

Abstract
Human antibody-ribonuclease (RNase) fusion proteins, referred to as immunoRNases, have been proposed as an alternative to heterologous immunotoxins, without their immunogenicity and unspecific toxicity issues. In this study, we investigated if human pancreatic RNase will be suitable as effector component in a therapeutic antibody development platform. We generated several fusion proteins consisting of tumor-specific human immunoglobulins (IgGs) and human pancreatic RNase. Transient mammalian cell production was efficient and IgG-RNases were purified to homogeneity. Antigen binding was comparable to the parental antibodies and RNase catalytic activity was retained even in the presence of 50-fold molar excess of human cytosolic RNase inhibitor (RI). Serum stability, cell binding and internalization of IgG-RNases were comparable to the parental IgGs. Despite these promising properties, none of the IgG-RNases revealed significant inhibition of tumor cell growth in vitro even when targeting different antigens putatively employing different endocytotic pathways. The introduction of different linkers containing endosomal protease cleavage sites into the IgG-RNase did not enhance cytotoxicity. Similarly, RI evasive human pancreatic RNase variants mediated only small inhibiting effects on tumor cell growth at high concentrations, potentially reflecting inefficient cytosolic translocation. Taken together, human pancreatic RNase and variants did not prove to be generally suitable as effector component for a therapeutic antibody drug development platform.
Keywords: antibodies, immunoglobulin, IgG, human pancreatic RNase, RNase inhibitor, immunoRNase, cancer therapy
Introduction
Recombinant antibodies represent the largest class of biological therapeutics for treatment of cancer diseases, but, to date, clinical benefit of therapies with monoclonal antibodies has not met the initial anticipations and hopes. Promising antibodies with novel mechanisms of action, such as programmed death 1 receptor antagonists lambrolizumab and nivolumab, are still in Phase 3 studies.1 Another promising area is use of antibodies to internalizing tumor-associated antigens as vehicles to deliver cytotoxic payloads inside target cells, although the results to date have been mixed.2,3 Highly toxic drugs and heterologous toxins conjugated to antibodies have been extensively tested as so-called immunotoxins,4 but this approach has been associated with severe adverse side effect, and even fatalities, due to unspecific toxicity and immunogenicity. The ADC gemtuzumab ozogamicin (Mylotarg®), approved by the US Food and Drug Administration for treatment of CD33+ acute myeloid leukemia was voluntarily withdrawn from the market because of increased occurrence of fatalities caused by hepato-occlusive disease upon treatment.5 A Phase 1 study of refractory B cell lymphoma patients with a combination of two different IgG::deglycosylated ricin A chain immunotoxin conjugates (Combotox) resulted in three deaths; at least two were directly related to the immunotoxin treatment. Subsequent evaluation revealed that one compound showed tendency of aggregation after thawing and fatalities appeared only in patients with low number circulating tumor cells.6
To overcome immunogenicity and non-specific toxicity issues, human effector enzymes have been studied as alternative to heterologous toxins. Particularly, ribonucleases (RNases) of the RNase A family have been investigated because they become cytotoxic only after translocation into the cytosol of target cells.7 Intracellular microinjection of pancreatic RNase into Xenopus oocytes revealed cytotoxicity similar to those of ricin whereas high extracellular concentrations are well-tolerated.8 Moreover, immunogenicity issues and unspecific toxicity are not expected because human RNases are ubiquitously distributed all over the human body and reside in plasma and most tissues. Indeed, even heterologous RNases like Rana pipiens-derived ranpirnase (Onconase®; Alfacell, Inc) were safely administered into human patients and immunologically well-tolerated.9 Numerous reports linked natural and engineered RNases with anti-tumor activity,10,11 and Onconase was already evaluated in advanced clinical study for treatment of malignant mesothelioma.12 Conjugation or fusion of RNases with antibodies or ligand targeting internalizing surface antigens, also termed “immunoRNase” or more generally “targeted RNases,”13 can increase cytotoxicity by several orders of a magnitude.14 Dimeric immunoRNases showed more efficient anti-tumor effects than monomeric variants.14
Unlike certain “cytotoxic” RNases, natural pancreatic RNase (also termed RNase 1) did not evolve any properties to invade host cells15 or to mediate any known special biological actions16,17 that would lead to unexpected adverse effects. The physiological function of human pancreatic RNase is still not known, but it can induce dendritic cell maturation and activation.18 All other secreted human RNase A members (RNase 2–8) are involved in host defense or other biological functions like angiogenesis.19 Properties that promote efficient cell binding and internalization can be endowed into pancreatic RNases by genetic engineering. Several independent studies have successfully demonstrated that human pancreatic RNase fused with cell-targeting ligands or antibodies can result in cytotoxic or antiproliferatory effects on the targeted cells.20-24 The design of immunoRNases also has effects on production and anti-tumor efficacy. The introduction of the human immunoglobulin (Ig) G Fc domain resulted in homodimeric IgG-like immunoRNase fusion proteins that showed excellent mammalian production and efficient anti-tumor properties.20,23 These previous studies indicated that human pancreatic RNase could also be a promising effector platform for development of anti-tumor antibodies.
In this study, human pancreatic RNase was tested as the potential effector component in a therapeutic antibody platform for cancer indications. IgG-RNase fusion proteins showed high expression levels in mammalian production cells, high serum stability, antigen and target cell binding as well as internalization comparable to the parental IgGs combined with wild type-like RNase catalytic activity.
Nevertheless, IgG-RNases did not mediate any significant tumor cell line growth-inhibitory effect even when RNase inhibitor (RI)-evasive RNases and putative endosomal cleavable linkers were introduced into these constructs. Therefore, at this stage of development human pancreatic RNase and variants thereof did not prove to be a generally suitable effector component in a therapeutic antibody platform.
Results
Construction and production of human IgG-RNases
Transient gene expression in HEK293–6E cells was employed for production of human IgG-RNases. To this end, suitable expression vectors were generated that had expression of light (L) and heavy (H) chain IgG fragments under the control of a CMV promoter. Human IgG1 sequences were taken from MN-IgG, which targets the internalizing tumor antigen MN (Carbonic anhydrase 9, CA9), and a cholera toxin (CTX) specific human IgG1, which was used as negative control in this study. The human IgG1 Fc fragment was carboxy terminally fused via an AAASSG linker either to the mature form of the wild type human pancreatic RNase (RNase) or to an enzymatically inactive RNase variant containing mutations in both active center histidines, termed as RNase(H12A, H119A).25 Both human pancreatic RNase variants were truncated at the carboxy terminus (ΔSVEDST) for enhanced stability.26 The IgG and IgG-RNase variants used in this study are illustrated in Figure 1 and all employed components are summarized in Table 1.

Figure 1. IgG and IgG-RNase constructs. (A) Schematic illustrations of IgG and IgG-RNase, as well as (B) the corresponding gene expression cassettes. In addition to IgG-RNase containing wild type human pancreatic RNase (RNase), a control construct is shown containing enzymatically inactive RNase variant RNase(H12A, H119A). The illustrations are not drawn to scale. attB1–2, BP recombination cloning sites; BGH Poly A, bovine growth hormone poly-adenylation signal; CMV, cytomegalovirus; RNase, human pancreatic ribonuclease; V, C, variable and constant regions of light (L) and heavy (H) IgG chain)
Table 1. Overview of the human IgGs and RNases used in this study.
| IgG | Antigen specificity | Remarks |
|---|---|---|
| MN | MN-antigen (tumor associated antigen carbonic anhydrase IX, CA9) | Integral plasma membrane glycoprotein |
| CTX | Cholera toxin (control antigen) | Soluble protein |
| Mesothelin | Mesothelin-antigen (tumor associated antigen; MSLN) | GPI-anchored plasma membrane glycoprotein |
| X | Undisclosed antigen X | GPI-anchored plasma membrane glycoprotein |
| RNase | ||
| RNase | Wild type human pancreatic RNase with truncated carboxy terminus (ΔSVEDST) for increased stability26 according to reference 23 | |
| QBI-119 | Wild type human pancreatic RNase with truncated carboxy terminus (ΔSVEDST) and with mutations providing RI evasion (R4C, G38R, R39D, L86E, N88R, G89D, R91D, V118C) according to patent WO2005115477 | |
| Jo2007 | Wild type human pancreatic RNase with truncated carboxy terminus (ΔSVEDST) and with mutations providing RI evasion (R39D, N67D, N88A, G89D, R91D)27 | |
Abbreviations: GPI, glycophosphatidylinositol
Transient production of MN-IgG, MN-IgG-RNase, CTX-IgG-RNase and MN-IgG-RNase(H12A, H119A), in HEK293–6E cells achieved volumetric yields between 10–180 mg/L. In general, there was no trend that fusing IgG with one of the RNase variants had a negative effect on protein expression levels.
Protein purification
Culture supernatants were purified by protein A affinity chromatography followed by size exclusion chromatography (SEC). All purified proteins had endotoxin levels below 0.05 units per milligram protein (EU/mg). SDS-PAGE under non-reducing conditions revealed single protein bands at the expected molecular weight of 150 to 200 kDa for all constructs, indicating the formation of disulfide bridges between the heavy chains and between heavy and light chain of the MN-IgG and the immunoRNase constructs (Fig. 2). Under reducing conditions, all constructs showed one small protein band of about 25 kDa corresponding to IgG light chains and one large protein bands of 50 to 75 kDa corresponding to IgG heavy chains or their fusion with the different RNase variants, respectively (Fig. 2).
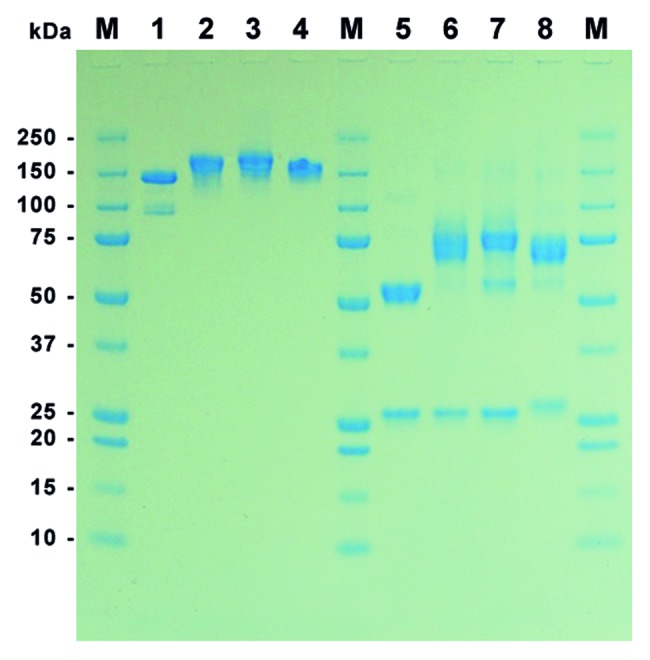
Figure 2. SDS-PAGE of purified MN-IgGs and IgG-RNase constructs. A total of 1 µg purified protein per lane was tested by SDS-PAGE under non-reducing (lane 1–4) and reducing (lane 5–8) conditions followed by Coomassie staining. Lane 1 and 5: MN-IgG; lane 2 and 6: MN-IgG-RNase; lane 3 and 7: MN-IgG-RNase(H12A, H119A); lane 4 and 8: CTX-IgG-RNase; M: protein standard All Blue (BioRad)
Analytical SEC revealed a single fraction peak for the MN-IgG, MN-IgG-RNase, MN-IgG-RNase(H12A, H119A), and the control CTX-IgG-RNase (Fig. 3). Product purity was in all cases more than 98% and aggregation was not observed for any of these constructs.

Figure 3. Analytical SEC of IgG and IgG-RNases. (A) MN-IgG, (B) MN-IgG-RNase, (C) MN-IgG-RNase(H12A, H119A) and (D) CTX-IgG-RNase. Base line is shown as gray dotted line.
Product homogeneity and aggregation was further tested by SEC with multi-angle laser light scattering (SEC-MALS) and dynamic light scattering (DLS). SEC-MALS was used for accurate sizing of MN-IgG-RNase, MN-IgG-RNase(H12A, H119A) and CTX-IgG-RNase yielding 271, 202 and 234 kDa, respectively. Higher glycosylation of constructs containing wild type RNase compared with that containing mutant RNase(H12A, H119) can explain the differences of the measured apparent molar mass in these experiments. DLS revealed a monodisperse molecule population (99.5–100%) with a hydrodynamic radius of 6–6.3 nm corresponding to 254 and 226 kDa for MN- and CTX-IgG-RNase and 231 kDa for the MN-IgG-RNase(H12A, H119A) variant.
Affinity determination
The MN-IgG and MN-IgG-RNase constructs had the same affinity with dissociation constants KD of 2.4–2.7 nM to their antigen measured by surface plasmon analysis (SPR, Table 2).
Table 2. Binding kinetic parameters from SPR calculated by 1:1 Langmuir binding model.
| Construct | ka (M−1 s−1) | kd (s−1) | KD (M) |
|---|---|---|---|
| MN-IgG1 | 2.6 × 105 | 7.1 × 10−4 | 2.7 × 10−9 |
| MN-IgG1-RNase | 2.7 × 105 | 6.5 × 10−4 | 2.4 × 10−9 |
| MN-IgG1-RNase(H12A, H119A) | 2.7 × 105 | 6.8 × 10−4 | 2.5 × 10−9 |
Ribonucleolytic activity
RNase activity was measured with the fluorescence-labeled substrate 6-FAM-dArUdAdA-BHQ1 containing a single RNase cleavage site per substrate molecule. After cleavage, the fluorescent dye is released from the quencher BHQ1. Time-dependent increase of fluorescence allows calculation of the catalytic efficiency kcat/Km. Data of all analyzed IgG-RNases, control constructs and free RNases are summarized in Table 3. The enzymatic efficiencies kcat/Km of IgG-RNases with the wild type RNase moiety were 0.7 to 1.9 × 108 M−1 s−1 calculated for single RNase moieties (i.e., one molecule IgG-RNase corresponds to two molecules free RNase) which were generally higher values than previously described for free wild type human pancreatic RNase produced in E. coli.27 The MN-IgG-RNase(H12A, H119A) construct with two active center histidine substitutions in the RNase moiety mediated no measurable ribonucleolytic activity at all concentrations tested.
Table 3. Catalytic efficiency of purified immunoRNases and control constructs.
| Construct | Production system | Catalytic efficiency kcat/Km per RNase moiety [M−1 s−1] |
|---|---|---|
| MN-IgG-RNase | HEK293 | 1.4 × 108 |
| MN-IgG-RNase(H12A, H119A) | HEK293 | < 104 * |
| CTX-IgG-RNase | HEK293 | 7.0 × 107 |
| Mesothelin-IgG-RNase | HEK293 | 1.1 × 108 |
| X-IgG-RNase | HEK293 | 1.9 × 108 |
| MN-IgG1-QBI119 | HEK293 | 5.2 × 107 |
| MN-IgG1-Jo2007 | HEK293 | 9.1 × 107 |
| QBI119 | E. coli | 4.1 × 106 |
| Onconase | E. coli | 4.6 × 103 |
*Catalytic efficiency below detection limit at the maximal tested concentration of 1010 M.
Inhibition of RNase-activity by human RNase inhibitor
Since all mammalian members of the RNase A superfamily are known to be efficiently inhibited by cytosolic RI, the catalytic activity of MN- and CTX-IgG-RNase was also tested in the presence of the RI RNasin. RNase activity of both IgG-RNases was not reduced even in the presence of up to 50-fold molar excess of RI (Fig. 4A and B). In contrast, bovine RNase used as a control showed a significant reduction of ribonucleolytic activity at equimolar concentration of RI, which further reduced with increasing RI ratio (Fig. 4C). This result is consistent with previous reports demonstrating decreased sensitivity of RNases toward RI upon dimerization.28,29

Figure 4. RNase activity in the presence of human placental RNase inhibitor. RNase activity of (A) MN-IgG-RNase and (B) CTX-IgG-RNase was measured in the presence of RNase inhibitor. 10−10 M RNase incubated with up to 50-fold molar excess of RI (RNasin). (C) Bovine RNase was tested as control.
Stability of the IgG and IgG-RNase constructs
To determine serum stability of IgG-RNases, purified proteins thereof were thawed at different points of time and incubated in mouse serum for up to 24 h at 37 °C. Antigen binding of all samples was measured by antigen ELISA in parallel (Fig. 5A). The sample freshly thawed just prior to ELISA was set to 100% and all other values were normalized. Antigen binding of MN-IgG, MN-IgG-RNase and CTX-IgG-RNase did not decrease within the first 24 h. Because classical antigen ELISA cannot reveal any information about RNase activity, a novel method that combines antigen binding and RNase activity in a single immunoassay was developed. Here, antigen binding was performed as in a classical ELISA, but detection was done by measuring RNase activity of the bound IgG-RNase using the hypersensitive fluorescence-labeled substrate 6-FAM-dArUdAdA-BHQ1. Unspecific background ribonucleolytic activity from the serum was not detected.

Figure 5. Stability tests. (A) IgG and IgG-RNase were incubated in 50% mouse serum at 37 °C for up to 24 h followed by testing of binding to the corresponding antigen by ELISA. Detection was done with an anti-human IgG-Fc specific secondary antibody HRP conjugate. (B) MN-IgG-RNase and (C) CTX-IgG-RNase were also tested by incubation in 50% human or mouse serum at 37 °C for up to 7 d followed by a novel immunoassay combining ELISA with an RNase activity assay. BSA was used as negative control antigen. All binding and activity data were normalized to values measured with samples freshly thawed (time 0 = 100%).
Since no anti-human IgG secondary antibody conjugate was necessary for detection, this method also allowed incubation of IgG-RNases in human serum. MN-IgG-RNase did not reveal any reduction of antigen-specific RNase activity in mouse or human serum after 24 h incubation at 37 °C (Fig. 5B). Notably, we observed a 1.5-fold increase of antigen-specific RNase activity of samples within the first 24 h after thawing compared with the freshly thawed samples. Then, the antigen-specific RNase activity remained stable at this level for at least 7 d. CTX-IgG-RNase incubated in human serum did not also show any reduction of antigen-specific RNase activity and also a slight increase of RNase activity compared with the freshly thawed sample in the first 24 h (Fig. 5C). However, CTX-IgG-RNase incubated in mouse serum showed a steadily ongoing decay of antigen-specific RNase activity over 7 d incubation at 37 °C. This effect seems to be caused by the mouse serum rather than by the instability of the IgG-RNase because mouse serum already interfered with the CTX antigen ELISA of CTX-IgG-RNase by reducing the overall absorbance compared with samples incubated without serum (data not shown).
Thermostability
The thermostability of MN- and CTX-IgG-RNase, as well as MN-IgG-RNase(H12A, H119A), was measured by differential scanning calorimetry (DSC) (Table 4). The transition of the IgG domains into an unfolded state was comparable between the IgG and the corresponding IgG-RNase constructs (Tm1, Tm2 and Tm3). Therefore, the carboxy terminal fusion of the RNase moiety to the IgG heavy chain did not affect the thermal stability of the IgG domains. Unfolding of the RNase moiety in IgG-RNases occurred at 54.1 °C (Tm4), which is comparable to the Tm of free wild-type human RNase.27,30 The construct containing the inactive RNase(H12A, H119A) with mutations in both active center histidines did not show any separate Tm4 peak.
Table 4. Thermostability of the IgG-RNases.
| Tm1 [°C] | Tm2 [°C] | Tm3 [°C] | Tm4 [°C] | |
|---|---|---|---|---|
| MN-IgG | 73.2 | 76.6 | 82.9 | - |
| MN-IgG-RNase | 72.4 | 76.5 | 82.7 | 54.1 |
| CTX-IgG | 70.2 | 80.7 | 83.0 | - |
| CTX-IgG-RNase | 69.8 | 79.7 | 82.2 | 54.1 |
| MN-IgG-RNase (H12A, H119A) | 71.8 | 76.6 | 82.7 | - |
Internalization of fluorescently-labeled immunoRNases by target cells
To study internalization of IgGs and immunoRNase, the constructs were chemically labeled with the pH-sensitive fluorescent dye CypHer 5E. Antigen binding was not changed by the chemical modification as tested by antigen ELISA and ribonucleolytic efficiency compared with non-labeled constructs (data not shown). MN-IgG, MN-IgG-RNase and IgG-RNase(H12A, H119A) were taken up by MN-antigen transfected MIAPaCa 2 (MIAPaca-MN+) cells in the same time-dependent manner (Fig. 6). Already after 3 h incubation, a significant number of fluorescent granules were detectable, indicating endocytosis of these constructs. After 24 h incubation, MN-IgG, MN-IgG-RNase and MN-IgG-RNase(H12A, H119A) were steadily further internalized, resulting in an increasing number of strongly fluorescent granula-like intracellular vesicles without reaching complete saturation. Incubation with the control CTX-IgG-RNase did not result in any fluorescence after 3 h and only very faint fluorescence was detectable after 24 h (Fig. 6).

Figure 6. Internalization of fluorescently labeled MN-specific IgG-RNases. IgGs, IgG-RNases and control constructs were chemically conjugated with CypHer 5E and incubated for up to 24 h on MIAPaCa-MN+ cell overexpressing MN antigen. CTX-IgG-RNase was used as control. (A) Fluorescence microscopy was performed after different time points, images after 3 and 24 h are shown as examples. Hoechst 33342 was used to counter stain for nuclei. (B) Internalization was quantified by counting of red fluorescent granules per cell for up to 24 h.
No significant tumor cell line growth inhibition of MN-IgG-RNases
After confirmation of the sufficiently high biochemical quality of the immunoRNases, the retention of antigen binding, the biochemical RNase activity, the potential escape from RI as well as the internalization of the immunoRNases into antigen-expressing tumor cell lines, and their potency in tumor cell line growth inhibition or killing was further characterized.
Surprisingly, MN-IgG-RNase incubated with MIAPaCa-MN+ tumor cells did not show any significant inhibitory effect on tumor cell line growth in vitro (Fig. 7). The control CTX-IgG-RNase and MN-IgG-RNase(H12A, H119A) also had no effect on tumor cell growth.

Figure 7. Growth inhibition of MN+ overexpressing tumor cell lines. MN+ overexpressing MIAPaCa 2 cells were incubated (A) with MN-IgG-RNase containing catalytic active human pancreatic RNase. MN-IgG-RNase(H12A, H119A) with a catalytic inactive RNase, and CTX-IgG-RNase, were used as negative controls, whereas Onconase was used as non-targeted positive control. B) Additionally, MN+ overexpressing MIAPaCa 2 were also incubated with (B) MN-IgG based immunoRNase constructs fused with RI evasive human pancreatic RNase variants (Jo2007, QBI-119) as well as MN-IgG-RNase and MN-IgG-QBI119 constructs containing other linker sequences (GGFKGG, GGGGGG, GGFLGG, GGAANG, and GALALAG), which are putatively cleavable in endosomes. Free QBI119 and Onconase were also tested.
To confirm the susceptibility of our test system toward RNase mediated growth inhibition, free Onconase from Rana pipiens was produced in E. coli as additional control. It exhibited a catalytic efficiency of 4.6 × 103 M−1 s−1 which is lower than previously described.31 This might be explained by an only partial conversion of N-terminal glutamine to pyroglutamine as evident by mass spectrometric analysis (data not shown). Nevertheless, free Onconase achieved an IC50 of 1.8 × 10−6 M on K562 cells, close to the published value.32,33 It also achieved a tumor cell line growth inhibition on MIAPaCa-MN+ cells with an IC50 of about 2.8 × 10−7 M (Fig. 7), confirming the susceptibility of MIAPaCa 2 cells toward RNase mediated toxicity.
Targeting alternative antigens with immunoRNase
The inability to inhibit tumor cell line growth by immunoRNase could be related to the specific antigen used for targeting. It is conceivable that different antigens might use different endocytotic pathways and therefore the immunoRNase might end or accumulate in different endocytotic compartments, possibly not all of them favoring cytosolic translocation and subsequent cytotoxic action. Therefore, alternative antigens were analyzed: MN antigen is an integral plasma membrane glycoprotein, whereas two alternative antigen test cases (mesothelin and undisclosed target X) were included, which are glycophosphatidylinositol (GPI) anchored proteins, and for which complex endocytotic sorting have been described.34,35
After production of mesothelin-IgG-RNase and X-IgG-RNase, the binding of these molecules to their cognate antigens was analyzed by SPR and found to be similar to their parental IgGs (data not shown). Internalization into their target cells was confirmed (Fig. 8, data for mesothelin-IgG-RNase not shown). Incubation of X-IgG and X-IgG-RNase with transfected A549-X+ cells overexpressing X-antigen on their surface also resulted in a specific internalization of both constructs in a time-dependent manner and similar intracellular vesicular distribution (Fig. 8). The uptake of the X-IgG-RNase was slightly faster compared with the corresponding IgG. Interestingly, the morphology of internal vesicular structures stained with X-IgG-RNase loaded with CypHer 5E was different to that in MIAPaCa-MN+ cells incubated with MN-IgG-RNase loaded with CypHer 5E (compare with Fig. 6). This is consistent with a different endocytotic fate or an accumulation in a different endocytotic compartment than in the case of MN antigen. In contrast, with MCF7 cells endogenously expressing X-antigen on lower level compared with A549-X+ cells, internalization was slowed down for X-IgG-RNase compared with the parental IgG (Fig. 8). After 24 h exposure time of the control construct, CTX-IgG-RNase was also slightly internalized, which is probably caused by interaction of the positively charged RNase moiety with the negatively charged cell surface31 (Fig. 8).

Figure 8. Internalization of fluorescently labeled X-antigen specific IgG-RNases. IgGs, IgG-RNases and control constructs were chemically conjugated with CypHer 5E and incubated for up to 24 h on A549-X+ cell or MCF7 cells which either overexpress or endogenously express X-antigen, respectively. CTX-IgG-RNase was used as control. (A) Fluorescence microscopy was performed at different time points, images after 3 and 24 h are shown. Hoechst 33342 was used as counter stain for nuclei. (B) Internalization was quantified by counting of red fluorescent granules per cell for up to 24 h.
Yet, even when different antigens were targeted, immunoRNases failed to show any significant inhibitory effect on tumor cell line growth in vitro (Fig. 9 and data not shown). Mesothelin-IgG chemically conjugated to a maytansinoid-based toxophore was used as positive control and inhibited tumor cell line growth of HT29 transfected with mesothelin antigen with an IC50 of 2 × 10−9 M.
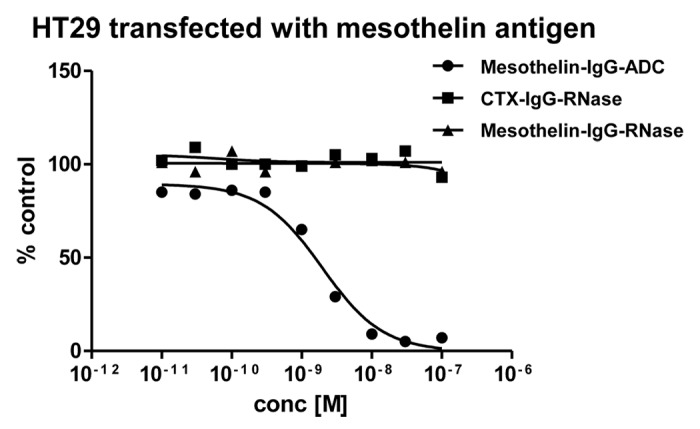
Figure 9. Growth inhibition of mesothelin-antigen expressing tumor cell lines. Mesothelin-antigen stably overexpressing HT29 cells were incubated with mesothelin-IgG based immunoRNase fusion protein. CTX-IgG-RNase was used as negative control. Mesothelin-IgG conjugated to a maytansinoid-based toxophore was used as positive control.
No significant tumor cell line growth inhibition of IgG-RNases with RI evasive RNase fusion variants or with variants containing endosomal cleavable linkers
Inefficient cytosolic delivery of cytotoxic payloads, including RNases, has been proposed to be one major limiting factor of cytotoxicity in immunoRNases as well as immunotoxins.36 However, the mechanism of the escape of RNases from endolysosomal system into the cytosol is not known in detail. Similarly, it is unknown, whether immunoRNases translocate into the cytosol as a whole or only after degradation of the antibody moiety. Moreover, the release of free RNase in intracellular compartments might be a prerequisite for efficient cytosolic translocation.31,37
On the other hand, free human pancreatic RNases might no longer be evasive to cytosolic RI due to their monomeric state. Therefore, two previously described RI-evasive “cytotoxic” human pancreatic RNase variants were also tested: QBI-119, which is RNase(R4C, G38R, R39D, L86E, N88R, G89D, R91D, V118C) (according to patent WO2005115477), and Jo2007 which is RNase(R39D, N67D, N88A, G89D, R91D).27 MN-IgG-Jo2007 revealed a similar catalytic efficiency per RNase molecule of 9.1 × 107 M−1 s−1 compared with MN-IgG-QBI-119 (5.2 × 107 M−1 s−1, Table 3). MN-IgG-Jo2007 did not show any anti-proliferative effect in vitro, MN-IgG-QBI-119 mediated marginal inhibitory effects on tumor cell growth at the highest tested concentration of 10−6 M (Fig. 7).
To test if generation of free RNases can be facilitated in the endolysosomal system resulting in a higher cytotoxicity, a set of five additional linkers that are putatively cleavable by endolysosomal enzymes were employed—GGFKGG,38 GGGGGG,39 GGFLGG,40 GGAANG,41 and GALALAG42—and combined them with MN-IgG-RNase (wild type) and MN-IgG-QBI-119 (RI evasive) fusions. However, we were unable to detect any enhanced cytotoxicity (Fig. 7).
Discussion
In this study, human pancreatic RNase was tested as the effector component in a therapeutic antibody platform. Several IgG-RNase fusion proteins were generated and produced in mammalian cells with high yields comparable to their parental IgGs. Therefore, the RNase moiety neither reduced secretion of the antibody fusion protein nor harmed the producer cell line, which confirms previous studies of single chain immunoRNase constructs.20,23 IgG-RNases were purified using standard IgG purification methods to homogeneity passing the quality control of pharmaceutical R&D standards, including no detectable endotoxin content and high product homogeneity. Moreover, antigen binding of the IgG-RNases was comparable to parental IgGs and catalytic activity was similar to human pancreatic RNase.27 RNase activity was not inhibited in the presence of a 50-fold molar excess of RNase inhibitor in vitro, which seems to be due to their forced dimeric state in the IgG-RNase constructs. In contrast, catalytic activity of monomeric bovine RNase A in parallel experiments was significantly reduced at equimolar ratios. IgG-RNases showed high serum stability regarding antigen binding and RNase activity. Cell binding and internalization of IgG-RNases was strictly dependent on the IgG targeting component. Internalization of IgG-RNases was time-dependent and intracellular distribution after uptake was not different compared with the parental antibodies except for X-IgG-RNase, which showed a slower uptake into MCF-7 cells with endogenous X-antigen expression compared with the parental X-IgG. Although all prerequisites for anti-tumor immunoRNases seem to be perfectly addressed by these IgG-RNases, they did not mediate any anti-proliferative or cytotoxic effects against several antigen positive tumor cells at concentration up to 10−6 M. These results were confirmed with several IgG-RNases targeting different classes of tumor antigens. Therefore, antigen-dependent effects, including different intracellular endocytosis pathways, are unlikely. Inefficient cytoplasmic release of the active component of immunoRNase, as well as of immunotoxins, is supposed to be the major limiting factor for their cytotoxic action.43 Therefore, alternative linker sequences that were previously described to be cleaved in endosomal environments were introduced.38-41,44 However, all linker variants of IgG-RNases did not show any enhanced tumor growth inhibition. Composite linker sequences consisting of a membrane-penetrating translocation signal with one or several flanking endosomal and cytoplasmic protease cleavage sites have been described to enhance cytotoxicity of immunotoxin fusion proteins but also reduce serum stability45 and were, therefore, not tested in this study.
Even though IgG-RNases with wild type RNase evade RI inhibition in a cell-free assay, the situation in a cellular setting might be different. For example, it cannot be excluded that the RNases are released from the antibody moiety and transform into a monomeric RI sensitive state. Therefore, previously described RI-evasive “cytotoxic” human pancreatic RNase variants27,46 were also tested, but they also did not increase anti-tumor effects of IgG-RNases. Only MN-IgG1-QBI119 showed low inhibition of tumor growth, but only at micromolar concentrations exceeding therapeutically useful concentrations. Again, introduction of different endosomal cleavable linkers did not improve tumor cell line growth inhibition.39,44 Free QBI-119 did not reveal any cytotoxicity, but previous studies with free “cytotoxic” human pancreatic RNase variants only reported inhibitory effects on tumor cells at high concentrations of more than 10 µM,27 which have not been tested in this study.
Taken together, neither human pancreatic RNase nor RI evasive variants thereof proved to be suitable as effector components for a therapeutic IgG antibody platform. The employment of different antibodies and antigen targets, as well as several tumor cell lines, cannot rule out all antigen and cell type depending effects, but it makes them unlikely to be the major limiting factors. Moreover, a mesothelin-specific IgG-ADC with the same IgG moiety inhibited the tested tumor cell lines in the subnanomolar range. In this study, it remained unclear if the human pancreatic RNase component of IgG-RNases translocated across the cytosolic lipid membrane bilayer and reached the cytosol in its active form. So far, the mechanism of cytosolic translocation of “cytotoxic” RNases has not been completely uncovered. For example, translocation of Onconase was strongly dependent of intracellular trafficking pathways.47 Moreover, retrograde translocation of secreted or extracellular proteins, as well as of heterologous toxins, often require passage through the endoplasmic reticulum (ER) and association with ER protein degradation pathway including de- and renaturation steps.48 The fate of retro-translocated unfolded proteins is usually immediate degradation in cytosolic proteasomes, but some toxins evolved mechanisms to evade these mechanisms.49 Onconase in combination with proteasome inhibitors mediates enhanced in vitro cytotoxicity.50 Nuclear localization sequences engineered into human pancreatic RNase have been shown to increase cytotoxicity that was supposed to enhance RI evasion,51 but it can also provide protection from proteasomal degradation.
There are also doubts about whether the effector mechanism of “cytotoxic” RNases is only dependent on their ribonucleolytic activity because there is no correlation between catalytic efficiency of different RNases and their cytotoxic properties, suggesting more complex mechanisms than unspecific RNA cleavage. Preference of certain RNAs has been proposed to be of importance. For example, Onconase prefers cleavage of tRNAs,52 but also targets micro-RNA precursors.53 For human pancreatic RNase, in vivo RNA substrates and effects on micro-RNAs are still unknown. Since free human pancreatic RNases and RI evasive mutants thereof mediate cytotoxic properties only at high micromolar concentrations, other processes have also to be considered like membrane disruption due to positive charge.54 Cationization of RNases enhanced cellular uptake and cytotoxicity.55 Positive charge distribution of Onconase effects translocation across the lipid bilayer,31 but could also cause inhibitory effects on cells. Engineering human pancreatic RNase to mimic such properties of “cytotoxic” heterologous RNases could offer potential paths forward to dramatically improve effector function in immunoRNases.
Material and Methods
Cell lines and cell culture
The HEK293–6E cell line is genetically modified with a truncated Epstein Barr virus (EBV) nuclear antigen 1 (EBNA1) and was licensed from National Research Council (NRC), Biotechnological Research Institute (BRI), Montreal, Canada.56 Cells were cultured according to the supplier’s description in the chemically defined F17 medium (Invitrogen, Life Technologies; Freestyle™ version of chemical defined medium CD17) supplemented with 1 g/L pluronic F68 (Applichem; prepare a 10% [w/v] stock solution in water, filter sterilize), 7.5 mM L-glutamine (PAA) and 25 mg/L G418 (PAA). HEK293–6E cells were cultured in 125 mL to 1 L polycarbonate shake flasks (Corning) in a MinitronTM CO2 orbital shaker with 50 mm orbital (Infors) at 37 °C and 5% CO2 atmosphere with 110 round per minute (rpm) without exceeding 2 × 106 cells/mL during maintenance. MIAPaCa 2 (ATCC CRL-1420) and derived clone #37 transfected with full-length MN antigen cDNA sequence (MIAPaCa-Mn+) were cultured in DMEM (high glucose, 4.5 g/L) supplemented with 10% (v/v) fetal calf serum (PAA) and Mycokill (PAA). A549 (ATCC CCL-185) tumor cells were transfected with X-antigen cDNA (A549-X+) and a high expressing clone B#4 was isolated and cultivated in F-12 Caighn’s medium (Invitrogen). MCF7 (ATCC HTB-22) cells were cultivated in MEM Earl’s medium (Invitrogen). A clone overexpressing Mesothelin-antigen was isolated after transfection of HT-29 tumor cells (ATCC HTB-38) with human mesothelin cDNA and was cultivated in RPMI 1640 supplemented with 10% FCS and 0.075% sodium bicarbonate solution. All tumor cell lines were incubated at 37 °C and 5% CO2 in static culture.
Construction of transient expression plasmid vectors and transient expression
To facilitate generation of different constructs the vector pCMV-hIgG1Fc-XP57 was modified. A new cassette flanked by attB1/2 recombination sites containing human IgG1 Fc fused to human pancreatic RNase was produced by gene synthesis (GeneArt, Regensburg, Germany) and introduced into the vector. Unique restriction sites were used for introduction and exchange of alternative IgGs, as well as alternative RNase gene sequences including different linkers. These expression cassettes were finally shuffled by Gateway cloning (Invitrogen) into a pTT5-derivate (NRC-BRI) that contained Gateway acceptor recombination sites. These final expression vectors had expression of light (L) and heavy (H) chain IgG fragments under the control of a CMV promoter. Transient production in suspension HEK293–6E cells was performed as previously described in detail.56-58 The extracellular domain of MN antigen was amended with a His Tag, cloned into pCEP4 expression vector (Invitrogen) and purified from transient HEK293–6E cell culture supernatants using standard techniques.58
Purification of IgG, RNases and IgG-RNase fusion constructs
Free Onconase and the human pancreatic RNase mutant QBI-119 (R4C, G38R, R39D, L86E, N88R, G89D, R91D, V118C) were produced in E. coli (BL21 (DE3); Invitrogen) using pET22b(+) (Novagen) as inclusion bodies and re-folded after purification as previously described.46,59 Onconase expression was induced by 0.5 mM IPTG for 3.5 h in Circlegrow medium (Q-Biogen), whereas QBI-119 expression was induced by 0.5 mM IPTG for 3.5 h in LB medium.
IgG and IgG-RNase fusion constructs were purified from HEK293–6E cell culture supernatants using protein A affinity chromatography using an Äkta prime (GE Healthcare) system at 4 °C. Therefore, supernatant from productions were loaded on a 1 mL MAbSelect SuReTM affinity column (GE Healthcare) at 1 mL/min. After loading, columns were washed with binding buffer (20 mM NaPO4 buffer + 150 mM NaCl, pH 7.2) and washing buffer (100 mM citrate buffer, pH 5.0) till A280 showed a stable signal lower than 5 mAU. Eluted proteins (100 mM acetate-buffer + 150 mM NaCl, pH 3.0) were collected in 1 mL factions and directly neutralized by the addition of 0.5 M TRIS-HCl, pH 9.0. Pooled fractions from protein A purification are further purified by size exclusion chromatography (SEC) using a Superdex 200 16/60 column (GE Healthcare). Fractions containing IgG or IgG-RNases are pooled and filter sterilized using a 0.22 µm filter. Endotoxin level was quantified using Limulus Amebocyte Lysate (LAL) QCL-1000 Assay kit (Lonza).
Analytical SEC
Analytical SEC was performed on an Äkta System (GE Healthcare) system using a Tricorn SuperdexTM200 300/10GL column (GE Healthcare), with a mobile phase of PBS (pH 7.4) at a flow rate of 0.8 ml/min for assessment of homogeneity. Size exclusion chromatography with multi-angle laser light scattering (SEC-MALS) was used to determine the molecular weight of the antibody RNase constructs. A Wyatt Treos Mini Dawn detector and a Optilab® rEX refractive Index Detector were coupled online to an Agilent 1200 series HPLC system. Samples (50 µl) were separated on a Tricorn SuperdexTM200 300/10GL column (GE Healthcare), with a mobile phase of PBS (pH 7.4) at a flow rate of 0.5 ml/min.
Sodiumdodecylsulfate PAGE
Continuous sodiumdodecysulfate (SDS) PAGE (4–12% gradient gel, NuPAGE, life sciences) was used for separation of protein samples using a SDS/2-(N-morpholino)ethanesulfonic acid (MES) buffer system. Protein samples were diluted in PBS and 4 × non-reducing (NuPage, Invitrogen) or reducing (Roth) SDS gel loading buffer and, in the case of the latter, heated at 95 °C for 5 min. Proteins were electrophoretically separated at 170 V for 40 min. Protein bands were visualized by staining with Simply Blue Safe Stain (Invitrogen).
Dynamic light scattering
Dynamic light scattering (DLS) was performed using a Wyatt DynaPro Plate Reader. Samples were diluted to 1–5 mg/ml and monodispersity and diffusion coefficient to deduce the hydrodynamic radius were determined using the DYNAMICS software.
Differential scanning calorimetry
Differential scanning calorimetry (DSC) was used to compare the thermal stability of 200–600 μg of the different RNase constructs on a VP-DSC Capillary Cell Micro Calorimeter (GE Healthcare) and the results were analyzed using Origin 7 software.
Measurement of binding affinities
Binding affinities were determined by SPR analysis on a BIAcore T100 instrument (GE Healthcare Biacore, Inc). Antibodies were immobilized onto a CM5 sensor chip through the indirect capturing reagent, anti-human IgG Fc. Reagents from the “Human Antibody Capture Kit” (BR-1008–39, GE Healthcare Biacore, Inc) were used as described by the manufacturer. Soluble extracellular domain of MN antigen in several concentrations was used as analyte. The dissociation equilibrium constant (KD) was calculated based on the ratio of association (kon) and dissociation rated (koff) constants, obtained by fitting sensograms with a first order 1:1 binding model using BIAevaluation Software (version 4.0).
Ribonucleolytic activity assay and inhibition by RNase inhibitor
RNase activity was tested using a hypersensitive fluorescence-labeled substrate 6-FAM-dArUdAdA-BHQ1 (modified from ref. 60). Cleavage after uridine (rU) resulted in an increase of fluorescence emission. Therefore, IgG-RNase and RNase constructs and the substrate were diluted in substrate buffer (100 mM MES, 100 mM NaCl, pH6.0), 150 µl of respective antibody dilution was added into one well of a high binding FLUORTRAC 600 (Greiner Bio-one) plate. For starting the reaction 50 µl 2.5 nM substrate was added and fluorescence was immediately measured at excitation of 488 nm and emission at 535 nm at 25 °C in a TECAN Ultra multi-well plate reader (TECAN). To measure the time-dependent conversion of the substrate, measurements were repeated each 3 min.
Optimal RNase concentration was tested (10−9 to 10−12 M RNase) and adapted for each RNase variant. RNase enzyme efficiency was calculated according to following formula: kcat/Km = v0/(ΔF × [RNase]) with v0 = f(t) = ΔF/Δt (at t = 0), Δf = Fmax-F0 (F0, background fluorescence; Fmax, maximal fluorescence at complete substrate cleavage; [RNase]; RNase concentration, i.e. for IgG-RNases containing two RNase moieties per molecules [RNase] = [IgG-RNase] × 2).
In addition, RNase activity of MN-IgG-RNase was also tested in the presence of a concentration series of up to 50 fold molar excess of human placental RNase inhibitor (RNasin Plus, Promega). Bovine RNase (Applichem) was used as positive control for RI mediated activity inhibition. In these inhibition assays, 5 mM DTT was added to the substrate buffer.
Serum stability tests by enzyme-linked immunosorbent assay and antigen specific RNase activity assay
The stability assay was performed by antigen binding in ELISA. Briefly, samples containing 1.3 × 10−7 M IgG-RNase in PBS supplemented with 50% mouse serum were shock frozen at -80 °C, thawed at different time points, then incubated at 37 °C for up to 24 h and finally tested for antigen binding and antigen specific RNase activity. The antigen ELISA was performed by coating 1 µg of the antigen in PBS onto MaxiSorp® 96 well plates (Nunc, Thermo Fisher) for 1 h at 37 °C. After coating, wells were washed three times with PBS containing 0.05% (v/v) tween-20 (PBST) and then blocked for 1 h with 2% (w/v) skim milk powder in PBST. IgG-RNase samples from the stability assay were diluted 1:10, added and then incubated for 1 h at room temperature. After another washing step, secondary goat-anti-human IgG antibody horseradish peroxidase conjugate (Sigma A0170) was incubated for 1 h at room temperature. The wells were washed again and then incubated with a substrate solution containing 3,3′,5,5′-tetramethylbenzidine (TMB). The color reaction was stopped with one volume 0.5 M sulfuric acid and absorbances were measured at a wavelength of 450 nm (reference wavelength 620 nm) using a Sunrise ELISA reader (TECAN).
Additionally, antigen-specific RNase activity was measured with a modified ELISA using the RNase assay for detection instead of the secondary antibody conjugate. Briefly, IgG-RNase samples from the stability assay were diluted 1:1000 in PBS prior incubation on antigen-coated black 96-well plates (LUMITRAC-600TM, Greiner Bio-one, Germany) that were blocked with acetylated RNase-free BSA. After three washing steps with RNase-free PBS, RNase activity assay was measured as described above using the hypersensitive fluorescence-labeled substrate 6-FAM-dArUdAdA-BHQ1. Assuming that active RNase moieties of the same construct have same activity the formula for RNase catalytic efficiency was transposed to calculate the active RNase concentrations. The concentrations of active RNase of all samples were normalized to the mean value of freshly thawed samples set to 100%.
Internalization studies
The pH-sensitive fluorescent dye CypHer 5E (GE Healthcare, PA15401) was conjugated to the antibody RNase constructs in order to detect internalization because this dye becomes fluorescent only at acidic pH, like in endosomes, while it remains non-fluorescent at neutral or basic pH values. For coupling, IgGs or IgG-RNase fusions were incubated for 1 h at 20 °C with a 2-molar excess of dye in PBS/Na-carbonate pH 8.3. On average, a lysine-coupled dye load of 1.6 was achieved. After CypHer 5E labeling, a negligible reduction in binding affinity and no effect on RNase activity was observed.
A total of 1 × 104 cells were used to investigate specific internalization upon antibody binding. Cells were treated with various concentrations of labeled IgG or IgG-RNase at 37 °C and 5% CO2. Internalization was measured for up to 24 h using the INCell Analyzer 1000 at several time points. Analysis of internalization was performed microscopically at 40 × magnification and by determination of granularity (granule count/cell; 620 nm excitation, and 700 nm emission for CypHer5E). Nuclei were visualized with DNA staining using cell permeable stain Hoechst 33342 (0.5 μg/ml, 30 min incubation, emission at 353–365 nm, detection at 480 nm). The corresponding empty vector control cells were used as specificity control in addition to CTX-IgG-RNase. Three independent experiments were performed; data points represent triplicate determinations.
Tumor cell line growth inhibition assay
A total of 1500–2500 cells per well (depending on cell type) were seeded into 96-well tissue culture plates (Corning CellBIND) using DMEM (high glucose: 4.5 g/L glucose, with Glutamax, Invitrogen) supplemented with 5% (v/v) fetal calf serum (PAA). A concentration series (final concentration: 10−7 to 10−12 M) of sterile compounds were added 24 h later to the cells. Incubation was pursued for 72 h at 37 °C, 5% CO2. For detection of cell viability, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, ATCC) assay was performed according to the manufacturer’s recommendations. An alternative detection procedure was performed with Cell Titer Glo Luminescent Cell Viability Assay (Promega) according to the manufacturer’s recommendations. Untreated controls were set to 100% and all other values were normalized.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Acknowledgments
We are gratefully to Dmitry Zubov for helpful discussions during choice of endosomal cleavable linkers and Andreas Schubel for technical assistance measuring RNase activities.
Footnotes
Previously published online: www.landesbioscience.com/journals/mabs/article/27830
References
- 1.Reichert JM. Antibodies to watch in 2014. MAbs. 2013;6:6. doi: 10.4161/mabs.27333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thie H, Toleikis L, Li J, von Wasielewski R, Bastert G, Schirrmann T, Esteves IT, Behrens CK, Fournes B, Fournier N, et al. Rise and fall of an anti-MUC1 specific antibody. PLoS One. 2011;6:e15921. doi: 10.1371/journal.pone.0015921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang J, Baskar S, Kwong KY, Kennedy MG, Wiestner A, Rader C. Therapeutic potential and challenges of targeting receptor tyrosine kinase ROR1 with monoclonal antibodies in B-cell malignancies. PLoS One. 2011;6:e21018. doi: 10.1371/journal.pone.0021018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Muirhead M, Martin PJ, Torok-Storb B, Uhr JW, Vitetta ES. Use of an antibody-ricin A-chain conjugate to delete neoplastic B cells from human bone marrow. Blood. 1983;62:327–32. [PubMed] [Google Scholar]
- 5.Giles FJ, Kantarjian HM, Kornblau SM, Thomas DA, Garcia-Manero G, Waddelow TA, David CL, Phan AT, Colburn DE, Rashid A, et al. Mylotarg (gemtuzumab ozogamicin) therapy is associated with hepatic venoocclusive disease in patients who have not received stem cell transplantation. Cancer. 2001;92:406–13. doi: 10.1002/1097-0142(20010715)92:2<406::AID-CNCR1336>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 6.Messmann RA, Vitetta ES, Headlee D, Senderowicz AM, Figg WD, Schindler J, Michiel DF, Creekmore S, Steinberg SM, Kohler D, et al. A phase I study of combination therapy with immunotoxins IgG-HD37-deglycosylated ricin A chain (dgA) and IgG-RFB4-dgA (Combotox) in patients with refractory CD19(+), CD22(+) B cell lymphoma. Clin Cancer Res. 2000;6:1302–13. [PubMed] [Google Scholar]
- 7.Rybak SM, Arndt MAE, Schirrmann T, Dübel S, Krauss J. Ribonucleases and immunoRNases as anticancer drugs. Curr Pharm Des. 2009;15:2665–75. doi: 10.2174/138161209788923921. [DOI] [PubMed] [Google Scholar]
- 8.Rybak SM, Saxena SK, Ackerman EJ, Youle RJ. Cytotoxic potential of ribonuclease and ribonuclease hybrid proteins. J Biol Chem. 1991;266:21202–7. [PubMed] [Google Scholar]
- 9.Mikulski SM, Costanzi JJ, Vogelzang NJ, McCachren S, Taub RN, Chun H, Mittelman A, Panella T, Puccio C, Fine R, et al. Phase II trial of a single weekly intravenous dose of ranpirnase in patients with unresectable malignant mesothelioma. J Clin Oncol. 2002;20:274–81. doi: 10.1200/JCO.20.1.274. [DOI] [PubMed] [Google Scholar]
- 10.Benito A, Ribó M, Vilanova M. On the track of antitumour ribonucleases. Mol Biosyst. 2005;1:294–302. doi: 10.1039/b502847g. [DOI] [PubMed] [Google Scholar]
- 11.Lee I. Ranpirnase (Onconase), a cytotoxic amphibian ribonuclease, manipulates tumour physiological parameters as a selective killer and a potential enhancer for chemotherapy and radiation in cancer therapy. Expert Opin Biol Ther. 2008;8:813–27. doi: 10.1517/14712598.8.6.813. [DOI] [PubMed] [Google Scholar]
- 12.Wu Y, Mikulski SM, Ardelt W, Rybak SM, Youle RJ. A cytotoxic ribonuclease. Study of the mechanism of onconase cytotoxicity. J Biol Chem. 1993;268:10686–93. [PubMed] [Google Scholar]
- 13.Schirrmann T, Krauss J, Arndt MAE, Rybak SM, Dübel S. Targeted therapeutic RNases (ImmunoRNases) Expert Opin Biol Ther. 2009;9:79–95. doi: 10.1517/14712590802631862. [DOI] [PubMed] [Google Scholar]
- 14.Krauss J, Arndt MAE, Vu BK, Newton DL, Seeber S, Rybak SM. Efficient killing of CD22+ tumor cells by a humanized diabody-RNase fusion protein. Biochem Biophys Res Commun. 2005;331:595–602. doi: 10.1016/j.bbrc.2005.03.215. [DOI] [PubMed] [Google Scholar]
- 15.Beintema JJ, Kleineidam RG. The ribonuclease A superfamily: general discussion. Cell Mol Life Sci. 1998;54:825–32. doi: 10.1007/s000180050211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boix E, Nogués MV. Mammalian antimicrobial proteins and peptides: overview on the RNase A superfamily members involved in innate host defence. Mol Biosyst. 2007;3:317–35. doi: 10.1039/b617527a. [DOI] [PubMed] [Google Scholar]
- 17.D’Alessio G. New and cryptic biological messages from RNases. Trends Cell Biol. 1993;3:106–9. doi: 10.1016/0962-8924(93)90166-X. [DOI] [PubMed] [Google Scholar]
- 18.Yang D, Chen Q, Rosenberg HF, Rybak SM, Newton DL, Wang ZY, Fu Q, Tchernev VT, Wang M, Schweitzer B, et al. Human ribonuclease A superfamily members, eosinophil-derived neurotoxin and pancreatic ribonuclease, induce dendritic cell maturation and activation. J Immunol. 2004;173:6134–42. doi: 10.4049/jimmunol.173.10.6134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gupta SK, Haigh BJ, Griffin FJ, Wheeler TT. The mammalian secreted RNases: mechanisms of action in host defence. Innate Immun. 2013;19:86–97. doi: 10.1177/1753425912446955. [DOI] [PubMed] [Google Scholar]
- 20.Borriello M, Laccetti P, Terrazzano G, D’Alessio G, De Lorenzo C. A novel fully human antitumour immunoRNase targeting ErbB2-positive tumours. Br J Cancer. 2011;104:1716–23. doi: 10.1038/bjc.2011.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huhn M, Sasse S, Tur MK, Matthey B, Schinköthe T, Rybak SM, Barth S, Engert A. Human angiogenin fused to human CD30 ligand (Ang-CD30L) exhibits specific cytotoxicity against CD30-positive lymphoma. Cancer Res. 2001;61:8737–42. [PubMed] [Google Scholar]
- 22.De Lorenzo C, Arciello A, Cozzolino R, Palmer DB, Laccetti P, Piccoli R, D’Alessio G. A fully human antitumor immunoRNase selective for ErbB-2-positive carcinomas. Cancer Res. 2004;64:4870–4. doi: 10.1158/0008-5472.CAN-03-3717. [DOI] [PubMed] [Google Scholar]
- 23.Menzel C, Schirrmann T, Konthur Z, Jostock T, Dübel S. Human antibody RNase fusion protein targeting CD30+ lymphomas. Blood. 2008;111:3830–7. doi: 10.1182/blood-2007-04-082768. [DOI] [PubMed] [Google Scholar]
- 24.Zewe M, Rybak SM, Dübel S, Coy JF, Welschof M, Newton DL, Little M. Cloning and cytotoxicity of a human pancreatic RNase immunofusion. Immunotechnology. 1997;3:127–36. doi: 10.1016/S1380-2933(97)00070-5. [DOI] [PubMed] [Google Scholar]
- 25.Park C, Schultz LW, Raines RT. Contribution of the active site histidine residues of ribonuclease A to nucleic acid binding. Biochemistry. 2001;40:4949–56. doi: 10.1021/bi0100182. [DOI] [PubMed] [Google Scholar]
- 26.Bal HP, Batra JK. Human pancreatic ribonuclease--deletion of the carboxyl-terminal EDST extension enhances ribonuclease activity and thermostability. Eur J Biochem. 1997;245:465–9. doi: 10.1111/j.1432-1033.1997.t01-1-00465.x. [DOI] [PubMed] [Google Scholar]
- 27.Johnson RJ, McCoy JG, Bingman CA, Phillips GN, Jr., Raines RT. Inhibition of human pancreatic ribonuclease by the human ribonuclease inhibitor protein. J Mol Biol. 2007;368:434–49. doi: 10.1016/j.jmb.2007.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murthy BS, Sirdeshmukh R. Sensitivity of monomeric and dimeric forms of bovine seminal ribonuclease to human placental ribonuclease inhibitor. Biochem J. 1992;281:343–8. doi: 10.1042/bj2810343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Riccio G, Borriello M, D’Alessio G, De Lorenzo C. A novel human antitumor dimeric immunoRNase. J Immunother. 2008;31:440–5. doi: 10.1097/CJI.0b013e31816bc769. [DOI] [PubMed] [Google Scholar]
- 30.Benito A, Bosch M, Torrent G, Ribó M, Vilanova M. Stabilization of human pancreatic ribonuclease through mutation at its N-terminal edge. Protein Eng. 2002;15:887–93. doi: 10.1093/protein/15.11.887. [DOI] [PubMed] [Google Scholar]
- 31.Turcotte RF, Lavis LD, Raines RT. Onconase cytotoxicity relies on the distribution of its positive charge. FEBS J. 2009;276:3846–57. doi: 10.1111/j.1742-4658.2009.07098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leland PA, Staniszewski KE, Kim B-M, Raines RT. Endowing human pancreatic ribonuclease with toxicity for cancer cells. J Biol Chem. 2001;276:43095–102. doi: 10.1074/jbc.M106636200. [DOI] [PubMed] [Google Scholar]
- 33.Leland PA, Schultz LW, Kim B-M, Raines RT. Ribonuclease A variants with potent cytotoxic activity. Proc Natl Acad Sci U S A. 1998;95:10407–12. doi: 10.1073/pnas.95.18.10407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mayor S, Pagano RE. Pathways of clathrin-independent endocytosis. Nat Rev Mol Cell Biol. 2007;8:603–12. doi: 10.1038/nrm2216. [DOI] [PubMed] [Google Scholar]
- 35.Mayor S, Riezman H. Sorting GPI-anchored proteins. Nat Rev Mol Cell Biol. 2004;5:110–20. doi: 10.1038/nrm1309. [DOI] [PubMed] [Google Scholar]
- 36.Leich F, Stöhr N, Rietz A, Ulbrich-Hofmann R, Arnold U. Endocytotic internalization as a crucial factor for the cytotoxicity of ribonucleases. J Biol Chem. 2007;282:27640–6. doi: 10.1074/jbc.M702240200. [DOI] [PubMed] [Google Scholar]
- 37.Notomista E, Mancheño JM, Crescenzi O, Di Donato A, Gavilanes J, D’Alessio G. The role of electrostatic interactions in the antitumor activity of dimeric RNases. FEBS J. 2006;273:3687–97. doi: 10.1111/j.1742-4658.2006.05373.x. [DOI] [PubMed] [Google Scholar]
- 38.Dubowchik GM, Firestone RA, Padilla L, Willner D, Hofstead SJ, Mosure K, Knipe JO, Lasch SJ, Trail PA. Cathepsin B-labile dipeptide linkers for lysosomal release of doxorubicin from internalizing immunoconjugates: model studies of enzymatic drug release and antigen-specific in vitro anticancer activity. Bioconjug Chem. 2002;13:855–69. doi: 10.1021/bc025536j. [DOI] [PubMed] [Google Scholar]
- 39.Harada M, Sakakibara H, Yano T, Suzuki T, Okuno S. Determinants for the drug release from T-0128, camptothecin analogue-carboxymethyl dextran conjugate. J Control Release. 2000;69:399–412. doi: 10.1016/S0168-3659(00)00321-7. [DOI] [PubMed] [Google Scholar]
- 40.Vasey PA, Kaye SB, Morrison R, Twelves C, Wilson P, Duncan R, Thomson AH, Murray LS, Hilditch TE, Murray T, et al. Cancer Research Campaign Phase I/II Committee Phase I clinical and pharmacokinetic study of PK1 [N-(2-hydroxypropyl)methacrylamide copolymer doxorubicin]: first member of a new class of chemotherapeutic agents-drug-polymer conjugates. Clin Cancer Res. 1999;5:83–94. [PubMed] [Google Scholar]
- 41.Mathieu MA, Bogyo M, Caffrey CR, Choe Y, Lee J, Chapman H, Sajid M, Craik CS, McKerrow JH. Substrate specificity of schistosome versus human legumain determined by P1-P3 peptide libraries. Mol Biochem Parasitol. 2002;121:99–105. doi: 10.1016/S0166-6851(02)00026-9. [DOI] [PubMed] [Google Scholar]
- 42.Schmid B, Chung D-E, Warnecke A, Fichtner I, Kratz F. Albumin-binding prodrugs of camptothecin and doxorubicin with an Ala-Leu-Ala-Leu-linker that are cleaved by cathepsin B: synthesis and antitumor efficacy. Bioconjug Chem. 2007;18:702–16. doi: 10.1021/bc0602735. [DOI] [PubMed] [Google Scholar]
- 43.Erickson HA, Jund MD, Pennell CA. Cytotoxicity of human RNase-based immunotoxins requires cytosolic access and resistance to ribonuclease inhibition. Protein Eng Des Sel. 2006;19:37–45. doi: 10.1093/protein/gzi073. [DOI] [PubMed] [Google Scholar]
- 44.Schmid B, Warnecke A, Fichtner I, Jung M, Kratz F. Development of albumin-binding camptothecin prodrugs using a Peptide positional scanning library. Bioconjug Chem. 2007;18:1786–99. doi: 10.1021/bc0700842. [DOI] [PubMed] [Google Scholar]
- 45.Hetzel C, Bachran C, Fischer R, Fuchs H, Barth S, Stöcker M. Small cleavable adapters enhance the specific cytotoxicity of a humanized immunotoxin directed against CD64-positive cells. J Immunother. 2008;31:370–6. doi: 10.1097/CJI.0b013e31816a2d23. [DOI] [PubMed] [Google Scholar]
- 46.Gaur D, Swaminathan S, Batra JK. Interaction of human pancreatic ribonuclease with human ribonuclease inhibitor. Generation of inhibitor-resistant cytotoxic variants. J Biol Chem. 2001;276:24978–84. doi: 10.1074/jbc.M102440200. [DOI] [PubMed] [Google Scholar]
- 47.Wu Y, Saxena SK, Ardelt W, Gadina M, Mikulski SM, De Lorenzo C, D’Alessio G, Youle RJ. A study of the intracellular routing of cytotoxic ribonucleases. J Biol Chem. 1995;270:17476–81. doi: 10.1074/jbc.270.29.17476. [DOI] [PubMed] [Google Scholar]
- 48.Hazes B, Read RJ. Accumulating evidence suggests that several AB-toxins subvert the endoplasmic reticulum-associated protein degradation pathway to enter target cells. Biochemistry. 1997;36:11051–4. doi: 10.1021/bi971383p. [DOI] [PubMed] [Google Scholar]
- 49.Worthington ZEV, Carbonetti NH. Evading the proteasome: absence of lysine residues contributes to pertussis toxin activity by evasion of proteasome degradation. Infect Immun. 2007;75:2946–53. doi: 10.1128/IAI.02011-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mikulski SM, Viera A, Deptala A, Darzynkiewicz Z. Enhanced in vitro cytotoxicity and cytostasis of the combination of onconase with a proteasome inhibitor. Int J Oncol. 1998;13:633–44. doi: 10.3892/ijo.13.4.633. [DOI] [PubMed] [Google Scholar]
- 51.Rodríguez M, Benito A, Tubert P, Castro J, Ribó M, Beaumelle B, Vilanova M. A cytotoxic ribonuclease variant with a discontinuous nuclear localization signal constituted by basic residues scattered over three areas of the molecule. J Mol Biol. 2006;360:548–57. doi: 10.1016/j.jmb.2006.05.048. [DOI] [PubMed] [Google Scholar]
- 52.Saxena SK, Sirdeshmukh R, Ardelt W, Mikulski SM, Shogen K, Youle RJ. Entry into cells and selective degradation of tRNAs by a cytotoxic member of the RNase A family. J Biol Chem. 2002;277:15142–6. doi: 10.1074/jbc.M108115200. [DOI] [PubMed] [Google Scholar]
- 53.Qiao M, Zu L-D, He X-H, Shen R-L, Wang Q-C, Liu M-F. Onconase downregulates microRNA expression through targeting microRNA precursors. Cell Res. 2012;22:1199–202. doi: 10.1038/cr.2012.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Torrent G, Ribó M, Benito A, Vilanova M. Bactericidal activity engineered on human pancreatic ribonuclease and onconase. Mol Pharm. 2009;6:531–42. doi: 10.1021/mp8001914. [DOI] [PubMed] [Google Scholar]
- 55.Futami J, Maeda T, Kitazoe M, Nukui E, Tada H, Seno M, Kosaka M, Yamada H. Preparation of potent cytotoxic ribonucleases by cationization: enhanced cellular uptake and decreased interaction with ribonuclease inhibitor by chemical modification of carboxyl groups. Biochemistry. 2001;40:7518–24. doi: 10.1021/bi010248g. [DOI] [PubMed] [Google Scholar]
- 56.Durocher Y, Perret S, Kamen A. High-level and high-throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Res. 2002;30:E9. doi: 10.1093/nar/30.2.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jäger V, Büssow K, Wagner A, Weber S, Hust M, Frenzel A, Schirrmann T. High level transient production of recombinant antibodies and antibody fusion proteins in HEK293 cells. BMC Biotechnol. 2013;13:52. doi: 10.1186/1472-6750-13-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tom R, Bisson L, Durocher Y. Transient expression in HEK293-EBNA1 cells. In: Dyson MR, Durocher, Yves, editors. Expression Systems: Methods Express. Scion; 2007. [Google Scholar]
- 59.Leland PA, Schultz LW, Kim B-M, Raines RT. Ribonuclease A variants with potent cytotoxic activity. Proc Natl Acad Sci U S A. 1998;95:10407–12. doi: 10.1073/pnas.95.18.10407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kelemen BR, Klink TA, Behlke MA, Eubanks SR, Leland PA, Raines RT. Hypersensitive substrate for ribonucleases. Nucleic Acids Res. 1999;27:3696–701. doi: 10.1093/nar/27.18.3696. [DOI] [PMC free article] [PubMed] [Google Scholar]