-558 using siRNA BD caused a G0/G1 phase arrest in all of the cell lines except for the NCI-H1703 cell line (Fig. 3). The depletion of SmgGDS-607 and SmgGDS-558 simultaneously, using siRNA I1, caused a G2/M phase arrest in all of the cell lines except the H23 (Fig. 3B, left) and MCF-7 (Fig. 3C, left) cell lines. In addition to this G2/M phase arrest, the simultaneous depletion of both SmgGDS-607 and SmgGDS-558 also caused a G0/G1 phase arrest in the H23 and H1703 NSCLC cell lines, and this response was not exhibited by the pancreatic or breast cancer cell lines (Fig. 3B). In every cell line we tested there was no change in the percent of the cells in the sub G1 phase (representative data shown in Fig. S1), indicating that SmgGDS does not play a role in apoptosis or cell death.

Figure 3. Silencing SmgGDS-558 alone or both SmgGDS-558 and SmgGDS-607 together causes a G1 or G2 cell cycle arrest. Pancreatic (A), NSCLC (B), or breast (C) cancer cell lines were transfected with 25 nM of the indicated siRNA and changes in cell cycle progression were determined by staining the cells with propidium iodide 72 h post-transfection, followed by fluorescence-activated cell sorter analysis. Results are the mean ± SE from 3 or more independent experiments. The symbol above a column indicates a statistical comparison of progression through each phase of the cell cycle by the indicated cells vs. the control cells transfected with Scramble #3 siRNA. (*P < 0.05)
Although some variance was found, the overall conclusion from these data is that SmgGDS-558 plays a larger role in the passage of the cancer cells through the phases of the cell cycle than does SmgGDS-607, with the outlier being H1703 cells. Another interesting finding is that the loss of both splice variants of SmgGDS produces a different effect than that of depleting only SmgGDS-558 alone. These data suggest that silencing SmgGDS-607, which has no effect alone, can cause an additive effect when silenced along with SmgGDS-558.
Silencing SmgGDS mediates cell cycle protein expression
We next tested the role of SmgGDS in regulating cell cycle proteins. There are multiple reports that SmgGDS regulates the NFκB pathway in NSCLC,22 prostate,23 and breast cancers,24 as indicated by the findings that NFκB activity is increased by overexpressing SmgGDS-558, and NFκB activity is decreased by depleting SmgGDS-558 alone, or both splice variants simultaneously.22 NFκB is a vital cellular protein that can be responsible for the transcription of many genes including cyclin D1, p21, and p27.38-41 Cyclin D1 is a cell cycle promoter, and both p27 and p21 are cell cycle inhibitors.42 The cell cycle is often regulated by the stability and degradation of multiple proteins involved in the pathway, and therefore we tested the hypothesis that the loss of SmgGDS splice variants in our cancer cell lines would affect the cyclin D1, p27, and p21 protein expression levels.
The expression of cyclin D1 was significantly decreased by silencing only SmgGDS-558 in all of the cell lines, with the exception of the H1703 NSCLC cells (Fig. 4A; Fig. S2). Since cyclin D1 is a G0/G1 phase promoter, these data correlated with the G0/G1 phase arrest seen after depleting SmgGDS-558 in all of the cell lines except the H1703 cells (Fig. 3). Similarly, the G0/G1 cell cycle inhibitor, p27, was significantly increased after silencing only SmgGDS-558 in all cancer cell lines except the H1703 NSCLC cell line (Fig. 4B; Fig. S2). Depleting SmgGDS-558 also increased the expression of p21 in the MiaPaca, Panc1, and H23 cancer cell lines (Fig. 4C; Fig. S2). The cell cycle inhibitor p21 has been shown to regulate multiple phases of the cell cycle.43 The participation of p21 in multiple phases of the cell cycle may contribute to our observation that changes in p21 expression (Fig. 4C) do not correlate with the arrest of the cells in a specific phase of the cell cycle (Fig. 3) following depletion of SmgGDS-558.

Figure 4. Expression of Cyclin D1, p27, and p21 is altered in pancreatic, NSCLC, and breast cancer cells after silencing SmgGDS splice variants. Cyclin D1 (A), p27Kip1 (p27) (B), and p21/WAF1 (p21) (C) protein expression was analyzed in the indicated pancreatic, NSCLC, and breast cancer cell lines 72 h after transfection of the indicated SmgGDS siRNA. An equal number of live cells were lysed in SDS sample buffer and immunoblotted using antibodies to the cell cycle proteins. Densitometric analysis of the immunoblotted proteins was conducted by comparing the O.D. of the indicated cell cycle protein normalized to the O.D. of GAPDH for each treatment, and expressed as a percent of normalized protein expression in the cells treated with the control Scramble #3 siRNA. Results are the mean ± SE from 3 independent experiments. (ns, not significant; *P < 0.05 by one-way ANOVA with Dunnett post-hoc multiple comparisons test.)
Unique changes in the expression of the cell cycle proteins provide an explanation for why cells arrest in different cell cycle phases when only SmgGDS-558 is lost vs. when both SmgGDS-558 and SmgGDS-607 are lost. The trend seen with the cell cycle analysis was that a depletion of SmgGDS-558 in the cell lines resulted in a G0/G1 phase arrest, whereas depletion of both splice variants simultaneously resulted in a G2/M arrest (Fig. 3). In every cancer cell line, we show that the loss of both splice variants simultaneously does not significantly affect cyclin D1 expression (Fig. 4A; Fig. S2). Additionally, we show that the loss of both splice variants together does not affect p27 protein expression, with the exception of the H1703 cells (Fig. 4B; Fig. S2). The p21 protein expression was significantly increased in all the cell lines after silencing SmgGDS-607 and SmgGDS-558 simultaneously, except the H1703 cell line (Fig. 4C; Fig. S2).
As expected, we show that the depletion of SmgGDS-607 had virtually no effect on the expression of the cell cycle proteins in all of the cancer cell lines, except for an increase in p21 expression seen in the MCF-7 breast cancer cell line (Fig. 4C; Fig. S2). Taken together, these data show that SmgGDS-558 is a more important regulator of the cell cycle when compared with SmgGDS-607. Interestingly, we show that depletion of both splice variants simultaneously, when compared with the depletion of SmgGDS-558 alone, results in a different effect on the cell cycle and expression of the proteins involved in this pathway. Since the depletion of SmgGDS-607 has no effect on the cell cycle compared with depletion of SmgGDS-558, we would expect that loss of both splice variants together would generate similar results as compared with the loss of SmgGDS-558 alone, yet there are differences. These data lead to the hypothesis that cancer cells can function normally after the loss of SmgGDS-607, but not after losing SmgGDS-558, and loss of both splice variants together provides an alternate/additive effect. It is intriguing to speculate that the cancer cells may rely more on the splice variant SmgGDS-607 for signaling when SmgGDS-558 is depleted.
Another interesting finding was the differences we saw in the H1703 NSCLC cell line compared with the other cancer cell lines, primarily the lack of effect that the loss off SmgGDS-558 has on the cell cycle in H1703 cells (Figs. 3 and 4). Our data show that loss of both splice variants together in H1703 cells results in strong G0/G1 and G2/M phase arrests, which correlates to previous data,22 yet the only effect that the loss of both splice variants had on the protein expression analysis was an increased expression of p27. It is interesting that the H23 cell line also had a G0/G1 phase arrest after silencing both splice variants simultaneously. It could be that these NSCLC cell lines have a unique protein signaling network, which varies slightly from the other cancer cell lines.
Depletion of SmgGDS-607 and SmgGDS-558 simultaneously decreases tumorigenesis of H1703 xenografts in SCID hairless outbred (SHO) mice
We next wanted to assess the role of the SmgGDS splice variants in tumor growth. Previous data show that depletion of SmgGDS-558 alone or both SmgGDS-607 and SmgGDS-558 simultaneously slows tumorigenesis of the MDA-MB-231 breast cancer cell line in SHO mice.24 Since the responses of the MDA-MB-231 cells are similar to most of the cell lines we tested except the H1703 NSCLC cells, and because there have been no published studies examining the role of SmgGDS in NSCLC tumorigenesis, we chose to examine tumorigenesis by the H1703 NSCLC cell line.
We generated sublines of H1703 cells stably transfected with a luciferase reporter, tet-repressor, and doxycycline-inducible shRNAs that targeted either SmgGDS-607 alone (C2), SmgGDS-558 alone (BD), or both SmgGDS-607 and SmgGDS-558 (BD + C2). H1703 cells stably transfected with a luciferase reporter, tet-repressor, and doxycycline-inducible non-targeting shRNA (Sc #3) served as a control subline (Fig. 5). In order to test the tetracycline-inducible depletion of SmgGDS in these stably transduced cell lines, we cultured each cell line with increasing concentrations of tetracycline in order to block the tet-repressor and allow for production of the indicated shRNA (Fig. 5A). As expected, tetracycline diminished the expression of the SmgGDS splice variant that was specifically targeted by the shRNA expressed in the cells (Fig. 5A). The immunoblot shown in Figure 5A indicates that tetracycline treatment of the H1703-TR-LUC-shRNA C2 cell line significantly diminished SmgGDS-607 expression, as expected, but also slightly diminished SmgGDS-558 expression, which would not be expected to occur, since the C2 shRNA targets only SmgGDS-607. It should be noted that this loss of SmgGDS-558 was not reproducibly observed, because in additional experiments tetracycline diminished only SmgGDS-607 expression without diminishing SmgGDS-558 expression in the H1703-TR-LUC-shRNA C2 line (Fig. S3).

Figure 5. Silencing both SmgGDS-607 and SmgGDS-558 together diminishes tumorigenesis by H1703 xenografts in mice. (A) H1703-LUC-TR cell lines stably expressing the indicated inducible shRNAs were cultured with increasing amounts of tetracycline (0, 2, or 5 μg) daily for 72 h. The cells were then lysed and subjected to ECL-western blotting using antibodies to SmgGDS and GAPDH. Results are representative of 3 independent experiments. (BandC) H1703-LUC-TR cell lines stably expressing the indicated inducible shRNAs were injected into the flank of SHO mice. At week 3, doxycycline (dox) was introduced via the diet of half of the animals in each group. Representative mouse images (B) are shown for each cell line (KD = knockdown of the indicated splice variant). The graphs (C) represent relative weekly growth of tumors: H1703-Sc #3 (top left; n = 7 mice without dox and n = 6 mice with dox), H1703-C2 (top right; n = 6 mice without dox and n = 6 mice with dox), H1703-BD (bottom left; n = 6 mice without dox and n = 6 mice with dox), and H1703-BD+C2 (bottom right; n = 6 mice without dox and n = 7 mice with dox.) Values are normalized to luminescence obtained at week 3 for each mouse and are the mean ± SE assessed by 2-way ANOVA with secondary Bonferroni multiple comparisons test (*P < 0.05).
After injecting the sublines into the flanks of SHO mice, we utilized in vivo biophotonic imaging to measure weekly tumor growth (Fig. 5B). When the tumors were established 3 weeks after the cell injections, the mice were divided into 2 groups based on normalized starting luciferase values (Fig. S4); one group received feed that was supplemented with doxycycline, whereas the other group received unsupplemented feed. We found that the doxycycline-inducible depletion of either SmgGDS-607 alone or SmgGDS-558 alone did not significantly affect tumor growth, similar to the control (Sc #3) mice (Fig. 5C). In contrast, the simultaneous depletion of both SmgGDS-607 and SmgGDS-558 significantly decreased tumorigenesis by week 6 (Fig. 5C, bottom right). At week 7, the number of mice in the control group fell below the 6-mouse threshold, but continuous biophotonic imaging through week 10 indicated trends in tumor growth that were similar to those occurring in the early part of the study (Fig. S4).
These data provide the first evidence that SmgGDS promotes tumorigenesis of NSCLC cells. Depleting SmgGDS expression produced similar effects on the tumorigenesis of H1703 xenografts in mice and the proliferation of cultured H1703 cells; we found that depleting SmgGDS-558 alone is not sufficient to diminish tumorigenesis of xenografts nor to cause cell cycle arrest of culture cells, but simultaneous loss of both splice variants resulted in both decreased tumorigenesis and a strong cell cycle arrest in cultured cells. These data provide insights into the unique SmgGDS signaling profiles that occur in different cancer cell lines, and validates SmgGDS-558 as therapeutic target for multiple types of cancer.
Discussion
This study demonstrates that SmgGDS-558 is a key regulator of the cell cycle in breast cancer, pancreatic cancer, and NSCLC. SmgGDS has been described as a “master regulator” of small GTPases, because it interacts with multiple PBR-containing GTPases, such as RhoA, Rac1, Rap1A, Rap1B, and K-Ras.15 Many of these GTPases have been implicated as either direct or indirect promoters of the cell cycle.25,26 SmgGDS controls the activities of these GTPases through several mechanisms, most notably by controlling their prenylation and trafficking to cell membranes.20,21 SmgGDS-558 also promotes GDP/GTP exchange by Rac1, Rap1A, Rap1B, and K-Ras,44-46 and acts as a true guanine nucleotide exchange factor (GEF) for RhoA and RhoC.24,47 This ability of SmgGDS to regulate many features of Ras and Rho family members, all of which can promote the cell cycle of malignant cells, undoubtedly contributes to our finding that SmgGDS promotes cell cycle progression in multiple types of cancer.
SmgGDS may promote the expression of cell cycle-regulatory proteins by activating many signaling cascades that are regulated by GTPases. These signaling cascades may ultimately converge to activate NFκB, since many of the GTPases that interact with SmgGDS promote NFκB activity,30,48-50 and NFκB activation is one of the most commonly reported functions of SmgGDS-558 in malignant cells.22-24 NFκB regulates the expression of all of the cell cycle proteins that we have examined in this study. NFκB directly binds to the promoter of cyclin D1 enhancing its transcription.38,51 NFκB can suppress p27 transcription through the actions of miR221 and miR222 in some cancers,41 and NFκB epigenetically suppresses p21 expression through HDAC1.39,40 Consistent with these functions of NFκB, we observed that depleting SmgGDS-558 decreased cyclin D1 expression and increased p27 expression in the majority of the cancer cell lines we tested, and increased p21 in 3 of the 6 cancer cell lines (Fig. 3). Taken together, these findings suggest that SmgGDS-558 controls the expression of cyclin D1, p27, and p21 in the cells by promoting the activity of the many GTPases that enhance NFκB activity.
Our recent study of the regulation of RhoA by SmgGDS-558 in breast cancer cells provides evidence that SmgGDS-558 activates GTPases that promote NFκB activity.24 We found that depleting SmgGDS-558 expression utilizing siRNA BD decreases both RhoA and NFκB activity in breast cancer cells, and overexpressing SmgGDS-558 promotes a RhoA-mediated increase in NFκB activity in breast cancer cells.24 Previous studies indicate that inhibition of RhoA elevates p27 levels, whereas active RhoA decreases p27 levels.52,53 These findings suggest that one mechanism by which SmgGDS-558 promotes cancer malignancy involves the activation of RhoA, which, in turn, promotes NFκB activity.48,49
In addition to RhoA, it is likely that other small GTPases also participate in cell cycle progression induced by SmgGDS-558. K-Ras is a probable participant, since it was previously reported that SmgGDS-558 enhances the transforming abilities of K-Ras.54,55 K-Ras can promote cell cycle progression through activation of NFκB as well as through other signaling cascades.50,56,57 For example, K-Ras can promote cyclin D1 expression by activating the RAF-MEK-ERK/MAPK pathway, which results in direct transcriptional control of the AP-1 site in the cyclin D1 promoter, with sustained K-Ras activity leading to increased cyclin D1 expression.25,58,59 Furthermore, the stability of cyclin D1 is controlled through the Ras-PI3K-PKB(AKT)-GSK3β pathway, with active K-Ras inhibiting the GSK3β-mediated degradation of cyclin D1.60 These K-Ras-mediated MAPK and PI3K pathways might be activated when SmgGDS-558 promotes the trafficking of prenylated K-Ras to the plasma membrane. In this model, the depletion of SmgGDS-558 would diminish the ability of active K-Ras to reach the cell membrane, where it activates the MAPK or PI3K signaling cascades, resulting in decreased cyclin D1 transcription and increased degradation of cyclin D1.
Our results indicate that cell cycle progression is promoted much more by SmgGDS-558 than by SmgGDS-607 in the majority of the cancer cell lines we examined. In almost all of the cell lines that we tested, depleting SmgGDS-607 had virtually no effect on progression through the cell cycle or expression of cell cycle proteins, as well as little effect on cell proliferation. In contrast, depleting SmGDS-558 in these cell lines significantly reduced cell proliferation, caused a cell cycle arrest, and changed cell cycle protein expression. Several mechanisms might cause SmgGDS-558 to play a greater role than SmgGDS-607 in the cell cycle. One mechanism might involve the different interactions of the SmgGDS splice variants with the prenylated and non-prenylated forms of GTPases. In cells, SmgGDS-607 interacts only with newly synthesized, non-prenylated GTPases, whereas SmgGDS-558 interacts only with prenylated GTPases.20 Since the prenylated forms of small GTPases are believed to be more actively involved in oncogenic signaling than the non-prenylated forms,24,61,62 it is reasonable to conclude that the unique preference of SmgGDS-558 for prenylated GTPases allows SmgGDS-558, but not SmgGDS-607, to uniquely control oncogenic signaling cascades activated by prenylated Ras and Rho family members.
Another mechanism that might contribute to the greater importance of SmgGDS-558 than SmgGDS-607 in the cell cycle is the greater ability of SmgGDS-558 than SmgGDS-607 to act as a GEF for RhoA in cells. Although both SmgGDS-558 and SmgGDS-607 act as RhoA GEFs in recombinant in vitro systems,47 SmgGDS-558 is more effective than SmgGDS-607 as a RhoA GEF when the SmgGDS splice variants are expressed in cells.24 Our recent studies indicate that the actions of SmgGDS-558 as a RhoA GEF contribute to SmgGDS-558 playing a more prominent role than SmgGDS-607 in breast cancer malignancy.24
Intriguingly, we found that the loss of SmgGDS-558 alone induced different cellular responses than the loss of both SmgGDS-558 and SmgGDS-607 simultaneously. In all cell lines tested, except the H1703 cells, cyclin D1 was decreased and p27 was increased when SmgGDS-558 alone was depleted, but surprisingly, these changes in cyclin D1 and p27 did not occur when both SmgGDS-558 and SmgGDS-607 were depleted simultaneously. These changes in the cell cycle proteins were mimicked to some extent by changes in cell cycle progression. A G0/G1 arrest was induced by depleting SmgGDS-558 alone in the MiaPaca, Panc1, and MDA-MB-231 cells, but this G0/G1 arrest did not occur in these cells when both SmgGDS-558 and SmgGDS-607 were depleted simultaneously. Instead, these cells arrested in G2/M when both SmgGDS-558 and SmgGDS-607 were depleted simultaneously. These findings suggest that different signaling cascades are disrupted by the loss of SmgGDS-558 alone vs. the loss of both SmgGDS-558 and SmgGDS-607 together. Intriguingly, even though different signaling cascades may be disrupted in these 2 conditions, we found that cell proliferation was significantly diminished in both conditions, as indicated by significant reductions in thymidine uptake and cell count when either SmgGDS-558 alone was depleted, or when both SmgGDS-558 and SmgGDS-607 were depleted together. These findings underscore the multi-factorial mechanisms that SmgGDS splice variants use to regulate the cell cycle, consistent with SmgGDS splice variants promoting multiple features of Ras and Rho family members, including their prenylation, membrane trafficking, and GDP/GTP exchange.
The H1703 NSCLC cell line differed from all other cell lines that we studied, because the loss of only SmgGDS-558 in H1703 cells did not cause a G0/G1 arrest, nor did it significantly decrease cyclin D1 or increase p27 expression, even though all of these events were induced by depleting SmgGDS-558 in the other cell lines. Furthermore, depleting both SmgGDS-558 and SmgGDS-607 together uniquely increased p27 expression and caused a strong G1 arrest in H1703 cells, which did not occur in the majority of the other cell lines when both SmgGDS-558 and SmgGDS-607 were depleted. The reasons for these unique responses of H1703 cells are unclear. Some of these responses might be due to key signaling molecules being uniquely mutated (or not mutated) in H1703 cells compared with the other cell lines. However, an examination of the mutation status of key signaling molecules in H1703 has not indicated candidates that might cause the unique responses of H1703 cells. For example, K-Ras has an oncogenic mutation in all of the cell lines we tested except for the H1703 and MCF-7 cell lines. It is tempting to speculate that the presence of wild-type K-Ras in H1703 cells contributes to these cells’ unique responses. However, this speculation is not supported by the fact that MCF-7 cells also have wild-type K-Ras and yet MCF-7 cells respond differently than the H1703 cells to the RNAi-mediated depletion of SmgGDS. Further studies of the signaling pathways mediated by SmgGDS in H1703 cells are needed to define why these cells have unique responses to depletion of the SmgGDS splice variants.
Due to the unique responses of cultured H1703 cells to SmgGDS depletion, we investigated how the doxycycline-inducible depletion of SmgGDS affects tumorigenesis of H1703 xenografts in mice. Consistent with the responses of cultured H1703 cells, we found that tumorigenesis was decreased by the loss of both SmgGDS splice variants together, but not by the loss of SmgGDS-558 alone, in the H1703 xenografts (Fig. 5). Our finding that the loss of SmgGDS-558 alone does not inhibit tumorigenesis of H1703 cells differentiates these cells from MDA-MB-231 cells, because previous studies indicated that the loss of SmgGDS-558 alone provides a strong enough signal to slow tumorigenesis of MDA-MB-231 xenografts in mice.24 Thus, the unique responses that are exhibited by H1703 cells in culture are also exhibited by H1703 xenografts, suggesting that the signaling events that regulate H1703 cell cycle progression in culture are also operating in the xenografts to regulate tumorigenesis. Taken together, these results demonstrate the importance of SmgGDS in tumor growth and highlight the need to develop inhibitors that target either SmgGDS-558 alone or both SmgGDS splice variants simultaneously as a therapeutic strategy for multiple types of cancer.
This study is the first to define the different roles of SmgGDS-607 and SmgGDS-558 in the regulation of the cell cycle. Our findings provide mechanistic insights into the functions of SmgGDS in NSCLC22 and breast cancer,24 and expand the list of cancers that are regulated by SmgGDS to include pancreatic cancer. The unique ability of SmgGDS to regulate the cell cycle in multiple cancer cell lines with different mutational profiles indicates the importance of this protein as a key regulator of malignancy, most likely due to its interactions with multiple small GTPases, and provides impetus to develop SmgGDS inhibitors as therapeutic agents in cancer.
Material and Methods
Cell lines and reagents
All cell lines were obtained from the American Type Culture Collection (ATCC). MDA-MB-231, PANC1, and MiaPaCa2 cell lines were maintained in DMEM high glucose with L-glutamine media supplemented with sodium pyruvate and 10% FBS. NCI-H23 and NCI-H1703 cell lines were maintain in RPMI 1640 media supplemented with 10% FBS. MCF-7 cells were maintained in MEM with Earle Salts and L-glutamine media supplemented with sodium pyruvate, non-essential amino acids, 10% FBS, and insulin (10 μg/ml). All culture media was supplemented with penicillin and streptomycin.
siRNA transfection
Previously optimized siRNA duplexes for SmgGDS depletion were used.20 In this study we used siRNA I1 target sequence 5′-GCAAAGATGT TATCAGCTG-3′ to deplete both splice variants of SmgGDS simultaneously, siRNA C2 target sequence 5′-GAACTATAGC AATGAGAAT-3′to deplete only SmgGDS-607, and siRNA BD targeting sequence 5′-ACGATAGCCA TTCGCTTCA-3′ to deplete only SmgGDS-558. We also used siRNA Scramble #3 as our nontargeting siRNA control (Dharmacon siControl 3). Cells were transfected with 25 nM siRNA using DharmaFECT-1 (MiaPaCa2, Panc1, MDA-MB-231, MCF-7) or DharmaFECT-3 (NCI-H23, NCI-H1703) transfection reagents (Dharmacon) according to the manufacturer's instruction.
Immunoblotting
Equal numbers of transfected cells were heated in Laemmli sample buffer and subjected to SDS-PAGE. The proteins were transferred to polyvinylidene difluoride and immunoblotted using the following antibodies: Mouse anti-SmgGDS (BD Transduction Laboratories, 612511), mouse anti-GAPDH (Santa Cruz Biotechnology, sc-32233), mouse anti-cyclin D1 (Cell Signaling, DCS6), rabbit anti-p27 (Cell Signaling, 2552), and rabbit anti-p21 (Cell Signaling, 12D1). Bound antibodies were visualized using horseradish peroxidase-linked anti-mouse IgG or anti-rabbit IgG (Amersham Biosciences), and ECL reagents (PerkinElmer Life Sciences) as previously described.63 Densitometry of blots was determined using ImageJ software.
[3H]-Thymidine uptake and cell counts
Cell proliferation was assessed by measuring [3H]-thymidine uptake by the cells as previously described.64 In brief, cells were transfected with the indicated siRNAs for 72 h and then incubated with [3H]-thymidine for 3 h. Cells were lifted, harvested on filters using an automatic cell harvester (Skatron), and radioactivity was measured by β-scintillation counting to determine the amount of [3H]-thymidine uptake by the cells. Cell counts were conducted using Trypan blue reagent 0.4% (Invitrogen) and a Countess automated cell counter (Invitrogen) after transfection with the indicated siRNAs for 72 h.
Cell cycle analysis
Cells were transfected with the indicated siRNA for 72 h and then lifted, washed with PBS, fixed in 50% ethanol, incubated with RNAase cocktail (Ambion) for 30 min, and stained with propidium iodide (100 μg/mL). The stained cells were subjected to flow cytometry.
NCI-H1703 cells stably expressing tet-inducible shRNA
NCI-H1703 cells were transduced with lentiviral vectors as described previously.20 Briefly, lentiviral vectors expressing inducible shRNA for SmgGDS-558 (shRNA BD), SmgGDS-607 (shRNA C2), or a nontargeting control (shRNA Sc #3) were generated using the “Block-iT” inducible H1 lentiviral RNAi system (Invitrogen, K4925-00) as described previously.20 Cells were maintained in RPMI 1640 triple selection media (100 μg/ml Zeocin, 6 μg/ml blasticidin, 200 ng/ml puromycin). Expression of shRNA was induced using 625 mg/Kg doxycycline in feed for in vivo studies.
Tumor xenograft study in SCID hairless outbred (SHO) mice
Animal studies were conducted according to protocols approved by the MCW Institutional Animal Care and Use Committee. Xenografts were established in the left flank of 6-wk-old female SHO mice (Charles River) by injecting 4 × 106 cells in a PBS solution. Mice were separated into 2 groups after 3 wk; one group remained on normal feed, while the other group was given doxycycline feed (Tekland-Harlen). Tumor size was measured weekly by bioluminescence imaging using a biophotonic imager (Xenogen). Mice were injected with 200 μL of 15 mg/mL luciferin five minutes before bioluminescent imaging.
Statistical analysis
The results are presented as the means ± SE from at least 3 independent experiments, unless noted otherwise. Symbols above a column indicate a statistical comparison between the control and experimental group by Student t test, by 2-way analysis of variance (ANOVA) with secondary Bonferroni multiple comparisons test, or by one-way or repeated measures ANOVA with Dunnett multiple comparison test as indicated in the figure legends. P values < 0.05 were considered significant.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to acknowledge Dr Jeffrey Woodliff for assistance with the FACs cell cycle analysis, and Marylou Mader for assistance with our animal study. This work is supported by NIH grants R01 CA136799 (C.L.W.); the Medical College of Wisconsin Cancer Center (C.L.W.), the Wisconsin Breast Cancer Showhouse (C.L.W.), and the Rock River Cancer Research Foundation (C.L.W.). J.W. is a member of the Medical Scientist Training Program at MCW, which is partially supported by a training grant from NIGMS T32-GM080202.
Glossary
Abbreviations:
- NSCLC
non-small cell lung cancer
- CA
constitutively active
- PBR
polybasic region
- FTase
farnesyltransferase
- GGTase
geranylgeranyltransferase
- FTI
farnesyltransferase inhibitor
- GGTI
geranylgeranyltransferase inhibitor
- p27
p27kip1
- p21
p21Cip1/Waf1
- ARM
armadillo
- MiaPaca
MiaPaCa-2
- Panc1
PANC-1
- H23
NCI-H23
- H1703
NCI-H1703
- Sc #3
scramble #3
- SHO
SCID hairless-outbred
- GEF
guanine nucleotide exchange factor
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/27804
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Garcea G, Neal CP, Pattenden CJ, Steward WP, Berry DP. Molecular prognostic markers in pancreatic cancer: a systematic review. Eur J Cancer. 2005;41:2213–36. doi: 10.1016/j.ejca.2005.04.044. [DOI] [PubMed] [Google Scholar]
- 3.Kusama T, Mukai M, Iwasaki T, Tatsuta M, Matsumoto Y, Akedo H, Nakamura H. Inhibition of epidermal growth factor-induced RhoA translocation and invasion of human pancreatic cancer cells by 3-hydroxy-3-methylglutaryl-coenzyme a reductase inhibitors. Cancer Res. 2001;61:4885–91. [PubMed] [Google Scholar]
- 4.Taniuchi K, Nakagawa H, Hosokawa M, Nakamura T, Eguchi H, Ohigashi H, Ishikawa O, Katagiri T, Nakamura Y. Overexpressed P-cadherin/CDH3 promotes motility of pancreatic cancer cells by interacting with p120ctn and activating rho-family GTPases. Cancer Res. 2005;65:3092–9. doi: 10.1158/0008.5472.CAN-04-3646. [DOI] [PubMed] [Google Scholar]
- 5.Liu Y, Wang Y, Zhang Y, Miao Y, Zhao Y, Zhang PX, Jiang GY, Zhang JY, Han Y, Lin XY, et al. Abnormal expression of p120-catenin, E-cadherin, and small GTPases is significantly associated with malignant phenotype of human lung cancer. Lung Cancer. 2009;63:375–82. doi: 10.1016/j.lungcan.2008.12.012. [DOI] [PubMed] [Google Scholar]
- 6.Tang Y, Olufemi L, Wang MT, Nie D. Role of Rho GTPases in breast cancer. Front Biosci. 2008;13:759–76. doi: 10.2741/2718. [DOI] [PubMed] [Google Scholar]
- 7.Clark GJ, Der CJ. Aberrant function of the Ras signal transduction pathway in human breast cancer. Breast Cancer Res Treat. 1995;35:133–44. doi: 10.1007/BF00694753. [DOI] [PubMed] [Google Scholar]
- 8.Shima F, Yoshikawa Y, Ye M, Araki M, Matsumoto S, Liao J, Hu L, Sugimoto T, Ijiri Y, Takeda A, et al. In silico discovery of small-molecule Ras inhibitors that display antitumor activity by blocking the Ras-effector interaction. Proc Natl Acad Sci U S A. 2013;110:8182–7. doi: 10.1073/pnas.1217730110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nussinov R, Tsai CJ, Mattos C. ‘Pathway drug cocktail’: targeting Ras signaling based on structural pathways. Trends Mol Med. 2013;19:695–704. doi: 10.1016/j.molmed.2013.07.009. [DOI] [PubMed] [Google Scholar]
- 10.Patel M, Côté JF. Ras GTPases’ interaction with effector domains: Breaking the families’ barrier. Commun Integr Biol. 2013;6:e24298. doi: 10.4161/cib.24298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cherfils J, Zeghouf M. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol Rev. 2013;93:269–309. doi: 10.1152/physrev.00003.2012. [DOI] [PubMed] [Google Scholar]
- 12.Hayes TK, Der CJ. Mutant and wild-type Ras: co-conspirators in cancer. Cancer Discov. 2013;3:24–6. doi: 10.1158/2159-8290.CD-12-0521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flemming A. Cancer: double-pronged approach to combat mutant KRAS. Nat Rev Drug Discov. 2013;12:188–9. doi: 10.1038/nrd3969. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y, Kaiser CE, Frett B, Li HY. Targeting mutant KRAS for anticancer therapeutics: A review of novel small molecule modulators. J Med Chem. 2013;56:5219–30. doi: 10.1021/jm3017706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Williams CL. The polybasic region of Ras and Rho family small GTPases: a regulator of protein interactions and membrane association and a site of nuclear localization signal sequences. Cell Signal. 2003;15:1071–80. doi: 10.1016/S0898-6568(03)00098-6. [DOI] [PubMed] [Google Scholar]
- 16.Marakasova E, Akhmatova N, Amaya M, Eisenhaber B, Eisenhaber F, van Hoek M, et al. Prenylation: From bacteria to eukaryotes. Mol Biol. 2013;47:622–33. doi: 10.1134/S0026893313050130. [DOI] [PubMed] [Google Scholar]
- 17.Nguyen UT, Goodall A, Alexandrov K, Abankwa D. Isoprenoid modifications. In: Post-Translational Modifications in Health and Disease: Springer, 2011:1-37. [Google Scholar]
- 18.Berndt N, Hamilton AD, Sebti SM. Targeting protein prenylation for cancer therapy. Nat Rev Cancer. 2011;11:775–91. doi: 10.1038/nrc3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Piwnica-Worms D. Elucidating Mechanisms of Farnesyltransferase Inhibitor Action and Resistance in Breast Cancer by Bioluminescence Imaging 2010. DTIC Document. [Google Scholar]
- 20.Berg TJ, Gastonguay AJ, Lorimer EL, Kuhnmuench JR, Li R, Fields AP, Williams CL. Splice variants of SmgGDS control small GTPase prenylation and membrane localization. J Biol Chem. 2010;285:35255–66. doi: 10.1074/jbc.M110.129916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ntantie E, Gonyo P, Lorimer EL, Hauser AD, Schuld N, McAllister D, Kalyanaraman B, Dwinell MB, Auchampach JA, Williams CL. An adenosine-mediated signaling pathway suppresses prenylation of the GTPase Rap1B and promotes cell scattering. Sci Signal. 2013;6:ra39. doi: 10.1126/scisignal.2003374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tew GW, Lorimer EL, Berg TJ, Zhi H, Li R, Williams CL. SmgGDS regulates cell proliferation, migration, and NFkappaB transcriptional activity in non-small cell lung carcinoma. J Biol Chem. 2008;283:963–76. doi: 10.1074/jbc.M707526200. [DOI] [PubMed] [Google Scholar]
- 23.Zhi H, Yang XJ, Kuhnmuench J, Berg T, Thill R, Yang H, See WA, Becker CG, Williams CL, Li R. SmgGDS is up-regulated in prostate carcinoma and promotes tumour phenotypes in prostate cancer cells. J Pathol. 2009;217:389–97. doi: 10.1002/path.2456. [DOI] [PubMed] [Google Scholar]
- 24.Hauser AD, Bergom C, Schuld NJ, Chen X, Lorimer EL, Huang J, et al. The SmgGDS splice variant SmgGDS-558 is a key promoter of tumor growth and RhoA signaling in breast cancer. Molecular cancer research. MCR. 2013 doi: 10.1158/1541-7786.MCR-13-0362. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coleman ML, Marshall CJ, Olson MF. RAS and RHO GTPases in G1-phase cell-cycle regulation. Nat Rev Mol Cell Biol. 2004;5:355–66. doi: 10.1038/nrm1365. [DOI] [PubMed] [Google Scholar]
- 26.Villalonga P, Ridley AJ, Ridley AJ. Rho GTPases and cell cycle control. Growth Factors. 2006;24:159–64. doi: 10.1080/08977190600560651. [DOI] [PubMed] [Google Scholar]
- 27.Bosco EE, Mulloy JC, Zheng Y. Rac1 GTPase: a “Rac” of all trades. Cell Mol Life Sci. 2009;66:370–4. doi: 10.1007/s00018-008-8552-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jung SM, Park SS, Kim WJ, Moon SK. Ras/ERK1 pathway regulation of p27KIP1-mediated G1-phase cell-cycle arrest in cordycepin-induced inhibition of the proliferation of vascular smooth muscle cells. Eur J Pharmacol. 2012;681:15–22. doi: 10.1016/j.ejphar.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 29.Chang F, Steelman LS, Shelton JG, Lee JT, Navolanic PM, Blalock WL, Franklin R, McCubrey JA. Regulation of cell cycle progression and apoptosis by the Ras/Raf/MEK/ERK pathway (Review) Int J Oncol. 2003;22:469–80. [review] [PubMed] [Google Scholar]
- 30.Gastonguay A, Berg T, Hauser AD, Schuld N, Lorimer E, Williams CL. The role of Rac1 in the regulation of NFκB activity, cell proliferation, and cell migration in non-small cell lung carcinoma. Cancer Biol Ther. 2012;13:647–56. doi: 10.4161/cbt.20082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9:153–66. doi: 10.1038/nrc2602. [DOI] [PubMed] [Google Scholar]
- 32.Lu Z, Hunter T. Ubiquitylation and proteasomal degradation of the p21(Cip1), p27(Kip1) and p57(Kip2) CDK inhibitors. Cell Cycle. 2010;9:2342–52. doi: 10.4161/cc.9.12.11988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shan J, Zhao W, Gu W. Suppression of cancer cell growth by promoting cyclin D1 degradation. Mol Cell. 2009;36:469–76. doi: 10.1016/j.molcel.2009.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Welsh CF, Roovers K, Villanueva J, Liu Y, Schwartz MA, Assoian RK. Timing of cyclin D1 expression within G1 phase is controlled by Rho. Nat Cell Biol. 2001;3:950–7. doi: 10.1038/ncb1101-950. [DOI] [PubMed] [Google Scholar]
- 35.Halilovic E, She QB, Ye Q, Pagliarini R, Sellers WR, Solit DB, Rosen N. PIK3CA mutation uncouples tumor growth and cyclin D1 regulation from MEK/ERK and mutant KRAS signaling. Cancer Res. 2010;70:6804–14. doi: 10.1158/0008-5472.CAN-10-0409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stacey DW. Three observations that have changed our understanding of cyclin D1 and p27Kip1 in cell cycle control. Genes Cancer. 2010;1:1189–99. doi: 10.1177/1947601911403475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cmielová J, Rezáčová M. p21Cip1/Waf1 protein and its function based on a subcellular localization [corrected] J Cell Biochem. 2011;112:3502–6. doi: 10.1002/jcb.23296. [DOI] [PubMed] [Google Scholar]
- 38.Guttridge DC, Albanese C, Reuther JY, Pestell RG, Baldwin ASNF., Jr. NFkappaB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol Cell Biol. 1999;19:5785–99. doi: 10.1128/mcb.19.8.5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ghantous A, Saikali M, Rau T, Gali-Muhtasib H, Schneider-Stock R, Darwiche N. Inhibition of tumor promotion by parthenolide: epigenetic modulation of p21. Cancer Prev Res (Phila) 2012;5:1298–309. doi: 10.1158/1940-6207.CAPR-12-0230. [DOI] [PubMed] [Google Scholar]
- 40.Zupkovitz G, Grausenburger R, Brunmeir R, Senese S, Tischler J, Jurkin J, Rembold M, Meunier D, Egger G, Lagger S, et al. The cyclin dependent kinase inhibitor p21 is a crucial target for histone deacetylase 1 as a regulator of cellular proliferation. Mol Cell Biol. 2010;30:1171–81. doi: 10.1128/MCB.01500-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.le Sage C, Nagel R, Egan DA, Schrier M, Mesman E, Mangiola A, Anile C, Maira G, Mercatelli N, Ciafrè SA, et al. Regulation of the p27(Kip1) tumor suppressor by miR-221 and miR-222 promotes cancer cell proliferation. EMBO J. 2007;26:3699–708. doi: 10.1038/sj.emboj.7601790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van den Heuvel S. Cell-cycle regulation. 2005; In: WormBook: The Online Review of C. elegans Biology [Internet]. Pasedena (CA): WormBook; 2005. Available from: http://www.ncbi.nlm.nih.gov/books/NBK19719/ [DOI] [PMC free article] [PubMed]
- 43.Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D. p21 is a universal inhibitor of cyclin kinases. Nature. 1993;366:701–4. doi: 10.1038/366701a0. [DOI] [PubMed] [Google Scholar]
- 44.Yamamoto T, Kaibuchi K, Mizuno T, Hiroyoshi M, Shirataki H, Takai Y. Purification and characterization from bovine brain cytosol of proteins that regulate the GDP/GTP exchange reaction of smg p21s, ras p21-like GTP-binding proteins. J Biol Chem. 1990;265:16626–34. [PubMed] [Google Scholar]
- 45.Kawamura S, Kaibuchi K, Hiroyoshi M, Hata Y, Takai Y. Stoichiometric interaction of smg p21 with its GDP/GTP exchange protein and its novel action to regulate the translocation of smg p21 between membrane and cytoplasm. Biochem Biophys Res Commun. 1991;174:1095–102. doi: 10.1016/0006-291X(91)91533-I. [DOI] [PubMed] [Google Scholar]
- 46.Mizuno T, Kaibuchi K, Yamamoto T, Kawamura M, Sakoda T, Fujioka H, Matsuura Y, Takai Y. A stimulatory GDP/GTP exchange protein for smg p21 is active on the post-translationally processed form of c-Ki-ras p21 and rhoA p21. Proc Natl Acad Sci U S A. 1991;88:6442–6. doi: 10.1073/pnas.88.15.6442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hamel B, Monaghan-Benson E, Rojas RJ, Temple BRS, Marston DJ, Burridge K, Sondek J. SmgGDS is a guanine nucleotide exchange factor that specifically activates RhoA and RhoC. J Biol Chem. 2011;286:12141–8. doi: 10.1074/jbc.M110.191122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gnad R, Kaina B, Fritz G. Rho GTPases are involved in the regulation of NFkappaB by genotoxic stress. Exp Cell Res. 2001;264:244–9. doi: 10.1006/excr.2001.5165. [DOI] [PubMed] [Google Scholar]
- 49.Kawanami D, Matoba K, Kanazawa Y, Ishizawa S, Yokota T, Utsunomiya K. Thrombin induces MCP-1 expression through Rho-kinase and subsequent p38MAPK/NFκB signaling pathway activation in vascular endothelial cells. Biochem Biophys Res Commun. 2011;411:798–803. doi: 10.1016/j.bbrc.2011.07.031. [DOI] [PubMed] [Google Scholar]
- 50.Bodemann BO, White MA. Ral GTPases and cancer: linchpin support of the tumorigenic platform. Nat Rev Cancer. 2008;8:133–40. doi: 10.1038/nrc2296. [DOI] [PubMed] [Google Scholar]
- 51.Hinz M, Krappmann D, Eichten A, Heder A, Scheidereit C, Strauss M. NFkappaB function in growth control: regulation of cyclin D1 expression and G0/G1-to-S-phase transition. Mol Cell Biol. 1999;19:2690–8. doi: 10.1128/mcb.19.4.2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rivard N, Boucher MJ, Asselin C, L’Allemain G. MAP kinase cascade is required for p27 downregulation and S phase entry in fibroblasts and epithelial cells. Am J Physiol. 1999;277:C652–64. doi: 10.1152/ajpcell.1999.277.4.C652. [DOI] [PubMed] [Google Scholar]
- 53.Laufs U, Marra D, Node K, Liao JK. 3-Hydroxy-3-methylglutaryl-CoA reductase inhibitors attenuate vascular smooth muscle proliferation by preventing rho GTPase-induced down-regulation of p27(Kip1) J Biol Chem. 1999;274:21926–31. doi: 10.1074/jbc.274.31.21926. [DOI] [PubMed] [Google Scholar]
- 54.Yoshida Y, Kawata M, Miura Y, Musha T, Sasaki T, Kikuchi A, Takai Y. Microinjection of smg/rap1/Krev-1 p21 into Swiss 3T3 cells induces DNA synthesis and morphological changes. Mol Cell Biol. 1992;12:3407–14. doi: 10.1128/MCB.12.8.3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fujioka H, Kaibuchi K, Kishi K, Yamamoto T, Kawamura M, Sakoda T, Mizuno T, Takai Y. Transforming and c-fos promoter/enhancer-stimulating activities of a stimulatory GDP/GTP exchange protein for small GTP-binding proteins. J Biol Chem. 1992;267:926–30. [PubMed] [Google Scholar]
- 56.Ramaswamy S, Nakamura N, Vazquez F, Batt DB, Perera S, Roberts TM, Sellers WR. Regulation of G1 progression by the PTEN tumor suppressor protein is linked to inhibition of the phosphatidylinositol 3-kinase/Akt pathway. Proc Natl Acad Sci U S A. 1999;96:2110–5. doi: 10.1073/pnas.96.5.2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kandel ES, Skeen J, Majewski N, Di Cristofano A, Pandolfi PP, Feliciano CS, Gartel A, Hay N. Activation of Akt/protein kinase B overcomes a G(2)/m cell cycle checkpoint induced by DNA damage. Mol Cell Biol. 2002;22:7831–41. doi: 10.1128/MCB.22.22.7831-7841.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Winston JT, Coats SR, Wang YZ, Pledger WJ. Regulation of the cell cycle machinery by oncogenic ras. Oncogene. 1996;12:127–34. [PubMed] [Google Scholar]
- 59.Aktas H, Cai H, Cooper GM. Ras links growth factor signaling to the cell cycle machinery via regulation of cyclin D1 and the Cdk inhibitor p27KIP1. Mol Cell Biol. 1997;17:3850–7. doi: 10.1128/mcb.17.7.3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Muise-Helmericks RC, Grimes HL, Bellacosa A, Malstrom SE, Tsichlis PN, Rosen N. Cyclin D expression is controlled post-transcriptionally via a phosphatidylinositol 3-kinase/Akt-dependent pathway. J Biol Chem. 1998;273:29864–72. doi: 10.1074/jbc.273.45.29864. [DOI] [PubMed] [Google Scholar]
- 61.Adjei AA. Blocking oncogenic Ras signaling for cancer therapy. J Natl Cancer Inst. 2001;93:1062–74. doi: 10.1093/jnci/93.14.1062. [DOI] [PubMed] [Google Scholar]
- 62.Winter-Vann AM, Casey PJ. Post-prenylation-processing enzymes as new targets in oncogenesis. Nat Rev Cancer. 2005;5:405–12. doi: 10.1038/nrc1612. [DOI] [PubMed] [Google Scholar]
- 63.Lanning CC, Daddona JL, Ruiz-Velasco R, Shafer SH, Williams CL. The Rac1 C-terminal polybasic region regulates the nuclear localization and protein degradation of Rac1. J Biol Chem. 2004;279:44197–210. doi: 10.1074/jbc.M404977200. [DOI] [PubMed] [Google Scholar]
- 64.Strassheim D, Shafer SH, Phelps SH, Williams CL. Small cell lung carcinoma exhibits greater phospholipase C-β1 expression and edelfosine resistance compared with non-small cell lung carcinoma. Cancer Res. 2000;60:2730–6. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.