

Abstract
Cancer cells adapt their metabolism to support proliferation and survival. A hallmark of cancer, this alteration is characterized by dysfunctional metabolic enzymes, changes in nutrient availability, tumor microenvironment and oncogenic mutations. Metabolic rewiring in cancer is tightly connected to changes at the epigenetic level. Enzymes that mediate epigenetic status of cells catalyze posttranslational modifications of DNA and histones and influence metabolic gene expression. These enzymes require metabolites that are used as cofactors and substrates to carry out reactions. This interaction of epigenetics and metabolism constitutes a new avenue of cancer biology and could lead to new insights for the development of anti-cancer therapeutics.
Keywords: cancer, epigenetics, metabolism, glucose flux, amino acid metabolism
1. Introduction
Conrad Waddington first established the concept of epigenetics in 1942 when he proposed that genes interact with their product to determine a phenotype [1]. This observation was later corroborated by findings from Barker and Osmond in 1986 that showed genes respond to environmental exposures during embryonic development [2]. They demonstrated that expectant mothers with poor eating habits give birth to children more susceptible to disease during childhood and well into their adulthood. This developmental response is characterized by changes in gene activity that is passed on to successive generations. Further evidence of transgenerational transmission of genetics emerged from the Bygren and Pembrey investigation into the possibility that some gene functions are not only passed on from mother to fetus during pregnancy but can be carried over from both male and female past exposures before conception [3; 4]. These findings shaped the definition of the term “epigenetics” to become the study of modifications in gene expression that do not involve changes in DNA nucleotide sequences [5]. Hence, the epigenetic layer of gene regulation controls both normal cellular processes and abnormal events associated with disease, notably cancer [6; 7].
It is widely recognized that cancer is a constellation of diseases manifested in various clinical subtypes, each characterized by distinct histopathological and biological features [8]. At the origin of all cancers remains abnormal cell proliferation, which has so far offered a useful but incomplete target for anticancer therapy [9; 10]. Chemotherapeutic agents exerting cytotoxic effects on rapidly dividing cells are commonly used as first line of therapy, but become inefficient when tumors acquire resistant phenotypes and progress into a refractory phase. There is considerable need to circumvent tumor resistance to conventional therapy to achieve a more successful treatment. This goal is potentially attainable by exploiting intrinsic and extrinsic factors that contribute to tumorigenesis. One emerging possibility of new cancer therapy is to target the alteration of cell metabolism [11; 12]. Over eighty years ago, Otto Warburg established that cancer cells metabolize increasing amounts of glucose through fermentation even in oxygen rich environments that originally suggested defective mitochondria [13]. Termed the Warburg Effect by Efraim Racker, this phenomenon was later shown to occur even with fully functional mitochondria [14]. It also has been observed that cancer cells utilize glutamine to support the synthesis of cellular building blocks (amino acids, ribonucleotides, and lipids) [15]. One mechanism of metabolic alteration in cancer cells is shown to occur at the epigenetic level [16]. Enzymes involved in epigenetic modulation necessitate a tightly regulated level of metabolic intermediates and cofactors, in addition to controlling genes implicated in metabolic reprogramming. In the context of cancer, these enzymes are dysregulated and promote functions conducive to tumor growth, namely, activation of oncogenes, and inactivation of tumor-suppressor genes [17]. In this review we will highlight the role of intermediate metabolites and cofactors in regulating epigenetic biochemical reactions. We will then address the significance of amino acid metabolism in mediating epigenetic changes. Furthermore, the role of environmental inputs such as nutrition in modulating the epigenome will be discussed. Finally, we will conclude with a discussion on pharmacologic intervention strategies for the reprogramming of metabolic pathways.
2. Metabolites and Cofactors mediate activity of epigenetic-associated enzymes
Chromatin restructuring is a dynamic event that regulates gene transcription. Chromatin is composed of a nucleosome core made of a histone octamer (histone 2A, 2B, 3 and 4) wrapped by DNA. Posttranslational modifications of the DNA and histone tails dictate the configuration of chromatin whether open (euchromatin) and generally conducive to gene transcription, or condensed (heterochromatin) that promotes gene repression. These covalent modifications are crucial to the accessibility of DNA to transcriptional machinery, hence determining which genes are turned “on” or “off” [18]. These modifications can be retained across generations conferring properties of epigenetics to their associated DNA. Epigenetic regulation of gene expression occurs at the level of DNA, histones, and RNA. The most well characterized are DNA methylation, histone methylation, acetylation, and phosphorylation, and microRNA-dependent gene silencing [19]. The activities of the many chromatin-modifying enzymes described herein are regulated in part by the concentrations of their required metabolic substrates or cofactors (Table 1) [16; 20].
Table 1.
Interface of metabolic pathways and epigenetic regulation.
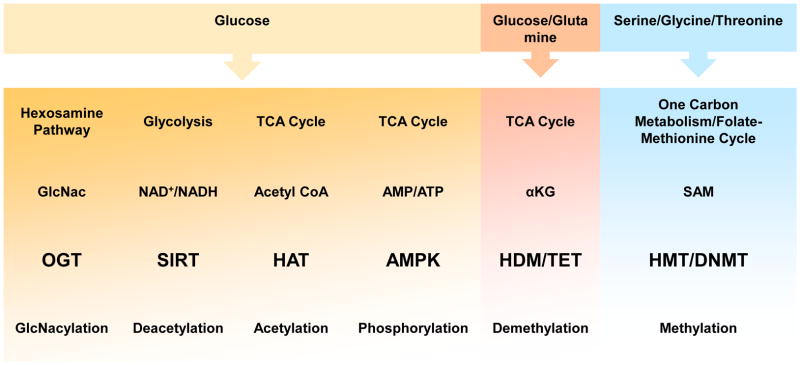
|
GlcNac, O-linked N-acetylglucosamine; OGT, O-linked N-acetylglucosamine transferase; SIRT, sirtuins; TCA cycle, tricarboxylic acid cycle; HAT, histone acetyltransferase; AMPK, 5′ adenosine monophosphate-activated protein kinase; αKG, alpha ketoglutarate; HDM, histone demethylase; SAM, S-Adenosyl methionine; HMT, histone methyltransferase; DNMT, DNA methyltransferase.
DNA methylation is mediated by DNA methyltransferase (DNMT) enzymes, which rely on the methyl donor S-Adenosyl methionine (SAM). The methyl group is transferred to the fifth position carbon of cytosine within cytosine guanine (CpG) dinucleotides. Methylation of CpG motifs in gene promoter sequences often results in gene silencing [21]. This event could be reversed through a multistep demethylation reaction mediated by ten-eleven translocation (TET) proteins [22; 23; 24]. Global DNA hypomethylation and site-specific CpG promoter hypermethylation are common epigenetic features of cancer [25].
Methylation of histones occurs at the lysine or arginine residue and is catalyzed by histone methyltransferase (HMT) enzymes [26]. Like DNMT, HMT utilizes SAM as a key methyl donor. Two family proteins have been identified to reverse histone methylation events. The first is a flavin adenine dinucleotide (FAD)-dependent oxidase known as LSD1, and the second is alpha-ketoglutarate (αKG) and ferrous ion-dependent oxygenase known as JmjC-domain containing histone demethylase (JHDM) [27; 28; 29; 30]. Histone demethylase activity is associated with context-dependent activation or repression of gene transcription [31; 32; 33].
Besides histone methylation, histone acetylation is another dynamic process that is regulated by two classes of enzymes: the histone acetyltransferases (HAT) and histone deacetylases (HDAC) [34]. HAT transfers the acetyl group of Acetyl-CoA to lysine residues of histones and is mostly associated with transcriptional activation [35; 36]. Histone deacetylation is frequently carried by two broad classes of deacetylases. The first is zinc-dependent and the second is a nicotinamide adenine dinucleotide (NAD+)-dependent family of proteins termed Sirtuins. The levels of these deacetylases have been shown to be elevated in several types of cancers and promote gene repression and silencing [37; 38].
Additional routes of epigenetic modification are gaining interest. Molecules involved in intracellular signaling pathways have been known to affect nuclear transcription through indirect mechanisms. However, it is now recognized that direct mechanisms also exist, as some kinases are capable of translocating to the nucleus to directly phosphorylate histones [39]. One such example is AMP-activated protein kinase (AMPK), a kinase that serves as a metabolic sensor of ATP/AMP ratio [40]. During metabolic stress and in response to low ATP/AMP, AMPK phosphorylates histone H2B on serine 36 that triggers the expression of genes necessary for cell survival and adaptation to metabolic changes [41]. Additionally, modifications of the O-linked N-acetylglucosamine (GlcNAc) type have been reported to occur on histone H2B at serine 112. The glycosylation reaction is catalyzed by O-GlcNAc transferase (OGT) [42; 43]. The implication of this type of epigenetic modification is not fully understood and warrants further investigation.
3. Genetic and epigenetic alteration of metabolic enzymes in cancer
Cancer initiation and progression are driven by alteration in gene expression as a result of specific activating mutations in oncogenes and prometastatic genes, or inactivating mutations in tumor suppressor genes. Compelling evidence implicates mutations in metabolic enzymes as a predisposition to tumorigenesis [44; 45; 46]. Among the metabolic enzymes reported to contribute to cancer pathogenesis we cite: NADP+-dependent isocitrate dehydrogenase (cytosolic IDH1 and mitochondrial IDH2) in gliomas [47; 48], acute myelogenous leukemia [49], and chondrosarcoma [50]; succinate dehydrogenase (SDH) in familial paragangliomas [51]; and fumarate hydratase (FH) in leiomyoma, leiomyosarcoma, and papillary renal cancers [52]. Mutational inactivation in these enzymes leads to the accumulation of 2-hydroxyglutarate, succinate, and fumarate, respectively [53]. At high concentrations, these metabolites inhibit the activity of histone and DNA demethylases and take on the role of oncometabolites [54] (Figure 1). In recent work by Killian et al., a characteristic DNA hypermethylation pattern was observed in gastrointestinal stromal tumors with mutations in SDH. Such alterations were shown to be sufficient to drive oncogenesis [55]. Similar observations of genomic hypermethylation were reported by Letouze et al. in paragangliomas and pheochromocytomas carrying mutations in SDH. This phenotype associated with aggressive clinical behavior of the disease [56].
Figure 1.

Metabolic pathways involved in epigenetic regulation through acetylation and methylation. Pyruvate (PYR), one of the end products of glycolysis, is converted to Acetyl-Co-EnzymeA (Ac-CoA) by pyruvate dehydrogenase (PDH) and serves as the acetyl donor for histone acetylation by histone acetyl transferases (HATs). Accumulated 2-hydroxyglutarate (2-HG) generated by mutated isocitrate dehydrogenase (IDH1mut, cytosol; IDH2mut, mitochondria) inhibits alpha-ketoglutarate (αKG)-dependent DNA- and histone-demethylases. Several additional mutated TCA cycle enzymes lead to accumulation of succinate (SUC), fumarate (FUM) and malate (MAL) which leads to a similar inhibition of demethylases. FAD-dependent Lysine (K)-specific demethylases (LSDs) are also involved in histone demethylation but are not subject to inhibition by 2-HG and TCA cycle intermediates. Phosphoglycerate dehydrogenase (PHGDH) diverts 3-phosphoglycerate (3PG) away from glycolysis into the tetrahydrofolate (THF) and methionine (MET) cycles. Dietary uptake of folate, cobalamine, choline and methionine also contribute to one-carbon metabolism. The methionine cycle intermediate S-adenosylmethionine (SAM) serves as the methyl donor during DNA methyl transferase (DNMT) and histone methyl transferase (HMT) reactions. Histone deacetylation is performed by either zink dependent histone deacetylases (HDACs) or NAD+ dependent Sirtuins. CIT, citrate; ICIT, isocitrate; OAA, Oxaloacetate; SER, Serine; GLY, Glycine; 5,10-MTHF, 5,10-methylene-THF; 5-MTHF, 5-methyl-THF; TET, ten-eleven translocation; JmjC-domain containing histone demethylases (JHDMs); homocysteine, hCYS; SAH, S-adenosylhomocysteine; TCA cycle, tricarboxylic acid cycle; FAD, Flavin adenine dinucleotide.
Phosphoglycerate dehydrogenase (PHGDH), a metabolic enzyme, is amplified in melanoma and breast cancer [57; 58]. Mutational amplification in PHGDH directs the metabolic flux toward the serine biosynthetic pathway, which regulates one-carbon metabolism (Figure 1). This event increases the concentration of the methyl donor methionine that can affect cellular epigenetics [59]. A current study by Ulanovskaya et al. uncovered a role for nicotinamide N-methyltransferase (NNMT) in regulating epigenetic events in cancer cells. NNMT is aberrantly expressed in various cancer types and is associated with increased cell invasive and migratory potentials. NNMT catalyzes the transfer of the methyl group from SAM to nicotinamide, a sequestration process that impairs SAM-mediated methylation of histone and DNA [60].
A number of metabolic enzymes is reported to be altered by epigenetic events rather than genetic mutations in cancer cells. Hexokinase isoform 2 (HK2) upregulation in liver cancer and glioblastoma is believed to be the result of hypomethylation of its promoter [61; 62]. Elevated levels of HK2 in cancers promote increased glycolytic flux. Along the same line, pyruvate kinase M2 (PKM2), the enzyme that carries out the final step in glycolysis, is subject to acetylation reactions that decrease its activity, favoring trafficking of glycolytic intermediates into biosynthetic processes of nucleic acids, lipids, and amino acids [63; 64]. This event appears to be required for proliferation in certain contexts. Fructose-1,6-bisphosphatase (FBP1) regulates gluconeogenesis and is silenced through promoter methylation in gastric, colon, and liver cancers [65; 66]. The inhibition of gluconeogenesis leads to higher glycolytic rates, which are advantageous to tumor cells.
4. Amino acid metabolic pathways in epigenetic regulation
One-carbon metabolism is a network of interrelated biological reactions with integral roles in DNA synthesis and methylation (Figure 1). A key metabolite in one-carbon metabolism is S-Adenosyl methionine (SAM). The ability to donate a methyl group gives SAM its role in cells as a universal methyl donor. Many enzymes that carry out nucleic acid modifications undergo a methyltransfer reaction. For example, DNA and lysine methylation enzymes that regulate DNA transcription during chromatin remodeling are methyl group acceptors. A number of metabolites feed into the one-carbon metabolism and facilitate the availability of SAM. Glucose and glutamine are the most studied factors contributing to the understanding of cancer cell metabolism [67]. Non-essential amino acids (NEAA) also play a significant role by feeding into pathways that generate the cell’s building blocks and provide substrates or cofactors for epigenetic processes [58; 68]. For example, serine and glycine are metabolized through the one-carbon metabolism pathway that has been reviewed elsewhere[69]. On the other hand, proline’s metabolic system is distinct in that it is catabolized by proline dehydrogenase (PRODH) to produce pyrroline-5-carboxylate (P5C) [68]. P5C is then sequentially converted to glutamate and αKG potentially influencing epigenetic mechanisms. Overexpression of PRODH in immunodeficient mice exhibited tumor-suppressive properties and could be negatively regulated by microRNA-23b [66]. Such crosstalk emphasizes the complexity of cancer cell metabolism. Further studies on NEAA metabolism and their regulation of epigenetic events in cancer will provide a new avenue for the development of anti-cancer therapeutics.
5. Bioactive food compounds targeting the epigenome
Metabolism is a regulatory interface shaped by the environment. Food exposure plays a direct role in resetting genes for wellness or illness by targeting different elements of the epigenetic machinery [70]. A variety of natural compounds from different sources have been shown to directly regulate metabolism related to epigenetics [71]. Of the micronutrients with direct effects on one-carbon metabolism, folate, cobalamine, choline, flavonoids, methionine, and betaine are metabolized in pathways that mediate epigenetic events [72]. Bioactive food compounds such as polyphenols found in plants are claimed to regulate DNMT expression and activity, increase HAT activity, and activate sirtuins to elicit anti-oxidative, anti-tumorigenic, and anti-metastatic properties [72; 73]. Fatty acids are also emerging as important regulators of post-translational modifications and have been implicated in cancer progression [74; 75]. We refer the reader to a detailed review by Ong et al. on the role of dietary compounds on epigenetic regulation in cancer[76]. There is increased interest in harnessing the benefits of these compounds to affect the epigenetic reprogramming of cancer cell metabolism. However, the field faces a technical challenge in determining metabolite bioavailability and dose-dependent effects in humans. It is not clear whether any of these factors affect metabolism at orally available doses. It is important to note concentrations used for studies in vitro do not recapitulate the levels of metabolites provided through diet. Primary factors that should be considered in future investigations include the effective doses and dose timing of bioactive food compounds to attain epigenetic effects [77; 78].
6. Epigenetic therapeutic targets for the treatment of cancer
Epigenetics and metabolism are highly interconnected in a reciprocal fashion. The importance of these relationships are accentuated by the reversibility of both processes[79]. This feature has attracted a significant amount of attention in the prevention and treatment of many illnesses including cancer. As epigenetic abnormalities have been shown to be both causative and contributing factors in cancer, chemical agents and natural compounds that are direct or indirect regulators of the epigenome constitute an excellent approach in cancer prevention and potentially in anti-cancer therapy [80; 81; 82; 83] (summarized in Table 2).
Table 2.
Pharmacological agents and natural compounds for epigenetic cancer therapy
| Target pathway | Pharmacological agents and Natural compounds | Metabolic/Epigenetic Mechanism of action | Stage of therapy development |
|---|---|---|---|
| Glucose metabolism | 2-deoxyglucose Mannoheptulose |
Reduce glycolysis | Clinical and preclinical data84, 85 Preclinical data86 |
|
| |||
| Glutamine/amino acid metabolism | L-asparagine | Depletion of asparagine and glutamine | Approved |
|
| |||
| Serine/Glycine metabolism | shRNA to PHGDH | Inhibitor of de novo serine synthesis | Preclinical data57,58 |
|
| |||
| Lipid metabolism | Betulin Hydroxycitrate Statins |
Inhibitor of SREBP Inhibitor of ATP Citrate lyase Inhibitor of HMG-CoA reductase |
Preclinical data87 Preclinical data88 Approved |
|
| |||
| Nucleotide metabolism | 5-fluorouracil Methotrexate |
Impair DNA synthesis | Approved Approved |
|
| |||
| Epigenetic modifications | Folate, choline, methionine, betaine, selected B vitamins, flavonoids, EGCG, genistein | Inhibitors of DNA methyltransferases | Lab studies |
| Butyrate, sulforaphane, allylmercaptan, 3,3-diindolylmethane | Inhibitors of Histone deacetylases | Lab studies | |
| Anacardic acid, garcinol, curcumin, EGCG, Genistein | Inhibitors of Histone acetyltransferases | Lab studies | |
| Butyrate, cambinol, dihydrocoumarin, genistein | Inhibitors of acetylation of non-histone proteins | Lab studies | |
PHGDH, phosphoglycerate dehydrogenase; SREBP, sterol regulatory element-binding protein; EGCG, epigallocatechin-3-gallate; HMG-CoA, 3-hydroxy-3-methylglutaryl coenzyme A.
With every potential therapy comes a challenge. Targeting cancer cell metabolism aims to reestablish normal cell metabolism, correct signaling cascades, and reverse epigenetic reactions. As our understanding of cancer cell metabolism expands, more combination therapy targeting different branches of metabolic pathways should be considered. In addition to targeting metabolic pathways that support biosynthesis [15], a new direction is to modulate metabolic pathways involved in epigenetic reprogramming that contributes to tumor progression. It is important to note that metabolic events occur in multiple cellular compartments, thus learning the transport of metabolites between cytoplasm, mitochondria and nucleus may be important for developing effective targeting. Of importance is also the artificial culturing conditions used experimentally to study cancer cell biology. These growth media are complex in composition and not fully defined. Changes in media composition are predicted to have dramatic effects on metabolism. It is therefore imperative to carefully interpret data pertaining to quantitative metabolic flux prior to extrapolating to human disease. Nevertheless it is anticipated that with rapid technological advances, refinement of current protocols, and the surge of interest in the field, the intervention of cancer cell metabolism may contribute to a breakthrough in the prevention and treatment of cancer in the near future.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Van Speybroeck L. From Epigenesis to Epigenetics. Annals of the New York Academy of Sciences. 2002;981:61–81. [PubMed] [Google Scholar]
- 2.Barker DJP, Osmond C. Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales. The Lancet. 1986;327:1077–1081. doi: 10.1016/s0140-6736(86)91340-1. [DOI] [PubMed] [Google Scholar]
- 3.Pembrey ME, Bygren LO, Kaati G, Edvinsson S, Northstone K, Sjostrom M, Golding J. Sex-specific, male-line transgenerational responses in humans. Eur J Hum Genet. 2005;14:159–166. doi: 10.1038/sj.ejhg.5201538. [DOI] [PubMed] [Google Scholar]
- 4.Kaati G, Bygren LO, Pembrey M, Sjostrom M. Transgenerational response to nutrition, early life circumstances and longevity. Eur J Hum Genet. 2007;15:784–790. doi: 10.1038/sj.ejhg.5201832. [DOI] [PubMed] [Google Scholar]
- 5.Morgan HD, Santos F, Green K, Dean W, Reik W. Epigenetic reprogramming in mammals. Human Molecular Genetics. 2005;14:R47–R58. doi: 10.1093/hmg/ddi114. [DOI] [PubMed] [Google Scholar]
- 6.Esteller M. Epigenetics in Cancer. New England Journal of Medicine. 2008;358:1148–1159. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- 7.Lillycrop KA, Burdge GC. Epigenetic mechanisms linking early nutrition to long term health. Best Practice & Research Clinical Endocrinology & Metabolism. 2012;26:667–676. doi: 10.1016/j.beem.2012.03.009. [DOI] [PubMed] [Google Scholar]
- 8.Marjanovic ND, Weinberg RA, Chaffer CL. Cell Plasticity and Heterogeneity in Cancer. Clinical Chemistry. 2013;59:168–179. doi: 10.1373/clinchem.2012.184655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Esteva FJ, Yu D, Hung M-C, Hortobagyi GN. Molecular predictors of response to trastuzumab and lapatinib in breast cancer. Nat Rev Clin Oncol. 2010;7:98–107. doi: 10.1038/nrclinonc.2009.216. [DOI] [PubMed] [Google Scholar]
- 10.Collecchi P, Baldini E, Giannessi P, Naccarato AG, Passoni A, Gardin G, Roncella M, Evangelista G, Bevilacqua G, Conte PF. Primary chemotherapy in locally advanced breast cancer (LABC): effects on tumour proliferative activity, bcl-2 expression and the relationship between tumour regression and biological markers. European Journal of Cancer. 1998;34:1701–1704. doi: 10.1016/s0959-8049(98)00213-5. [DOI] [PubMed] [Google Scholar]
- 11.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 13.Warburg O. On the origin of cancer cells. Science. 1956 doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 14.Racker E. Bioenergetics and the problem of tumor growth. Am Sci. 1972;60:56–63. [PubMed] [Google Scholar]
- 15.Dang C. MYC, miRNA, and glutamine addiction in cancers. Cell Cycle. 2009:3243–3245. doi: 10.4161/cc.8.20.9522. [DOI] [PubMed] [Google Scholar]
- 16.Yun J, Johnson J, Hanigan C, Locasale J. Interactions between epigenetics and metabolism. Frontiers in Oncology. 2012;2 doi: 10.3389/fonc.2012.00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Esteller M. Epigenetics provide a new generation of oncogenes and tumor-suppressor genes. British Journal of Cancer. 2006:179–183. doi: 10.1038/sj.bjc.6602918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsukiyama T, Wu C. Chromatin remodeling and transcription. Current Opinion in Genetics & Development. 1997;7:182–191. doi: 10.1016/s0959-437x(97)80127-x. [DOI] [PubMed] [Google Scholar]
- 19.Margueron R, Reinberg D. Chromatin structure and the inheritance of epigenetic information. Nat Rev Genet. 2010;11:285–296. doi: 10.1038/nrg2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Katada S, Imhof A, Sassone-Corsi P. Connecting Threads: Epigenetics and Metabolism. Cell. 2012;148:24–28. doi: 10.1016/j.cell.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 21.Ulrey CL, Liu L, Andrews LG, Tollefsbol TO. The impact of metabolism on DNA methylation. Human Molecular Genetics. 2005;14:R139–R147. doi: 10.1093/hmg/ddi100. [DOI] [PubMed] [Google Scholar]
- 22.Bhutani N, Burns DM, Blau HM. DNA Demethylation Dynamics. Cell. 2011;146:866–872. doi: 10.1016/j.cell.2011.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.He Y-F, Li B-Z, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, Sun Y, Li X, Dai Q, Song C-X, Zhang K, He C, Xu G-L. Tet-Mediated Formation of 5-Carboxylcytosine and Its Excision by TDG in Mammalian DNA. Science. 2011;333:1303–1307. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y. Tet Proteins Can Convert 5-Methylcytosine to 5-Formylcytosine and 5-Carboxylcytosine. Science. 2011;333:1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sandoval J, Esteller M. Cancer epigenomics: beyond genomics. Current Opinion in Genetics & Development. 2012;22:50–55. doi: 10.1016/j.gde.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 26.Varier RA, Timmers HTM. Histone lysine methylation and demethylation pathways in cancer. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer. 2011;1815:75–89. doi: 10.1016/j.bbcan.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 27.Teperino R, Schoonjans K, Auwerx J. Histone methyl-transferases and demethylases; can they link metabolism and transcription ? Cell Metabolism. 2010;12:321–327. doi: 10.1016/j.cmet.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shi Y-J, Matson C, Lan F, Iwase S, Baba T, Shi Y. Regulation of LSD1 Histone Demethylase Activity by Its Associated Factors. Molecular Cell. 2005;19:857–864. doi: 10.1016/j.molcel.2005.08.027. [DOI] [PubMed] [Google Scholar]
- 29.Klose RJ, Kallin EM, Zhang Y. JmjC-domain-containing proteins and histone demethylation. Nat Rev Genet. 2006;7:715–727. doi: 10.1038/nrg1945. [DOI] [PubMed] [Google Scholar]
- 30.Tsukada Y-i, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, Zhang Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006;439:811–816. doi: 10.1038/nature04433. [DOI] [PubMed] [Google Scholar]
- 31.Vakoc CR, Mandat SA, Olenchock BA, Blobel GA. Histone H3 Lysine 9 Methylation and HP1gamma Are Associated with Transcription Elongation through Mammalian Chromatin. Molecular Cell. 2005;19:381–391. doi: 10.1016/j.molcel.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 32.Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 33.Bernstein BE, Meissner A, Lander ES. The Mammalian Epigenome. Cell. 2007;128:669–681. doi: 10.1016/j.cell.2007.01.033. [DOI] [PubMed] [Google Scholar]
- 34.Shahbazian MD, Grunstein M. Functions of Site-Specific Histone Acetylation and Deacetylation. Annual Review of Biochemistry. 2007;76:75–100. doi: 10.1146/annurev.biochem.76.052705.162114. [DOI] [PubMed] [Google Scholar]
- 35.Racey L, Byvoet P. Histone acetyltransferase in chromatin. Evidence for in vitro enzymatic transfer of acetate from acetyl-coenzyme A to histones. Exp. Cell Res. 1971:366–370. doi: 10.1016/0014-4827(71)90089-9. [DOI] [PubMed] [Google Scholar]
- 36.Cai L, Sutter BM, Li B, TBP Acetyl-CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Molecular Cell. 2011;42:426–437. doi: 10.1016/j.molcel.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Imai S-i, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 38.Finkel T, Deng C-X, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009;460:587–591. doi: 10.1038/nature08197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baek SH. When Signaling Kinases Meet Histones and Histone Modifiers in the Nucleus. Molecular Cell. 2011;42:274–284. doi: 10.1016/j.molcel.2011.03.022. [DOI] [PubMed] [Google Scholar]
- 40.Hardie DG. Adenosine Monophosphate-Activated Protein Kinase: A Central Regulator of Metabolism with Roles in Diabetes, Cancer, and Viral Infection. Cold Spring Harbor Symposia on Quantitative Biology. 2011;76:155–164. doi: 10.1101/sqb.2011.76.010819. [DOI] [PubMed] [Google Scholar]
- 41.Bungard D, Fuerth BJ, Zeng P-Y, Faubert B, Maas NL, Viollet B, Carling D, Thompson CB, Jones RG, Berger SL. Signaling Kinase AMPK Activates Stress-Promoted Transcription via Histone H2B Phosphorylation. Science. 2010;329:1201–1205. doi: 10.1126/science.1191241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fujiki R, Hashiba W, Sekine H, Yokoyama A, Chikanishi T, Ito S, Imai Y, Kim J, He HH, Igarashi K, Kanno J, Ohtake F, Kitagawa H, Roeder RG, Brown M, Kato S. GlcNAcylation of histone H2B facilitates its monoubiquitination. Nature. 2011;480:557–560. doi: 10.1038/nature10656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wellen KE, Lu C, Mancuso A, Lemons JMS, Ryczko M, Dennis JW, Rabinowitz JD, Coller HA, Thompson CB. The hexosamine biosynthetic pathway couples growth factor-induced glutamine uptake to glucose metabolism. Genes & Development. 2010;24:2784–2799. doi: 10.1101/gad.1985910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Soga T. Cancer metabolism: Key players in metabolic reprogramming. Cancer Science. 2013;104:275–281. doi: 10.1111/cas.12085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Losman J-A, Kaelin WG. What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes & Development. 2013;27:836–852. doi: 10.1101/gad.217406.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kaelin WG, Jr, McKnight SL. Influence of Metabolism on Epigenetics and Disease. Cell. 2013;153:56–69. doi: 10.1016/j.cell.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, Deimling A. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol. 2008;116:597–602. doi: 10.1007/s00401-008-0455-2. [DOI] [PubMed] [Google Scholar]
- 48.Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, Friedman H, Friedman A, Reardon D, Herndon J, Kinzler KW, Velculescu VE, Vogelstein B, Bigner DD. IDH1 and IDH2 Mutations in Gliomas. New England Journal of Medicine. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rakheja D, Konoplev S, Medeiros LJ, Chen W. IDH mutations in acute myeloid leukemia. Human Pathology. 2012;43:1541–1551. doi: 10.1016/j.humpath.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 50.Kerr D, Lopez H, Deshpande V, Hornicek F, Duan Z, Zhang Y, Rosenberg A, Borger D, Nielsen G. Molecular Distinction of Chondrosarcoma From Chondroblastic Osteosarcoma Through IDH1/2 Mutations. Am J Surg Pathol. 2013;37:787–795. doi: 10.1097/PAS.0b013e31827ab703. [DOI] [PubMed] [Google Scholar]
- 51.Vandy FC, Sisk G, Berguer R. Synchronous carotid body and thoracic paraganglioma associated with a germline SDHC mutation. Journal of Vascular Surgery. 2011;53:805–807. doi: 10.1016/j.jvs.2010.09.064. [DOI] [PubMed] [Google Scholar]
- 52.Adam J, Yang M, Bauerschmidt C, Kitagawa M, O’Flaherty L, Maheswaran P, Ozkan G, Sahgal N, Baban D, Kato K, Saito K, Iino K, Igarashi K, Stratford M, Pugh C, Tennant DA, Ludwig C, Davies B, Ratcliffe PJ, El-Bahrawy M, Ashrafian H, Soga T, Pollard PJ. A Role for Cytosolic Fumarate Hydratase in Urea Cycle Metabolism and Renal Neoplasia. Cell Reports. 2013;3:1440–1448. doi: 10.1016/j.celrep.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, Marks KM, Prins RM, Ward PS, Yen KE, Liau LM, Rabinowitz JD, Cantley LC, Thompson CB, Vander Heiden MG, Su SM. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2010;465:966–966. doi: 10.1038/nature09132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.King A, Selak MA, Gottlieb E. Succinate dehydrogenase and fumarate hydratase: linking mitochondrial dysfunction and cancer. Oncogene. 2006;25:4675–4682. doi: 10.1038/sj.onc.1209594. [DOI] [PubMed] [Google Scholar]
- 55.Killian JK, Kim SY, Miettinen M, Smith C, Merino M, Tsokos M, Quezado M, Smith WI, Jahromi MS, Xekouki P, Szarek E, Walker RL, Lasota J, Raffeld M, Klotzle B, Wang Z, Jones L, Zhu Y, Wang Y, Waterfall JJ, O’Sullivan MJ, Bibikova M, Pacak K, Stratakis C, Janeway KA, Schiffman JD, Fan J-B, Helman L, Meltzer PS. Succinate Dehydrogenase Mutation Underlies Global Epigenomic Divergence in Gastrointestinal Stromal Tumor. Cancer Discovery. 2013;3:648–657. doi: 10.1158/2159-8290.CD-13-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Letouze E, Martinelli C, Loriot C, Burnichon N, Abermil N, Ottolenghi C, Janin M, Menara M, Nguyen AT, Benit P, Buffet A, Marcaillou C, Bertherat J, Amar L, Rustin P, De Reynies A, Gimenez-Roqueplo A-P, Favier J. SDH Mutations Establish a Hypermethylator Phenotype in Paraganglioma. Cancer Cell. 2013;23:739–752. doi: 10.1016/j.ccr.2013.04.018. [DOI] [PubMed] [Google Scholar]
- 57.Locasale JW, Grassian AR, Melman T, Lyssiotis CA, Mattaini KR, Bass AJ, Heffron G, Metallo CM, Muranen T, Sharfi H, Sasaki AT, Anastasiou D, Mullarky E, Vokes NI, Sasaki M, Beroukhim R, Stephanopoulos G, Ligon AH, Meyerson M, Richardson AL, Chin L, Wagner G, Asara JM, Brugge JS, Cantley LC, Vander Heiden MG. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet. 2011;43:869–874. doi: 10.1038/ng.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K, Sethumadhavan S, Woo H-K, Jang HG, Jha AK, Chen WW, Barrett FG, Stransky N, Tsun Z-Y, Cowley GS, Barretina J, Kalaany NY, Hsu PP, Ottina K, Chan AM, Yuan B, Garraway LA, Root DE, Mino-Kenudson M, Brachtel EF, Driggers EM, Sabatini DM. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature. 2011;476:346–350. doi: 10.1038/nature10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lu C, Thompson CB. Metabolic Regulation of Epigenetics. Cell Metabolism. 2012;16:9–17. doi: 10.1016/j.cmet.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ulanovskaya OA, Zuhl AM, Cravatt BF. NNMT promotes epigenetic remodeling in cancer by creating a metabolic methylation sink. Nat Chem Biol. 2013;9:300–306. doi: 10.1038/nchembio.1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wolf A, Agnihotri S, Munoz D, Guha A. Developmental profile and regulation of the glycolytic enzyme hexokinase 2 in normal brain and glioblastoma multiforme. Neurobiology of Disease. 2011;44:84–91. doi: 10.1016/j.nbd.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 62.Goel A, Mathupala SP, Pedersen PL. Glucose Metabolism in Cancer: Evidence that demethylation events play a role in activating type II hexokinase gene expression. Journal of Biological Chemistry. 2003;278:15333–15340. doi: 10.1074/jbc.M300608200. [DOI] [PubMed] [Google Scholar]
- 63.Mazurek S. Pyruvate kinase type M2: A key regulator of the metabolic budget system in tumor cells. The International Journal of Biochemistry & Cell Biology. 2011;43:969–980. doi: 10.1016/j.biocel.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 64.Lv L, Li D, Zhao D, Lin R, Chu Y, Zhang H, Zha Z, Liu Y, Li Z, Xu Y, Wang G, Huang Y, Xiong Y, Guan K-L, Lei Q-Y. Acetylation Targets the M2 Isoform of Pyruvate Kinase for Degradation through Chaperone-Mediated Autophagy and Promotes Tumor Growth. Molecular Cell. 2011;42:719–730. doi: 10.1016/j.molcel.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen M, Zhang J, Li N, Qian Z, Zhu M, Li Q, Zheng J, Wang X, SG Promoter hypermethylation mediated downregulation of FBP1 in human hepatocellular carcinoma and colon cancer. PLoS One. 2011;6:e25564. doi: 10.1371/journal.pone.0025564. doi:25510.21371/journal.pone.0025564 Epub 0022011 Oct 0025519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu W, Zabirnyk O, Wang H, Shiao YH, Nickerson ML, Khalil S, Anderson LM, Perantoni AO, Phang JM. miR-23b targets proline oxidase, a novel tumor suppressor protein in renal cancer. Oncogene. 2010;29:4914–4924. doi: 10.1038/onc.2010.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Locasale JW. The consequences of enhanced cell-autonomous glucose metabolism. Trends in Endocrinology & Metabolism. 2012;23:545–551. doi: 10.1016/j.tem.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 68.Phang JM, Liu W, Hancock C. Bridging epigenetics and metabolism: Role of non-essential amino acids. Epigenetics. 2013;8:231–236. doi: 10.4161/epi.24042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Locasale JW. Serine, Glycine, and one-carbon units: cancer metabolism full circle. Nature Reviews Cancer. 2013 doi: 10.1038/nrc3557. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Landecker H. Food as exposure: Nutritional epigenetics and the new metabolism. BioSocieties. 2011;6:167–194. doi: 10.1057/biosoc.2011.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gerhauser C. Cancer cell metabolism, epigenetics and the potential influence of dietary components. A perspective. Biomedical Research. 2012;23 [Google Scholar]
- 72.Stefanska B, Karlic H, Varga F, Fabianowska-Majewska K, Haslberger AG. Epigenetic mechanisms in anti-cancer actions of bioactive food components – the implications in cancer prevention. British Journal of Pharmacology. 2012;167:279–297. doi: 10.1111/j.1476-5381.2012.02002.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yang CS, Wang X, Lu G, Picinich SC. Cancer prevention by tea: animal studies, molecular mechanisms and human relevance. Nat Rev Cancer. 2009;9:429–439. doi: 10.1038/nrc2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Du J, Zhou Y, Su X, Yu JJ, Khan S, Jiang H, Kim J, Woo J, Kim JH, Choi BH, He B, Chen W, Zhang S, Cerione RA, Auwerx J, Hao Q, Lin H. Sirt5 Is a NAD-Dependent Protein Lysine Demalonylase and Desuccinylase. Science. 2011;334:806–809. doi: 10.1126/science.1207861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E, Buchou T, Cheng Z, Rousseaux S, Rajagopal N, Lu Z, Ye Z, Zhu Q, Wysocka J, Ye Y, Khochbin S, Ren B, Zhao Y. Identification of 67 Histone Marks and Histone Lysine Crotonylation as a New Type of Histone Modification. Cell. 2011;146:1016–1028. doi: 10.1016/j.cell.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ong TP, Moreno FS, Ross SA. Targeting the Epigenome with Bioactive Food Components for Cancer Prevention. Journal of Nutrigenetics and Nutrigenomics. 2011;4:275–292. doi: 10.1159/000334585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ross JA, Kasum CM. DIETARY FLAVONOIDS: Bioavailability, Metabolic Effects, and Safety. Annual Review of Nutrition. 2002;22:19–34. doi: 10.1146/annurev.nutr.22.111401.144957. [DOI] [PubMed] [Google Scholar]
- 78.Crozier A, Jaganath IB, Clifford MN. Dietary phenolics: chemistry, bioavailability and effects on health. Natural Product Reports. 2009;26:1001–1043. doi: 10.1039/b802662a. [DOI] [PubMed] [Google Scholar]
- 79.Henikoff S, Matzke MA. Exploring and explaining epigenetic effects. Trends in Genetics. 1997;13:293–295. doi: 10.1016/s0168-9525(97)01219-5. [DOI] [PubMed] [Google Scholar]
- 80.Gerhauser C. Cancer Chemoprevention and Nutri-Epigenetics: State of the Art and Future Challenges. In: Pezzuto JM, Suh N, editors. Natural Products in Cancer Prevention and Therapy. Springer; Berlin Heidelberg: 2013. pp. 73–132. [Google Scholar]
- 81.Teicher BA, Linehan WM, Helman LJ. Targeting Cancer Metabolism. Clinical Cancer Research. 2012;18:5537–5545. doi: 10.1158/1078-0432.CCR-12-2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dawson MA, Kouzarides T. Cancer Epigenetics: From Mechanism to Therapy. Cell. 2012;150:12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 83.Robles-Fernández I, Rodríguez-Serrano F, Alvarez P, Ortiz R, Rama A, Prados J, Melguizo C, Alvarez-Manzaneda E, Aránega A. Antitumor Properties of Natural Compounds and Related Molecules. Recent Pat Anticancer Drug Discov. 2012;16:16. doi: 10.2174/1574891x113089990034. [DOI] [PubMed] [Google Scholar]
- 84.Mohanti BK, Rath GK, Anantha N, Kannan V, Das BS, Chandramouli BAR, Banerjee AK, Das S, Jena A, Ravichandran R, Sahi UP, Kumar R, Kapoor N, Kalia VK, Dwarakanath BS, Jain V. Improving cancer radiotherapy with 2-deoxy-d-glucose: phase I/II clinical trials on human cerebral gliomas. International Journal of Radiation Oncology*Biology*Physics. 1996;35:103–111. doi: 10.1016/s0360-3016(96)85017-6. [DOI] [PubMed] [Google Scholar]
- 85.Mathupala SP, Ko YH, Pedersen PL. Hexokinase-2 bound to mitochondria: Cancer’s stygian link to the Warburg effect and a pivotal target for effective therapy. Seminars in Cancer Biology. 2009;19:17–24. doi: 10.1016/j.semcancer.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rasschaert J, Kadiata MM, Malaisse WJ. Effects of D-mannoheptulose upon D-glucose metabolism in tumoral pancreatic islet cells. Mol Cell Biochem. 2001;226:77–81. doi: 10.1023/a:1012737803088. [DOI] [PubMed] [Google Scholar]
- 87.Alakurtti S, Makela T, Koskimies S, Yli-Kauhaluoma J. Pharmacological properties of the ubiquitous natural product betulin. European Journal of Pharmaceutical Sciences. 2006;29:1–13. doi: 10.1016/j.ejps.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 88.Hanai Ji, Doro N, Seth P, Sukhatme VP. ATP citrate lyase knockdown impacts cancer stem cells in vitro. Cell Death Dis. 2013;4:e696. doi: 10.1038/cddis.2013.215. [DOI] [PMC free article] [PubMed] [Google Scholar]