

Abstract
While glyco-engineered monoclonal antibodies (mAbs) with improved antibody-dependent cell-mediated cytotoxicity (ADCC) are reaching the market, extensive efforts have also been made to improve their pharmacokinetic properties to generate biologically superior molecules. Most therapeutic mAbs are human or humanized IgG molecules whose half-life is dependent on the neonatal Fc receptor FcRn. FcRn reduces IgG catabolism by binding to the Fc domain of endocytosed IgG in acidic lysosomal compartments, allowing them to be recycled into the blood. Fc-engineered mAbs with increased FcRn affinity resulted in longer in vivo half-life in animal models, but also in healthy humans. These Fc-engineered mAbs were obtained by alanine scanning, directed mutagenesis or in silico approach of the FcRn binding site. In our approach, we applied a random mutagenesis technology (MutaGenTM) to generate mutations evenly distributed over the whole Fc sequence of human IgG1. IgG variants with improved FcRn-binding were then isolated from these Fc-libraries using a pH-dependent phage display selection process. Two successive rounds of mutagenesis and selection were performed to identify several mutations that dramatically improve FcRn binding. Notably, many of these mutations were unpredictable by rational design as they were located distantly from the FcRn binding site, validating our random molecular approach. When produced on the EMABling® platform allowing effector function increase, our IgG variants retained both higher ADCC and higher FcRn binding. Moreover, these IgG variants exhibited longer half-life in human FcRn transgenic mice. These results clearly demonstrate that glyco-engineering to improve cytotoxicity and protein-engineering to increase half-life can be combined to further optimize therapeutic mAbs.
Keywords: ADCC, Fc-engineering, FcRn, glycoengineering, phage display, pharmacokinetics, random mutagenesis
Introduction
Therapeutic monoclonal antibodies (mAbs) of the immunoglobulin G (IgG) isotype are in widespread use for the treatment of a variety of disorders, including cancers, autoimmune and infectious diseases. Since the approval of the first-generation mAbs in the late 1990s, tremendous improvement of their efficacy has been achieved by molecular engineering. The second-generation of therapeutic mAbs usually included optimized variable domains (Fv) such as human/humanized Fv domains or higher antigen affinity. Over the past decade, a third-generation of mAbs has emerged1 with improved Fc region because it was clearly established that the success of mAbs as cancer therapeutics substantially relied on their ability to engage the immune system via this Fc domain.2,3 This Fc-dependent engagement is mediated by activation of the lytic complement pathway (classical pathway) and crosslinking of Fcγ receptors (FcγRs) widely expressed on many innate immune effector cells such as natural killer (NK) cells, monocytes, macrophages and neutrophils.4 Therefore, current knowledge of the mechanisms of action of therapeutic mAbs involve direct (blockade of intracellular signaling, deprivation of growth hormone or pro-apoptotic effect) as well as indirect (complement- or cell-mediated cytotoxicity) killing of the tumor cells via Fv and Fc domains respectively.5 With the exception of T cells, immune effector cells express one or more activating FcγRs (FcγRI, FcγRIIA, and FcγRIIIA) and, for the majority, co-express the inhibitory FcγR (FcγRIIB). Following engagement of their activating FcγRs, these cells kill tumor cells through antibody-dependent cell-mediated cytotoxicity (ADCC) or through phagocytosis (ADCP).
The rationale that improved mAb-mediated effector functions will be clinically beneficial is supported by the correlation of clinical outcomes of mAb therapies in patients with natural polymorphisms of FcγRs. The allelic FcγR variants show different affinities for the Fc domain of IgG1 and more favorable clinical responses were observed in patients who were homozygous for the higher-affinity allele of the activating receptor FcγRIIIA (V158). This has been observed for treatment with the anti-CD20 rituximab in follicular lymphoma,6 the anti-HER2 trastuzumab in metastatic breast cancer7 and the anti-EGFR cetuximab in metastatic colorectal cancer.8 These findings, obtained for various specificities and malignancies, suggest that the low-affinity receptor FcγRIIIA is crucial for the anti-tumor activity of mAbs in vivo and that increasing crosslinking of this receptor should translate into enhanced therapeutic activity. To this aim, glyco-engineered mAbs have been designed. Indeed, the absence of α1,6 innermost fucose residues on the Fc glycan moiety of mAbs has long been associated with enhanced affinity for FcγRIIIA, and there is strong evidence that such afucosylated or low fucosylated mAbs translate into enhanced therapeutic activity in mouse models in vivo.9 To date, more than 20 third-generation glyco-engineered mAbs, with higher ADCC, are being evaluated in clinical studies. Two of these mAbs have already been approved, mogamulizumab in March 2012 and obinutuzumab in November 2013, confirming the success of this approach.10
In addition to increasing mAbs cytotoxicity, extensive efforts have also been made to improve mAbs pharmacokinetic (PK) properties.11,12 Despite the relatively long half-lives of mAbs, improved molecules with longer half-lives and conserved efficacy would permit less frequent dosing schedules and provide higher patient convenience. IgG half-life is dependent on the neonatal Fc receptor (FcRn), a member of the MHC class I superfamily, which is expressed by many cell types including endothelial cells, macrophages, monocytes and monocyte-derived dendritic cells in human.13,14 FcRn is a high affinity Fc receptor that binds IgG in a strictly pH-dependent manner. IgGs are internalized by non-specific pinocytosis into the endosomes of cells where the low pH (pH 5–6) promotes binding to FcRn with nanomolar affinity. Bound IgG-FcRn complexes are recycled back to the cell surface and dissociate at the neutral pH of the extracellular fluid, returning to the blood circulation. IgGs that do not bind to FcRn traffic into the lysosomes where they are degraded by proteases. Moreover, FcRn was shown to be involved in IgG transport across the feto-maternal barrier and across endothelial and epithelial cellular barriers increasing their overall bioavailability.15,16 This property has been successfully exploited for pulmonary delivery, as well as intranasal immunization with Fc-fusion proteins.17,18 Lastly, FcRn was also reported to transport IgG-antigen complexes, thus favoring antigen-presentation,19-21 and to enable phagocytosis of IgG-opsonized bacteria by neutrophils,22 demonstrating a wide range of functions for this receptor in several immunological and non-immunological mechanisms.
Consistent with its role in IgG catabolism, several Fc-engineered mAbs with increased FcRn affinity and conserved pH dependency were designed and resulted in longer half-life in vivo in human FcRn transgenic mice (hFcRn mice) and cynomolgus monkeys.23,24 Hence, Fc-engineered variants (Fc-LS) of the anti-VEGF bevacizumab and the anti-EGFR cetuximab demonstrated half-lives extended up to almost 5-fold in hFcRn mice and 3-fold in cynomolgus monkeys.25 These mAbs exhibited greater anti-tumor activity when tested in hFcRn/Rag1–/– tumor xenografted mice, validating that prolonged exposure due to FcRn-mediated enhancement of half-life should also improve therapeutic mAbs efficacy in human. Moreover, for the first time in healthy humans an Fc-engineered anti-RSV (motavizumab-YTE) recently demonstrated a significant increase in serum half-life of up to 100 days, validating the usefulness of increasing FcRn binding and fully confirming the results previously obtained in animal models.26
For the past 20 years, the Fc/FcRn interaction has been extensively studied by directed mutagenesis and crystallography using the respective human, murine and rat proteins.27,28 The FcRn binding site is structurally conserved among species and comprises three distinct zones in the CH2 and CH3 domains that are localized in the CH2/CH3 interdomain region. Fc-engineered mAbs with increased FcRn affinity were obtained by alanine scanning,27 but also directed mutagenesis29 or in silico approaches25,30,31 focused on the FcRn binding site. Therefore, the resulting Fc variants usually include 2–3 substitutions in or close to the FcRn binding site. To identify new mutations that could be distant from the FcRn binding site but have an indirect, and possibly cooperative, positive impact on the Fc/FcRn complex, we randomly mutated the entire Fc domain and selected several FcRn affinity enhanced Fc variants at pH 6.0. To improve both mAb cytotoxicity and half-life, we produced the selected IgG variants as low fucosylated molecules and demonstrated increased serum persistence in hFcRn mice, as well as conserved enhanced ADCC.
Results
First round of mutagenesis and selection (MS1)
Construction of random libraries
The human IgG1 Fc gene encoding amino acid residues 226–447 (EU index as in Kabat32), referred to as Fc wild type (Fc-WT), was cloned into our modified phagemid vector.33 This Fc fragment includes the entire CH2 and CH3 domains and the last 5 amino acids of the hinge region. To set up the experiments, the double variant T250Q/M428L (Fc-QL)30,31 and the triple mutant M252Y/S254T/T256E (Fc-YTE)29 were constructed as positive controls. In the phagemid vector, several fully randomized libraries were generated using the MutaGenTM technology that uses low fidelity human DNA polymerases (mutases) to introduce random mutations homogeneously throughout the target sequence.34 Three distinct human mutases (pol. β, pol. η and pol. ι) were used in different conditions to create complementary mutational patterns. Four different sub-libraries were constructed for this first round of mutagenesis and selection (MS1). A first sub-library was obtained using pol. β on the Fc-WT gene and contained 3.2 × 106 clones (called Mut1.1). The DNA of this first sub-library was used to generate the second and the third sub-libraries, using respectively pol. β (3.8 × 106 clones, Mut1.2) and pol. η plus ι (3.0 × 106 clones, Mut1.3). The fourth library was generated with pol. η alone on the Fc-WT gene (1.0 × 106 clones, Mut1.4). These four sub-libraries were proportionally mixed to obtain the final library Mut1, representing 1.1 × 107 clones (Table 1).
Table 1. Nucleotide mutations analysis of the four sub-libraries Mut1.1 to Mut1.4 and the Mut1 and Mut2 final libraries.
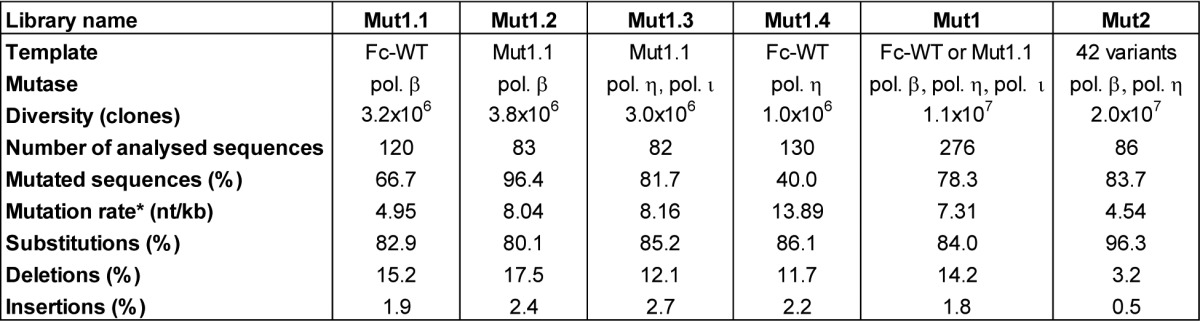
*excluding the unmutated sequences
For each sub-library and the final pooled library Mut1, 48 to 192 clones were sequenced after PCR on cells and sequences were analyzed using MilleGen’s proprietary software Mutanalyse4Fc adapted for the Fc molecule from the Mutanalyse 2.5 software described previously. Analysis of the four sub-libraries’ sequences showed that the use of two sequential mutagenesis steps (Fc-WT to Mut1.1 and Mut1.1 to either Mut1.2 or Mut1.3) permitted an increase in the mutation rate (Table 1). The mutation rate of Mut1.1 is 4.95 nucleotide mutations per kilo base (kb), whereas Mut1.2 and Mut1.3 had mutation rates of 8.04 and 8.16 mutations per kb, respectively. The percentage of mutated sequences was also increased from Mut1.1 (66.7%) to the two sub-libraries (96.4 and 81.7%, respectively). The mutation rate for Mut1.4, constructed with pol. η alone, is high (13.89 mutations per kb) but this library contains only 40% of mutated sequences. For the four sub-libraries, the mutations correspond mainly to substitutions (between 80 and 86% of the mutations), with a minority of deletions (~15%) and insertions (~2%). The analysis of proportionally pooled sequences of the 4 sub-libraries and of 35 sequences for the Mut1 library (276 sequences, i.e., 70 sequences for Mut1.1, 83 sequences for Mut1.2, 66 sequences for Mut1.3, 22 sequences for Mut1.4 and 35 sequences for Mut1) showed that the frequency of nucleotide mutations of Mut1 library is 7.31 mutations per kb, which corresponds to 4.89 mutations per Fc gene [666 nucleotides (nt)] (Table 1). Among these mutations, 84.0% are substitutions, 14.2% are deletions and 1.8% are additions, these last two categories introducing frame shifts in the gene. When considering only the sequences mutated in frame, which represent 42.0% of the clones (excluding the unmutated sequences and the sequences with a stop codon other than a TAG), the mutation frequency is 4.05 mutations per kb, corresponding to 2.63 mutations per gene (1 to 6 mutated nucleotides per gene), with mainly 1 mutation per gene (Fig. 1A). The mutation analysis was also performed at the protein level. The Mut1 library contains 27.5% of clones expressing the Fc-WT [unmutated or with silent mutation(s)], 37.3% of clones containing a sequence out of frame or with a stop codon (different from the amber stop codon TAG expressed in our system) and 35.1% of clones with a mutated sequence (Fc variants). These last clones represent the active part of the library, comprising ~3.8 × 106 mutated clones with on average 2.35 mutated amino acids per molecule. The distribution of the mutations on the Fc sequence was also analyzed and confirmed that the random mutations are evenly distributed along the entire gene without any hot spot (Fig. 1B).

Figure 1. Analysis of the mutated sequences in frame in the Mut1 library. (A) Distribution of the number of nucleotide mutations per Fc. (B) Distribution of the mutations along the Fc region (amino acids).
Selection of Fc variants on human FcRn and characterization by phage-ELISA
The Fc variants of the Mut1 library were displayed on the M13 bacteriophage using the classical technique of phage display.35 Selection was performed using three different strategies, two using immunoplates coated with either biotinylated FcRn or the FcRn-p3 fusion molecule and one using biotinylated FcRn in solution trapped with streptavidin-coated magnetic bead, to increase the chance to select for affinity-enhanced FcRn binders. FcRn binding and washing steps were performed at pH6.0 whereas bound Fc-phages were eluted at pH 7.4 to preserve Fc/FcRn interaction pH-dependency, rendering the selection process highly specific as well. Three to eight rounds of selection were performed for each strategy. Retrieved clones were isolated, sequenced and analyzed with internally developed bioinformatics tools.
During the MS1 process, a total of 227 different Fc variants were isolated. These 227 mutated Fc-phages were then ranked for their FcRn binding using a comparative phage-ELISA assay at pH6.0 on FcRn-coated plates with the Fc-WT as reference. In this phage-ELISA assay, the Fc-QL and Fc-YTE positive controls, expressed on the M13 bacteriophage, showed a better binding than Fc-WT and similar pH-dependent binding properties, i.e., strong binding at pH6.0, decreased binding at higher pH and no binding at pH7.4, validating our phage display system (data not shown). Fc-QL and Fc-YTE had a specific signal that was on average 3-fold stronger than the Fc-WT (ratio Fc-QL/Fc-WT = 3.2 and Fc-YTE/Fc-WT = 3.5). From the 227 mutants isolated during MS1, 139 were considered positive with a ratio/Fc-WT > 2 and 73 were better than the positive control Fc-QL (ratio/Fc-WT > 3.2). The best clone, S5A-41, had a ratio/Fc-WT of 9.0 (Fig. 2A). Sequence analyses of these positive Fc variants showed that they contain on average 2.7 mutated amino acids per Fc and permitted to identify 4 key positions, which are positions mutated in more than 10% of the improved Fc variants (Fig. 2B). Three key positions are located outside the FcRn binding site: P230 in the hinge domain, V264 in the CH2 domain and A378 in the CH3 domain. The last position, N434 is in the CH3 domain included in the FcRn binding site. This position is predominant and has been largely described before by others,27 validating our selection system. Finally, the positive Fc variants isolated are combinations of at least 1 or 2 mutations of these key positions, as shown for the 10 best Fc variants (Fig. 2C).

Figure 2. Characterization of the clones selected during MS1. (A) Ratio obtained by phage-ELISA for the positive controls and the best clone of MS1 (S5A-41). (B) Sequence analysis of the 139 positive clones (ratio/Fc-WT > 2), distribution of the mutations along the Fc domain revealing 4 key positions, which are mutated in more than 10% of the positive clones. (C) Sequences of the 10 best clones isolated during MS1. Key positions as identified in Figure 2B are in bold.
Second round of mutagenesis and selection (MS2)
Construction of random libraries
Based on the results obtained during MS1, we performed a second round of mutagenesis and selection (MS2) to further improve the Fc/FcRn binding. As previously described for MS1, randomized libraries were constructed with the MutaGenTM technology. For these new libraries, the DNA template used was a DNA pool of 42 variants isolated during MS1 and having improved FcRn binding by phage-ELISA. To minimize the number of mutations of the final variants, only the variants with one or two amino acid mutations were included in this second mutagenesis process. In addition, variants mutated on position N434 were also excluded as the mutations N434Y or S were too predominant and not novel, our aim being to identify new mutations. At the protein level, our pool of 42 single and double variants has a mutation rate of 1.45 amino acids per Fc. As many variants have silent mutations (15 variants with one silent mutation, 4 variants with 2 and 4 variants with 3 silent mutations), the nucleotide mutation rate of the pool is on average of 3.43 mutations per kb, i. e., 2.28 mutations per gene (1 to 5 nucleotide mutations, Fig. 3B). Two libraries were constructed with mild conditions to achieve a low mutation rate. A first library was obtained using pol. β (1.9x107 clones, Mut2.1) and a second library with pol. η (1 × 106 clones, Mut2.2). These two libraries were proportionally mixed to obtain the final library Mut2, representing 2 × 107 different clones (Table 1). The frequency of mutations of the Mut2 library is 4.54 nucleotide mutations per kb, i.e., 3.02 mutations per gene (666 nt). Among these mutations, 96.3% are substitutions, 3.2% are deletions and 0.5% are additions (Table 1). When considering only the sequences in frame, the mutation frequency increases to 4.34 mutations per kb, i.e., 2.89 mutations per gene (1 to 7 mutated nucleotides per gene, Fig. 3B). At the protein level, the Mut2 library contains 17.4% of clones expressing the Fc-WT fragment (unmutated or with silent mutations), 9.3% of clones with a sequence out of frame or with a stop codon (not expressed) and 73.3% of clones with a mutated sequence (Fc variants). These last clones represent the active part of the library comprising 1.5 × 107 different clones with an average of 1.87 mutated amino acids per molecule.

Figure 3. Analysis of the mutated sequences in frame in the Mut2 library. (A) Nucleotide analysis of the 42 starting variants. (B) Nucleotide analysis of Mut2.
Selection of Fc variants on human FcRn and characterization by phage-ELISA
As previously described for MS1, the Fc variants of Mut2 library were displayed on M13 bacteriophages and were selected on human recombinant FcRn using the three different strategies described above (solid and liquid phases). During the MS2 process, 223 different mutated clones were isolated and ranked by phage-ELISA on FcRn coated plates at pH 6.0. Because the difference between the signals of the Fc-WT and the Fc variants was too great to be compared on the same ELISA plate, the Fc variants were directly compared with the Fc-QL variant as reference. Moreover, the best Fc variant isolated during MS1, S5A-41, was used as positive control on each phage-ELISA plate. Among the 223 Fc variants tested, 209 Fc variants were better than the Fc-QL (ratio/Fc-QL at least > 1.1) and 39 Fc variants were better than the S5A-41 variant. The best clone isolated during MS2, C6A-69, had a ratio/Fc-QL of 9.0 (Fig. 4A). These results emphasize the importance of performing this second round of random mutagenesis to further improve the binding. To compare the variants with the Fc-WT, an estimated ratio/Fc-WT was calculated by multiplying the ratio/Fc-QL of the variants with the ratio/Fc-WT of the Fc-QL ( = 3.2) determined during MS1 (ratio/Fc-WT = 3.2 × ratio/Fc-QL). Overall, 219 Fc variants were considered positive with an estimated ratio greater than 2 compared with the Fc-WT. Sequence analysis of these positive Fc variants showed that they contain on average 3.9 mutated amino acids per Fc and permitted identification of 9 key positions (P230, F241, V264, T307, N315, A330, A378, N389, and N434), including the 4 positions previously identified and 5 new positions (Fig. 4B). One position is in the FcRn binding site and is well-characterized (T307), the other positions are in the CH2 and CH3 domains but not directly in the FcRn binding site. Three positions are nonetheless close to the FcRn binding site (N315, A378 and N389). Finally, we found the position N434 mutated in the majority of the positive variants, as exemplified for the 10 best Fc variants isolated (Fig. 4C).

Figure 4. Characterization of the clones selected during MS2. (A) Ratio obtained by phage-ELISA for the best clones of MS1 and MS2 compared with the positive control Fc-QL. (B) Sequence analysis of the 219 positive clones (ratio/Fc-WT > 2), distribution of the mutations along the Fc region revealing 9 key positions, which are mutated in more than 10% of the positive clones. (C) Sequences of the 10 best variants isolated during MS2. Key positions as identified in Figure 4B are in bold.
Production and functional characterization of IgG variants
Choice and production of IgG variants
To validate the results obtained by phage display, we first produced Fc variants as Fc fragments in E. coli. For that purpose, we chose 15 Fc variants with distinct enhanced FcRn binding properties as shown by phage-ELISA. Overall, FcRn binding analysis of Fc fragments by ELISA and SPR confirmed the phage-ELISA ranking and pH-dependent binding of the selected Fc variants (data not shown). We then chose six among the ten best Fc variants selected by phage display (Fig. 4C; Table 2). These Fc variants correspond to the 5 best variants plus the C6A-66 variant, which has the originality of containing an amino acid deletion (named E294Del) that we were keen on characterizing. Given the relatively high number of mutations of these Fc variants, we then tried to ensure that each mutation had a real positive effect on the FcRn binding. To that aim, we constructed a database to store and analyze all the DNA sequences and phage-ELISA results obtained for the Fc variants isolated during MS1 and MS2 processes (~450 clones). Analysis of this database showed that many of the Fc variants characterized were combinations of 2–3 mutations also found in our 6 preferred variants. We therefore identified mutations that could be dispensable for improved FcRn binding. In an attempt to reduce the final number of mutations of the IgG variants, we constructed several new variants by removing these mutations and tested them by phage-ELISA as previously described. Most of the new variants showed a decreased FcRn binding compared with the starting variants. Finally, only one new variant was retained, C6A-78A, because the removal of one mutation from the C6A-78 variant (N383S) seemed to have no effect on FcRn binding (data not shown). The C6A-78A variant, the 6 chosen variants (Table 2), the IgG-WT and the positive control IgG-YTE were produced as full-length IgGs with the variable regions of the chimeric anti-human CD20 CAT 13.6E12.36 To combine the advantages of glyco- and protein-Fc engineering, the IgG variants were produced in YB2/0 cells (EMABling® platform), permitting low fucosylation of the glycan moiety present in the mAb and thus enhanced ADCC as previously described for the anti-CD20 mAb ublituximab37-39 and the anti-RhD mAb roledumab.40,41 Before any subsequent biological test, the quality of the purified IgGs was assessed using several biochemical techniques ensuring that the biophysical behavior of the IgGs were not modified due to the mutations introduced (i.e., similar pI and molecular weights, aggregation rates < 5% and endotoxin levels < 7UI/mg).
Table 2. Comparison of binding of IgG variants to FcRn using 3 different techniques: ELISA on coated FcRn, FcRn binding on cells and SPR on immobilised FcRn.
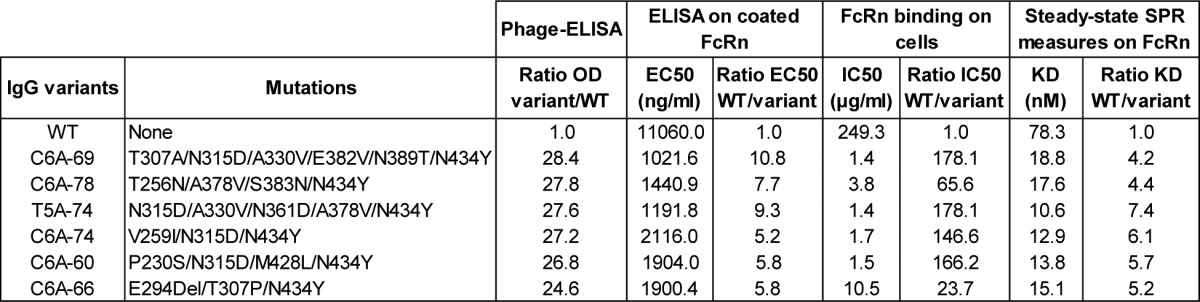
EC50: Concentration at 50% saturation obtained from the ELISA binding curve of each IgG variant. The ratio is the EC50 of the IgG-WT divided by the EC50 of the variant. IC50: Concentration at 50% inhibition of the MFI signal obtained with Alexa-conjugated Rituximab alone (100%). The ratio is the IC50 of the IgG-WT divided by the IC50 of the variant.
FcRn binding characterizations
The 6 IgG variants were compared with the IgG-WT for their FcRn binding properties using 3 different techniques: (1) ELISA using microtiter plates coated with FcRn, (2) binding to cells stably expressing a membrane version of FcRn, and (3) SPR on immobilized FcRn (Table 2; Fig. 5A and B). Whatever the technique used, all the IgG variants exhibited improved FcRn binding at pH6.0. By ELISA, the EC50 of IgG variants were at least 5.2-fold lower than that of the IgG-WT. The best results were obtained for the variants C6A-69 and T5A-74 variants (ratio EC50 variant/WT = 10.8 and 9.3, respectively). These data are in good correlation with the phage-ELISA results obtained previously to rank the variants isolated during MS1 and MS2, the C6A-69 variant being the best variant characterized during MS2. In the ELISA tests, the IgG variants displayed similar results, with ratios over IgG-WT ranging from 5.2 to 10.8, corresponding to a maximum ratio difference of 2.1. As expected, using the same protocol, no binding was detected at pH7.4 for any of the IgG variants (data not shown).

Figure 5. FcRn binding of the IgG variants. (A) FcRn binding of IgG variants on FcRn expressing cells. (B) Sensorgrams of FcRn binding at pH 6.0 for IgG-WT and C6A-66. Concentrations tested were 0, 2, 10, 25 (2 times), 50, 100, and 200 nM. (C) Complementary SPR measures for IgG-WT, IgG-YTE, C6A-78, and C6A-78A.
In a second series of tests, the IgG variants were assayed for their ability to bind FcRn expressed on cells. To this aim, we used Jurkat T cells stably expressing a truncated form of the human FcRn α-chain at their surface, similarly to what was recently described.42 This competitive assay clearly revealed the dramatic FcRn binding increase of the IgG variants compared with the IgG-WT (Fig. 5A). The measured IC50 for the IgG variants are significantly lower than that of the IgG-WT (Table 2). The IC50 decrease for the IgG variants compared with the IgG-WT ranged from 23.7 (for C6A-66) to 178.1 (C6A-69 and T5A-74). While similar results for all IgG variants were obtained in ELISA and SPR assays, this competitive cell-based assay revealed a different behavior for the C6A-66 deletion variant (factor 7.5 between C6A-69 and C6A-66). Currently, we have no explanation for this finding.
Lastly, human FcRn affinities of the IgG variants were measured by SPR at pH6.0. To rapidly compare several variants, we choose an experimental set up with recombinant human FcRn immobilized on the chip. As a result, IgG-FcRn affinities were higher than in the opposite format with immobilized mAbs, as previously described.43,44 In this format, the enhanced affinity of the IgG variants for human FcRn relative to the IgG-WT were predominantly driven by increased association kinetics as illustrated by the sensorgrams (Fig. 5B). In addition, as variable results were obtained when using the 1:2 model (included in the BIAevaluation software), we measured binding affinities at steady-state (KD values). As illustrated in Table 2, the binding affinity of the IgG-WT for human FcRn was 78.3 nM and the KD values of the 6 IgG variants ranged from 10.6 to 18.8 nM. Thus, in this test, the IgG variants exhibited similarly improved affinity, with increase ratio (WT KD divided by variant KD) between 4.2 and 7.4-fold. Finally, the positive control IgG-YTE, as well as the C6A-78 and C6A-78A variants, were tested in comparison with IgG-WT in a second complementary SPR experiment (Fig. 5C). This experiment confirmed that the C6A-78A variant has a similar affinity as C6A-78, albeit having one mutation less (N383S). Moreover, the IgG-YTE has a lower affinity increase than our IgG variants (ratio KD WT/KD YTE = 1.9), a result in line with the data obtained previously by phage-ELISA (Fig. 2A).
Binding to hFcγRIIIA and ADCC activity
The glycosylation patterns of our IgG variants produced in YB2/0 were determined as described before,37 and it was confirmed that they contain ~35% of fucosylated glyco-forms. Moreover, all the IgG produced have conserved the same antigen binding properties (data not shown). The binding of the IgG variants to hFcγRIIIA (F158 allotype) was assessed by ELISA, which showed that the IgG-WT and five out six IgG variants exhibited greatly improved hFcγRIIIA binding compared with rituximab (RTX; Fig. 6A). Only the C6A-66 variant completely failed to bind hFcγRIIIA. Analogous results were obtained for hFcγRIIIA V158 allotype (data not shown). The IgG variants were also tested for their capacity to elicit ADCC with purified NK cells against CD20-expressing target cells (Fig. 6B). All IgG variants display similarly high ADCC, compared with RTX, except for the C6A-66 variant that has no ADCC activity. This variant contains three mutations, two substitutions directly located in the FcRn binding site (T307 and N434) and one deletion (E294 deleted), the latter most probably being responsible for the loss of hFcγRIIIA binding and ADCC activity. Overall, the ADCC results are in total agreement with the hFcγRIIIA binding capacity, showing altogether that five IgG variants have retained glyco-engineered improved ADCC activity.

Figure 6. hFcγRIIIA (F158 allotype) ELISA binding and ADCC measures for the IgG variants and IgG-WT in comparison with rituximab (RTX). (A) hFcγRIIIA ELISA binding curves. (B) ADCC on CD20+ Raji cells.
Serum persistence in human FcRn transgenic mice
We then tested whether the improved FcRn binding properties of the IgG variants observed in vitro resulted in longer serum persistence in vivo. We therefore performed PK experiments in hFcRn mice that are homozygous for a knockout allele of murine FcRn and heterozygous for a human FcRn transgene (mFcRn−/− hFcRnTg 276 heterozygote on a B6 background). For these PK studies, each animal received a single intravenous injection of mAbs at 5 mg/kg via retro-orbital sinus, in a protocol similar to the one described previously.23 Blood samples were collected from the retro-orbital sinus at multiple time points and titrated by ELISA. The IgG variants were systematically compared with the IgG-WT and to IgG-YTE, a positive variant whose half-life has been largely studied in hFcRn mice earlier25 and was recently confirmed in humans.26
Three different PK studies were performed (PK1 to PK3, parameters in Table 3). The IgG variants tested were those of Table 2 except for the C6A-78 variant replaced by the C6A-78A variant, which exhibited similar FcRn binding and ADCC properties but had one mutation less. In all three experiments, the IgG-WT exhibited a half-life of ~1 d (23 h), a value lower than those published for other mAbs. This fast clearance in mice could be due to the high isoelectric point of the mAb (pI = 9.2), a parameter that has been shown to negatively affect half-life and clearance.45 In this set-up, all IgG variants displayed substantial serum half-life increase with ratios (half-life variant/WT) between 1.8 and 2.8.25 Similarly, IgG-YTE had a ratio between 2.3 and 3.3, slightly lower than the published values of ~3.8. The IgG T5A-74 variant was tested in two different experiments (PK2 and PK3) and similar results were obtained (ratio 2.5 and 2.2, respectively). In the second experiment (PK2), the C6A-66 variant is slightly more persistent than IgG-YTE (ratio 2.8 vs. 2.3), whereas the C6A-74 variant demonstrates similar persistence as IgG-YTE (ratio of 2.3 for both).
Table 3. PK parameters in hFcRn mice for the 3 different studies.
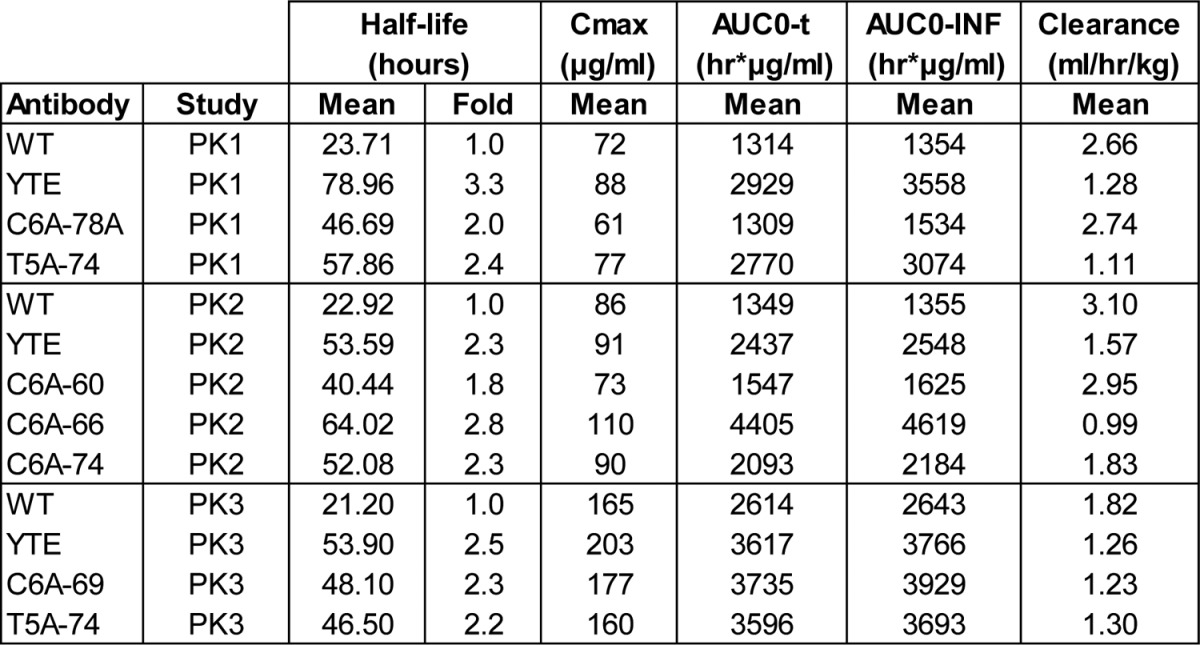
Half-life (T1/2): Final half-life of elimination. Cmax: maximum plasma concentration extrapolated at T0. AUCO-t: Area under the time x concentrations curve calculated from 0 to the last time (t) with quantifiable concentration. AUC0-INF: Area under the time x concentrations curve calculated from 0 to infinity (AUC0-t + extrapolation to infinity).
Discussion
Given the remarkable role of FcRn in IgG serum persistence, IgG/FcRn interactions have long been thoroughly studied at the molecular level by crystallography and directed mutagenesis.27,28,46-49 The FcRn binding site in the Fc domain comprises key residues located at the CH2/CH3 domain interface: residues 252–254, 307 and 309–311 in the CH2 domain, and residues 433–436 in the CH3 domain. In addition, the pH dependence has been attributed to two histidines (H310 and H435) involved in salt bridges with acidic FcRn residues. Without the help of this molecular knowledge, we identified several mutations that have a positive impact on the IgG/FcRn complex using a fully random mutagenesis approach of the entire human Fc fragment combined with a robust selection process. Interestingly, most of the best variants isolated include one mutation in the FcRn binding site, N434Y or N434S, combined with original mutations that are close to or distant from the FcRn binding site. These mutations, mainly P230S/T, F241L, V264A/E, N315D, A330V, N361D and A378V/T, probably have a positive long-range effect on the overall structure of the Fc domain favoring the IgG/FcRn complex. Using molecular modeling, impact of these mutations was difficult to interpret so they would have been impossible to be predicted (data not shown). Furthermore, the effect of the combination of several mutations would be even more difficult to predict, as opposite, additive or synergistic effects can be obtained. All these results validate our engineering approach involving two successive rounds of random mutagenesis and selection.
On the other hand, the precise knowledge of the IgG/FcRn interactions have been successfully used before by others to design and isolate various Fc variants with improved FcRn affinity.25,27,29,30 For instance, residues 250 and 428 have been targeted for mutation due to their proximity to the CH2/CH3 interface and resulted in the isolation of the double variant T250Q/M428L (Fc-QL) with a ~27-fold binding increase on human FcRn expressed on cells.31 We successfully used this Fc-QL variant as a positive control for our phage display process. Nevertheless, contradictory results were obtained with this IgG-QL variant in PK studies in rhesus and cynomolgus monkeys.30,31,50 We therefore choose the IgG-YTE variant (M252Y/S254T/T256E) as a positive control for PK studies.29 This triple variant was obtained by phage display selection against murine FcRn of rationally designed libraries targeting Fc residues in or close to the FcRn binding site (residues 251–256, 308–314, 385–389, and 428–436). Introduced in a humanized IgG1, these mutations increased the binding to human FcRn by about 10-fold, resulting in a nearly 4-fold increase in serum half-life in cynomolgus monkeys.24 More importantly, when introduced into the anti-RSV motavizumab, the IgG-YTE variant resulted in a 2- to 4-fold longer half-life than the WT motavizumab in healthy humans. This is a crucial result as it confirms for the first time in humans the concept of half-life extension using Fc-engineered antibodies.26 On the other hand, in silico analysis was also used to design several variants combining mutations in or close to the FcRn binding site. Two variants, M428L/N434S (IgG-LS, Xtend) and V259I/V308F/M428L (IgG-IFL), showed respectively a 1.6- and 2.7-fold improved binding to human FcRn by SPR compared with the IgG-YTE.25 Nonetheless, similarly to what we observed in PK studies in hFcRn mice, these variants exhibited only moderate half-life increase compared with the IgG-YTE, at best a 1.16-fold increase for the IgG-LS and a 1.30-fold increase for the IgG-IFL in one experiment. However, as we also observed, results can be variable since in a second experiment of this study no half-life increase was observed for the IgG-IFL compared with the IgG-YTE. Similarly, a lack of direct linear correlation between increased FcRn affinity and improved half-life had been observed earlier with the two variants N434A and T307A/E380A/N434A. These variants were isolated by alanine scanning mutagenesis and resulted in a 2.2 and 2.5-fold half-life increase in hFcRn mice, whereas affinity differences were greater, respectively 3.4 and 11.8-fold increase compared with the IgG-WT.23,27 All these results seem to demonstrate that Fc engineering may have reached a plateau for FcRn improvement designed to increase half-life.
This in vivo limit could be due to the critical need to keep the pH dependency property of the IgG/FcRn interaction. Indeed, increased affinity at high pH could be potentially problematic by preventing the release of FcRn-bound IgGs. In accordance with this mechanism, augmented clearance was observed for IgG engineered for improved FcRn affinity at both pH 6.0 and pH7.4, where pH dependence is reduced.51 Furthermore, recent studies clearly showed the difficulty of conserving the pH dependency because globally when a variant’s affinity to FcRn is increased at pH6.0, its binding at higher pH increases accordingly.44 Several mutations which resulted in a slight increase in FcRn binding at pH7.4 were described, including M252Y, M252W, V308P, N434Y, and N434W. This last mutation, N434W, induces the highest FcRn affinity at pH7.4 and despite an 80-fold increase binding to cynomolgus monkey FcRn at pH6.0, serum half-life was similar to the IgG-WT.44,52 On the other hand, the mutation M252Y is included in the IgG-YTE variant but clearly resulted in increased half-life for various mAbs.25 Likewise, our Fc variants include the mutation N434Y and herein we show that several variants have increased half-life in hFcRn mice. Combinations of several mutations may result in different pH dependency and very fine differences could have unpredictable effects on serum half-life. Therefore, pH dependency being difficult to assess and interpret we choose to directly test several variants for extended half-life in hFcRn mice. These FcRn-humanized mice, unlike wild-type mice, have been shown to be a reliable surrogate for studying human IgG serum half-life.23,53 Despite reduced circulating murine IgGs, these humanized mice reliably predicted antibody half-lives in primates when challenged with clinical mAbs for which PK data were available.54 Moreover, whereas human IgG binding and pH dependency are highly similar for human and monkey FcRn, human and murine FcRn have significant molecular differences rendering wild-type mice an inadequate model for studying half-life of engineered human IgGs.55,56 For instance, the N434A variant has increased serum half-life in monkeys, in correlation with its pH dependent increased binding to monkey FcRn, whereas in wild-type mice this variant has the same PK profile as the IgG-WT, due to unchanged binding properties to murine FcRn.57 Likewise, the N434Y variant has increased binding to murine FcRn at both pH6.0 and 7.4 resulting in comparable half-life to the IgG-WT in mice.58 Nevertheless, we show in this study that several variants including this mutation N434Y result in half-life extension in hFcRn mice, emphasizing the limitations of using wild-type mice as preclinical models for the analysis of engineered mAbs.
In addition, other factors than FcRn binding have been shown to influence PK of natural mAbs: overall protein structure, glycosylation status, isoelectric point,45 methionine oxidation,59 interaction with the target but also specificity. Indeed, mAbs with the same wild-type human Fc sequences but different Fv domains were shown to bind FcRn with considerable differences.60,61 Conversely, the Fc region has been shown to contribute to affinity and specificity of mAbs.62 For all these reasons, we didn’t focus our study on only one variant but several combinations that are stored and documented in a database, to be able to propose Fc variants that fit best for a given mAb. Preliminary results on two other specificities confirm that several combinations are directly transferrable to other mAbs, resulting in increased half-life in hFcRn mice (data not shown).
Finally, for the first time we clearly show that glyco-engineering to improve mAbs functional activity and Fc-mutations to increase half-life can be combined to further optimize mAbs. Beside glyco-engineering, protein engineering has been successfully employed as well to generate antibodies with enhanced binding to activating FcγRs. Alanine scanning,27 in silico approach,63 and random mutagenesis strategy64 allowed identification of distinct IgG1 variants (combinations of 2–5 mutations) with strongly enhanced binding to FcγRIIIA or FcγRIIA resulting in increased ADCC or ADCP in vitro and better anti-tumor activities in mouse models in vivo. We observed that these mutations have little or no effect on FcRn binding (our unpublished data). On the contrary, produced as low fucosylated mAbs, our IgG variants display both advantages: increased ADCC and longer serum persistence. Because we used a random mutagenesis approach, identified mutations could have resulted in a decreased binding to FcγRIIIA. Only one variant, C6A-66 showed poor affinity for this receptor, probably due to an unusual mutation: the deletion of amino acid 294. This variant is nonetheless of great interest as for certain clinical applications no effector function is preferable,65,66 and for this variant ADCC is completely abolished while half-life is largely enhanced.
Materials and Methods
Expression and purification of human FcRn
The expression of soluble human FcRn was performed by GTP Technology (Labège, France) using the baculovirus system as previously described.46 The α chain cDNA encoding the leader peptide and extracellular domains (codons 1–290) was tagged with a TEV sequence and a 6 × polyhistidine tag. The derivative α-chain and the β2-microglobulin chain were cloned into pFastBacDual under the P10 and polyhedrine promoters, respectively. A biotinylated version of FcRn (FcRn-biot) was prepared by chemical coupling with the FluoReporter® Biotin-XX Protein Labeling Kit, F2610 (Molecular Probes) according to the manufacturer’s protocol. A fusion protein containing the β2-microglobulin chain and the α-chain fused to the N-terminal part of the bacteriophage p3 protein and the CVDE protein (FcRn-p3) was also produced. Proteins with more than 90% purity were obtained after IgG-Sepharose and IMAC purification steps.
Construction of the Fc libraries using MutaGenTM
The human Fc gene encoding amino acid residues 226–447 (EU index as in Kabat), derived from a human IgG1 heavy chain (G1m1,17 allotype), was cloned into the phagemid vector pMG58 (pMG58-Fc226) as a BamHI/EcoRI fragment using standard PCR protocols. Several fully randomized libraries were then generated using the MutaGenTM procedure that uses low fidelity human DNA polymerases (pol. or mutases) to introduce random mutations homogeneously distributed over the whole target sequence. Three distinct mutases (pol. β, pol. η and pol. ι), produced and purified as described previously,34,67 were used in different conditions to create complementary mutational patterns. The human Fc gene was replicated with mutases using the 5′ primer MG-619: 5′-AGTACTGACT CTACCTAGGA TCCTGCCCAC CGTGC-3′ and the 3′ primer MG-621: 5′-ACTGCTCGAT GTCCGTACTA TGCGGCCGCG AATTC-3′ (BamHI and EcoRI restriction sites are underlined and italic characters correspond to the non-specific tails). A mixture containing 0.6 µg of the pMG58-Fc226 plasmid as template (wild type Fc for the Mut1 library or Fc variants for the Mut2 library), primers MG-619 and MG-621 (250 nM each) and the appropriate replication buffer (detailed below) was treated for 5 min at 95 °C and immediately cooled down to 4 °C to denature DNA strands. For pol. β, replication buffer was 50 mM TRIS-HCl pH 8.8, 10 mM MgCl2, 10 mM KCl, 1 mM DTT and 1% (v/v) glycerol. Replication buffer for pol. η (or pol. η and pol. ι) was 25 mM TRIS-HCl pH 7.2, 5 mM MgCl2, 10 mM KCl, 1 mM DTT, and 2.5% (v/v) glycerol. After the denaturation step, mutagenic replications were performed by adding 50μM dATP/dCTP, 100 μM dTTP/dGTP and 1 μg of pol. β or 100μM dNTPs and 1μg of pol. η (or pol. η and pol. ι, 1 μg of each mutase). The replication reaction was performed at 37 °C for two hours. The replication products were then desalted and concentrated on Microcon® centrifugal filters (Millipore) before amplification through a selective PCR with tail primers. The primers (MG-619 and MG-621) contain a tail that is non-complementary to the template allowing specific amplification of the DNA fragments synthesized by the mutases. A fraction of the replication products was added to a mixture containing the PCR buffer (20 mM TRIS-HCl pH 8.4, 50 mM KCl), 1.5 mM MgCl2, 10 pmol of the 5′ and 3′ primers, 200 μM dNTPs and 1.25 U Platinum Taq DNA polymerase (InvitroGen). The PCR cycles were as follows, first cycle: 2 min. at 94 °C, 10 s at 64 °C, 30 s at 7 5 °C, 1 min. at 94 °C, and then 30 selective cycles: 20 s at 94 °C and 30 s at 75 °C. The amplified replication products were purified on 1% (w/v) agarose gels, digested with BamHI and EcoRI restriction enzymes and cloned into the pMG58 vector. The ligation mixtures were used to transform electrocompetent E. coli XL1-Blue cells and subsequently plated on solid 2YT medium supplemented with 100 µg/ml ampicillin and 1% (w/v) glucose. After growth, the number of colonies was determined to estimate the size of the libraries and cells were scrapped in 2YT medium with 15% glycerol, frozen and kept at -80 °C. The quality of the different libraries was assessed by PCR on cells to amplify the Fc gene (with the 5′ primer 5′-CAGGAAACAG CTATGACC-3′ and the 3′ primer 5′- TCACGTGCAA AAGCAGCGGC -3′) and high throughput sequencing (with the 5′ primer 5′- TGATTACGCC AAGCTTGC -3′, MilleGen sequencing Department).
Phage display expression of Fc libraries and selection of variants with improved FcRn binding
Fc libraries were expressed on the surface of the bacteriophage M13 using standard procedures.35 E. coli XL1-Blue bacteria containing the Fc library (pMG58 vector) were grown in 60 ml of 2YT supplemented with 100 μg/ml ampicillin, 15 μg/ml tetracycline and 1% (w/v) glucose at 30 °C, 230rpm until OD600nm = 0.6 is reached. Cells were then infected with M13 helper phage (M13KO7, New England Biolabs, ratio bacteria: phage = 1:3) at 37 °C for 20 min and phage-Fc production was continued overnight at 26 °C, 230 rpm in 2YT/ampicillin/glucose with 0.5 mM IPTG and 30 μg/ml kanamycin. The following day, phages were precipitated with PEG6000 using standard protocols, resuspended in 1ml phosphate buffer pH6.0 (100 mM sodium phosphate, 50 mM sodium chloride pH6.0, called P6) and titrated by infecting XL1-Blue bacteria. For solid phase selections, 4 × 1011 phages in P6/5% skimmed milk/0.1%Tween-20 were incubated into 8 wells of Maxisorp immunoplates previously coated with 0.5 μg neutravidin and 0.5 μg biotinylated FcRn or 0.5 μg FcRn-p3 and blocked with 5% skimmed milk in P6. After incubation for 2 h at 37 °C, wells were washed 20 times with P6/0.1% Tween-20 and eluted by incubation in 100 μl phosphate buffer pH 7.4 (100 mM sodium phosphate, 50 mM sodium chloride, pH 7.4) per well for 2 h at 37 °C. After titration, eluted phages were used to reinfect 10 ml of exponentially growing XL1-Blue bacteria and subsequently plated on solid 2YT medium/ampicillin/glucose. On the following day, cells were scrapped in 2YT medium with 15% glycerol, frozen and kept at -80 °C until the next round of selection. For liquid phase selection, 4 × 1011 phages were first incubated with 250nM or 100nM biotinylated FcRn in 1ml P6/5% skimmed milk/0.1%Tween-20 for 1 h at room temperature (RT) under low agitation. Streptavidin-coated magnetic beads (Dynal), previously blocked with 5% skimmed milk in P6 were then added to the phages for 30 min. at RT. Phage-bead complexes were washed 15 times with P6/0.1% Tween-20 using a magnet (magnetic particle concentrator, Dynal). Phages were eluted by incubation in 500μl phosphate buffer pH 7.4 (100 mM sodium phosphate, 50 mM sodium chloride, pH 7.4) for 2 h at RT. Beads were discarded using the magnet and eluted phages in the supernatants were collected. After titration, eluted phages were used to reinfect 10ml of exponentially growing XL1-Blue bacteria and subsequently plated on solid 2YT medium/ampicillin/glucose. The following day, cells were scrapped in 2YT medium with 15% glycerol, frozen and kept at -80 °C until the next round of selection. During selection processes, for each strategy, from round 3 to round 8, 48 to 96 clones were sequenced as described above. Sequence analysis was performed using MilleGen’s proprietary software AnalyseFc internally developed to rapidly analyze the selected Fc variants. Fc variants were named according to the round of selection from which they were isolated. For Mut1 screening: B3A to B6A for solid phase selection on FcRn-p3, S3A to S6A for solid phase selection on biotinylated FcRn and L3A/B to L6A-F for liquid phase selection, for Mut2 screening: C3A to C8A for solid phase selection on FcRn-p3, T3A to T8A for solid phase selection on biotinylated FcRn and M3A/B liquid phase selection. Numbers (1 to 96) refer to the localization of the clone on the PCR plate used for sequencing.
Phage-ELISA assays of the selected variants
The binding characteristics of the variants isolated during selection displayed on the phage were determined using an ELISA test at pH 6.0 with FcRn-p3 coated on wells. Briefly, phage-Fc variants were produced as separate clones on a 96-well plate in 800 μl cultures in 2YT medium/ampicillin/glucose infected with helper phage M13K07 (as described previously). Phages produced overnight at 26 °C were recovered in the supernatants after centrifugation for 30 min at 3000 g. These supernatants were directly diluted (1/2 and 1/4) in P6/5% skimmed milk/0.1%Tween-20 and tested on Maxisorp immunoplates previously coated with 0.25 μg FcRn-p3/well and blocked with 5% skimmed milk in P6. After incubation for 2 h at 37 °C, wells were washed 3 times with P6/0.1% Tween-20 and bound phages were detected with an HRP anti-M13 antibody (GE Healthcare).
Directed mutagenesis
Directed mutagenesis was performed to construct positive controls and remove point mutations using standard PCR protocols. The first positive control: Fc-QL30,31 T250Q/M428L was obtained with two long primers (the 5′ primer 5′-CGGGATCCTG CCCACCGTGC CCAGCACCTG AACTCCTGGG GGGACCGTCA GTCTTCCTCT TCCCCCCAAA ACCCAAGGAC CAACTCATGA TCTCCCGGAC-‘3 and the 3′ primer 5′- GCGAATTCTT TACCCGGAGA CAGGGAGAGG CTCTTCTGCG TGTAGTGGTT GTGCAGAGCC TCATGCAGCA CGGAGCATGA GAAG-‘3 where BamHI and EcoRI restriction sites are underlined and mutated nucleotides are in italic). The second positive control: Fc-YTE29 M252Y/S254T/T256E was obtained by PCR overlap using the primers MG-619 and MG-621 for cloning (described for the construction of the libraries) and overlapping primers containing the mutations (the 5′ primer 5′- CAGGTTCCCG GGTGATGTAG AGGGTGTCCT TGGGTTTTGG-‘3 and the 3′ primer 5′- ACCCTCTACA TCACCCGGGA ACCTGAGGTC ACATGCGTGG TG-‘3 where overlapping regions are underlined and mutated nucleotides are in italic).
Production and purification of IgG variants
The Fc variants were produced in IgG format with an anti-CD20 specificity (based on the Fv domain of CAT 13.6E1236) using the YB2/0 cell line (ATCC, CRL-1662). For comparative purposes, IgGs based on wild-type Fc (IgG-WT) and Fc-YTE (IgG-YTE) were also produced. To maximize the productivity in the YB2/0 cell line, the full-length light and heavy chain cDNAs including the mutations in the Fc fragment were neo-synthetized with codon optimisation for Rattus norvegicus (GeneArt, Life-Technologies). Unwanted features such as cryptic splicing sites or restriction sites were removed. Only one restriction site (ApaI) was present at the junction of variable and constant region. In a first step, wild-type heavy chain was cloned between NheI and AscI in the expression vector CHK622–08, optimized for expression in YB2/0, resulting in the intermediate construct HCD20-Opti-GA. The optimized light chain was then cloned between SpeI and XbaI restriction sites resulting in the final construct HKCD20-Opti-GA for expression of the IgG-WT. Fc variants were prepared by replacing the wild-type IgG1 Fc fragment present in HKCD20-Opti-GA by the different mutant versions which were cloned between ApaI and AscI restriction sites. These cloning steps were done by classical digestion/ligation procedures, followed by bacterial transformation. Expression constructs were screened by enzymatic digestion and PCR and validated by sequencing. Each linearized expression construct was introduced by electroporation into 5.106 YB2/0 cells. Cells were then diluted to 25 000 cells/ml in RPMI 1640 medium + 5% v/v dialysed FCS (Invitrogen) and dispensed in 1 ml/well within 24-well plates. After 3 d, selection pressure was applied by adding geneticin (Invitrogen) and methotrexate (Sigma) to obtain final concentrations of 0.5g/l and 25 mM respectively, in 2ml/well. After 11 d, resistant cells were pooled for each construct (encoding for the selected IgG variants, IgG-YTE and IgG-WT) and progressively diluted with DMEM medium + 5% v/v Ultra-low IgG FCS (Invitrogen) until two 2 L-roller bottles, each containing 0.9 L of cell suspension, can be incubated at 2rpm. Cells were allowed to grow and die (4 to 5 d) before supernatant collection, clarification by low-speed centrifugation and volume reduction by ultra-filtration on Pellicon XL Filter (Millipore). The concentrated culture supernatants were injected into a HiTrap protein A FF column (GE Healthcare). Bound antibodies were eluted with 0.1 M sodium citrate, pH 3.0 and fractions were neutralized using 100 µl of 1M TRIS-HCl pH 7.5 per ml of elution buffer. Fractions containing the antibodies were pooled and dialyzed against PBS pH 6.0, and the samples were sterile-filtered (0.22 µm) and stored at 4 °C. The purified IgGs were analyzed using SDS-PAGE under non-reducing and reducing conditions. Coomassie Blue-stained gels indicated that the IgGs, whatever the mutations, had greater than 95% purity and displayed the characteristic heavy and light chain bands. Low aggregate levels and absence of contaminants were verified by analytical gel filtration in PBS on superdex 200 10/300 GL with an AKTA prime system (GE Healthcare). Purified IgGs were considered suitable for biological tests when aggregate rates were below 5%. None of the mutations tested induced IgG aggregation, aggregation rates of the purified IgGs were between 0.1 and 4.5%. Final IgG concentrations were between 3.5 and 8.2mg/ml. LAL endotoxin test (Limulus Amebocyte Lysate) gel clot method was further used to test purified IgGs for the presence of endotoxins. Endotoxin levels of the purified IgGs were below 7 UI/mg.
FcRn binding of the IgG variants
Three distinct tests were set up to characterize the interaction of the IgG variants with FcRn: (1) an ELISA test, (2) a competitive binding assays on FcRn expressing Jurkat cells, and (3) SPR. (1) The binding properties of the IgG variants, in comparison with the IgG-WT, were determined using an ELISA test at pH 6.0 with FcRn-p3 coated on wells. Purified IgG variants serially diluted in P6/5% skimmed milk/0.1% Tween-20 were tested on Maxisorp immunoplates previously coated with 0.1 μg FcRn-p3/well and blocked with 5% skimmed milk in P6. After incubation for 2 h at 37 °C, wells were washed 3 times with P6/0.1% Tween-20 and bound IgG variants were detected with an HRP F(ab’)2 goat anti-human F(ab’)2 (Interchim). For each IgG variant, the percentage of bound FcRn was plotted vs. the log of the concentration of IgG variant. For each resulting binding curve, the IgG concentration corresponding to 50% saturation of the curve (EC50) was determined and compared with the EC50 of IgG-WT. (2) Competitive immunofluorescence assays were performed to evaluate the ability of IgG-WT and IgG variants to interact with FcRn by a method adapted from that described elsewhere.68 Briefly, IgG-WT and IgG variants were diluted in PBS pH6.0 at a final concentration ranging from 0.06 to 2 mg/ml and incubated with 1.5 × 105 Jurkat cells expressing on the cell membrane a truncated form of FcRn in the presence of Alexa-conjugated Rituximab (50 µg/ml). After 20 min., the cells were analyzed by flow cytometry to quantify the fluorescence signal. The results were expressed as a percentage of the mean fluorescence intensity (MFI), 100% refers to the MFI obtained with Alexa-conjugated Rituximab alone (i.e., without competitor) and 0% refers to the MFI value measured when Jurkat FcRn cells were not incubated with Alexa-conjugated Rituximab. Each experiment was done in triplicate. For each IgG, the MFI was plotted vs. the log of IgG concentration. The concentration of each IgG tested which provides an inhibition of 50% of the MFI signal (IC50) was determined. (3) The interaction of IgG-WT and IgG variants with recombinant, immobilized human-FcRn was monitored by SPR detection on a BIAcore X100 instrument using a CM5 sensor chip (BIAcore, GE Healthcare). The methodology was similar to that previously described for analyzing Fc-FcRn interactions.46 Recombinant soluble FcRn was coupled to flow cell 2 of the sensor chip using amine-coupling chemistry. The flow cells were activated for 3 min with a 1:1 mixture of 0.1M N-hydroxysuccinimide and 0.1M 3-(N,N-dimethylamino)propyl-N-ethylcarbodiimide at a flow rate of 30 µl/min. Recombinant human FcRn (5µg/ml in 10 mM sodium acetate, pH 5.0) was injected over flow cell 2 for 8 min at 10µl/min, which resulted in a surface density of 1200 to 1300 response units (RU). Surfaces were blocked with a 3 min injection of 1 M ethanolamine-HCl, pH 8.5. Flow cell 1 was used as a control surface without FcRn and was prepared similarly to sample flow cell. The data from this blank flow cell were subtracted from the sample data. IgGs were diluted in PBS/Tween-20 (50 mM sodium phosphate, pH 6.0, 150 mM NaCl, 0.02% NaN3, 0.01% Tween-20) which is used as running buffer in equilibrium binding experiments. All measurements were performed at 25 °C with IgG concentrations typically ranging from 1 to 200nM at a flow rate of 10 µl/min. Data were collected for 10 min. and a 1 min pulse of PBS, pH 8 containing 0.05% Tween-20 was used to regenerate the surfaces. Sensorgrams were generated and analyzed by using the steady-state affinity model included in BIAevaluation software version 3.1. The equilibrium RU observed for each injection was plotted against the IgG concentration. The equilibrium KD values were determined using the steady-state affinity model included in the BIAevaluation software.
hFcγRIIIA binding of the IgG variants
The human recombinant FcγRIIIA (F158 allotype) made by PX’Therapeutics was biotinylated with the EZ-link NHS-PEO kit (Pierce), diluted to 1 µg/ml in assay buffer (25 mM Tris, 150 mM NaCl, pH 7.35, 0.05% Tween-20, 0.1% BSA) and coated onto react-bind™ streptavidin ELISA plates (Pierce) for 2 h at RT. During this incubation time, IgG-F(ab’)2 anti-F(ab’)2 complexes were prepared in assay buffer by mixing 5 µg/ml of IgG and 2 µg/ml F(ab’)2 anti-human F(ab’)2 labeled with horseradish peroxidase (Jackson ImmunoResearch) for 2 h at RT. Serial dilutions of complexes were added to plates and incubated for 2 h at RT under gentle shaking. After washing plates with assay buffer, bound complexes to hFcγRIIIA were detected with TMB (Pierce). Absorbance at 450 nm was read using a plate reader (Tecan). For each IgG variant, the percentage of bound hFcγRIIIA (which is obtained from OD450nm) was plotted against the concentration of IgG variants.
ADCC activity
Natural killer cells (NK cells) were purified from the peripheral blood of healthy volunteer donors by the negative depletion technique developed by Miltenyi. The ADCC test comprises incubating the NK cells with CD20-expressing Raji target cells, in the presence of different concentrations of anti-CD20 antibodies. After 16 h of incubation, the cytotoxicity induced by the anti-CD20 antibodies was measured by quantifying in cell supernatants the level of the intracellular enzyme lactate dehydrogenase (LDH) released from lysed target cells. The specific lysis results are expressed as the percentage of lysis as a function of antibody concentration. EC50 values (antibody concentration inducing 50% of maximum lysis) were calculated using PRISM software.
PK study in human FcRn mice (hFcRn mice)
Animals were housed in the animal center of the Faculty of Pharmacy, University of Paris-Sud 11 and all studies were approved by the responsible Animal Care and Use Committee (authorization number 2012_121, CEEA 26). hFcRn mice (mFcRn−/− hFcRnTg 276 heterozygote on a B6 background) for PK studies were obtained by breeding of homozygous Tg276 human FcRn mice with FcRn−/− mice obtained from The Jackson Laboratory. These mice are deficient in the mouse FcRn α-chain and carry a human FcRn α-chain gene instead.23 For PK studies, each animal (4 animals per group) received a single dose of 5 mg/kg of antibody intravenously via the retro-orbital sinus. Blood samples were collected from the retro-orbital sinus at the following time points: 1 h, 4 h, 6 h, 24 h, 48 h, 96 h, 168 h, 216 h, 264 h, 336 h, 384 h, 432 h, and 504 h, and transferred into tubes. Three to five mice per group underwent retro-orbital bleeding for each time point, with 100 μl of blood collected per mouse (5 punctures distributed over 3 wk for each mouse). The blood samples were left at RT for at least 1 h, processed to serum and stored at -80 °C until analysis. A qualified anti-human IgG immunoassay was used to determine all mAb levels in the serum samples and was performed by LFB biotechnologies or Bertin Pharma. Briefly, an affiniPure Donkey Anti-Human IgG (H+L) polyclonal antibody was used for capture, and an affiniPure Donkey Anti-Human IgG (H+L) peroxydase polyclonal antibody was used for detection (both from Jackson ImmunoResearch Laboratories). The mAb concentrations were extrapolated from standard curves. Samples were diluted at least 40-fold. The assay range was established with spiked quality control samples. The lower limit of quantification was 60 ng/ml. Sample results were corrected for dilution; values within the standard curve range were averaged. The serum concentration was used for non-compartmental PK analysis using WinNonlin (Enterprise version 5.01). The mAb elimination phase terminal half-life (t1/2) was determined using data points from the terminal phase. These data points generally fitted well to a monoexponential decay function.
Disclosure of Potential Conflicts of Interest
C.M., S.J., A.J., N.F., C.D.R., R.U., C.K.B., P.M., and A.F are employees of LFB Biotechnologies and N.S., O.Z., F.C. and K.B. are employees of MilleGen; both companies financially supported the study.
Acknowledgments
Our sincere thanks to Alain Longue, Anne-Sophie Dezetter, Aurélie Dehenne, Virginie Pochet, Cécile Beghin and Aurélie Terrier for antibody production and characterization, Véronique Devos for SPR analyses, Sylvie le Ver, Bianca Boussier and Annie Regenman for in vivo studies, Annick Sauger and Isabelle Sans for bio-analyses and Pierre-Olivier Guillaumat for PK modeling. This work was supported by a French governmental grant (FUI 10, HuMabFc project) financed by DGCIS and Oséo.
Glossary
Abbreviations:
- ADCC
antibody-dependent cell-mediated cytotoxicity
- ADCP
antibody-dependent cell-mediated phagocytosis
- FcRn
neonatal Fc receptor
- FcγR
Fc gamma receptor
- IgG
immunoglobulin G
- mAb
monoclonal antibody
- MS
cycle of mutagenesis and selection
- NK
natural killer
- PK
pharmacokinetics
- RT
room temperature
- SPR
surface plasmon resonance
Footnotes
Previously published online: www.landesbioscience.com/journals/mabs/article/27854
References
- 1.Beck A, Wurch T, Bailly C, Corvaia N. Strategies and challenges for the next generation of therapeutic antibodies. Nat Rev Immunol. 2010;10:345–52. doi: 10.1038/nri2747. [DOI] [PubMed] [Google Scholar]
- 2.Strohl WR. Optimization of Fc-mediated effector functions of monoclonal antibodies. Curr Opin Biotechnol. 2009;20:685–91. doi: 10.1016/j.copbio.2009.10.011. [DOI] [PubMed] [Google Scholar]
- 3.Nimmerjahn F, Ravetch JV. Translating basic mechanisms of IgG effector activity into next generation cancer therapies. Cancer Immun. 2012;12:13. [PMC free article] [PubMed] [Google Scholar]
- 4.Albanesi M, Daëron M. The interactions of therapeutic antibodies with Fc receptors. Immunol Lett. 2012;143:20–7. doi: 10.1016/j.imlet.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 5.Weiner LM, Surana R, Wang S. Monoclonal antibodies: versatile platforms for cancer immunotherapy. Nat Rev Immunol. 2010;10:317–27. doi: 10.1038/nri2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, Watier H. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood. 2002;99:754–8. doi: 10.1182/blood.V99.3.754. [DOI] [PubMed] [Google Scholar]
- 7.Musolino A, Naldi N, Bortesi B, Pezzuolo D, Capelletti M, Missale G, Laccabue D, Zerbini A, Camisa R, Bisagni G, et al. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J Clin Oncol. 2008;26:1789–96. doi: 10.1200/JCO.2007.14.8957. [DOI] [PubMed] [Google Scholar]
- 8.Bibeau F, Lopez-Crapez E, Di Fiore F, Thezenas S, Ychou M, Blanchard F, Lamy A, Penault-Llorca F, Frébourg T, Michel P, et al. Impact of FcgammaRIIa-FcgammaRIIIa polymorphisms and KRAS mutations on the clinical outcome of patients with metastatic colorectal cancer treated with cetuximab plus irinotecan. J Clin Oncol. 2009;27:1122–9. doi: 10.1200/JCO.2008.18.0463. [DOI] [PubMed] [Google Scholar]
- 9.Shields RL, Lai J, Keck R, O’Connell LY, Hong K, Meng YG, Weikert SH, Presta LG. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. J Biol Chem. 2002;277:26733–40. doi: 10.1074/jbc.M202069200. [DOI] [PubMed] [Google Scholar]
- 10.Beck A, Reichert JM. Marketing approval of mogamulizumab: a triumph for glyco-engineering. MAbs. 2012;4:419–25. doi: 10.4161/mabs.20996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol. 2007;7:715–25. doi: 10.1038/nri2155. [DOI] [PubMed] [Google Scholar]
- 12.Kuo TT, Aveson VG. Neonatal Fc receptor and IgG-based therapeutics. MAbs. 2011;3:422–30. doi: 10.4161/mabs.3.5.16983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu X, Meng G, Dickinson BL, Li X, Mizoguchi E, Miao L, Wang Y, Robert C, Wu B, Smith PD, et al. MHC class I-related neonatal Fc receptor for IgG is functionally expressed in monocytes, intestinal macrophages, and dendritic cells. J Immunol. 2001;166:3266–76. doi: 10.4049/jimmunol.166.5.3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuo TT, Baker K, Yoshida M, Qiao SW, Aveson VG, Lencer WI, Blumberg RS. Neonatal Fc receptor: from immunity to therapeutics. J Clin Immunol. 2010;30:777–89. doi: 10.1007/s10875-010-9468-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ward ES, Ober RJ. Chapter 4: Multitasking by exploitation of intracellular transport functions the many faces of FcRn. Adv Immunol. 2009;103:77–115. doi: 10.1016/S0065-2776(09)03004-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tesar DB, Björkman PJ. An intracellular traffic jam: Fc receptor-mediated transport of immunoglobulin G. Curr Opin Struct Biol. 2010;20:226–33. doi: 10.1016/j.sbi.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bitonti AJ, Dumont JA, Low SC, Peters RT, Kropp KE, Palombella VJ, Stattel JM, Lu Y, Tan CA, Song JJ, et al. Pulmonary delivery of an erythropoietin Fc fusion protein in non-human primates through an immunoglobulin transport pathway. Proc Natl Acad Sci U S A. 2004;101:9763–8. doi: 10.1073/pnas.0403235101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ye L, Zeng R, Bai Y, Roopenian DC, Zhu X. Efficient mucosal vaccination mediated by the neonatal Fc receptor. Nat Biotechnol. 2011;29:158–63. doi: 10.1038/nbt.1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoshida M, Claypool SM, Wagner JS, Mizoguchi E, Mizoguchi A, Roopenian DC, Lencer WI, Blumberg RS. Human neonatal Fc receptor mediates transport of IgG into luminal secretions for delivery of antigens to mucosal dendritic cells. Immunity. 2004;20:769–83. doi: 10.1016/j.immuni.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 20.Qiao SW, Kobayashi K, Johansen FE, Sollid LM, Andersen JT, Milford E, Roopenian DC, Lencer WI, Blumberg RS. Dependence of antibody-mediated presentation of antigen on FcRn. Proc Natl Acad Sci U S A. 2008;105:9337–42. doi: 10.1073/pnas.0801717105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baker K, Qiao SW, Kuo TT, Aveson VG, Platzer B, Andersen JT, Sandlie I, Chen Z, de Haar C, Lencer WI, et al. Neonatal Fc receptor for IgG (FcRn) regulates cross-presentation of IgG immune complexes by CD8-CD11b+ dendritic cells. Proc Natl Acad Sci U S A. 2011;108:9927–32. doi: 10.1073/pnas.1019037108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vidarsson G, Stemerding AM, Stapleton NM, Spliethoff SE, Janssen H, Rebers FE, de Haas M, van de Winkel JG. FcRn: an IgG receptor on phagocytes with a novel role in phagocytosis. Blood. 2006;108:3573–9. doi: 10.1182/blood-2006-05-024539. [DOI] [PubMed] [Google Scholar]
- 23.Petkova SB, Akilesh S, Sproule TJ, Christianson GJ, Al Khabbaz H, Brown AC, Presta LG, Meng YG, Roopenian DC. Enhanced half-life of genetically engineered human IgG1 antibodies in a humanized FcRn mouse model: potential application in humorally mediated autoimmune disease. Int Immunol. 2006;18:1759–69. doi: 10.1093/intimm/dxl110. [DOI] [PubMed] [Google Scholar]
- 24.Dall’Acqua WF, Kiener PA, Wu H. Properties of human IgG1s engineered for enhanced binding to the neonatal Fc receptor (FcRn) J Biol Chem. 2006;281:23514–24. doi: 10.1074/jbc.M604292200. [DOI] [PubMed] [Google Scholar]
- 25.Zalevsky J, Chamberlain AK, Horton HM, Karki S, Leung IW, Sproule TJ, Lazar GA, Roopenian DC, Desjarlais JR. Enhanced antibody half-life improves in vivo activity. Nat Biotechnol. 2010;28:157–9. doi: 10.1038/nbt.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robbie GJ, Criste R, Dall’acqua WF, Jensen K, Patel NK, Losonsky GA, Griffin MP. A novel investigational Fc-modified humanized monoclonal antibody, motavizumab-YTE, has an extended half-life in healthy adults. Antimicrob Agents Chemother. 2013;57:6147–53. doi: 10.1128/AAC.01285-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shields RL, Namenuk AK, Hong K, Meng YG, Rae J, Briggs J, Xie D, Lai J, Stadlen A, Li B, et al. High resolution mapping of the binding site on human IgG1 for Fc gamma RI, Fc gamma RII, Fc gamma RIII, and FcRn and design of IgG1 variants with improved binding to the Fc gamma R. J Biol Chem. 2001;276:6591–604. doi: 10.1074/jbc.M009483200. [DOI] [PubMed] [Google Scholar]
- 28.Martin WL, West AP, Jr., Gan L, Bjorkman PJ. Crystal structure at 2.8 A of an FcRn/heterodimeric Fc complex: mechanism of pH-dependent binding. Mol Cell. 2001;7:867–77. doi: 10.1016/S1097-2765(01)00230-1. [DOI] [PubMed] [Google Scholar]
- 29.Dall’Acqua WF, Woods RM, Ward ES, Palaszynski SR, Patel NK, Brewah YA, Wu H, Kiener PA, Langermann S. Increasing the affinity of a human IgG1 for the neonatal Fc receptor: biological consequences. J Immunol. 2002;169:5171–80. doi: 10.4049/jimmunol.169.9.5171. [DOI] [PubMed] [Google Scholar]
- 30.Hinton PR, Johlfs MG, Xiong JM, Hanestad K, Ong KC, Bullock C, Keller S, Tang MT, Tso JY, Vásquez M, et al. Engineered human IgG antibodies with longer serum half-lives in primates. J Biol Chem. 2004;279:6213–6. doi: 10.1074/jbc.C300470200. [DOI] [PubMed] [Google Scholar]
- 31.Hinton PR, Xiong JM, Johlfs MG, Tang MT, Keller S, Tsurushita N. An engineered human IgG1 antibody with longer serum half-life. J Immunol. 2006;176:346–56. doi: 10.4049/jimmunol.176.1.346. [DOI] [PubMed] [Google Scholar]
- 32.Poul MA, Cerutti M, Chaabihi H, Ticchioni M, Deramoudt FX, Bernard A, Devauchelle G, Kaczorek M, Lefranc MP. Cassette baculovirus vectors for the production of chimeric, humanized, or human antibodies in insect cells. Eur J Immunol. 1995;25:2005–9. doi: 10.1002/eji.1830250731. [DOI] [PubMed] [Google Scholar]
- 33.Renaut L, Monnet C, Dubreuil O, Zaki O, Crozet F, Bouayadi K, Kharrat H, Mondon P. Affinity maturation of antibodies: optimized methods to generate high-quality ScFv libraries and isolate IgG candidates by high-throughput screening. Methods Mol Biol. 2012;907:451–61. doi: 10.1007/978-1-61779-974-7_26. [DOI] [PubMed] [Google Scholar]
- 34.Emond S, Mondon P, Pizzut-Serin S, Douchy L, Crozet F, Bouayadi K, Kharrat H, Potocki-Véronèse G, Monsan P, Remaud-Simeon M. A novel random mutagenesis approach using human mutagenic DNA polymerases to generate enzyme variant libraries. Protein Eng Des Sel. 2008;21:267–74. doi: 10.1093/protein/gzn004. [DOI] [PubMed] [Google Scholar]
- 35.Smith GP. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science. 1985;228:1315–7. doi: 10.1126/science.4001944. [DOI] [PubMed] [Google Scholar]
- 36.Polyak MJ, Deans JP. Alanine-170 and proline-172 are critical determinants for extracellular CD20 epitopes; heterogeneity in the fine specificity of CD20 monoclonal antibodies is defined by additional requirements imposed by both amino acid sequence and quaternary structure. Blood. 2002;99:3256–62. doi: 10.1182/blood.V99.9.3256. [DOI] [PubMed] [Google Scholar]
- 37.de Romeuf C, Dutertre CA, Le Garff-Tavernier M, Fournier N, Gaucher C, Glacet A, Jorieux S, Bihoreau N, Behrens CK, Béliard R, et al. Chronic lymphocytic leukaemia cells are efficiently killed by an anti-CD20 monoclonal antibody selected for improved engagement of FcgammaRIIIA/CD16. Br J Haematol. 2008;140:635–43. doi: 10.1111/j.1365-2141.2007.06974.x. [DOI] [PubMed] [Google Scholar]
- 38.Le Garff-Tavernier M, Decocq J, de Romeuf C, Parizot C, Dutertre CA, Chapiro E, Davi F, Debré P, Prost JF, Teillaud JL, et al. Analysis of CD16+CD56dim NK cells from CLL patients: evidence supporting a therapeutic strategy with optimized anti-CD20 monoclonal antibodies. Leukemia. 2011;25:101–9. doi: 10.1038/leu.2010.240. [DOI] [PubMed] [Google Scholar]
- 39.Ben Abdelwahed R, Donnou S, Ouakrim H, Crozet L, Cosette J, Jacquet A, Tourais I, Fournès B, Gillard Bocquet M, Miloudi A, et al. Preclinical study of Ublituximab, a Glycoengineered anti-human CD20 antibody, in murine models of primary cerebral and intraocular B-cell lymphomas. Invest Ophthalmol Vis Sci. 2013;54:3657–65. doi: 10.1167/iovs.12-10316. [DOI] [PubMed] [Google Scholar]
- 40.Sibéril S, de Romeuf C, Bihoreau N, Fernandez N, Meterreau JL, Regenman A, Nony E, Gaucher C, Glacet A, Jorieux S, et al. Selection of a human anti-RhD monoclonal antibody for therapeutic use: impact of IgG glycosylation on activating and inhibitory Fc gamma R functions. Clin Immunol. 2006;118:170–9. doi: 10.1016/j.clim.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 41.Beliard R, Waegemans T, Notelet D, Massad L, Dhainaut F, Romeuf Cd, Guemas E, Haazen W, Bourel D, Teillaud JL, et al. A human anti-D monoclonal antibody selected for enhanced FcgammaRIII engagement clears RhD+ autologous red cells in human volunteers as efficiently as polyclonal anti-D antibodies. Br J Haematol. 2008;141:109–19. doi: 10.1111/j.1365-2141.2008.06985.x. [DOI] [PubMed] [Google Scholar]
- 42.Mathur A, Arora T, Liu L, Crouse-Zeineddini J, Mukku V. Qualification of a homogeneous cell-based neonatal Fc receptor (FcRn) binding assay and its application to studies on Fc functionality of IgG-based therapeutics. J Immunol Methods. 2013;390:81–91. doi: 10.1016/j.jim.2013.01.011. [DOI] [PubMed] [Google Scholar]
- 43.Vaughn DE, Bjorkman PJ. High-affinity binding of the neonatal Fc receptor to its IgG ligand requires receptor immobilization. Biochemistry. 1997;36:9374–80. doi: 10.1021/bi970841r. [DOI] [PubMed] [Google Scholar]
- 44.Yeung YA, Leabman MK, Marvin JS, Qiu J, Adams CW, Lien S, Starovasnik MA, Lowman HB. Engineering human IgG1 affinity to human neonatal Fc receptor: impact of affinity improvement on pharmacokinetics in primates. J Immunol. 2009;182:7663–71. doi: 10.4049/jimmunol.0804182. [DOI] [PubMed] [Google Scholar]
- 45.Igawa T, Tsunoda H, Tachibana T, Maeda A, Mimoto F, Moriyama C, Nanami M, Sekimori Y, Nabuchi Y, Aso Y, et al. Reduced elimination of IgG antibodies by engineering the variable region. Protein Eng Des Sel. 2010;23:385–92. doi: 10.1093/protein/gzq009. [DOI] [PubMed] [Google Scholar]
- 46.Popov S, Hubbard JG, Kim J, Ober B, Ghetie V, Ward ES. The stoichiometry and affinity of the interaction of murine Fc fragments with the MHC class I-related receptor, FcRn. Mol Immunol. 1996;33:521–30. doi: 10.1016/0161-5890(96)00004-1. [DOI] [PubMed] [Google Scholar]
- 47.Kim JK, Tsen MF, Ghetie V, Ward ES. Localization of the site of the murine IgG1 molecule that is involved in binding to the murine intestinal Fc receptor. Eur J Immunol. 1994;24:2429–34. doi: 10.1002/eji.1830241025. [DOI] [PubMed] [Google Scholar]
- 48.Ghetie V, Popov S, Borvak J, Radu C, Matesoi D, Medesan C, Ober RJ, Ward ES. Increasing the serum persistence of an IgG fragment by random mutagenesis. Nat Biotechnol. 1997;15:637–40. doi: 10.1038/nbt0797-637. [DOI] [PubMed] [Google Scholar]
- 49.Kim JK, Firan M, Radu CG, Kim CH, Ghetie V, Ward ES. Mapping the site on human IgG for binding of the MHC class I-related receptor, FcRn. Eur J Immunol. 1999;29:2819–25. doi: 10.1002/(SICI)1521-4141(199909)29:09<2819::AID-IMMU2819>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 50.Datta-Mannan A, Witcher DR, Tang Y, Watkins J, Wroblewski VJ. Monoclonal antibody clearance. Impact of modulating the interaction of IgG with the neonatal Fc receptor. J Biol Chem. 2007;282:1709–17. doi: 10.1074/jbc.M607161200. [DOI] [PubMed] [Google Scholar]
- 51.Vaccaro C, Zhou J, Ober RJ, Ward ES. Engineering the Fc region of immunoglobulin G to modulate in vivo antibody levels. Nat Biotechnol. 2005;23:1283–8. doi: 10.1038/nbt1143. [DOI] [PubMed] [Google Scholar]
- 52.Igawa T, Maeda A, Haraya K, Tachibana T, Iwayanagi Y, Mimoto F, Higuchi Y, Ishii S, Tamba S, Hironiwa N, et al. Engineered monoclonal antibody with novel antigen-sweeping activity in vivo. PLoS One. 2013;8:e63236. doi: 10.1371/journal.pone.0063236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Proetzel G, Roopenian DC. Humanized FcRn mouse models for evaluating pharmacokinetics of human IgG antibodies. Methods. 2014;65:148–53. doi: 10.1016/j.ymeth.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tam SH, McCarthy SG, Brosnan K, Goldberg KM, Scallon BJ. Correlations between pharmacokinetics of IgG antibodies in primates vs. FcRn-transgenic mice reveal a rodent model with predictive capabilities. MAbs. 2013;5:5. doi: 10.4161/mabs.23836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ober RJ, Radu CG, Ghetie V, Ward ES. Differences in promiscuity for antibody-FcRn interactions across species: implications for therapeutic antibodies. Int Immunol. 2001;13:1551–9. doi: 10.1093/intimm/13.12.1551. [DOI] [PubMed] [Google Scholar]
- 56.Vaccaro C, Bawdon R, Wanjie S, Ober RJ, Ward ES. Divergent activities of an engineered antibody in murine and human systems have implications for therapeutic antibodies. Proc Natl Acad Sci U S A. 2006;103:18709–14. doi: 10.1073/pnas.0606304103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Deng R, Loyet KM, Lien S, Iyer S, DeForge LE, Theil FP, Lowman HB, Fielder PJ, Prabhu S. Pharmacokinetics of humanized monoclonal anti-tumor necrosis factor-alpha antibody and its neonatal Fc receptor variants in mice and cynomolgus monkeys. Drug Metab Dispos. 2010;38:600–5. doi: 10.1124/dmd.109.031310. [DOI] [PubMed] [Google Scholar]
- 58.Deng R, Meng YG, Hoyte K, Lutman J, Lu Y, Iyer S, DeForge LE, Theil FP, Fielder PJ, Prabhu S. Subcutaneous bioavailability of therapeutic antibodies as a function of FcRn binding affinity in mice. MAbs. 2012;4:101–9. doi: 10.4161/mabs.4.1.18543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang W, Vlasak J, Li Y, Pristatsky P, Fang Y, Pittman T, Roman J, Wang Y, Prueksaritanont T, Ionescu R. Impact of methionine oxidation in human IgG1 Fc on serum half-life of monoclonal antibodies. Mol Immunol. 2011;48:860–6. doi: 10.1016/j.molimm.2010.12.009. [DOI] [PubMed] [Google Scholar]
- 60.Wang W, Lu P, Fang Y, Hamuro L, Pittman T, Carr B, Hochman J, Prueksaritanont T. Monoclonal antibodies with identical Fc sequences can bind to FcRn differentially with pharmacokinetic consequences. Drug Metab Dispos. 2011;39:1469–77. doi: 10.1124/dmd.111.039453. [DOI] [PubMed] [Google Scholar]
- 61.Suzuki T, Ishii-Watabe A, Tada M, Kobayashi T, Kanayasu-Toyoda T, Kawanishi T, Yamaguchi T. Importance of neonatal FcR in regulating the serum half-life of therapeutic proteins containing the Fc domain of human IgG1: a comparative study of the affinity of monoclonal antibodies and Fc-fusion proteins to human neonatal FcR. J Immunol. 2010;184:1968–76. doi: 10.4049/jimmunol.0903296. [DOI] [PubMed] [Google Scholar]
- 62.Torres M, Casadevall A. The immunoglobulin constant region contributes to affinity and specificity. Trends Immunol. 2008;29:91–7. doi: 10.1016/j.it.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 63.Lazar GA, Dang W, Karki S, Vafa O, Peng JS, Hyun L, Chan C, Chung HS, Eivazi A, Yoder SC, et al. Engineered antibody Fc variants with enhanced effector function. Proc Natl Acad Sci U S A. 2006;103:4005–10. doi: 10.1073/pnas.0508123103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stavenhagen JB, Gorlatov S, Tuaillon N, Rankin CT, Li H, Burke S, Huang L, Vijh S, Johnson S, Bonvini E, et al. Fc optimization of therapeutic antibodies enhances their ability to kill tumor cells in vitro and controls tumor expansion in vivo via low-affinity activating Fcgamma receptors. Cancer Res. 2007;67:8882–90. doi: 10.1158/0008-5472.CAN-07-0696. [DOI] [PubMed] [Google Scholar]
- 65.Chan AC, Carter PJ. Therapeutic antibodies for autoimmunity and inflammation. Nat Rev Immunol. 2010;10:301–16. doi: 10.1038/nri2761. [DOI] [PubMed] [Google Scholar]
- 66.Labrijn AF, Aalberse RC, Schuurman J. When binding is enough: nonactivating antibody formats. Curr Opin Immunol. 2008;20:479–85. doi: 10.1016/j.coi.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 67.Mondon P, Souyris N, Douchy L, Crozet F, Bouayadi K, Kharrat H. Method for generation of human hyperdiversified antibody fragment library. Biotechnol J. 2007;2:76–82. doi: 10.1002/biot.200600205. [DOI] [PubMed] [Google Scholar]
- 68.Dall’Ozzo S, Tartas S, Paintaud G, Cartron G, Colombat P, Bardos P, Watier H, Thibault G. Rituximab-dependent cytotoxicity by natural killer cells: influence of FCGR3A polymorphism on the concentration-effect relationship. Cancer Res. 2004;64:4664–9. doi: 10.1158/0008-5472.CAN-03-2862. [DOI] [PubMed] [Google Scholar]