

SUMMARY
There is significant demand for experimental approaches to aid protein localization in electron microscopy micrographs and ultimately in three-dimensional reconstructions of macromolecular assemblies. We report preparation and use of a reagent consisting of tris-nitrilotriacetic acid (tris-NTA) conjugated with a monofunctional gold nanoparticle (AuNPtris-NTA) for site-specific, non-covalent labeling of protein termini fused to a histidine-tag (His-tag). Multivalent binding of tris-NTA to a His-tag via complexed Ni(II) ions results in subnanomolar affinity and a defined 1:1 stoichiometry. Precise localization of AuNPtris-NTA labeled proteins by electron microscopy is further ensured by the reagent’s short conformationally restricted linker. We have employed AuNPtris-NTA to localize His-tagged proteins in an oligomeric ATPase and in the bacterial 50S ribosomal subunit. AuNPtris-NTA can specifically bind to the target proteins in these assemblies and is clearly discernible. Our new labeling reagent should find broad application in non-covalent site-specific labeling of protein termini to pinpoint their location in macromolecular assemblies.
INTRODUCTION
Single-particle electron microscopy (EM) is a valuable methodological approach to elucidate structure of large biological assemblies, particularly if an assembly or one or more of its functional states is refractory to crystallization. Significantly, single-particle cryo-EM density maps of symmetric virus particles and other assemblies have been acquired at resolutions and of such quality where it has been possible to trace a Cα-backbone as well as build models that include protein side chains (Zhang et al. 2010, Grigorieff and Harrison, 2011, Hryc et al., 2011). In the case of the most well studied asymmetric assembly, the ribosome, resolutions 4.5–5.5 Å have been reported from particles frozen in vitreous ice (Bai et al., 2013). However, the vast majority of single-particle EM-derived three-dimensional reconstructions of asymmetric biological assemblies are at resolutions much lower than that of the ribosome, such as those of the spliceosome (van der Feltz et al., 2012). But even at 5.5 Å resolution, α-helices and β-sheets appear as tubes and slabs of density respectively (Muirhead and Perutz, 1963, Pomeranz Krummel et al., 2009). The docking of atomic models determined using X-ray crystallography, NMR or homology modeling into a low-resolution three-dimensional EM density map is error-prone in the absence of positional constraints, especially in the context of a compositionally and structurally diverse assembly. For this reason, there is significant demand for approaches that aid the localization of subunits in a macromolecular assembly.
A label can aid protein localization optimally if it: (1) binds fast and with high affinity to a specific site on the target protein, thus insuring high occupancy and low non-specific binding; (2) is unambiguously identifiable i.e., generates high-contrast image features; (3) is of uniform size, structure and conformation; and is (4) close in proximity to the target protein. A broad range of approaches has been employed to serve as labels to aid localization of protein(s) in EM micrographs, but they have not satisfied the listed criteria for an optimal label. Colloidal gold clusters or nanoparticles (AuNPs) provide excellent contrast for visualization by transmission EM and antibodies conjugated with a AuNP have been applied as labels to localize protein(s) (Hacker et al., 2008, Ackerson et al., 2010). But there are fundamental shortcomings of immunogold labeling for precise localization of individual proteins in macromolecular complexes, including: (1) the large size of antibodies (~10 nm diameter); (2) the spatially undefined, multidisperse attachment of the AuNP to an antibody; and (3) the bivalent binding of the typically used IgG molecule.
To overcome the limitations of such biopolymer based labeling approaches, several small, purely chemical functional groups have been employed for conjugating AuNP with proteins via amine or thiol groups (Büchel et al., 2001, Ackerson et al., 2010). However, these approaches not only suffer from a lack of specificity due to multiple lysine and cysteine residues within macromolecular assemblies; but also compared to non-covalent labeling, these chemical reactions are 2–3 orders of magnitude slower, thus requiring higher concentrations and longer incubation times, which increase the probability of non-specific modifications. In an attempt to combine the advantages of a small chemical recognition unit with fast and specific non-covalent interaction, AuNPs conjugated to mono-nitrilotriacetic acid (mono-NTA) (Figure 1A) have been employed to bind proteins via a oligohistidine-tag (His-tag) (Hu et al., 2007, Hu et al., 2008). Compared to chemical coupling, capturing via Ni-NTA/His-interaction is much faster (~1×105 M−1s−1), i.e. similar to an antigen-antibody interaction (Lata et al., 2005). However, a drawback to this approach is the low affinity of NTA to a His-tag (~10 μM) due to the fast dissociation rate constant of ~1 s−1 of this complex (Lata et al., 2005). Thus, high reagent concentrations are necessary to ensure full labeling and rapid wash-out can be encountered, in the absence of further stabilizing interactions, by proximal amino acid side chains such as cysteine or lysine directly interacting with the AuNP surface. Moreover, the specificity of mono-NTA is low since only two histidine residues in close proximity are required for optimum binding (Figure 1A).
Figure 1.

Structure and preparation of AuNPtris-NTA. (A) Charged mono-nitrilotriacetic acid (NTA) (left) and scheme for its binding to a His-tagged protein (right). Complexed nickel ions are indicated as green diamonds and histidines as gray pentagons. Ni-NTA possesses two sites (each indicated by an ‘x’) for coordination of a His-tagged protein. (B) Charged tris-nitrilotriacetic acid (tris-NTA) (left) and its binding to a His-tagged protein (right). Tris-NTA has six sites (each indicated by an ‘x’) for stably coordinating a His-tagged protein. (C) Reaction of tris-NTA-C6-NH2 with mono-NHS-Nanogold® comprising a 1.4 nm gold nanocluster (orange) coated with a via a ~0.6 nm phosphine layer (blue) to produce AuNPtris-NTA.
Functionalization of AuNP with multiple NTA moieties can overcome the problem of low affinity and stability. However, multifunctionalized NTA-AuNP can interact with multiple His-tagged proteins and therefore is prone to promote intermolecular crosslinking. In order to control the highly desirable multivalent recognition of His-tagged proteins, we designed a molecular entity comprising three NTA moieties grafted onto a cyclic scaffold (tris-NTA) to recognize His-tagged proteins with high affinity and specificity (Figure 1B) (Lata et al., 2005). Tris-NTA binds to hexahistidine (H6)-tagged proteins in a 1:1 stoichiometry with ~2 nM affinity and a half-life of several hours, an affinity and half-life four orders of magnitude higher than that of mono-NTA (Lata et al., 2005). Since at least six histidine residues in close proximity are needed to achieve high-affinity binding, the specificity of tris-NTA towards His-tagged proteins is much higher compared to mono-NTA. While proteins having a H6-tag can be efficiently used for labeling, increasing the tag length to a decahistidine (H10)-tag further increases the affinity of the tris-NTA for its target protein to ~0.2 nM (Lata et al., 2005). We have previously used tris-NTA conjugated to fluorescent dyes as well as quantum dots, to efficiently label His-tagged proteins both in vitro and in cell culture (Lata et al., 2006, Reichel et al., 2007, Roullier et al., 2009, You et al., 2010).
Here, we report the conjugation of tris-NTA with a monodisperse gold cluster (AuNPtris-NTA, see Figure 1C) to non-covalently and site-specifically label protein termini genetically fused with a His-tag for the purpose of visualization by transmission EM. To establish the effectiveness of AuNPtris-NTA as a label, we have employed it to determine the location of the His-tagged N-terminus of Thermus thermophilus (Tth) RuvB when it forms an oligomer, in the absence of DNA. E. coli RuvB has been reported to assemble into both homo-hexameric (Chen et al., 2002) as well as homo-heptameric rings in the absence of DNA and into homo-hexamers in the presence of DNA (Miyata et al., 2000). We have also used AuNPtris-NTA to label a single protein subunit in the large and compositionally diverse Escherichia coli 50S ribosomal subunit, thus challenging the reagents ability to specifically and efficiently identify a His-tagged protein amongst a broad array of other protein subunits. We show that our new gold labeling reagent acts as a high-affinity gold nanoparticle ‘pin’ to label the His-tagged proteins in both these challenging assemblies.
RESULTS
AuNPtris-NTA preparation and characterization
Specific binding of AuNPtris-NTA to His-tagged proteins was first assessed by analytical size exclusion chromatography (SEC) using the maltose binding protein fused with a C-terminal deca-histidine-tag (MBP-H10). AuNPtris-NTA bound to MBP-H10 was monitored by AuNPtris-NTA’s absorption at 350 nm, since the absorption cut-off of MBP-H10 is at 300 nm (Figure S1A). While AuNPtris-NTA alone eluted at 11.5 min (Figure 2A and 2B), a second peak appeared with a retention time of 9.8 min after MBP-H10 was incubated with AuNPtris-NTA, corresponding to AuNPtris-NTA bound with MBP-H10. Accordingly, a shoulder around 9.8 min beside the MBP-H10 peak appeared in a SEC trace monitored at 280 nm (Figure S1B). From the intensity ratio of the two peaks, the fraction of functional AuNPtris-NTA was estimated to be 0.31. As a control for specificity, Ni(II) ions were removed by addition of 10 mM EDTA, reducing the peak of AuNPtris-NTA bound with MBP-H10 to less than ten percent and the sample eluted as AuNPtris-NTA alone (Figure 2A and 2B). These studies confirmed specific binding of AuNPtris-NTA to MBP-H10 with high affinity and stability, and with a defined stoichiometry.
Figure 2.
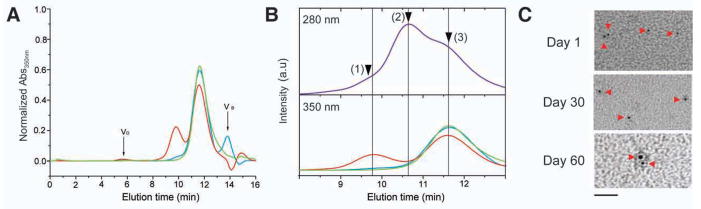
Functional characterization of AuNPtris-NTA. (A) Specific binding of AuNPtris-NTA to a deca-histidine-tagged maltose binding protein (MBP-H10) detected by analytical size exclusion chromatography monitored at 350 nm. Integral intensities of each run were normalized to AuNPtris-NTA alone (green line); MBP-H10 incubated with AuNPtris-NTA (red line); and MBP-H10 incubated with AuNPtris-NTA in the presence of 10 mM EDTA (blue line). Vo, void volume and VB, bed volume. (B) The details of (A) (shown in the bottom chromatogram) and a comparison with the chromatogram of MBP-H10 incubated with AuNPtris-NTA recorded at 280 nm (top chromatogram). The peaks corresponding to MBP-H10 bound to AuNPtris-NTA (1), MBP-H10 (2) and AuNPtris-NTA (3) are marked by arrows. (C) AuNPtris-NTA (8 μM, 1.4 nm diameter AuNP) visualized by transmission electron microscopy (TEM) after storage for 1, 30, and 60 days. Scale bar is 20 nm. Images acquired using an FEI Morgani TEM operating at 80 keV and 56000x magnification. See also Figure S1.
AuNPtris-NTA (8 μM) that is not charged with Ni(II) ions appears predominantly monodisperse, as visualized by TEM, when stored at 4°C for up to 60 days (Figure 2C). Over time, however, the AuNPtris-NTA aggregates to form larger spherical clusters (Figure S1C). Based on the observation that uncharged AuNPtris-NTA was kinetically stable for up to 60 days, the AuNPtris-NTA was used during this time frame. Following charging with Ni(II) ions, the AuNPtris-NTA did not appear to aggregate during the course of our experiments, usually spanning an hour. However, we observed by EM and SEC that after four hours following nickel-charging, the colloidal properties of the gold coupled to tris-NTA were destabilized and aggregates were formed. We attribute the aggregation to complexed Ni(II) ions interacting with the surface of another nanoparticle.
Labeling T. thermophiles RuvB with AuNPtris-NTA
To establish utility of the AuNPtris-NTA to label a macromolecular assembly for visualization by EM, we first explored labeling of the Thermus thermophiles (Tth) RuvB protein. The Tth RuvB used in our studies was fused with an N-terminal deca-histidine-tag (H10) and a three amino acid linker. Tth RuvB-H10 over-expressed in E. coli and purified to homogeneity retains a His-tag, as demonstrated by staining of the protein on an SDS gel with InVision™ His-tag stain (Invitrogen) (Figure 3A). In the absence of ATP, RuvB-H10 particles do not form clear ring-shaped aggregates (Figure 3B). But upon addition of ATP, RuvB-H10 oligomers do form ring-shaped oligomers - although not all RuvB-H10 aggregates appear competent to do so (Figure 3B). AuNPtris-NTA binding in solution may compromise the solubility of RuvB-H10 oligomers and therefore labeling was carried out after RuvB-H10 adsorption to an EM grid. Under these conditions, labeling of RuvB-H10 subunits was highly efficient, with nearly all RuvB-H10 particles that appeared to form rings having five AuNPs bound - as evidenced by visual examination of particles on the EM grid (Figure 3B). The AuNPtris-NTA bound to all ring-shaped oligomers present but did not appear to bind to other, non-ring shaped aggregates. The labeling of RuvB-H10 occurs in a time-dependent manner (Figure 3C). After incubating AuNPtris-NTA for 20 min, five AuNPs are observed bound per RuvB-H10 oligomer (Figure 3C). Strikingly, each of the five AuNPs bound to the RuvB-H10 oligomer are in similar orientations and equidistant from each other as a result of the short and conformationally restricted linker of the tris-NTA reagent. Incubation times longer than 20 min, however, did not increase the number of AuNPs bound per RuvB-H10 ring. Purified RuvB monomer has been reported to assemble into both homo-heptamer and hexamers in the presence of ATP (Miyata et al., 2000, Chen et al., 2002). One possible explanation as to why we observe only five AuNPs bound per RuvB-H10 ring-shaped oligomer is an inaccessibility of the His-tag of one monomer, either due to it being buried or “face down” once adsorbed onto the EM grid and therefore inaccessible for labeling. It is also possible that RuvB-H10 does not form a hexamer efficiently in the absence of DNA. There is no structure of an intact RuvB ring in the absence of DNA. Only crystal structures of a RuvB monomer have been reported, both in a free state and in complex with a RuvA monomer. We, however, produced a model of a RuvB hexamer based on the crystal structure of its human homolog Tip48b, whose hexa-oligomeric structure has been determined (Petukhov et al., 2012). The AuNP labels on RuvB-H10 approximate nicely the more extended N-terminus of Tip48b monomers (Figure 3C).
Figure 3.

Gold labeling of deca-histidine-tagged Thermus thermophiles RuvB oligomers with multivalent AuNPtris-NTA. (A) Purified deca-histidine-tagged (H10) RuvB electrophoresed on a 12% SDS gel. Shown are a protein molecular weight ladder, in kilodaltons (lane 1); purified RuvB-H10 monomer stained with coomassie (lane 2); and RuvB-H10 stained with InVision™ His-tag stain (Invitrogen) (lane 3). (B) Raw negative-stain EM micrographs of RuvB-H10: in the absence of ATP and unlabeled (left); in the presence of ATP and unlabeled (middle); and in the presence of ATP and labeled with AuNPtris-NTA (right). (C) EM images of single RuvB-H10 particles in the presence of AuNPtris-NTA over time. Gold nanoparticles (AuNPs) are indicated by red arrowheads. Scale bar is 10 nm. A model is shown of a homo-hexameric RuvB-H10 oligomer produced by superposition of six molecules of RuvB-H10 onto the human homo-hexameric oligomer Tip48b (Petukhov et al., 2012). N-termini of the monomers are shown bound to AuNP (yellow spheres). Monomers of the Tth RuvB (PDB code 1HQC) (Yamada et al., 2001) were superimposed individually onto the functionally equivalent domains of the structure of Tip48b (PDB code 3UK6) (Petukhov et al., 2012). Superposition was produced using the program MatchMaker and the Needleman-Wunsch alignment algorithm within the Chimera suite of programs. Molecular graphics image of the model was rendered using the UCSF Chimera package (University of California, San Francisco; http://www.cgl.ucsf.edu/chimera). Micrographs acquired using FEI Morgani TEM microscope operating at 80 keV at 45000x magnification. Sample was stained with 1.5% uranyl acetate.
Labeling the E. coli 50S ribosomal subunit with AuNPtris-NTA
In addition to labeling Tth RuvB-H10, we explored the binding of AuNPtris-NTA to the E. coli 50S ribosomal subunit - a particle significantly more compositionally diverse than RuvB. Over-expression of the human 50S ribosomal subunit Rpl3 protein fused with an N-terminal deca-histidine (H10) in E. coli yielded a 50S-H10 subunit that contained Rpl3-H10 protein in place of its endogenous bacterial homolog, L3, (Figure 4A) and migrated on a native polyacrylamide gel as an intact complex free of the smaller 30S subunit (Figure 4B). In contrast to the method used to successfully label RuvB-H10, the 50S-H10 subunit could be labeled with AuNPtris-NTA not only by first absorbing it onto a grid but also by incubating it with AuNPtris-NTA in solution and subsequently absorbing the complex onto an EM grid. Our initial observations indicated that a subset of 50S-H10 particles had two AuNPs bound per particle (Figure S2A), which is not consistent with the known stoichiometry of the protein L3. We therefore took several precautions in labeling of the 50S-H10 subunit: (1) its pre-treatment with 2-iodoacetamide to alkylate free cysteines; and (2) conducting post-labeling washes of the EM grid with up to 50 mM imidazole in a wash buffer so as to remove non-specific binding of AuNPtris-NTA to histidine-rich 50S ribosomal protein subunits. We incubated the bacterial 50S sample that was alkylated with iodoacetamide that lacked the His-tagged protein and observed no non-specific binding of Au to the 50S when treated with the AuNPtris-NTA (Figure S2B and C). We observed, however, that alkylated 50S-H10 particles bound only single AuNPs and that these bound AuNPs were not dislodged when the labeled sample was washed with up to 50 mM imidazole - single electron dense AuNPs appeared evident when compared to the 50S-H10 subunit not incubated with AuNPtris-NTA (Figure 4C). Not all 50S-H10 particles were labeled with a AuNPtris-NTA, as one would expect if the 50S-H10 particles adsorbed to an EM grid without a significant bias in orientation. In addition, not all 50S-H10 particles may have the larger human Rpl3-H10 in place of its endogenous bacterial protein homolog, L3. Indeed, the replacement of an exogenously over-expressed protein for the endogenous variant in the ribosome has been shown to be protein-dependent and yield a successful incorporation of 25–50% (Uhlein et al., 1998). It is not critical, however, that all 50S particles contain the exogenous His-tagged protein for the purpose of localization. Of course, it is important that the protein binds stoichiometrically to the correct site on the 50S and that the AuNP recognizes and binds to only this protein. To highlight the successful AuNPtris-NTA labeling of the human protein Rpl3-H10 incorporated into the bacterial 50S, we selected those particles with the AuNP bound and generated two-dimensional classifications of these 50S-H10 particles (Figure 4D). As shown in a class average, the AuNP is clearly apparent as a distinct dense body - absent in the class average of the same view of 50S-H10. Importantly, the AuNP position reliably co-localized with the site of the E. coli L3 protein (Figure 4D), confirming the high targeting fidelity of AuNPtris-NTA.
Figure 4.

Labeling of Escherichia coli 50S ribosomal subunit with AuNPtris-NTA. (A) Purified E. coli 50S subunit containing the deca-His-tagged (H10) protein Rpl3 (50S-H10) electrophoresed on a 12% SDS gel. Shown are a protein molecular weight ladder, in kilodaltons (lane 1); E. coli 50S-H10 silver stained (lane 2); and E. coli 50S-H10 stained with InVision™ His-tag stain (Invitrogen), highlighting the presence of Rpl3-H10 (lane 3). (B) Purified E. coli 50S-H10 electrophoresed on a native polyacrylamide gradient (3–12%) gel and silver stained. Shown are a molecular weight ladder, in kilodaltons (lane 1); purified E. coli 50S-H10 subunit (lane 2); purified E. coli 30S subunit (lane 3). (C) Raw EM micrographs showing a lawn of negative-stained E. coli 50S-H10 particles not incubated with AuNPtris-NTA and E. coli 50S-H10 particles incubated with the AuNPtris-NTA. 50S-H10 particles in complex with AuNPs are boxed in red. Scale bar is 20 nm. (D) Selected class averages of unlabeled 50S-H10 and of the 50S-H10 labeled with AuNPtrisNTA. Shown is a crystal structure of the E. coli 50S subunit (green; PDB code 3R8S; Dunkle et al., 2011) with the exception of L3, which is colored blue with its N-terminus shown bound to a 1.4 nm diameter sphere (yellow) to represent the gold nanoparticle. The crystal structure is fitted into an EM envelope (grey; EMD code 1019; Matadeen et al., 1999). Images for pane C acquired using an FEI Morgani TEM microscope operating at 80 keV at 36000x magnification. Data for preparation of class averages shown in pane D was collected on a Phillips CM12 operating at 120 keV at 31000x magnification. Sample negative-stained with 1.5% uranyl acetate. The molecular graphics image was rendered using the UCSF Chimera package (University of California, San Francisco; http://www.cgl.ucsf.edu/chimera). See also Figure S2.
DISCUSSION
We have here introduced tris-NTA conjugated with a monodisperse 1.4 nm diameter gold nanoparticle, present in a 1:1 ratio, as a powerful tool for localization of His-tagged proteins within macromolecular assemblies studied by EM. Tris-NTA is attractive as a scaffold for labeling as it has a subnanomolar affinity and defined 1:1 stoichiometry for a His-tag, thus ensuring fast binding and high specificity for its ‘target’ protein and low non-specific binding. Compared to protein-based recognition, tris-NTA is a very small compound - we therefore expected that it would bring the AuNP close in proximity to a ‘target’ protein’s terminus to accurately pinpoint its location. We successfully employed AuNPtris-NTA to localize His-tagged proteins in the T. thermophiles ATPase RuvB and in the E. coli 50S ribosomal subunit. Efficient labeling with AuNPtris-NTA was possible both in solution and after particle adsorption onto an EM grid. Yet as we observed for RuvB, the binding of multiple AuNPtris-NTA to a single protein complex in solution may affect macromolecular solubility. The order of labeling and adsorbing one’s sample onto an EM grid is thus a point to consider in optimizing the labeling strategy of a particle. Moreover, non-specific binding of the AuNP to cysteine residues has to be considered as a potential side reaction, which can be blocked by alkylation prior to the labeling reaction. We also washed the ribosomal 50S-H10 subunit with imidazole at a concentration as high as 50 mM to ensure that the AuNPtris-NTA did not complex with unintended histidine-rich protein(s). Following the pre- and post-labeling treatment and wash of the ribosomal 50S-H10 subunit, we obtained particles that had only a single AuNP bound. Class averages of the AuNP labeled 50S-H10 indicated the location of the His-tagged protein L3.
Exhibiting rapid, site-specific labeling with high affinity, specificity and defined stoichiometry in combination with its small size, this novel AuNPtris-NTA reagent ideally fulfills the requirements for precisely pinpointing protein termini in macromolecular assemblies by EM. We here applied AuNPtris-NTA after protein adsorption to an EM grid as relatively fragile assemblies were involved. However, purification of protein complexes after incubation with AuNPtris-NTA as demonstrated for MBP-H10 and 50S-H10 is also possible, opening exciting possibilities for cryo-EM applications, which has been shown to be compatible with such 1.4 nm AuNP. Thus, we believe that AuNPtris-NTA should find broad application in non-covalent site-specific labeling of protein termini to pinpoint their location in macromolecular assemblies as well as in challenging applications such as in situ electron tomography that requires protein localization in a complex and crowded milieu. We also envision other possible roles for the AuNPtris-NTA once bound to its intended target, including serving as a fiducial marker by providing high-contrast image features for particle identification in vitrified ice and aiding particle classification and/or alignment (Jensen and Kornberg, 1998).
EXPERIMENTAL PROCEDURES
Conjugation of tris-NTA with a monofunctional gold nanoparticle
Tris-NTA modified by an aminocaproic acid (tris-NTA-C6-NH2, 1046 Da) was synthesized as previously described (Lata et al., 2005). The overall synthesis of AuNPtris-NTA is outlined in Figure 1C. One milligram of the tris-NTA-C6-NH2 was dissolved in 100 μL dry dimethyl sulfoxide (DMSO) containing 5 μL ethyldiisopropylamine. Six nmoles of mono-Sulfo-N-hydroxy-Succinimido-Nanogold® (mono-NHS- Nanogold®, 1.4 nm diameter; Nanoprobes) was dissolved in 100 μL dry DMSO and then suspended in a 2 mL glass reactor. After reaction at room temperature for 20 hours under N2 protection in the dark, 100 μL water was added for 30 min to quench the NHS ester. The mixture was subsequently loaded onto a 2 mL NAP-5 column (G25, DNA grade; GE Healthcare) for purification. The relevant fractions were concentrated to approximately 150 μL using an Amicon Ultra centrifugal filter (Ultracel®, 0.5 mL, 10,000 MWCO). The sample was then washed three times with 500 μL of 20 mM HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), pH 7.5 to further remove the un-coupled tris-NTA compounds. The gold nanoparticle concentration was determined by UV-Vis spectrometric reading at 420 nm, applying an extinction coefficient of 156,000 cm−1M−1 from the manufacturer. A typical yield of 36% based on the total mono-NHS- Nanogold® was obtained from the preparation. Without loading with nickel(II) ions, AuNPtris-NTA can be stored at 4°C for at least two months in 20 mM HEPES, pH 7.5.
Nickel loading was done according to a previously published protocol with modifications (You et al., 2010). In brief, for labeling of the RuvB-H10 and 50S-H10 samples, 20 μL of 8 μM AuNPtris-NTA stock solution was mixed with 100 μL 1 mM and 100 μL 100 μM NiCl2, respectively, in 20 mM HEPES, pH 7.5 for 5 minutes, then loaded onto an Amicon Ultra centrifugal filter (Ultracel®, 0.5 mL, 10000 MWCO) which was centrifuged at 10,000 revolutions per minute (rpm) for 5 minutes. The sample was buffer exchanged on the concentrator with addition of 900 μL of phosphate buffered saline (PBS) to remove residual nickel ions. The AuNPtris-NTA was then immediately (within 1 hour) used for protein labeling at a concentration of 500 nM.
Functional characterization of AuNPtris-NTA
Specific interaction of AuNPtris-NTA with His-tagged proteins in solution was explored by SEC using maltose binding protein with a C-terminal deca-histidine-tag (MBP-H10) as a model protein. Labeling was done according to a previously published protocol (You et al., 2010). The freshly prepared Ni(II)-loaded AuNPtris-NTA was incubated with MBP-H10 in HEPES buffered saline (HBS) for 30 minutes with final concentrations of AuNPtris-NTA and MBP-H10 at approximately 100 nM and 3 μM, respectively. The sample was fractionated by a Superdex 200 5/150 SEC column (GE Healthcare) using a high-pressure liquid chromatography (HPLC) system (Jasco) monitored by a diode array detector (MD-2015 plus; Jasco) at a flow rate of 0.2 mL/min.
Preparation of Tth RuvB and the E. coli 50S ribosomal subunit
The Thermus thermophiles (Tth) RuvB gene coding for full-length RuvB protein (324 residues; 37 kDa) was cloned into the pET3a expression vector (Qiagen). Tth RuvB was cloned with an N-terminal deca-histidine-tag (H10) followed by a three amino acid (Ser-Ser-Gly) linker. BL21 (DE3) cells transformed with RuvB-H10 were grown in Luria Broth (LB) at 37°C. Cells were induced with 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) at OD600 = 0.8 and grown for 5 hours post-induction at 37°C. Cells were harvested by centrifugation at 5,000 rpm for 20 minutes at 4°C. The cell pellet was suspended and cells lysed in 20 mM HEPES, pH 7.5; 0.5 M NaCl; 50 mM imidazole, with EDTA-free protease inhibitor cocktail (Roche) present. Soluble fraction(s) were heated at 75°C for 15 minutes and placed at 4°C for 30 minutes to denature and precipitate non-thermophilic proteins. Polyethyleneimine (0.01% final concentration) was added to the remaining soluble protein while mixing on ice for 1 hour to remove DNA, an approach described previously (Burgess, 1991). DNA was then pelleted and the remaining protein was loaded onto a gravity column containing Ni-NTA resin (Qiagen) for affinity purification of full-length RuvB-H10. Protein was eluted using a linear gradient from 0–500 mM imidazole in 20 mM HEPES, pH 7.5; 0.5 M NaCl. RuvB-H10 was further purified and buffer exchanged into a storage buffer (20 mM tris-HCl, pH 7.8; 0.2 M KCl) by SEC using a Superdex 200 16/60 column (GE Healthcare). Purified protein was kept at 4°C. To assemble RuvB-H10 in the absence of DNA, purified RuvB-H10 at 5 μM was incubated with 2 mM ATP/10 mM MgCl2 for 30 minutes at 37°C, as described previously (Tong and Wetmur, 1996).
The gene coding for full-length human 60S ribosomal subunit protein Rpl3 (403 residues; 46 kDa) was cloned into the pET15b expression vector, such that its natural initiator methionine position is preceded by a deca-histidine-tag, a SerSerGly linker, and a thrombin cleavage site (Kelly et al., 2008) (gift of Dukovski and Walz). Rpl3-H10 is the human homolog of E. coli 50S ribosomal subunit protein L3. Rpl3-H10 was over-expressed in E. coli BL21 (DE3) cells, essentially as previously described (Kelly et al., 2008). BL21 (DE3) cells transformed with Rpl3-H10 were grown in LB at 37°C. Cells were induced with 0.5 mM IPTG at OD600 = 0.8 and grown overnight at 16°C. The cells were harvested by centrifugation at 5,000 rpm for 20 minutes at 4°C. The cell pellet was suspended in ribosomal buffer (20 mM HEPES-K, pH 7.5; 6 mM magnesium acetate; 30 mM ammonium acetate; 4 mM β-mercaptoethanol (BME)) and centrifuged at 4,000 rpm for 20 minutes at 2°C. The cell pellet was flash frozen in liquid nitrogen and stored at −80°C. Cells were lysed using a bead-beater and aluminum oxide. Lysed cells were centrifuged for 15 minutes at 12,300 rpm followed by centrifugation at 17,600 rpm for 55 minutes at 2°C. The supernatant was centrifuged for an additional 18 hours at 2°C and 40,700 rpm. The top layer was removed from the pellet, while the bottom layer was rinsed with and suspended in dissociation buffer (20 mM HEPES-K, pH 7.5; 1 mM magnesium acetate; 200 mM ammonium acetate; 4 mM BME). The sample was centrifuged at 14,000 rpm for 15 minutes at 2°C to pellet insoluble aggregate. Approximately 150 OD260 units of sample were layered onto a 10–30% sucrose gradient prepared with ribosomal dissociation buffer. Gradients were centrifuged at 19,000 rpm for 17 hours at 2°C. Each gradient was fractionated by pipetting 1 mL fractions starting at the top of the gradient until the bottom was reached. Every second fraction was run on a 1.2% agarose gel. Fractions containing 50S-H10 ribosomal RNA were pooled and 10% of the total volume of dissociation buffer was added. 50S-H10 subunit fractions were centrifuged at 70,000 rpm at 2°C for 24 hours to pellet the 50S-H10 ribosomal subunit. 50S-H10 subunit pellets were then suspended in dissociation buffer. Purified 50S-H10 subunit was flash frozen in liquid nitrogen and stored at −80°C until use.
Visualization of unlabeled Tth RuvB and the E. coli 50S ribosomal subunit
Approximately 4 μL of either 5 μM Tth RuvB-H10 or E. coli 50S with or without Rpl3-H10 were applied to glow-discharged carbon-coated copper 400 mesh EM grids (SPI Supplies). Samples were incubated on the grid for 1 minute and then blotted using filter paper. Grids with bound RuvB-H10 or E. coli 50S subunit variants were washed once to remove aggregates with either RuvB-H10 storage buffer or ribosomal dissociation buffer, respectively. Grids were negatively stained by addition of three drops of 1.5% uranyl acetate, blotting between drops. Grids were dried for at least 5 minutes prior to visualization using an FEI Morgani TEM operating at 80 keV or a Phillips CM12 operating at 120 keV.
Labeling of Tth RuvB and E. coli 50S subunit with AuNPtris-NTA
The AuNPtris-NTA was visualized in the absence of substrate by dispensing 4 μL of the charged label (500 nM) onto a glow-discharged copper-coated 400 mesh EM grid, allowing the sample to dry, staining the grid with 1.5% uranyl acetate, and then visualized by TEM as described above. To label RuvB-H10, the purified and assembled complex was similarly applied to glow-discharged carbon-coated EM grids at 5 μM. Grids were washed once with RuvB-H10 storage buffer and blotted. Subsequently, 4 μLof charged AuNPtris-NTA (500 nM) was dispensed onto the grid containing the washed sample and incubated for either 1, 5, 10, 15 or 20 minutes. Following, grids were washed twice with RuvB-H10 storage buffer and blotted to remove unbound AuNPtris-NTA. Labeled RuvB-H10 was then stained with 1.5% uranyl acetate and visualized by TEM, as described above for the unlabeled complex.
To label E. coli 50S-H10 subunit, purified sample was either labeled directly in solution or first alkylated to prevent thiol-mediated interaction with the AuNP. To alkylate the 50S-H10 subunit, 4 μL of 5 OD260 units of 50S-H10 subunit was incubated with 1 μL 15 mM 2-Iodoacetamide (5 mM final) for 15 minutes and then buffer exchanged to remove free 2-Iodoacetamide. E. coli 50S-H10 subunit was then labeled either on a grid or in solution by incubation with 500 nM charged AuNPtris-NTA and incubation for 1 to 3 minutes. Labeled 50S-H10 was applied to the grid for 1 minute and then blotted using filter paper. Grids were washed twice with ribosomal dissociation buffer containing 20–50 mM imidazole to remove possible non-specific interactions of the AuNPtris-NTA and sample. Between washes, grids were blotted to remove any aggregate and unbound AuNPtris-NTA. Labeled 50S-H10 subunit was then stained with 1.5% uranyl acetate and visualized by TEM, as described above for the unlabeled particle.
Preparation of E. coli 50S class averages
Grids were prepared of E. coli 50S-H10 ribosomal subunits containing the deca-His-tagged Rpl3, with or without bound AuNPtris-NTA - as described above. Data for preparation of class averages was collected on a Phillips CM12 operating at 120 keV at 31,000 times magnification. 2,036 and 217 particles were picked for the unlabeled and AuNPtris-NTA labeled 50S-H10 subunits, respectively, using e2boxer (Tang et al., 2007). 50S-H10 particles class averages were produced and further refined using the IMAGIC image processing software package (van Heel et al., 1996).
Supplementary Material
Highlights.
Synthesis of a novel Au conjugated reagent possessing high affinity for a His-tag
Site-specific, non-covalent labeling of protein with high positional accuracy
Novel reagent pinpoints protein termini in challenging macromolecular assemblies
Acknowledgments
This work was supported by the National Science Foundation Materials Research Science and Engineering Center at Brandeis (D.A.P.K.), an investigator grant to D.A.P.K. from the National Science Foundation (Award Number 1157892), and funding to J.P. by the Deutsche Forschungsgemeinschaft (SFB944). We are grateful to Nikolaus Grigorieff for advice and support; Tom Walz and Danijela Dukovski for the His-tagged Rpl3 clone; Axel Brilot for guidance in preparation of the E. coli 50S ribosomal subunit; Clarisse van der Feltz and Mike Rigney for discussion; and Chen Xu for EM guidance and advice. D.A.P.K. is very grateful for the inspiration during the later stages of this work provided by Amita Sarai Pomeranz, a ‘golden girl’. The Brandeis EM facility is supported by National Institutes of Health Grant P01 GM62580.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ackerson CJ, Powell RD, Hainfeld JF. Site-specific biomolecule labeling with gold clusters. Methods Enzymol. 2010;481:195–230. doi: 10.1016/S0076-6879(10)81009-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai X-C, Fernandez IS, McMullan G, Scheres SH. Ribosome structures to near-atomic resolution from thirty thousand cryo-EM particles. eLife Sciences. 2013:2. doi: 10.7554/eLife.00461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Büchel C, Morris E, Orlova E, Barber J. Localisation of the PsbH subunit in photosystem II: a new approach using labelling of his-tags with a Ni2+-NTA gold cluster and single particle analysis. J Mol Biol. 2001;312:371–379. doi: 10.1006/jmbi.2001.4951. [DOI] [PubMed] [Google Scholar]
- Burgess RR. Use of polyethyleneimine in purification of DNA-binding proteins. Methods Enzymol. 1991;208:3–10. doi: 10.1016/0076-6879(91)08003-z. [DOI] [PubMed] [Google Scholar]
- Chen YJ, Yu X, Egelman EH. The hexameric ring structure of the Escherichia coli RuvB branch migration protein. J Mol Biol. 2002;319:587–591. doi: 10.1016/S0022-2836(02)00353-4. [DOI] [PubMed] [Google Scholar]
- Dunkle JA, Wang L, Feldman MB, Pulk A, Chen VB, Kapral GJ, Noeske J, Richardson JS, Blanchard SC, Cate JH. Structures of the bacterial ribosome in classical and hybrid states of tRNA binding. Science. 2011;332:981–984. doi: 10.1126/science.1202692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigorieff N, Harrison SC. Near-atomic resolution reconstructions of icosahedral viruses from electron cryo-microscopy. Curr Opin Struct Biol. 2011;21:265–273. doi: 10.1016/j.sbi.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacker I, Sander B, Golas MM, Wolf E, Karagoz E, Kastner B, Stark H, Fabrizio P, Luhrmann R. Localization of Prp8, Brr2, Snu114 and U4/U6 proteins in the yeast tri-snRNP by electron microscopy. Nat Struct Mol Biol. 2008;15:1206–1212. doi: 10.1038/nsmb.1506. [DOI] [PubMed] [Google Scholar]
- Hainfeld JF, Liu W, Halsey CM, Freimuth P, Powell RD. Ni-NTA-gold clusters target His-tagged proteins. J Struct Biol. 1999;127:185–198. doi: 10.1006/jsbi.1999.4149. [DOI] [PubMed] [Google Scholar]
- Hryc CF, Chen DH, Chiu W. Near-atomic-resolution cryo-EM for molecular virology. Curr Opin Virol. 2011;1:110–117. doi: 10.1016/j.coviro.2011.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M, Qian L, Briñas RP, Lymar ES, Hainfeld JF. Assembly of nanoparticle-protein binding complexes: from monomers to ordered arrays. Angewandte Chemie. 2007;46:5111–5114. doi: 10.1002/anie.200701180. [DOI] [PubMed] [Google Scholar]
- Hu M, Qian L, Briñas RP, Lymar ES, Kuznetsova L, Hainfeld JF. Gold nanoparticle-protein arrays improve resolution for cryo-electron microscopy. J Struct Biol. 2008;161:83–91. doi: 10.1016/j.jsb.2007.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen GJ, Kornberg RD. Single-particle selection and alignment with heavy atom cluster-antibody conjugates. Proc Natl Acad Sci USA. 1998;95:9262–9267. doi: 10.1073/pnas.95.16.9262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly DF, Dukovski D, Walz T. Monolayer purification: A rapid method for isolating protein complexes for single-particle electron microscopy. Proc Natl Acad Sci USA. 2008;105:4703–4708. doi: 10.1073/pnas.0800867105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lata S, Gavutis M, Tampé R, Piehler J. Specific and stable fluorescence labeling of histidine-tagged proteins for dissecting multi-protein complex formation. J Am Chem Soc. 2006;128:2365–2372. doi: 10.1021/ja0563105. [DOI] [PubMed] [Google Scholar]
- Lata S, Reichel A, Brock R, Tampé R, Piehler J. High-affinity adaptors for switchable recognition of histidine-tagged proteins. J Amer Chem Soc. 2005;127:10205–10215. doi: 10.1021/ja050690c. [DOI] [PubMed] [Google Scholar]
- Lawson CL, Baker ML, Best C, Bi C, Dougherty M, Feng P, van Ginkel G, Devkota B, Lagerstedt I, Ludtke SJ, Newman RH, Oldfield TJ, Rees I, Sahni G, Sala R, Velankar S, Warren J, Westbrook JD, Henrick K, Kleywegt GJ, Berman HM, Chiu W. EMDataBank.org: unified data resource for CryoEM. Nucl Acids Res. 2011;39:D456–D464. doi: 10.1093/nar/gkq880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matadeen R, Patwardhan A, Gowen B, Orlova EV, Pape T, Cuff M, Mueller F, Brimacombe R, van Heel M. The Escherichia coli large ribosomal subunit at 7.5 Å resolution. Structure. 1999;7:1575–1583. doi: 10.1016/s0969-2126(00)88348-3. [DOI] [PubMed] [Google Scholar]
- Miyata T, Yamada K, Iwasaki H, Shinagawa H, Morikawa K, Mayanagi K. Two Different Oligomeric States of the RuvB Branch Migration Motor Protein as Revealed by Electron Microscopy. J Struct Biol. 2000;131:83–89. doi: 10.1006/jsbi.2000.4290. [DOI] [PubMed] [Google Scholar]
- Muirhead H, Perutz MF. Structure of Haemoglobin. A three-dimensional fourier synthesis of reduced human Haemoglobin at 5.5 Å resolution. Nature. 1963;199:633–638. doi: 10.1038/199633a0. [DOI] [PubMed] [Google Scholar]
- Nishino Y, Yasunaga T, Miyazawa A. A Genetically Encoded Metallothionein Tag Enabling Efficient Protein Detection by Electron Microscopy. J Electron Micr. 2007;56:93–101. doi: 10.1093/jmicro/dfm008. [DOI] [PubMed] [Google Scholar]
- Petukhov M, Dagkessamanskaja A, Bommer M, Barrett T, Tsaneva I, Yakimov A, Queval R, Shvetsov A, Khodorkovskiy M, Kas E, Grigoriev M. Large-Scale Conformational Flexibility Determines the Properties of AAA+ TIP49 ATPases. Structure. 20:1321–1331. doi: 10.1016/j.str.2012.05.012. [DOI] [PubMed] [Google Scholar]
- Pomeranz Krummel DA, Oubridge C, Leung AKW, Li J, Nagai K. Crystal structure of human spliceosomal U1 snRNP at 5.5 Å resolution. Nature. 2009;458:475–480. doi: 10.1038/nature07851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichel A, Schaible D, Al Furoukh N, Cohen M, Schreiber G, Piehler J. Noncovalent, Site-Specific Biotinylation of Histidine-Tagged Proteins. Anal Chem. 2007;79:8590–8600. doi: 10.1021/ac0714922. [DOI] [PubMed] [Google Scholar]
- Roullier V, Clarke S, You C, Pinaud F, Gouzer G, Schaible D, Marchi-Artzner V, Piehler J, Dahan M. High-affinity labeling and tracking of individual histidine-tagged proteins in live cells using Ni2+ tris-nitrilotriacetic acid quantum dot conjugates. Nano Lett. 2009;9:1228–1234. doi: 10.1021/nl9001298. [DOI] [PubMed] [Google Scholar]
- Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, Ludtke SJ. EMAN2: An extensible image processing suite for electron microscopy. J Structural Biol. 2007;157:38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- Tong J, Wetmur JG. Cloning, sequencing, and expression of ruvB and characterization of RuvB proteins from two distantly related thermophilic eubacteria. J Bact. 1996;178:2695–2700. doi: 10.1128/jb.178.9.2695-2700.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlein M, Weglöhner W, Urlaub H, Wittmann-Liebold B. Functional implications of ribosomal protein L2 in protein biosynthesis as shown by in vivo replacement studies. Biochem J. 1998;331:423–430. doi: 10.1042/bj3310423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Feltz C, Anthony K, Brilot A, Pomeranz Krummel DA. Architecture of the Spliceosome. Biochem. 2012;51:3321–3333. doi: 10.1021/bi201215r. [DOI] [PubMed] [Google Scholar]
- van Heel M, Harauz G, Orlova EV, Schmidt R, Schatz M. A New Generation of the IMAGIC Image Processing System. J Structural Biol. 1996;116:17–24. doi: 10.1006/jsbi.1996.0004. [DOI] [PubMed] [Google Scholar]
- Yamada K, Kunishima N, Mayanagi K, Ohnishi T, Nishino T, Iwasaki H, Shinagawa H, Morikawa K. Crystal structure of the Holliday junction migration motor protein RuvB from Thermus thermophilus HB8. Proc Natl Acad Sci USA. 2001;98:1442–1447. doi: 10.1073/pnas.031470598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You C, Wilmes S, Beutel O, Löchte S, Podoplelowa Y, Roder F, Richter C, Seine T, Schaible D, Uzé G, Clarke S, Pinaud F, Dahan M, Piehler J. Self-controlled monofunctionalization of quantum dots for multiplexed protein tracking in live cells. Angew Chem Int Ed Engl. 2010;49:4108–4112. doi: 10.1002/anie.200907032. [DOI] [PubMed] [Google Scholar]
- Zhang J, Baker ML, Schröder GF, Douglas NR, Reissmann S, Jakana J, Dougherty M, Fu CJ, Levitt M, Ludtke SJ, Frydman J, Chiu W. Mechanism of folding chamber closure in a group II chaperonin. Nature. 2010;463:379–383. doi: 10.1038/nature08701. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.