

Abstract
Ch-mAb7F9, a human-mouse chimeric monoclonal antibody (mAb) designed to bind (+)-methamphetamine (METH) with high affinity and specificity, was produced as a treatment medication for METH abuse. In these studies, we present the preclinical characterization that provided predictive evidence that ch-mAb7F9 may be safe and effective in humans. In vitro ligand binding studies showed that ch-mAb7F9 is specific for and only binds its target ligands (METH, (+)-amphetamine, and 3,4-methylenedioxy-N-methylamphetamine) with high affinity. It did not bind endogenous neurotransmitters or other medications and was not bound by protein C1q, thus it is unlikely to stimulate in vivo complement-dependent cytotoxicity. Isothermal titration calorimetry potency studies showed that METH binding by ch-mAb7F9 is efficient. Pharmacokinetic studies of METH given after ch-mAb7F9 doses in rats demonstrated the in vivo application of these in vitro METH-binding characteristics. While METH had little effect on ch-mAb7F9 disposition, ch-mAb7F9 substantially altered METH disposition, dramatically reducing the volume of distribution and clearance of METH. The elimination half-life of METH was increased by ch-mAb7F9, but it was still very fast compared with the elimination of ch-mAb7F9. Importantly, the rapid elimination of unbound METH combined with previous knowledge of mAb:target ligand binding dynamics suggested that ch-mAb7F9 binding capacity regenerates over time. This finding has substantial therapeutic implications regarding the METH doses against which ch-mAb7F9 will be effective, on the duration of ch-mAb7F9 effects, and on the safety of ch-mAb7F9 in METH users who use METH while taking ch-mAb7F9. These results helped to support initiation of a Phase 1a study of ch-mAb7F9.
Keywords: methamphetamine, human, monoclonal antibody, bioequivalence, pharmacokinetics, chimeric antibody, addiction
Introduction
(+)-Methamphetamine (METH) abuse causes devastating acute and chronic medical effects, which are mediated via multiple neurotransmitter sites.1,2 Small molecule medications that target a single site of action (e.g., the dopamine transporter) have proven ineffective and in some cases addictive. The addiction treatment community thus has no US Food and Drug Administration (FDA)-approved medications for the physiological or psychiatric components of METH addiction.3
A monoclonal antibody (mAb) with high affinity binding to METH could provide a novel therapeutic strategy for METH abuse (e.g., see refs. 4 and 5). In rats, anti-METH mAbs bind METH in the blood away from its sites of action in the brain, thereby reducing its central nervous system effects.6,7 Through this pharmacokinetic antagonism of METH effects, it is anticipated that anti-METH mAbs will reduce the pleasurable reinforcing effects of METH in humans, resulting in extinction of METH use behavior over time when combined with behavioral modification therapy. In addition, the use of a very long-acting anti-METH mAb antagonist, with a 2–4 wk half-life, could be a major breakthrough for patient compliance and provide support for a patient’s willpower to reduce his or her vulnerability to METH-induced relapse during treatment.
Ch-mAb7F9, developed for use in humans, is a human-mouse chimeric mAb designed to bind METH with high affinity and specificity. It was created based on the murine anti-METH mAb7F9 that is effective in preclinical studies at reducing METH-induced pharmacological effects in rats.8 The binding characteristics of the original murine mAb were preserved by combining the mAb7F9 variable region with human IgG2 constant domains chosen to minimize the risk of effector function compared with IgG1 or IgG3 isotypes.9 While IgG4 also have low effector function risk, it has been shown that monoclonal IgG4 can participate in Fab-arm exchange with endogenous IgG4 molecules, which makes them unpredictable as therapeutics.10 Further, the antibody was designed as a chimeric to avoid a potential loss of affinity during humanization; for proof-of-concept studies with a first anti-METH mAb, this design was more time- and cost-effective.
In preparation for a first-in-human clinical study, a series of preclinical studies was performed. The binding characteristics of ch-mAb7F9 to a variety of targets were determined. In vitro and in vivo studies were performed using potential target ligands, including METH, to predict safety and efficacy of the mAb in humans.
In vitro studies examined ligand cross-reactivity to a variety of stimulants, neurotransmitters, over-the-counter medications, and other drugs of abuse. Studies of antibody binding to protein C1q were conducted to determine the potential for complement activation, an undesired effector function. Finally, capacity studies of ch-mAb7F9 for METH were done with isothermal titration calorimetry (ITC) as a measure of mAb potency.
A pharmacokinetic study of ch-mAb7F9 was performed in rats that included characterization of the effects of METH on ch-mAb7F9 pharmacokinetics, and of ch-mAb7F9 on METH pharmacokinetics. In addition, the effect of murine mAb7F9 on METH pharmacokinetics was examined in a separate group of rats to determine if the in vivo METH binding characteristics of murine mAb7F9 and chimeric mAb7F9 were the same.
Taken together, the in vitro and in vivo results demonstrated that ch-mAb7F9 is pharmacologically similar to its murine counterpart, and they helped to support a Phase 1a study of ch-mAb7F9 in humans.
Results
Ligand cross-reactivity
The ability of ch-mAb7F9 to bind to target ligands or other endogenous and exogenous molecules was determined and compared with binding characteristics of the murine antibody, mAb7F9. Similar affinities of both murine and chimeric mAb forms for METH, (+)-amphetamine (AMP) and (+)-3,4-methylenedioxy-N-methylamphetamine [(+)-MDMA] demonstrate that the design of ch-mAb7F9 was successful in maintaining the binding characteristics inherent to the parent mAb7F9 (Table 1). These data also suggest that ch-mAb7F9 will effectively bind METH in vivo, since other anti-METH mAbs with similar in vitro characteristics, including mAb7F9, exhibit METH binding in vivo.5,8,11 Affinity results for mAb7F9 have been previously reported.12 These match well with the current results for METH and (+)-MDMA. It is important to note, however, that an error made in preparation of the inhibitor stocks in the original AMP binding study caused the earlier result12 to be reported incorrectly.
Table 1. Affinity of anti-METH mAbs for stimulants.
| Ligand | ch-mAb7F9 | mAb7F9 |
|---|---|---|
| (+)-METH | KD = 6.9 nM | KD = 7.7 nM |
| (+)-AMP | KI = 350 nM | KI = 370 nM |
| (+)-MDMA | KI = 6.7 nM | KI = 7.9 nM |
Among the other compounds tested (Table 2), only (-)-MDMA was capable of inhibiting [3H]-METH binding at greater than 50% when tested individually; therefore, it was the only ligand with a KI (concentration of inhibitor that prevents 50% of the [3H]-METH from binding) less than 1 µM. All others had to be present at concentrations greater than 1 µM to have a significant effect on the [3H]-METH binding capacity of ch-mAb7F9. Ch-mAb7F9 therefore does not bind any of these compounds well enough to have a clinically significant effect. Ecstasy is a racemic mixture of the (+) and (-)-isoforms of MDMA. They are structural analogs of (±)-METH; therefore, some cross reactivity is expected. The ability of ch-mAb7F9 to bind both forms of MDMA may actually improve its utility as a potential treatment for MDMA abuse in the future.
Table 2. Ligands tested in Ch-mAb7F9 cross-reactivity study.
| Related stimulants | Neurotransmitters | Medications | Drugs of abuse |
|---|---|---|---|
| (+)-Methamphetamine | Dopamine | (+)-Pseudoephedrine | Cocaine |
| (+)-Amphetamine | (-)-Norepinephrine | (+)-Norpseudoephedrine | Morphine |
| (+)-MDMA | (-)-Epinephrine | (-)-Phenylephrine | Phencyclidine |
| (-)-MDMA | Serotonin | (± )-Ephedrine | |
| (+)-MDA | γ-aminobutyric acid | 2-Phenylethylamine | |
| L-Glutamate | Tyramine |
C1q binding
Ch-mAb7F9 was designed with human IgG2 constant domains to avoid unnecessary effector activities such as the ability to stimulate complement-dependent cytotoxicity (CDC), which is inherent to other IgG subtypes, e.g., IgG1.9 A requisite first step in antibody stimulation of CDC is C1q protein binding. An ELISA-like assay was developed to examine the ability of ch-mAb7F9 to bind C1q protein. The results showed that only the positive control IgG1 bound C1q, and it did so in a concentration-dependent manner (Fig. 1). Neither the ch-mAb7F9 nor the negative control IgG2 bound C1q over a wide range of antibody concentrations. These data argue that ch-mAb7F9 is unlikely to stimulate CDC in vivo. While these results appear predictable, the FDA required a study to indicate a lack of unintended Fc effector function.
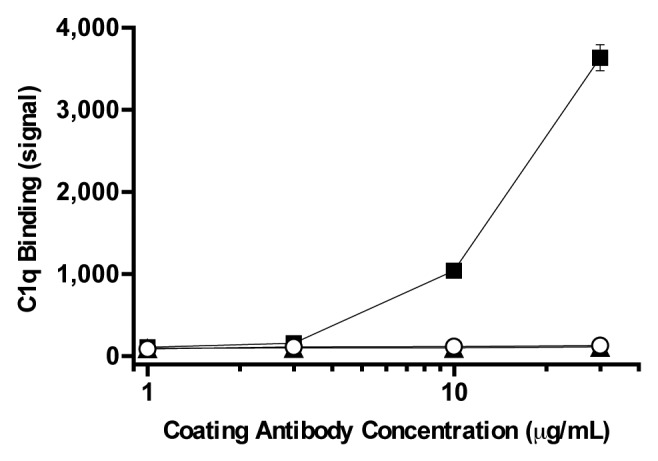
Figure 1. C1q binding as a function of ch-mAb7F9 (open circles) and control antibody concentration. The positive control was purified IgG1 (squares) and the negative control was IgG2 (triangles). Results shown are the average of four replicates ± standard deviation. Error bars not visible are smaller than the symbol.
ITC analysis of METH binding to ch-mAb7F9
Isothermal titration calorimetry analysis of ch-mAb7F9 binding to METH provided thermodynamic and stoichiometry measurements as shown in Table 3 and Figure 2. Data reported for three therapeutic anti-epidermal growth factor receptor (EGFR) mAbs13 and an anti-cocaine murine antibody14 (mAb08) are shown for comparison.
Table 3. Antibody thermodynamic values and stoichiometry for target binding.
| Antibody | ΔG (kJ/mol) | ΔH (kJ/mol) | -TΔS (kJ/mol) | Stoichiometry (N) |
|---|---|---|---|---|
| Ch-mAb7F9 | -43 | -45 | 1 | 1.89 |
| Matuzumaba | -47 | -77 | 30 | 1.78 |
| Cetuximaba | > -50 | -70 | < 20 | 1.76 |
| Panitumumaba | > -49 | -48 | < -1 | 1.68 |
| mAb08b | -49 | -117 | 68 | 0.84 |
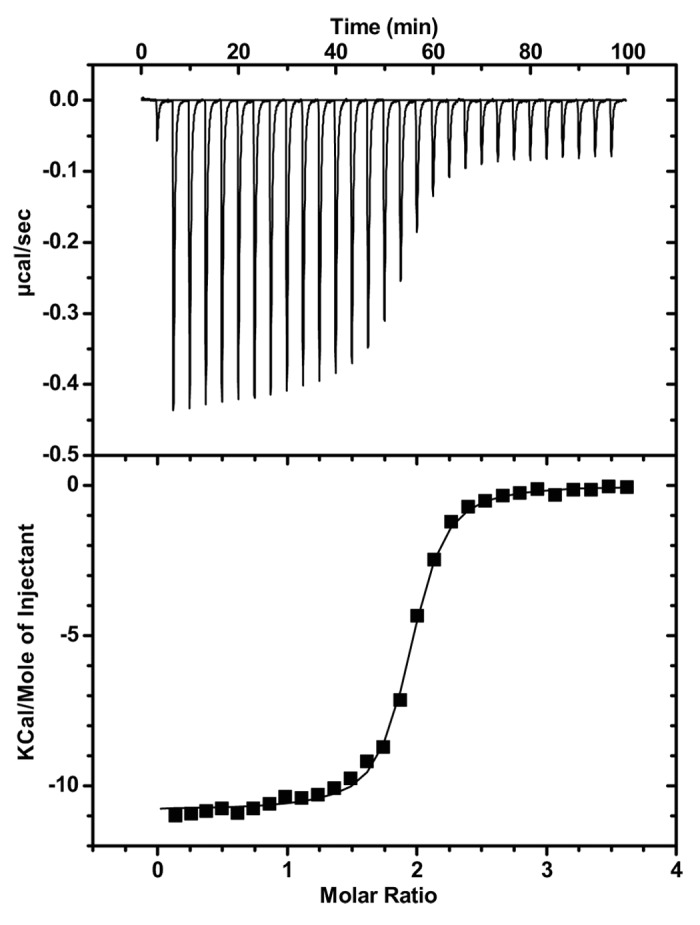
Figure 2. Study of METH binding to ch-mAb7F9 using ITC. METH was titrated into a solution of ch-mAb7F9 as described in the Methods section. The top half shows differential power signals as recorded over the experiment; the lower half shows the same data integrated over time and fitted to the model for a single binding site.
The Gibbs free energy change (ΔG) upon ch-mAb7F9 binding to METH is very similar to the other antibodies shown. Like the other antibodies, binding is mostly driven by a favorable enthalpy (ΔH) change that compensates for an almost nonexistent entropy (ΔS) penalty. The stoichiometry of an antibody is expected to be 2 binding sites per antibody molecule. Like the antibodies against EGFR, ch-mAb7F9 is almost completely active. Errors in the estimated concentrations of the mAb or ligand, or modifications of the protein during manufacture or storage, may be the cause of the slight discrepancy.
For comparison of antibody potency over time (stability) and between manufactured lots, the stoichiometry was converted to an activity value to show the capacity for binding METH per quantity of mAb. The average activity from the first good manufacturing practice-compliant lot of ch-mAb7F9 was 1.96 μg METH bound per mg mAb.
ITC also provides the affinity of the binding reaction, which is the inverse of the dissociation constant (KD). For ch-mAb7F9, the average experimentally determined KD was 49 nM. This result differs somewhat from that determined by ligand cross-reactivity studies using a modified radioimmunoassay (RIA; see Table 1); however, ITC measures the heats of binding in solution, while the RIA requires precipitation of the METH:mAb complex. Further, the RIA and ITC experiments were conducted at different temperatures and pH, both of which influence binding.
In vivo studies
Clinical observations, body weights, and hematocrit values
In these studies, each rat was given a single antibody dose followed by a single METH (or saline placebo) dose 24 h later as outlined in Table 4. All rats in all groups survived to their scheduled euthanasia. Body weights remained consistent or increased slightly in all groups throughout the study and did not differ among the groups. Hematocrit values decreased slightly (e.g., from a mean of 51% to 47%) throughout the study, but this was not physiologically significant because the total decrease was less than 10% from baseline. No rat exhibited evidence of chromodacryorrhea (a stress indicator) or other health problem. No abnormalities were noted on gross pathologic examination.
Table 4. Study groups: Antibody and METH doses.
| Study Group | Antibody and Dose | Challenge Medication | No. of Rats |
|---|---|---|---|
| 1 | Ch-mAb7F9 15 mg/kg | Saline | 12 |
| 2 | Ch-mAb7F9 15 mg/kg | METH 1 mg/kg | 12 |
| 3 | Ch-mAb7F9 150 mg/kg | Saline | 12 |
| 4 | Ch-mAb7F9 150 mg/kg | METH 1 mg/kg | 12 |
| 5 | MAb7F9 150 mg/kg | METH 1 mg/kg | 12 |
Pharmacokinetic analysis
Pharmacokinetic parameter estimates for ch-mAb7F9 were consistent between ch-mAb7F9 doses (Table 5). The presence of METH did not substantially alter the pharmacokinetics of ch-mAb7F9. Because ch-mAb7F9 concentrations were not obtained until 24 h after ch-mAb7F9 dosing, compartmental analysis was not possible; therefore, the distribution phase of ch-mAb7F9 disposition could not be characterized.
Table 5. Ch-mAb7F9 pharmacokinetic values with and without METH.
| Treatment Group | Vdss (mL/kg) | ClT (mL/kg/d) | t1/2λz (d) |
|---|---|---|---|
| Ch-mAb7F9 15 mg/kg | 108.4 | 6.9 | 10.9 |
| Ch-mAb7F9 15 mg/kg with METH | 115.1 | 6.1 | 13.1 |
| Ch-mAb7F9 150 mg/kg | 108.3 | 6.5 | 11.5 |
| Ch-mAb7F9 150 mg/kg with METH | 108.2 | 7.6 | 9.8 |
Analysis of the METH concentrations in the presence of ch-mAb7F9 and mAb7F9 suggests that both mAbs substantially altered the concentration history of METH. Both antibodies substantially decreased Vdss and ClT for METH, resulting in an increased METH elimination half-life compared with METH alone (Table 6; Fig. 3). At the 150 mg/kg mAb dose, ch-mAb7F9 decreased clearance of METH by approximately 50% compared with mAb7F9, roughly doubling the t1/2λz of METH.
Table 6. METH pharmacokinetic values after SC METH administration and ch-mAb7F9 treatment.
| Treatment Group | Vdss (mL/kg) | ClT (mL/kg/hr) | t1/2λz (hr) |
|---|---|---|---|
| Ch-mAb7F9 15 mg/kg | 1881 | 575 | 2.3 |
| Ch-mAb7F9 150 mg/kg | 365 | 38.6 | 6.5 |
| MAb7F9 150 mg/kg | 391 | 75.2 | 3.6 |
| METH alonea | 9000 | 7560 | 1.1 |
a METH pharmacokinetic values are from Riviere et al.21
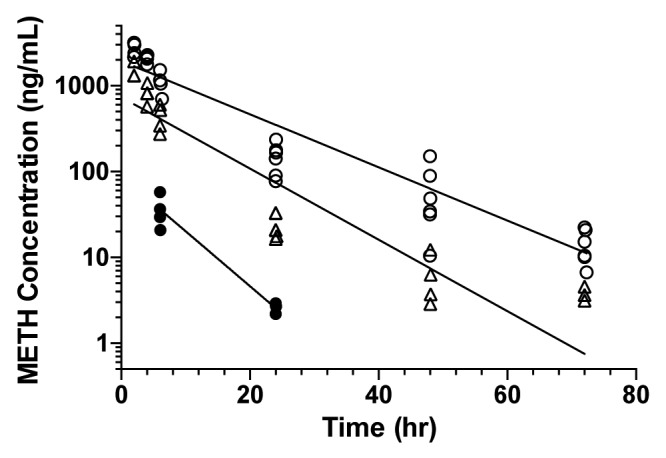
Figure 3. METH concentrations vs. time in the presence of anti-METH mAbs. Data points from individual rats are graphed; lines were fit to the average concentration at each time point because each animal was typically sampled only every other time. Rats received 15 mg/kg ch-mAb7F9 (closed circles), 150 mg/kg ch-mAb7F9 (open circles), or 150 mg/kg mAb7F9 (open triangles).
In addition, METH pharmacokinetic values were altered dose-dependently by ch-mAb7F9. Ch-mAb7F9 decreased METH Vdss ~5- and 25-fold at the 15 and 150 mg/kg doses, respectively. Ch-mAb7F9 decreased ClT to a greater degree in comparison to METH alone at 13- and 196-fold at the low and high doses, respectively. Because t1/2λz is a function of Vdss and ClT (t1/2λz = (0.693*Vdss)/ClT), it was expected that t1/2λz would increase. However, these changes resulted in increases in METH t1/2λz of only 2.1- and 5.9-fold, respectively, which indicate that METH elimination was still rapid compared with ch-mAb7F9 elimination (2–7 h vs. 10–13 d). This finding is critically important to the understanding of ch-mAb7F9 action in attenuating METH effects.
Discussion
This preclinical characterization predicted that ch-mAb7F9 would be safe in humans. Safety is predicted by the specificity of ch-mAb7F9 only for its target ligands, the lack of significant binding to other possible targets (e.g., neurotransmitters), and the lack of binding by C1q. Ch-mAb7F9 should not, therefore, interfere with normal neurological function or affect the medical use of structurally-related medications, and the risk for undesired effector function is expected to be minimal. Additionally, rats in the in vivo studies exhibited no significant health changes as evidenced by normal eating behavior and preservation of body weight throughout the study, providing further evidence that ch-mAb7F9 will be safe.
Separate human tissue cross-reactivity and in vivo rat toxicology studies performed under good laboratory practice (GLP) conditions also indicate ch-mAb7F9 should be safe in humans (results not shown). Briefly, the tissue cross-reactivity studies showed ch-mAb7F9 staining in a variety of tissues, but the staining was cytoplasmic. It is unlikely that the cytoplasm and cytoplasmic structures would be accessible to ch-mAb7F9 in vivo because of the molecular size of the antibody. In the toxicology study, ch-mAb7F9 was well-tolerated at single doses up to 400 mg/kg (no-observed-adverse-effect level, NOAEL), which is well above possible human doses. No ch-mAb7F9-related mortality or evidence of systemic toxicity was observed and no target organs were identified. In this GLP study, the elimination half-life of ch-mAb7F9 appeared to be 8–12 d. Together, these safety studies of ch-mAb7F9 were sufficient to support a Phase 1a study of this medicine.
In the current studies, ch-mAb7F9 substantially altered METH disposition in a dose-dependent manner. Given the greater relative reduction in METH ClT compared with Vdss, the increase in t1/2λz was expected. The elimination of METH, however, remained high compared with antibody elimination (Table 5 and 6). The rapid elimination of METH in the presence of ch-mAb7F9 suggests that clearance of unbound METH was not affected as much as would be predicted by high affinity binding by the mAb alone. In other words, if ch-mAb7F9 bound METH tightly, but did not release it, the METH half-life would approach that of the antibody. The fact that antibody affinity reflects both the ligand on rate and off rate means that even high affinity antibodies may release a ligand very quickly if the rate of ligand binding is sufficiently rapid. In support, preliminary surface plasmon resonance studies of the on-off rates of METH binding to ch-mAb7F9 indicate that both reactions are fast (data not shown). An explanation for the preserved elimination of METH is that it is quickly released from the mAb into the blood from which it is eliminated.
Ch-mAb7F9 was able to effect a change in METH disposition when there was an excess of METH relative to antibody binding sites. Based on a calculation of the predicted numbers of ch-mAb7F9 METH binding sites at the time of METH dosing in this study, the ratio of METH molecules to ch-mAb7F9 binding sites in the body was greater than 3:1 for the 150 mg/kg mAb dose, and greater than 30:1 for the 15 mg/kg mAb dose. The observation that anti-drug antibodies effectively redistribute their target ligands in the face of target ligand excess has been shown in other studies of anti-drug antibodies (see refs. 5 and 15). These findings indicate that the observed capacity of ch-mAb7F9 to alter METH disposition is greater than predicted by the relationship of the number of METH molecules to ch-mAb7F9 binding sites in the body. The in vitro binding characteristics of ch-mAb7F9 for METH and the continued rapid elimination of METH in the presence of ch-mAb7F9 indicate that METH bound to ch-mAb7F9 is released to regenerate METH binding sites that can subsequently act on additional METH molecules. This could explain the ability of ch-mAb7F9 to redistribute METH in the face of doses in excess of the mAb’s binding capacity.
Regeneration of binding capacity is relevant to the clinical safety concern that users will take METH during treatment with ch-mAb7F9. The possibility that a user will attempt to surmount the blocking effects on reinforcement of ch-mAb7F9 by taking high METH doses is substantial based on recidivism rates.16 Each mAb molecule does not appear to bind irreversibly to METH, but rather alters the equilibrium of METH through reversible binding. Therefore, the risk of reaching a threshold or “ceiling” effect of ch-mAb7F9 function in which METH effects suddenly spike due to an acute increase in unbound METH seems unlikely. Furthermore, the METH that dissociates from the antibody likely does not all distribute to pharmacologically “active” sites, since not all of cardiac output goes to active sites, and since the METH clearance remains rapid. This hypothesis will be tested in future studies.
In the current studies, a t1/2λz of 10–13 d for ch-mAb7F9 in rats was observed. We predict that ch-mAb7F9 will have a long t1/2λz in humans, as much as 3 wk. This is because the Vdss of IgG molecules in both species is similar (due to the large size of the IgG molecule), but their ClT in humans is approximately one-third that observed in rats, resulting in t1/2λz values in humans of approximately 2- to 3-fold that of the rat, or 3 wk.17,18 This is important because a long half-life suggests that the duration of effects from a single ch-mAb7F9 dose will protect a METH user during a period in which the user is very vulnerable to recidivism to METH use, i.e., the first several days after initiation of abstinence. Furthermore, the long duration of action will allow for infrequent dosing, improving compliance with the treatment regimen.
The final goal of this study was to determine the pharmacokinetic bioequivalence of ch-mAb7F9 to mAb7F9 as an in vivo test that binding characteristics were preserved on production of the chimeric antibody. The in vivo binding study of METH by ch-mAb7F9 indicates ch-mAb7F9 is as effective, if not more so, than the murine mAb parent in altering METH disposition. Evaluation of the differences between the effects of ch-mAb7F9 and mAb7F9 on METH disposition show that ch-mAb7F9 (150 mg/kg dose) decreased METH Vdss slightly and ClT substantially more than the murine mAb7F9 at the same dose (Table 6). One pharmacokinetic explanation for this finding is that ch-mAb7F9 may have smaller Vdss and ClT values than the murine mAb7F9. It is also possible that in vivo binding of METH by ch-mAb7F9 is more effective than by mAb7F9, perhaps due to higher in vivo affinity for METH. Finally, the on-off rates of METH binding by mAb7F9 compared with ch-mAb7F9 may be slightly different and could also have contributed to the differences in METH pharmacokinetics. Further biophysical comparison of ch-mAb7F9 and murine mAb7F9 characteristics is needed to confirm this hypothesis. While the pharmacokinetics of mAb7F9 and its METH binding characteristics have not been evaluated, ch-mAb7F9 was at least as effective as mAb7F9 in altering METH disposition, proving the hypothesis of bioequivalence.
The primary limitations of this preclinical characterization relate to the pharmacokinetic study and analysis of the pharmacokinetic data. Blood samples were obtained for only 21 d from the rats. Given that the observed t1/2λz of approximately 10–13 d would require sampling for 40–70 d (4–7 half-lives) to completely describe the elimination phase of ch-mAb7F9 disposition, the duration of blood sampling was too short. Furthermore, because blood sampling for ch-mAb7F9 concentrations did not start until 24 h after antibody dosing, the distribution phase of ch-mAb7F9 disposition could not be described, and a non-compartmental approach to the analysis was used. In addition, the sampling schedule did not allow determination of individual pharmacokinetic fits for each rat, so a pooled approach was used. Despite these limitations, the results were similar to those obtained in the GLP toxicology study for ch-mAb7F9 in which samples were obtained for 56 d; the antibody t1/2λz determined in that study was 8–12 d (results not shown).
In the current study, only a single dose of METH (1 mg/kg, SC) was given as a challenge for the antibody. This is certainly less than the amount METH users would self-administer in attempts to surmount the effects of ch-mAb7F9. Future studies are planned to address the safety of the interaction between ch-mAb7F9 and large doses of METH.
In summary, these results showed that binding characteristics of the murine mAb7F9 and the desired pharmacologic characteristics of an IgG2 were preserved in the design of ch-mAb7F9 from its murine counterpart, mAb7F9. These studies confirm that the capacity of ch-mAb7F9 to alter METH disposition is greater than predicted by the relationship of the number of METH molecules to ch-mAb7F9 binding sites in the body. This is a critically important finding because it suggests that ch-mAb7F9 will be effective in the face of large METH doses. If this is true, the risk that a user can effectively surmount the effects of ch-mAb7F9 by taking very large (binge) doses of METH and even experience METH toxicity may be clinically insignificant. The combination of high affinity and specificity for METH with a lack of other antibody effects suggests that ch-mAb7F9 may be safe and effective in humans. Only when studies in humans are performed will the real effect of this new medication for the treatment of METH abuse be realized.
Materials and Methods
Test and control articles
Murine mAb7F9 was developed, produced and formulated as described previously.12 The antigen binding site of murine mAb7F9 served as the starting template for creating anti-METH ch-mAb7F9. To make the chimeric antibody, RNA from the murine mAb7F9 hybridoma cell line was extracted and used in reverse transcription to obtain the heavy and light chain coding sequences of the mAb7F9 variable region. These were then joined to the coding sequences for the constant domains of a human IgG2κ antibody and inserted into a Chinese hamster ovary cell line for expression. Catalent Pharma Solutions generated the cell line and manufactured ch-mAb7F9 for these studies.
METH was obtained as solid (+)-methamphetamine hydrochloride and as tritiated METH in liquid formulation. Other controlled substances including (+)-amphetamine sulfate (AMP), (+)-3,4-methylenedioxy-N-methylamphetamine hydrochloride (MDMA), (-)-MDMA, (+)-norpseudoephedrine hydrochloride, (+)-3,4-methylenedioxyamphetamine hydrochloride (MDA), cocaine hydrochloride, morphine sulfate, and phencyclidine hydrochloride were received as powders. All came from the Research Triangle Institute through the National Institute on Drug Abuse. Other chemicals for ligand binding studies were purchased from Sigma Aldrich.
Control IgGs for the C1q binding studies were obtained from Sigma Aldrich (Cat #:I5154, I5404); the C1q protein itself came from Abcam, Inc. (Cat #:ab96363).
In vitro ligand binding
Non-GLP in vitro ligand binding studies were performed to determine the ability of ch-mAb7F9 to bind its intended targets and other endogenous and exogenous molecules. The assay used a modified radioimmunoassay (RIA) in a competitive binding format with [3H]-METH as a tracer.12 For (+)-METH (+)-AMP and (+)-MDMA, a complete inhibition curve was constructed to determine the dissociation constant (KD, which is the inverse of the affinity constant) or KI (concentration of inhibitor which prevents 50% of the [3H]-METH from binding) for mAb7F9 and ch-mAb7F9. For other potential ligands with a lower probability of binding to the mAb, pools of compounds (each ligand at 1 μM) were tested as potential inhibitors of [3H]-METH binding. Ligands were retested individually if the pooled test results revealed significant (>50%) inhibition.
In vitro C1q binding
The potential effector function of ch-mAb7F9 was determined using a non-GLP interaction assay to measure ch-mAb7F9 binding to C1q, the first protein in the complement cascade. Commercially available purified human IgG1 and IgG2 were used as positive and negative controls, respectively. Assay plates were coated with ch-mAb7F9, IgG1, or IgG2 in the range from 0.3 to 30 µg/mL. After blocking to prevent non-specific binding, C1q (labeled for detection) was applied to each well at 1 µg/mL. Assay wells were analyzed by electrochemiluminescence detection to determine the relative amount of antibody binding to C1q, a requisite step for complement activation in vivo.
Isothermal titration calorimetry (ITC)
The thermodynamics, affinity and stoichiometry of ch-mAb7F9 binding to METH were measured using isothermal titration calorimetry (ITC) with a VP-ITC instrument from GE Healthcare. For a review of ITC and its use in antibody quality control, see ref. 19 and www.microcal.com. ITC runs were conducted by titrating an 86 μM solution of METH into a cell containing 5 μM ch-mAb7F9 with a stirring rate of 307 rpm. The sample and reference cells were held at the same constant temperature. Upon binding between METH and ch-mAb7F9, heat was given off in proportion to the amount of binding. The instrument measures the difference in heat required to maintain the same temperature in the sample cell as that in the reference cell with no titration occurring. As METH is incrementally injected into the cell, the antibody becomes saturated and only the heats of dilution are measured. Over the course of each run, 28 individual injections of 10 μL were completed with a 200 s delay between each to reach equilibrium. All runs were conducted at 37 °C in phosphate-buffered saline at pH 6.5.
ITC data were analyzed using the customized Origin 7 software (OriginLab) provided with the instrument using the included model for one binding site. A binding curve was constructed from the plot of heats from each injection against the ratio of METH and ch-mAb7F9 in the cell. From this curve, the stoichiometry of binding (N) was determined by the ITC software. Because N should be 2 METH:ch-mAb7F9 for a 100% active antibody solution, the calculated N value and the molecular weights of METH and ch-mAb7F9 can be used to calculate the amount of METH bound per quantity of antibody, with 2.07 being the μg METH bound per mg mAb from a 100% active antibody. Further, the software calculates enthalpic and entropic contributions to binding and the association constant, which is the reciprocal of KD.
In vivo pharmacokinetics study
The objectives of this study were to (1) determine the pharmacokinetics of ch-mAb7F9 using the concentration history of ch-mAb7F9 beginning one day after its administration; (2) determine the effect of ch-mAb7F9 on the concentration history of METH given 24 h after ch-mAb7F9 administration; and (3) determine the relative pharmacokinetic bioequivalence of ch-mAb7F9 and murine mAb7F9.
Animals
Male, Sprague Dawley (Charles River Laboratories) rats were used. The number of animals in each dose group (n = 12) was selected to allow collection of at least 4 serum samples from different rats at each time point over the 3 wk period of the study without inflicting undue stress on the animals due to tail vein blood draws or anemia. The animals were acclimated to their housing for at least 1 wk before antibody dosing and were housed individually throughout the study. Housing and care were as specified in the USDA Animal Welfare Act (9 CFR, Parts 1, 2, and 3) and as described in the Guide for the Care and Use of Laboratory Animals from the National Research Council. These studies were reviewed and approved by the University of Arkansas for Medical Sciences Animal Care and Use Committee. The disposition of all animals was documented in the study records. Rats in all the studies were euthanized by exsanguination and decapitation while under isoflurane anesthesia. Veterinary care was available throughout the study and the veterinary staff examined the animals as warranted by clinical signs or other changes. No veterinary medicinal treatments were administered during the study.
Dose formulation
Ch-mAb7F9 and mAb7F9 doses were prepared in 10 mL/kg volumes for each group. The day before administration to the rats, an aliquot of stock antibody solution was centrifuged at ~100,000 times gravity for 1 h at 5 °C and the supernatant was refrigerated overnight. On the day of administration, the supernatant was diluted with filtered phosphate-buffered saline (15 mM sodium phosphate, 150 mM NaCl) to obtain the final dose for each group. All procedures were performed using aseptic techniques in a laminar flow hood.
METH prepared as a 1 mg/mL solution was made using 0.9% NaCl for injection. The stock solution was filtered and placed in sterilized, capped vials, and stored at 2 to 8 °C.
Experimental design
Using 5 study groups with 12 rats per group, each rat received a single dose of mAb on Day 1 (Monday, see Table 4). The mAb dose was administered through a tail vein over approximately 2 min. The dose volume for each animal was calculated based on the most recent body weight measurement. A single METH dose (or saline as placebo) was administered subcutaneously (SC) on Day 2 (Tuesday).
Six tail vein blood samples (350 or 400 μL each) were taken from each rat over a 3 wk period, with a maximum of 1.2 mL withdrawn in a 24 h period (see Table 7). Hematocrits were measured at baseline, at 3 or 4 d, and at 5 or 8 d (depending on the group) to ensure the percentage of red cells did not decrease more than 10% over the course of the study. Body weights and general health observations were recorded for each animal, and gross pathologic observations were made at euthanasia.
Table 7. Dosing and sampling times for each study group.
| Study Day | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Baseline | 1 | 2 | 3 | 4 | 5 | 8 | 15 | 22 | |||||||
| Wed-Fri | Mon | Tues | Wed | Thur | Fri | Mon | Mon | Mon | |||||||
| Rat No. | * | 1min | 5min | 1hr | 2hr | 4hr | 6hr | 24hr | 48hr | 3d | 7d | 14d | 21d | ||
| 1–4 | X | Y | * | X | X | X | X | X | |||||||
| 5–8a | X | Y | * | X | X | X | X | X | |||||||
| 9–12 | X | Y | * | X | X | X | X | X | |||||||
X, Blood samples were taken via a tail vein. The baseline sample was taken during the week prior to antibody dosing. Blood sample volumes were 400 μL at baseline, day 3 or 4, and day 8 or 15. The other sample volumes were 350 μL. Y, Antibody doses were given in the morning of study day 1. The doses were given in a volume of approximately 3 mL over 2 min via the tail vein. *The METH dose was given SC in a dose of 1 mg/mL in a volume of 1 mL/kg. All subsequent blood sampling times were relative to the METH dosing time. For the 1 and 5 min time points, rats were anesthetized with isoflurane prior to METH administration to ensure timely blood sampling. aRats 5 and 6 in each group had blood samples taken on day 3, while rats 7 and 8 had blood samples taken on day 4. Similar sampling occurred on days 5 and 8, and 15 and 22. Thus, there were 6 samples at each time point on days 3 through 22. Four (4) samples were taken at each time point on day 2. Hematocrits were measured at baseline, on days 3 or 4, and days 5 or 8, depending on when each animal’s blood sample was drawn on these days.
METH concentrations
Solid phase extraction combined with an Acquity Ultra Performance Liquid Chromatography (UPLC®; Waters Corp) and tandem mass spectrometer (MS) was used for the detection of METH in rat serum.20 The method utilizes solid phase extraction via Oasis® MCX sorbent technology (Waters Corp). The UPLC-MS is a Waters Acquity system interfaced with a Waters Quattro Premier XE mass spectrometer. For analysis, the following parameters were used: capillary voltage, 2.5 kV; cone voltage, 18 V; source block temp, 150 °C; solvent gas (nitrogen) delivered at 600 L/hr gas flow rate at 500 °C. Argon gas pressure was used to dissociate the precursor ions [(+)-METH] and the collision energy was optimized to enhance the signal of the product ions used in quantification. The ion transition was m/z 150.1 → 90.8 for (+)-METH. A standard curve was formulated using the calibration standards. The lower limit of quantification was the lowest that met acceptance criteria on the standard curve, and was either 0.3 or 1 ng/mL. The upper limit of quantitation was 1000 ng/mL. Reproducibility was determined through repeated extractions of standards and all predicted values were within ± 20% of their expected concentrations.
Ch-mAb7F9 concentrations
The concentration of ch-mAb7F9 in rat serum samples was determined using a Meso Scale Discovery® (MSD) 96-well microtiter format by direct electrochemiluminescence detection. Plates coated with 1 µg/mL donkey anti-human (Fc-specific) antibody (Jackson Labs Cat #:709-005-098) for 60 min were washed with phosphate-buffered saline containing 0.05% Tween 20 (PBS-T) and incubated an additional 60 min with SuperBlock (Pierce Biotechnology, Inc. Cat #: 37535). Rat serum samples, or different amounts of ch-mAb7F9 for a standard curve and quality control samples, were added to wells of each plate in duplicate. After an hour, plates were washed with PBS-T to remove unbound protein and incubated for 60 min with donkey anti-human (Fc-specific) antibody labeled with MSD Sulfo-Tag (0.5 µg/mL) for ch-mAb7F9 detection. MSD Read Buffer T was added to the wells and each plate was read with the MSD Sector PR 100 instrument. The entire method was conducted at room temperature with shaking on a plate shaker.
Serum samples were diluted 1:10 with SuperBlock and then further dilutions made as necessary using SuperBlock containing 10% naïve rat serum so that the final concentration of serum in all samples was 10%. The standard curve of ch-mAb7F9 ranged from 5 to 1000 ng/mL. Two sets of quality control samples per plate were prepared with ch-mAb7F9 at 0 (blank), 15, 75, and 350 ng/mL. The standard curve and quality control samples were prepared in SuperBlock containing 10% naïve rat serum.
The average experimentally derived concentration value and percent difference (%Diff) for each set of replicates were calculated. If the %Diff of a set was ≤ 25%, the average value was accepted. A minimum of 6 acceptable standards was required to generate a 4 parameter logistic standard curve with R2 ≥ 0.990. If acceptance criteria for the standards and quality control samples were met, average sample values that met the %Diff requirement from that plate were accepted. Final values were calculated as the average of all accepted values obtained from each sample.
Pharmacokinetic analysis
Log concentration-vs.-time curves for all time points in each study group were analyzed by model-independent methods using the fitting software, WinNonLin (Pharsight Corporation). Non-compartmental analysis was done to estimate pharmacokinetic parameters using the following equations: Vdss = (D*AUMC0-∞)/AUC0-∞2; ClT = D/AUC0-∞; and t1/2λz = 0.693*MRT, where Vdss is the volume of distribution at steady-state, D is the injected dose, AUMC0-∞ is the area under the first moment curve from time 0 to infinity, AUC0-∞ is the area under the serum concentration time curve from time 0 to infinity, ClT is the total systemic clearance, MRT is the mean residence time, and t1/2λz is the elimination half-life. The non-compartmental analysis was performed using the sparse sampling routine, which utilizes the mean concentration at each time point for analysis. Input for the mAb doses were intravenous infusions of 2 min duration, and the extravascular input function was used for the METH doses.
Statistical analysis
The interval data for body weights and hematocrit values were determined as mean ± standard deviations. These were compared across groups with a one-way ANOVA. For post-hoc analysis, a Dunnett test was used for comparison to baseline values of the hematocrits, and a test for linear trends was used for the body weight data. For all experiments, statistical significance was achieved at a level of P < 0.05. Because of the sparse sampling approach, statistical analysis of the pharmacokinetic data was not performed.
Disclosure of Potential Conflicts of Interest
Owens SM and Gentry WB are full time faculty members at the University of Arkansas for Medical Sciences, and Henry RL is a full time faculty member at the University of Arkansas. They have financial interests in and serve as Chief Scientific Officer (Owens SM), Chief Medical Officer (Gentry WB), and Vice President of Biopharmaceutics (Henry RL) for InterveXion Therapeutics, LLC (Little Rock, AR), a pharmaceutical biotechnology company focused on treating human drug addiction with antibody-based therapy.
Acknowledgments
The authors would like to thank Michael Watson, Melinda Gunnell, and Sherri Wood for their technical assistance during the in vivo studies.
This work was supported by a grant from the National Institute on Drug Abuse (RC2DA028915) of the National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
We also thank the Arkansas Statewide Mass Spectrometry and Protein Core Facilities for their assistance. Based on this assistance, we also acknowledge that Grant Number P30 GM103450 from the National Institute of General Medical Sciences of the National Institutes of Health supported this publication.
Glossary
Abbreviations:
- AMP
(+)-amphetamine
- CDC
complement-dependent cytotoxicity
- ClT
total clearance
- GLP
good laboratory practice
- ITC
isothermal titration calorimetry
- KD
dissociation constant
- KI
concentration of inhibitor which prevents 50% of the target ligand from binding
- mAb
monoclonal antibody
- (+)-MDMA
(+)-3,4-methylenedioxy-N-methylamphetamine
- METH
(+)-methamphetamine
- MSD
MesoScale Discovery
- RIA
radioimmunoassay
- t1/2λz
elimination half-life
- SC
subcutaneously
- Vdss
volume of distribution at steady state
Footnotes
Previously published online: www.landesbioscience.com/journals/mabs/article/27620
References
- 1.Darracq L, Blanc G, Glowinski J, Tassin JP. Importance of the noradrenaline-dopamine coupling in the locomotor activating effects of D-amphetamine. J Neurosci. 1998;18:2729–39. doi: 10.1523/JNEUROSCI.18-07-02729.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Auclair A, Drouin C, Cotecchia S, Glowinski J, Tassin J-P. 5-HT2A and alpha1b-adrenergic receptors entirely mediate dopamine release, locomotor response and behavioural sensitization to opiates and psychostimulants. Eur J Neurosci. 2004;20:3073–84. doi: 10.1111/j.1460-9568.2004.03805.x. [DOI] [PubMed] [Google Scholar]
- 3.Vocci FJ, Appel NM. Approaches to the development of medications for the treatment of methamphetamine dependence. Addiction. 2007;102(Suppl 1):96–106. doi: 10.1111/j.1360-0443.2007.01772.x. [DOI] [PubMed] [Google Scholar]
- 4.Gentry WB, Rüedi-Bettschen D, Owens SM. Anti-(+)-methamphetamine monoclonal antibody antagonists designed to prevent the progression of human diseases of addiction. Clin Pharmacol Ther. 2010;88:390–3. doi: 10.1038/clpt.2010.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Laurenzana EM, Hendrickson HP, Carpenter D, Peterson EC, Gentry WB, West M, Che Y, Carroll FI, Owens SM. Functional and biological determinants affecting the duration of action and efficacy of anti-(+)-methamphetamine monoclonal antibodies in rats. Vaccine. 2009;27:7011–20. doi: 10.1016/j.vaccine.2009.09.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Byrnes-Blake KA, Laurenzana EM, Carroll FI, Abraham P, Gentry WB, Landes RD, Owens SM. Pharmacodynamic mechanisms of monoclonal antibody-based antagonism of (+)-methamphetamine in rats. Eur J Pharmacol. 2003;461:119–28. doi: 10.1016/S0014-2999(03)01313-X. [DOI] [PubMed] [Google Scholar]
- 7.Gentry WB, Laurenzana EM, Williams DK, West JR, Berg RJ, Terlea T, Owens SM. Safety and efficiency of an anti-(+)-methamphetamine monoclonal antibody in the protection against cardiovascular and central nervous system effects of (+)-methamphetamine in rats. Int Immunopharmacol. 2006;6:968–77. doi: 10.1016/j.intimp.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 8.Owens SM, Atchley WT, Hambuchen MD, Peterson EC, Gentry WB. Monoclonal antibodies as pharmacokinetic antagonists for the treatment of (+)-methamphetamine addiction. CNS Neurol Disord Drug Targets. 2011;10:892–8. doi: 10.2174/187152711799219370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hamilton RG. The Human IgG Subclasses. Baltimore, MD: Calbiochem-Novabiochem Corporation; 2001. [Google Scholar]
- 10.Labrijn AF, Buijsse AO, van den Bremer ETJ, Verwilligen AYW, Bleeker WK, Thorpe SJ, Killestein J, Polman CH, Aalberse RC, Schuurman J, et al. Therapeutic IgG4 antibodies engage in Fab-arm exchange with endogenous human IgG4 in vivo. Nat Biotechnol. 2009;27:767–71. doi: 10.1038/nbt.1553. [DOI] [PubMed] [Google Scholar]
- 11.Byrnes-Blake KA, Laurenzana EM, Landes RD, Gentry WB, Owens SM. Monoclonal IgG affinity and treatment time alters antagonism of (+)-methamphetamine effects in rats. Eur J Pharmacol. 2005;521:86–94. doi: 10.1016/j.ejphar.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 12.Carroll FI, Abraham P, Gong PK, Pidaparthi RR, Blough BE, Che Y, Hampton A, Gunnell M, Lay JO, Jr., Peterson EC, et al. The synthesis of haptens and their use for the development of monoclonal antibodies for treating methamphetamine abuse. J Med Chem. 2009;52:7301–9. doi: 10.1021/jm901134w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alvarenga ML, Kikhney J, Hannewald J, Metzger AU, Steffens K-J, Bomke J, Krah A, Wegener A. In-depth biophysical analysis of interactions between therapeutic antibodies and the extracellular domain of the epidermal growth factor receptor. Anal Biochem. 2012;421:138–51. doi: 10.1016/j.ab.2011.10.039. [DOI] [PubMed] [Google Scholar]
- 14.Ramakrishnan M, Alves De Melo F, Kinsey BM, Ladbury JE, Kosten TR, Orson FM. Probing cocaine-antibody interactions in buffer and human serum. PLoS One. 2012;7:e40518. doi: 10.1371/journal.pone.0040518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pitas G, Laurenzana EM, Williams DK, Owens SM, Gentry WB. Anti-phencyclidine monoclonal antibody binding capacity is not the only determinant of effectiveness, disproving the concept that antibody capacity is easily surmounted. Drug Metab Dispos. 2006;34:906–12. doi: 10.1124/dmd.105.005934. [DOI] [PubMed] [Google Scholar]
- 16.Brecht ML, von Mayrhauser C, Anglin MD. Predictors of relapse after treatment for methamphetamine use. J Psychoactive Drugs. 2000;32:211–20. doi: 10.1080/02791072.2000.10400231. [DOI] [PubMed] [Google Scholar]
- 17.Bazin-Redureau MI, Renard CB, Scherrmann JM. Pharmacokinetics of heterologous and homologous immunoglobulin G, F(ab’)2 and Fab after intravenous administration in the rat. J Pharm Pharmacol. 1997;49:277–81. doi: 10.1111/j.2042-7158.1997.tb06795.x. [DOI] [PubMed] [Google Scholar]
- 18.Joos B, Trkola A, Kuster H, Aceto L, Fischer M, Stiegler G, Armbruster C, Vcelar B, Katinger H, Günthard HF. Long-term multiple-dose pharmacokinetics of human monoclonal antibodies (MAbs) against human immunodeficiency virus type 1 envelope gp120 (MAb 2G12) and gp41 (MAbs 4E10 and 2F5) Antimicrob Agents Chemother. 2006;50:1773–9. doi: 10.1128/AAC.50.5.1773-1779.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pierce MM, Raman CS, Nall BT. Isothermal titration calorimetry of protein-protein interactions. Methods. 1999;19:213–21. doi: 10.1006/meth.1999.0852. [DOI] [PubMed] [Google Scholar]
- 20.Hendrickson H, Laurenzana E, Owens SM. Quantitative determination of total methamphetamine and active metabolites in rat tissue by liquid chromatography with tandem mass spectrometric detection. AAPS J. 2006;8:E709–17. doi: 10.1208/aapsj080480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rivière GJ, Byrnes KA, Gentry WB, Owens SM. Spontaneous locomotor activity and pharmacokinetics of intravenous methamphetamine and its metabolite amphetamine in the rat. J Pharmacol Exp Ther. 1999;291:1220–6. [PubMed] [Google Scholar]