

Abstract
Germline mutations in DNA mismatch repair (MMR) genes, such as MSH2, cause Lynch syndrome, an autosomal dominant predisposition to colorectal as well as other cancers. Our research clinic focuses on hereditary colorectal cancer, and over the past 9 years we have identified germline mutations in DNA MMR genes in 101 patients using commercial genetic reference laboratories. We also collected samples from twelve patients with absent MSH2 protein expression and microsatellite instability in tumor tissue, with a family history suggestive of Lynch syndrome, but negative germline test results. The most likely explanation for this set of results is that the germline testing did not detect true germline mutations in these patients. Two of our patients with failed commercial testing were later found to have deletions in the 3′ region of EPCAM, the gene just upstream of MSH2, but no explanation could be found for inactivation of MSH2 in the other ten patients. We used allelic dropout in long PCR to look for potential regions of rearrangement in the MSH2 gene. This method detected a potential rearrangement breakpoint in the same region of MSH2 where one breakpoint of a 10 Mb inversion was reported previously. We tested these ten patients for this inversion. Six of 10 patients had the inversion, indicating the importance of including testing for this inversion in patients suspected of having MSH2-type Lynch syndrome in our population. Additionally, this method could be further developed to look for inversions in other genes where current methods of testing fail to find a causative mutation.
Keywords: Lynch syndrome, Inversion, MSH2, Colon cancer
Introduction
Lynch syndrome is the familial cancer syndrome caused by a germline mutation in one of the DNA mismatch repair (MMR) genes [1]. Cancers in this syndrome characteristically have microsatellite instability (MSI) and abnormal immunohistochemistry (IHC), in which the MMR gene with the germline mutation is not expressed in the tumor [2, 3]. MSH2 is one of the most important genes causing Lynch syndrome, but traditional exon-by-exon sequencing will not always detect the germline mutation. MSH2 incurs frequent Alu-mediated large deletions, which cannot be detected by exon sequencing [4]. Additionally, silencing of MSH2 can occur due to deletion of the polyadenylation signal of the EPCAM gene located 5′ to MSH2. Deletions of the 3′ end of the EPCAM gene abolish transcription termination, which results in transcription read-through into the MSH2 gene, and subsequent methylation-induced silencing of the MSH2 gene in tissues that express EPCAM [5]. The presence of MSI with the absence of MSH2 expression in a colorectal cancer (CRC) is highly suggestive of Lynch syndrome-MSH2 type, but in some instances, no germline mutation can be found in the MSH2 gene, even when testing for large deletions in MSH2 or EPCAM.
Our referral clinic focuses on hereditary CRC, and over the past 9 years we have identified germline mutations in DNA MMR genes in 101 patients using commercial genetic reference laboratories. We have also collected samples from twelve patients who had tumors with absent MSH2 protein expression, MSI, a family history suggestive of Lynch syndrome, but negative germline test results. The most likely explanation for this combination is that the germline testing was unable to detect true germline mutations in these patients. Even the best commercial testing methods do not detect mutations deep within introns or copy number-neutral inversions whose breakpoints are located in noncoding regions of the gene. Two of our patients with failed commercial testing were later found to have deletions in the 3′ region of EPCAM, but no explanation could be found for inactivation of MSH2 in the other ten [5].
One important type of mutation not examined by current testing methods is the presence of large inversions which result in rearrangement of the order of the exons of the gene. We sought to identify locations of potential inversion breakpoints in MSH2 by looking for allelic drop-out of single nucleotide polymorphisms (SNPs) in a series of long overlapping (~10 kb) PCR products. Starting with one patient with suspected MSH2-type Lynch syndrome, we found a long PCR product which was completely homozygous at all SNPs sequenced. This product encompassed exon 8 and part of intron 7 of MSH2. Upon review of the literature, we found two publications which had previously described a 10 Mb inversion in the MSH2 gene, in which the 3′ breakpoint was located in the same region as our long PCR amplicon [6, 7]. Thus, the objective of our study was to determine if this inversion of the MSH2 gene previously described was present in a group of patients with suspected MSH2-type Lynch syndrome of unknown origin.
Methods
Patients
Patients were selected from our familial CRC registry based upon suspicion of having MSH2-type Lynch syndrome. Patients previously had tested negative for MSH2 germline mutations by commercial genetic testing services. All patients had exhibited loss of MSH2 expression by IHC. Germline testing, IHC, and MSI results listed in the table were provided by CLIA certified labs. Family members of patients who tested positive for the inversion were subsequently enrolled in our study and tested for the inversion when possible. All patients provided written informed consent, and the study was approved by the Baylor Research Institute institutional Review Board.
Control Patients
Five control patients with no known family history of CRC were tested using the primers designed by Wagner et al. [6] for the 5′ inversion breakpoint. 22 controls with no known history of familial CRC were tested using the primers designed to amplify across the 3′ breakpoint.
Inversion PCR
Patients and controls were tested for the 5′ inversion breakpoint using primers F3 and R3 published by Wagner et al. [6]. Primers F4MV and B3MV were designed to amplify across the 3′ inversion breakpoint using MacVector (Cary, NC, USA). The forward primer sequence was 5′-GGGAGGGGAAAATGACTTACAAAG-3′. The reverse primer sequence was 5′-GCAAAAGGAACAGTCAGCAG AAGG-3′. PCR was performed using HotStar Taq (Qiagen, Valencia, CA, USA). Both inversion primer pairs only amplify a product in carriers of the inversion. Inversion PCR products were sequenced on an ABI 3100-Avant sequencer (Applied Biosystems, Foster City, CA, USA). An additional 1.6 kb PCR that amplifies exons 12–13 of MSH2 was included on all patient samples to exclude the possibility of false negative results due to poor DNA integrity (Fig. 1).
Fig. 1.
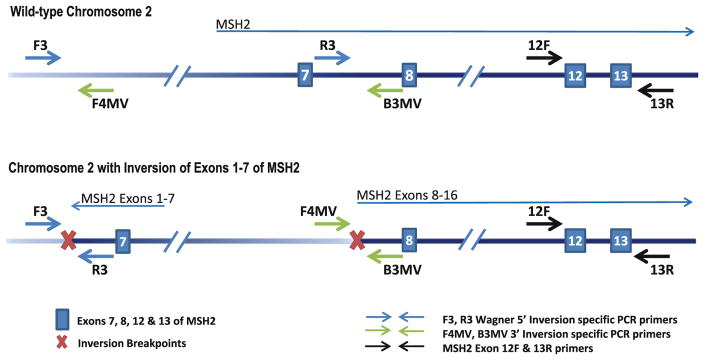
Inversion-specific PCR. Representation of PCR assays used to detect the MSH2 inversion. Primers F3 and R3 were described by Wagner et al. [6] and are used to amplify the 5′ inversion breakpoint. Primers F4MV and B3MV were designed in our lab to amplify the 3′ inversion breakpoint. Amplification is achieved in carriers of the inversion due to re-orientation of the primer directionality. No amplification occurs in patients with wild-type MSH2. Amplification of exons 12–13 of MSH2 using 12F and 13R was used to verify DNA integrity
SNP genotyping
Patients were genotyped at multiple SNPs in MSH2 by PCR and DNA sequencing and/or denaturing high performance liquid chromatography (dHPLC). Primers and dHPLC conditions are available upon request.
Allelic drop out PCR
Two PCRs were designed to look for allelic drop out in a long PCR product from inversion carriers. A short PCR product was designed to amplify and genotype SNP rs7607076, which is located in MSH2 intron 7 downstream of the 3′ inversion breakpoint. A second set of primers anneal to each side of the 3′ inversion breakpoint and only amplify the wild type allele. This results in allelic drop out in the long PCR product in carriers of the inversion who are heterozygous at SNP rs7607076. PCR products from both the short and long PCR products were sequenced and genotyped at rs7607076 (Fig. 2).
Fig. 2.
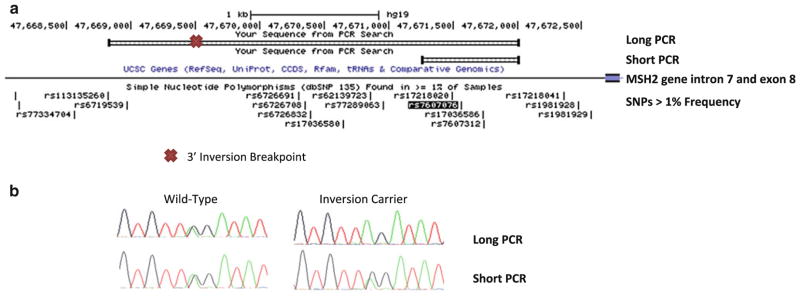
Design of PCR analyses used to detect allelic drop out in carriers of the MSH2 inversion. a Long and short PCR amplicons and their positions on chromosome 2 relative to the 3′ inversion breakpoint, MSH2 intron 8, and SNP rs7607076 are depicted. b Sequencing results of rs7607076 in the long and short amplicons for patients tested for the MSH-2 inversion by use of inversion-specific PCR. Only one allele is amplified in the long PCR of inversion carriers which results in the SNP appearing to be homozygous
Results and discussion
Starting with one patient with suspected MSH2-type Lynch syndrome, we found a long PCR product which was completely homozygous at all SNPs sequenced. Sequencing of short PCR products containing multiple SNPs confirmed allelic drop out in the long PCR product. This product encompassed exon 8 and part of intron 7 of MSH2. Upon review of the literature, we found a previously described 10 Mb inversion in the MSH2 gene, in which the 3′ breakpoint was located in the same region as our long PCR amplicon. PCR and sequencing using inversion-specific primers described by Wagner revealed that this patient carried the inversion previously reported [6].
We next performed PCR of the 5′ inversion breakpoint on our remaining suspected MSH2-type Lynch syndrome patients, and six patients were positive for the inversion (Table 1; Fig. 3). We designed additional primers to amplify the 3′ inversion breakpoint using the sequence data provided by Chen [7]. PCR products from both the 5′ and 3′ breakpoints were sequenced for confirmation (Fig. 1). No amplification of the 3′ inversion PCR product was seen in 22 controls. A 1.6 kb PCR amplification was included in all samples to exclude the possibility of false negative results due to problems with DNA integrity.
Table 1.
Clinical characteristics of unexplained MSH2-type Lynch syndrome patients
| Diagnosis | Age at diagnosis | MSH2/MSH6 IHC | MSI testing | Germline testing | Amsterdam I criteria | Amsterdam II criteria | Bethesda criteria | PREMM 1,2,6 | Inversion (%) | |
|---|---|---|---|---|---|---|---|---|---|---|
| 213 | Endometrial adenocarcinoma | 55 | Absent | ND | MSH2-VUS L800P | Negative | Positive | Positive | 27 | No |
| 314 | Synchronous endometrial & ovarian adenocarcinoma | 46 | Absent | MSI-H | NMD | Negative | Positive | Negative | 38 | Yes |
| 328 | Endometrial adenocarcinoma | 44 | Absent | MSI-H | NMD | Negative | Negative | Negative | <5 | No |
| 370 | Endometrial adenocarcinoma Mucinous cecal adenocarcinoma |
41 47 |
Absent | MSI-H | NMD | Positive | Positive | Positive | 36.90 | Yes |
| 372 | Muir-Torre syndrome Stage I cecal poorly differentiated adenocarcinoma |
41 47 |
Absent | MSI-H | NMD | Positive | Positive | Positive | 65.80 | Yes |
| 381 | Colon adenocarcinoma Endometrial adenocarcinoma Squamous cell kidney cancer |
41 47 48 |
Absent | MSI-H | NMD | Positive | Positive | Positive | 94 | Yes |
| 431 | Stage II mucinous hepatic flexure adenocarcinoma | 57 | Absent | ND | NMD | Negative | Positive | Positive | 29 | No |
| 474 | Stage III cecal adenocarcinoma | 40 | Absent | ND | MSH2-VUS V163D | Positive | Positive | Positive | 62 | No |
| 538 | Rectal adenocarcinoma Terminal ileum adenocarcinoma |
41 43 |
Absent | MSI-H | NMD | Positive | Positive | Positive | 89.80 | Yes |
| 572 | CIS in a large colonic polyp | 40 | Absent | ND | ND | Positive | Positive | Positive | 25.20 | Yes |
For six of the patients, the positive inversion test results are highlighted. For two of the remaining four patients who are negative for the inversion, the VUS identified in their germline testing results is indicatd in bold. Of the remaining two unsolved cases, the negative Amsterdam and Bethesda criteria as well as low PREMM score are indicated in bold. Only one unexplained case remains
CIS Carcinoma in situ (high grade dysplasia), MTS Muir-Torre syndrome, ND not done, NMD no mutation detected, VUS variant of uncertain significance
Fig. 3.

Pedigrees of patients who are carriers of the MSH2 gene inversion. Cancer history is illustrated as reported by the probands. Only Lynch syndrome-related cancers are shown. Inconsistencies in cancer incidence and ages of onset were found when comparing related kindreds, emphasizing the necessity for caution when interpreting family histories provided by patients. CIS carcinoma-in situ. Plus sign indicates the individual is a carrier of the inversion. Minus sign indicates the individual tested does not carry the inversion
After identifying the six initial patients, we tested additional family members where possible, and 21 family members from five families revealed an additional seven inversion carriers (Fig. 3). To confirm our initial results we designed an additional PCR amplification to test for allelic drop-out in carriers of the 10 Mb inversion. Similar to the original long PCR, this PCR amplification relies upon a common SNP located near one of the inversion breakpoints. Primers were designed which amplified a region of the MSH2 gene encompassing both the common SNP and the neighboring region where the 3′ breakpoint is located. This strategy amplifies only the wild type allele of MSH2, and therefore patients who are heterozygous for the internal SNP are homozygous in the PCR product if they also carry the inversion in MSH2. Only carriers of the inversion displayed allelic drop out in the long PCR, and no inversion carriers had amplification of both alleles (Fig. 2).
Initial screening for mutations in the MSH2 gene had indicated that all carriers of the inversion also carried the C allele at SNP rs2303428. Genotyping of several additional SNPs within the MSH2 locus revealed that all inversion carriers may have had a common haplotype, suggesting the possibility that the inversion derived from a common ancestor (Table 2). Upon additional in-depth analysis of the pedigrees of the five affected families, we discovered that two of the carriers were first cousins. Based upon information in the pedigrees, we believe a third family may be related to these two families; however an exact connection could not be determined. No additional connections could be made among these three families and the other two families. None of these families was previously known to be related to the others.
Table 2.
Patient genotypes at multiple SNPs within the MSH2 locus
| PatinetlD# | rs10191478 | rs2347794 | rs17224360 | rs6726691 | rs6726832 | rs7607076 | rs7607312 | rs1981928 | rs1981929 | rs7602094 | rs6711675 | rs3732182 | rs12998837 | rs3732183 | rs2303428 | rs2042649 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 314 | GT | AG | TT | CG | AG | AG | AC | AT | AG | CT | AG | GT | AA | AG | CT | CT |
| 370 | GT | AG | TT | CG | AG | AG | AC | AT | AG | AG | AA | AG | CT | |||
| 372 | CG | AG | AG | AC | AT | AG | CT | GT | AA | AG | CT | CT | ||||
| 372 Tumor | A | C | ||||||||||||||
| 381 | TT | AG | TT | CG | AG | AG | AC | AT | AA | CT | AG | GT | AA | AG | CT | CT |
| 538 | TT | AA | TT | CG | AA | AG | CC | AT | AA | CT | GG | GT | AA | AG | CT | CT |
| 572 | n | AA | TT | CC | AA | GG | CC | AA | AA | CC | GG | TT | AA | AA | CC | CC |
| Inferred Inversion Carrier Haplotype | T | A | T | C | A | G | C | A | A | C | G | T | A | A | C | C |
It is possible to assign one common haplotype to all carriers. The retained SNP genotype in the tumor tissue of 372 is assumed to represent LOH of the wild-type allele and retention of the allele carrying the inversion of MSH2. The inferred haplotype of the MSH2 allele carrying the inversion is shown at the bottom of the table
Based upon our current information we are unable to determine if the mutation we have found represents a founder effect in our local population, or if it applies to a wider population of suspected Lynch syndrome cases. In order to address this issue, we are currently in collaboration with multiple groups to collect DNA samples from mutation negative Lynch syndrome patients throughout the United States.
Our results resolved a considerable proportion of previously unexplained MSH2-type Lynch syndrome families in our registry. Of 10 patients tested, six carried this specific inversion. Of the remaining four unexplained cases, two cases (213,474) had non-synonymous alterations in the coding region of the MSH2 gene which were categorized as “variants of uncertain significance” by the reference laboratory, which may have accounted for Lynch syndrome in these two patients, as both are predicted to be deleterious by SIFT analysis and “probably damaging” by PolyPhen analysis. Of the remaining two cases, one case (328) was negative for standard diagnostic criteria for Lynch syndrome (Amsterdam II criteria, Bethesda guidelines, and a low PREMM 1,2,6 score [8], but was included in the study because of loss of MSH2 expression in a Lynch syndrome-associated cancer and a family history of multiple cancers. Although it is generally assumed that loss of MSH2 expression occurs only in the setting of Lynch syndrome, it remains possible that in some cases the loss is due to somatic mutation, especially in cases with weak or no family history of CRC [9]. Our study did not include exhaustive testing for other inversions in MSH2 in the remaining patients. It is possible that other inversions may explain additional cases of Lynch syndrome in which genetic testing fails to find a causative mutation. Current genetic testing methodology uses convenient approaches and only looks for mutations which can be identified by direct exon sequencing and copy number variation. Even more labor intense methods such as Southern blots and sequencing of full length RT-PCR transcripts may fail to find more obscure mutations such as inversions. This limitation is exemplified in our study. Although Wagner initially discovered the 10 Mb inversion using Southern blotting, a commercial testing service performed Sothern blotting on one of our patients and failed to find this MSH2 gene inversion. New techniques need to be developed to capture all mutations causing Lynch syndrome, and other diseases in which the culpable mutation cannot be identified by current methods. In our analysis, we used allelic drop out in long PCR analysis as a method to find regions of possible inversions. This method could be further developed to look for other inversion in the MSH2 gene, as well as in other genes where current methods of testing fail to find a causative mutation. In the meantime, clinical testing services should add an analysis for the 10 Mb inversion to their clinical testing repertoire. This study also demonstrates the importance of enrolling patients with suspected Lynch syndrome, but negative germline testing results, into research registries where their DNA can be tested until an answer is found.
Acknowledgments
This work supported by the National Institutes of Health grant CA72851 and funds from the Baylor Research Institute, Baylor University Medical Center at Dallas, and the Charles A. Sammons Cancer Center. We also thank Linda Robinson, MS, CGC, Assistant Director, Clinical Cancer Genetics, Simmons Comprehensive Cancer Center, UT Southwestern Medical Center at Dallas, Texas, USA for patient referrals, Margaret M. Hinshelwood, Ph.D., manager, Office of Scientific Publications at the Sammons Cancer Center for her suggestions and the preparation of the manuscript, and Thomas Jascur, Ph.D., for helpful discussion on PCR primer design.
Footnotes
Conflict of interest The authors declare no competing financial interests.
Contributor Information
Jennifer Rhees, Email: jennifrh@baylorhealth.edu, Gastrointestinal Cancer Research Laboratory, Division of Gastroenterology, Department of Internal Medicine, Baylor University Medical Center, Dallas, TX, USA. Baylor Research Institute and Sammons Cancer Center, Baylor University Medical Center, 250 Hoblitzelle, 3500 Gaston Ave, Dallas, TX 75246, USA.
Mildred Arnold, Gastrointestinal Cancer Research Laboratory, Division of Gastroenterology, Department of Internal Medicine, Baylor University Medical Center, Dallas, TX, USA. Baylor Research Institute and Sammons Cancer Center, Baylor University Medical Center, 250 Hoblitzelle, 3500 Gaston Ave, Dallas, TX 75246, USA.
C. Richard Boland, Email: rickbo@baylorhealth.edu, Gastrointestinal Cancer Research Laboratory, Division of Gastroenterology, Department of Internal Medicine, Baylor University Medical Center, Dallas, TX, USA. Baylor Research Institute and Sammons Cancer Center, Baylor University Medical Center, 250 Hoblitzelle, 3500 Gaston Ave, Dallas, TX 75246, USA.
References
- 1.Boland CR. Evolution of the nomenclature for the hereditary colorectal cancer syndromes. Fam Cancer. 2005;4(3):211–218. doi: 10.1007/s10689-004-4489-x. [DOI] [PubMed] [Google Scholar]
- 2.Boland CR, Thibodeau SN, Hamilton SR, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58(22):5248–5257. [PubMed] [Google Scholar]
- 3.Hampel H, Frankel WL, Martin E, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer) N Engl J Med. 2005;352(18):1851–1860. doi: 10.1056/NEJMoa043146. [DOI] [PubMed] [Google Scholar]
- 4.Gylling A, Ridanpaa M, Vierimaa O, et al. Large genomic rearrangements and germline epimutations in Lynch syndrome. Int J Cancer. 2009;124(10):2333–2340. doi: 10.1002/ijc.24230. [DOI] [PubMed] [Google Scholar]
- 5.Lynch HT, Riegert-Johnson DL, Snyder C, et al. Lynch syndrome-associated extracolonic tumors are rare in two extended families with the same EPCAM deletion. Am J Gastroenterol. 2011;106(10):1829–1836. doi: 10.1038/ajg.2011.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wagner A, van der Klift H, Franken P, et al. A 10-Mb paracentric inversion of chromosome arm 2p inactivates MSH2 and is responsible for hereditary nonpolyposis colorectal cancer in a North-American kindred. Genes Chromosomes Cancer. 2002;35(1):49–57. doi: 10.1002/gcc.10094. [DOI] [PubMed] [Google Scholar]
- 7.Chen JM. The 10-Mb paracentric inversion of chromosome arm 2p in activating MSH2 and causing hereditary nonpolyposis colorectal cancer: re-annotation and mutational mechanisms. Genes Chromosomes Cancer. 2008;47(6):543–545. doi: 10.1002/gcc.20556. doi:10.1002/gcc.20556. [DOI] [PubMed] [Google Scholar]
- 8.Kastrinos F, Steyerberg EW, Mercado R, et al. The PREMM(1, 2, 6) model predicts risk of MLH1, MSH2, and MSH6 germline mutations based on cancer history. Gastroenterology. 2011;140(1):73–81. doi: 10.1053/j.gastro.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rodriguez-Soler M, Perez-Carbonell L, Guarinos C, et al. Risk of cancer in cases of suspected Lynch syndrome without germline mutation. Gastroenterology. 2013;144(5):926 e1–932 e1. doi: 10.1053/j.gastro.2013.01.044. (quiz e13–e14) [DOI] [PubMed] [Google Scholar]