

Abstract
Acidic pH is an important feature of tumor microenvironment and a major determinant of tumor progression. We reported that cancer cells upregulate autophagy as a survival mechanism to acidic stress. Inhibition of autophagy by administration of chloroquine (CQ) in combination anticancer therapies is currently evaluated in clinical trials. We observed in 3 different human cancer cell lines cultured at acidic pH that autophagic flux is not blocked by CQ. This was consistent with a complete resistance to CQ toxicity in cells cultured in acidic conditions. Conversely, the autophagy-inhibiting activity of Lys-01, a novel CQ derivative, was still detectable at low pH. The lack of CQ activity was likely dependent on a dramatically reduced cellular uptake at acidic pH. Using cell lines stably adapted to chronic acidosis we could confirm that CQ lack of activity was merely caused by acidic pH. Moreover, unlike CQ, Lys-01 was able to kill low pH-adapted cell lines, although higher concentrations were required as compared with cells cultured at normal pH conditions. Notably, buffering medium pH in low pH-adapted cell lines reverted CQ resistance. In vivo analysis of tumors treated with CQ showed that accumulation of strong LC3 signals was observed only in normoxic areas but not in hypoxic/acidic regions. Our observations suggest that targeting autophagy in the tumor environment by CQ may be limited to well-perfused regions but not achieved in acidic regions, predicting possible limitations in efficacy of CQ in antitumor therapies.
Keywords: autophagy, chloroquine, tumor acidosis, cancer therapy, pH
Introduction
Tumor tissues are characterized by high heterogeneity in terms of physical, biochemical, and biological properties. Tumor cells localized in proximity of blood vessels are supplied with oxygen and nutrients which contribute to support growth and proliferation. As tumor mass increases, dysplastic growth will result in the formation of poorly oxygenated areas with reduced perfusion capacity, due to the lack of an organized and sufficient vasculature. Tumor cells located in these regions are exposed to low oxygen tension (hypoxia) and lack of nutrients, responsible for a metabolic shift toward sustained glycolysis with consequent extracellular accumulation of metabolic acids.1-3 Such acidic microenvironment exerts a selective pressure and represents an important mechanism for malignant progression and for intrinsic therapy resistance of many tumors.4-7 Moreover, the acidic environment also protects cancer cells from immune system control, thus favoring immune escape and tumor progression.8 For these reasons, counteracting tumor acidic pH is being evaluated as a feasible therapeutic strategy in clinical settings using different approaches.8-11
Recently, we and others have reported that autophagy is an important mechanism for cancer cells’ adaptation to acidosis.12,13 This has been shown for metastatic breast carcinoma and human melanoma, suggesting that inhibition of autophagy and buffering of tumor pH may synergize in reducing tumor growth. Inhibition of autophagy is currently considered as a promising approach to treat cancer in combination therapies.14,15 Autophagy sustains tumor growth and progression by providing essential survival signals in metabolically and therapeutically stressed cells14-17 and by contributing to the maintenance of the cancer stem cell phenotype.18-20 Inhibition of autophagy has been demonstrated to increase chemosensitivity and radiosensitivity and cause tumor regression in several preclinical models.15,16,21-24 Chloroquine and its derivative hydroxychloroquine (HCQ) are the only autophagy-inhibiting agents currently investigated in clinical trials in solid tumors.14,25 CQ is the most widely used antimalaria drug and it has also been used in the treatment of other human diseases (rheumatoid arthritis and lupus erythematosus) due to its anti-inflammatory properties. Because of its chemical properties as a weak base, CQ acts as lysosomotropic agent and inhibits lysosomal activity.25 For this reason, CQ inhibits degradation of the autophagic cargo and induces a block of autophagic flux.26 Of note, compounds with similar mechanism of action as CQ are able to restore drug sensitivity in several models of human cancers by inhibiting the ion-trapping mechanism and increasing the intracellular targeting of specific drugs.27-29 We report here that tumor acidic conditions interfere with the autophagy-inhibiting activity of CQ, suggesting that CQ may be ineffective as autophagy inhibitor in solid tumors characterized in vivo by acidic regions.
Results and Discussion
CQ is unable to block autophagic flux in melanoma cells under acidic stress
Tumor acidosis represents an important mechanism involved in malignant progression and resistance to therapy.30-32 We and others have recently reported that human tumor cells activate autophagy as a protective and adaptive response to acidic stress,12,13 suggesting that autophagy inhibition may represent a therapeutic strategy to selectively kill cells adapted to low pH conditions. While CQ as a single-agent treatment has shown antitumor effects in some models,16 CQ is also reported to have no effect or even slightly increase tumor growth,33,34 suggesting that efficacy of CQ to inhibit autophagy may also depend on dosage, tumor types, and tumor microenvironment factors. Acidification of tumor pH is known to reduce entry of many weakly basic chemotherapeutic compounds,35-37 including CQ,38 which may represent an obstacle to obtain proper antiautophagic activity in vivo. Thus, we first measured in vivo and in vitro the effects of CQ on tumor cell growth and viability. Administration of CQ as single agent did not affect tumor growth in 2 different models of human colon carcinoma xenografts (Fig. 1A). Interestingly, we observed that colon carcinoma (HCT116) and melanoma (Me30966) cells cultured under acidic conditions were completely resistant to the cytotoxic activity of CQ (Fig. 1B). Since cancer cells under acidic stress are sensitive to autophagy inhibition,12,13,39 we further evaluated the ability of CQ to block autophagy under acidic conditions.

Figure 1. Lack of cytotoxic and autophagy-inhibiting activity of CQ in acidic conditions. (A) HCT116 and HT29 human colon carcinoma xenografts were treated with vehicle (n = 8) or with CQ (20 mg/kg, n = 8) administered i.p. every second day and tumor volume was monitored for 16 d. (B) The effects of CQ treatment on cell viability were analyzed in HCT116 and Me30966 cells cultured at pH 7.4 or pH 6.8 for 48 h. Data are presented as means ± standard deviations from 2 independent experiments. (C) Me30966 cells were plated in normal RPMI medium and the next day the medium was replaced with media at the indicated pH. Cells were incubated with fresh media for 24 h and BafA1 (50 nM) or CQ (50 μM) added during the last 4 h before collection of cells for WB analysis of LC3 expression. Representative WB analysis shows that CQ does not cause LC3-II accumulation at acidic pH. LC3-II signal in the histograms was quantified by densitometric analysis of 3 independent experiments using Adobe Photoshop and the ratio LC3-II/ACTB was calculated, using the values of the control cells at pH 7.4 for data normalization.
Autophagy is a dynamic process and the amount of LC3-II detected by western blot (WB) reflects both synthesis and consumption of the protein by the lysosomal compartment. Inhibition of lysosomal degradation mediated by bafilomycin A1 (BafA1) or CQ results in accumulation of LC3-II. The ratio between LC3-II levels in the presence and absence of lysosomal inhibitors can be used to estimate the autophagic flux, i.e., the amount of LC3-II delivered from autophagosomes to lysosomes for autophagic degradation.26 In order to analyze the status of autophagic flux in melanoma cells under acidic stress, we cultured Me30966 cells at physiological pH (pH 7.4) and acidic pH (pH 6.8 and 6.5) for 24 h, in the presence or absence of BafA1 or CQ during the last 4 h of incubation, and monitored the accumulation of LC3-II by WB. BafA1 induced massive accumulation of LC3-II at all pH culture conditions, indicating a sustained autophagic flux at acidic pH (Fig. 1C), as previously reported by us.13 Interestingly, CQ was able to induce accumulation of LC3-II only at the physiological pH 7.4 but not at acidic pH conditions (Fig. 1C). Quantification of LC3-II accumulation showed that CQ did not induce changes in LC3-II levels at acidic pH (Fig. 1C). A similar finding was observed in the human melanoma cell lines Sk-Mel-28 and A375 (Fig. S1A and S1B).
Acidic conditions inhibit the autophagy-blocking activity of CQ in colon carcinoma and osteosarcoma cells
CQ and HCQ are currently used in clinical trials in anticancer combination therapies as autophagy inhibitors.14 Based on the findings reported in Figure 1 for CQ, we further investigated the autophagy-inhibiting activity of both compounds in acidic conditions on additional cell lines, HCT116 and HOS cells. We determined that in both HCT116 and HOS cells the autophagy-blocking activity of CQ and HCQ was saturated at 50 μM (Fig. S2A and S2B).
HCT116 cells under acidic conditions showed a detectable and unchanged autophagic flux as determined in the presence of BafA1 (Fig. 2A). However, the activity of CQ dramatically decreased at lower medium pH and it was not restored even at the highest concentrations (100 μM), as shown by quantification of LC3-II expression (Fig. 2A). A similar observation was made by using HCQ in HCT116 and HOS cells (Fig. S2C and S2D).

Figure 2. CQ is unable to block autophagy in acidic conditions. (A) HCT116 cells were cultured for 24 h at different pH conditions. BafA1 (50 nM) and CQ (25, 50, 100 μM) were added during the last 4 h of incubation. (B and C) WB analysis of LC3-II accumulation in HCT116 and HOS cells was performed on cells cultured at different pH medium in presence of BafA1 (50 nM), CQ (50 μM) and a combination of BafA1 + CQ. Data in (A–C) are representative from 2 independent experiments. (D) HOS-GFP-LC3 cells were cultured at pH 7.4 and 6.8 for 24 h and BafA1 and CQ were added during the last 4 h in culture. Cells were treated with saponin as described in Materials and Methods and analyzed by flow cytometry to quantify autophagosome-associated GFP-LC3 (data are from 3 independent experiments).
To confirm this observation, we performed experiments by culturing cells in the presence of CQ, BafA1 and the combination of CQ and BafA1 as an additional control. As clearly depicted in Figure 2B and C, all 3 combinations blocked the autophagic flux at pH 7.4 in both HCT116 and HOS cells. As expected, the autophagy-inhibiting activity of CQ was pH-dependent, being dramatically reduced at pH 6.8 and totally abolished at pH 6.5. Moreover, the autophagic flux in the CQ-treated cells was blocked in the acidic pH conditions only when BafA1 was added together with CQ. To further support this observation, we investigated the cleavage of pro-CTSB (cathepsin B) to mature CTSB, a lysosomal cysteine protease whose mature form is detected in acidic lysosomes.40 In HCT116 cells at pH 7.4, the addition of CQ and BafA1 induced disappearance of mature CTSB, indicating that both CQ and BafA1 cause inhibition of lysosomal activity (Fig. S3A). At acidic pH only cells treated with BafA1 but not with CQ showed strong reduction of mature CTSB, indicating that in these conditions CQ does not affect lysosomal activity.
Accumulation of autophagosomes in the presence of lysosomal inhibitors can be monitored in HOS cells stably transfected with GFP-LC3 plasmid. Mild saponin extraction of GFP-LC3-I allows quantification of autophagosome-associated GFP-LC3-II by flow cytometry.41 In order to reduce potential quenching of GFP fluorescence at acidic cytosolic pH when cells are cultured at pH 6.5,42 this experiment was performed at the conditions of pH 7.4 and 6.8. As shown in Figure 2D, BafA1 and CQ treatment induced a comparable increase in autophagosome accumulation indicated by an overlapping GFP fluorescence signal in cells cultured at pH 7.4. Conversely, in cells cultured at pH 6.8, BafA1 treatment induced accumulation of autophagosomes similarly to the pH 7.4 condition whereas treatment with CQ failed to do so (Fig. 2D).
Autophagy inhibition by Lys-01 is detectable at acidic conditions
We have so far shown that CQ is unable to block the autophagic flux in cancer cells cultured at acidic pH, suggesting that CQ may be a poorly effective inhibitor of autophagy in the acidic tumor microenvironment in vivo. Recently, a dimeric CQ derivative named Lys-01 (or its water-soluble form Lys-05) has been described as a much more potent autophagy inhibitor with a strong single-agent antitumor activity.34 Interestingly, this compound accumulates at much higher concentrations as compared with HCQ in whole-cell homogenates and lysosomes within tumors grown in mice. This activity is partially explained by the increased capacity of Lys-01 to deacidify lysosomes, although the authors do not exclude targeting of unknown lysosomal proteins.34
Therefore, we compared the autophagy-blocking activity of CQ and Lys-01 under different pH culture conditions. Since the activity of CQ was undetectable already at pH 6.8 we restricted our analysis to pH 7.4 and pH 6.8 conditions. We observed that Lys-01 induced accumulation of LC3-II at pH 7.4 in the range 5 to 50 μM (data not shown). However, in contrast to CQ the autophagy-blocking activity of Lys-01 was maintained in cells cultured at pH 6.8 (Fig. 3A and B). These results were confirmed using HOS-GFP-LC3 cells cultured at pH 7.4 or 6.8 in presence of BafA1, CQ and Lys-01. In line with its reported activity and the data in Figure 3B, Lys-01 showed a higher autophagy-inhibiting capacity as compared with CQ, since even at pH 6.8 Lys-01 could efficiently induce autophagosomes accumulation with the same extent as BafA1 treatment (Fig. 3C and D). However, the GFP-LC3 signal detected in cells treated with Lys-01 significantly decreased from pH 7.4 to pH 6.8, suggesting that also the activity of Lys-01 is pH-dependent.

Figure 3. Autophagy inhibition by Lys-01 is detectable at acidic pH. HCT116 (A) and HOS (B) cells were cultured at different pH conditions and the ability of BafA1, CQ and Lys01 to induce LC3-II accumulation was analyzed by WB. Data in (A and B) are representative from 2 independent experiments. (C) HOS-GFP-LC3 cells were exposed to media at pH 7.4 and pH 6.8 for 24 h and different autophagy inhibitors were added during the last 4 h of incubation. Cells were collected and permeabilized as described in Materials and Methods and the accumulation of GFP-LC3+ autophagosomes was analyzed by flow cytometry. (D) The histograms show GFP-LC3 fluorescence from the experiments described in (C). Data are reported as means ± standard deviations from 3 independent experiments.
CQ and Lys-01 cellular uptake
Considering the reported action of Lys-01 in vivo where tumors develop in hypoxic and acidic conditions, we reckoned that the increased capacity to enter cancer cells also at low pH and block autophagy might contribute to the stronger antitumor activity reported for Lys-01. In order to test this possibility, we performed high-performance liquid chromatography (HPLC) analysis of intracellular CQ and Lys-01 concentration in HCT116 cells cultured at pH 7.4 and pH 6.8. Cells were exposed to the different medium and treated for 4 h with 50 μM CQ or Lys-01. We observed a clear pH-dependent intracellular accumulation of CQ (Table 1). In fact, CQ concentrations in the whole-cell lysate were 7-fold lower at pH 6.8 as compared with pH 7.4 (P < 0.05). This finding suggests that the lack of autophagy inhibition at acidic pH may be due to the dramatic reduction of CQ uptake in acidic conditions. As previously reported,34 intracellular Lys-01 concentration was significantly higher than CQ at pH 7.4 (P < 0.05, Table 1), consistent with its higher basicity. Lys-01 also showed a pH-dependent cellular accumulation. However, the amount of compound entering cells at acidic pH was significantly higher with respect to CQ (P < 0.05), which may explain its ability to block autophagy at pH 6.8. In line with this, analysis of CTSB in HCT116 cells at pH 6.8 showed that mature CTSB was present in CQ-treated cells but strongly reduced in Lys-01-treated cells (Fig. S3B). This suggests that, likely because of a higher intracellular concentration, Lys-01 still targets the lysosomes at pH 6.8.
Table 1. Influence of pH on intracellular CQ and Lys-01 concentration.
| pH 7.4 | pH 6.8 | |
|---|---|---|
| CQ | 34 ± 6 μM | 5 ± 1 μM |
| Lys-01 | 89 ± 15 μM | 52 ± 4 μM |
HCT116 cells were exposed for 4 h to CQ (50 μM) or Lys-01 (50 μM) in medium buffered at pH 7.4 and 6.8. Whole cell lysate was subjected to HPLC analysis. Data are expressed as means ± standard deviations.
Being a weak base CQ may be further protonated in the acidic extracellular space surrounding cancer cells, thus significantly reducing its capacity to freely cross the plasma membrane.25 It is reported that the intracellular accumulation of CQ is pH-dependent and that CQ accumulates at lower concentrations in cancer cells cultured at acidic pH,38 as also shown by our HPLC analysis. This may explain the lack of autophagy-inhibiting activity by CQ in cancer cells cultured in acidic conditions and might potentially hinder the clinical efficacy of CQ in combination anticancer therapies. In line with this observation, the activity of CQ in combination therapies may be dependent on both intrinsic cancer cell sensitivity to drugs and on whether the CQ levels achieved within the tumor are effective in blocking autophagy. Despite the expected anticancer effects of CQ, CQ-mediated autophagy inhibition does not increase sensitivity to radiation therapy in a model of breast cancer.33 Moreover, some published data indicate that CQ per se might slightly increase tumor growth,33,34 suggesting that efficacy of CQ to inhibit autophagy may also depend on tumor types and tumor microenvironment factors.
CQ does not block autophagy during chronic acidosis
The results described so far have been obtained in cells exposed for relatively short time (24 h) to an acidic environment and demonstrate the lack of autophagy inhibition by CQ in acidic conditions. However, cancer cells in vivo have the ability to adapt to chronic acidosis and acquire a more aggressive phenotype that may per se affect drug sensitivity.5,10,31,32,43 Moreover, it has been reported that acidosis increases the activity of P-glycoprotein, thus inducing multidrug resistance in cancer cells.44,45 In order to evaluate the activity of CQ and Lys-01 in cancer cells exposed to chronic acidosis, we used the low pH-adapted HCT116pH6.8 and Me30966pH6.8 cells. To exclude major involvement of factors other than pH, the activity of different lysosomal inhibitors was tested in HCT116pH6.8 and Me30966pH6.8 cells exposed to medium at pH 6.8 or medium at pH 7.4. As illustrated in Figure 4A and B, these cells maintain a normal autophagic flux detected as LC3-II accumulation in presence of BafA1. As expected, while the autophagy-blocking activity of CQ and HCQ was totally abolished at pH 6.8, Lys-01 was still able to reduce the autophagic flux in cells adapted to chronic acidosis, although to a lesser extent with respect to BafA1. On the other hand, culturing HCT116pH6.8 and Me30966pH6.8 cells at pH 7.4 restored completely the sensitivity to the autophagy inhibition mediated by CQ and HCQ, suggesting that pH is a major factor modulating CQ and Lys-01 activity in acidic conditions.
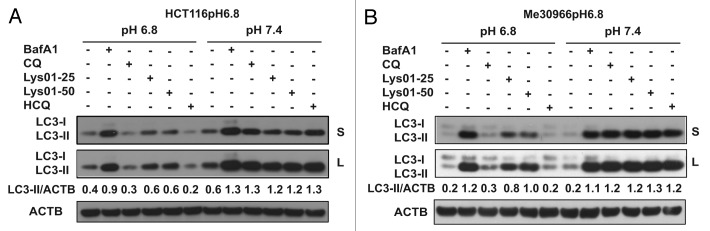
Figure 4. Autophagy-blocking activity of CQ and Lys-01 in low pH-adapted cells. The sublines HCT116pH6.8 (A) and Me30966pH6.8 (B) were used to evaluate accumulation of LC3-II by WB upon treatment with BafA1 (50 nM), CQ (50 μM), HCQ (50 μM) and Lys-01 (25 to 50 μM). Cells were exposed either to their medium buffered at pH 6.8 or to medium at pH 7.4 during the 4 h treatment with the different inhibitors. Data shown are representative from 2 independent experiments.
Low pH-adapted cancer cells are resistant to CQ cytotoxicity
In order to correlate the autophagy-inhibiting activity of Lys-01/CQ with its effects on cell viability, we used HCT116pH6.8 and Me30966pH6.8 cells and their parental cell lines (Table S1). As previously reported,34 Lys-01 showed an increased cytotoxicity in cells cultured at pH 7.4 with respect to CQ (Fig. 5A and B). However, while cells adapted to acidic pH showed total resistance to CQ, they were sensitive to the cytotoxic effects of Lys-01, although to a lesser extent as compared with cells at pH 7.4 (Fig. 5A and B). Since in vivo CQ is administered at repeated doses we tested also whether CQ and Lys-01 treatment repeated with a one-day interval improved their cytotoxicity. As shown in Figure S4 we could not observe any major difference between the 2 treatment schedules in all cell lines tested.

Figure 5. Cytotoxic activity of CQ and Lys-01 is pH-dependent. (A and B) Cell viability after 48 h exposure to CQ and Lys-01 was evaluated in HCT116 and HCT116pH6.8 cells (A) and Me30966 and Me30966pH6.8 cells (B). (C) Cell viability was evaluated in HCT116pH6.8 cells and Me30966pH6.8 cells cultured at their acidic pH or at pH 7.4. Data are expressed as means and standard deviations from 3 independent experiments.
Therapeutic strategies aiming at buffering tumor pH before administration of standard chemotherapy are under investigation in both preclinical11,29,30,46 and clinical settings (Clinical trials NCT01069081, NCT01198821). In order to simulate pH modulation in vivo we exposed low pH-adapted cancer cells to medium buffered at pH 7.4 and analyzed cell viability following exposure to CQ and Lys-01. Interestingly, as predicted by data shown in Figure 4, when low pH-adapted cells were exposed to medium at pH 7.4 the cytotoxicity of CQ was almost completely restored while that of Lys-01 was further increased (Fig. 5C). These data suggest that buffering tumor pH may significantly potentiate the autophagy-blocking activity of CQ and Lys-01.
Analysis of CQ effects on autophagy in vivo
In order to understand whether the observations reported might have significance in vivo, we performed IHC staining of HCT116 tumor sections from control mice and mice treated with CQ. Since markers specifically detecting acidic regions in tissues are not available we used the expression of hypoxia-induced CA9 (carbonic anhydrase IX) to identify tumor regions with expected low pH.12,47 Notably, CA9 is a major mediator of the extracellular acidification in tumors.10,48 Sequential tumor sections were immunostained for CA9 and LC3 and the presence of blood vessels was identified from the hematoxylin staining. As expected, CA9 expression was absent or very mild in the close proximity of vessels but its expression was nicely and strongly detected in the outer regions characterized by hypoxia (Fig. 6A). To quantify LC3 expression in the vascularized viable tumor regions we applied a method recently described.12 Briefly, we performed a pixel intensity analysis in the normoxic areas close to vessels (inner region) and in the hypoxic area delimiting the tumor tissue (outer region). First, the analysis of tumor tissues from untreated animals indicated that the expression of LC3 was significantly increased in the outer regions as compared with the inner regions by 7-fold (medians 1169 vs 7038, Fig. 6B). This observation suggests an increased autophagic compartment or the presence of a higher number of autophagic vesicles in cells in the hypoxic/acidic areas and confirms results recently reported by Wojtkowiak and colleagues.12 The autophagy-inhibiting activity of CQ may be detected in vivo by an increased LC3 positivity.26 In CQ-treated tumors, we observed a 5-fold increase in LC3 signal intensity in inner regions with respect to untreated tumors (medians 5191 vs 1169), indicating the ability of CQ to inhibit autophagy in areas with normal oxygen tension and an expected physiological pH (Fig. 6B). Interestingly, only a 1.4-fold increase in LC3 signal intensity was observed in outer regions of CQ-treated vs. untreated tumors (medians 9602 vs 7038), suggesting a dramatically reduced ability of CQ treatment to inhibit autophagy in hypoxic/acidic regions (Fig. 6B). Given the high volume distribution of CQ49 the effects reported are unlikely to be due to different perfusion to tumor regions.

Figure 6. Effects of CQ treatment on LC3 expression in HCT116 tumors in vivo. (A) Histological analysis of CA9 and LC3 expression in sequential sections of tumor regions. HE staining was used to identify viable and necrotic regions. The expression of CA9 identifies hypoxic regions about 100 μm away from the blood vessels while necrotic areas surrounding the CA9+ ring are indicated by N. The right panel shows a positive pixel analysis performed by using the Aperio ImageScope with the algorithm Positive Pixel Count v9 where strong positive pixels are shown in red. Blood vessels are shown as a light red shape with a blue outline. The white boxes and the black boxes represent inner regions and outer regions, respectively. The figure shows representative images from a tumor region for control and CQ-treated tumors. (B) The number of strong LC3 positive pixels in inner and outer regions from untreated and CQ-treated tumors is shown. Data were obtained from a total of 50 regions randomly selected from each group. Data are expressed as box plots showing medians and 25% to 75% percentiles. Differences between groups were analyzed by Mann-Whitney test and P values are shown.
Inhibition of autophagy is currently being evaluated as a potent tool in anticancer therapies and CQ/HCQ are used as autophagy inhibitor in combination with chemotherapy in ongoing clinical trials. We and others have reported recently that autophagy is a mechanism for tumor cell adaptation to an acidic environment, suggesting that targeting autophagy and buffering pH in these malignant cells may be a therapeutic strategy.12,13 However, CQ and HCQ are unable to block autophagy in cancer cells in acidic conditions in vitro and in hypoxic/acidic tumor regions in vivo. Moreover, cancer cells under acidic conditions are totally resistant to CQ cytotoxic activity, thus questioning its in vivo efficacy. Such results are somehow predicted by the chemical properties of CQ, a weak base whose protonation equilibrium is shifted toward the fully protonated form in acidic conditions, thus limiting its penetration through the plasma membrane. A similar pH-dependent mechanism is responsible for the lack of efficacy of many chemotherapeutic drugs that are chemically weak bases.29,32,35,36,43,50 Moreover, cancer cells exposed to chronic acidosis may acquire mechanisms of resistance to CQ (or Lys-01) other than those merely dependent on extracellular acidic pH or to the chemical properties of drugs. It should also be considered that CQ effects on increased drug sensitivity may occur through cellular functions other than autophagy, as reported in vitro for breast cancer cells.51 CQ may affect drug sequestration and distribution, and promote cell death through other mechanisms,25 which make more complex the interpretation of data from combination therapies in cancer patients. For instance, the first study testing CQ in combination therapy in cancer patients grounded its rationale on the DNA-intercalating activity of CQ.52 In addition, although the concentrations of CQ needed to inhibit tumor growth are achievable in humans, very little is known about the actual autophagy inhibition in tumors, also because of the lack of reliable methods to measure autophagic flux in tumor tissues.53 In line with this, there is a general consensus that CQ/HCQ are not the ideal drugs to inhibit autophagy.54 For instance, suboptimal CQ concentrations may indeed have dramatic effects on lysosomal functions, leading to increased lysosomal activity and possibly to reactivation of autophagy. This might explain why low pH-adapted cell lines grow slightly faster at low CQ concentrations and why in some tumor models CQ treatment may increase tumor growth. We found that the newly reported CQ-derivative Lys-01, despite being still characterized by a pH-dependent activity, is more active than CQ as inhibitor of autophagy in cancer cells exposed to acidosis and also exerts a better cytotoxicity on these cells, supporting the notion that this compound might also target tumor cells in acidic areas in vivo. Unfortunately, we were unable to confirm this hypothesis in HCT116 xenografts because of the high toxicity of Lys-05 (the water soluble salt of Lys-01) in mice. We found that cells adapted to acidosis restore their sensitivity to CQ and increase sensitivity to Lys-01 after buffering of the growth medium. This observation suggests that the in vivo efficacy of CQ (or other similar drugs) as autophagy inhibitor might be improved by pretreatment with tumor pH-modulating agents.
Materials and Methods
Cell cultures
Me30966, A375, SK-Mel-28, HCT116 and HOS cell lines were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (HyClone, SV30160.03) and antibiotics. HOS cells were kindly provided by Sofia Avnet (IOR, Bologna, Italy). The HOS cell line stably expressing the GFP-LC3 plasmid (HOS-GFP-LC3) was a kind gift from Gerry McInerney (MTC, Karolinska Institute, Stockholm). All cells lines were grown at 37 °C in presence of 5% CO2. The low pH adapted HCT-116 (HCT116pH6.8) and Me30966 (Me30966pH6.8) cell lines were obtained by growing the parental cells in RPMI-1640 medium buffered at pH 6.8 for 3 mo. The different pH in media was achieved by adding a different concentration of NaHCO3 and letting the media equilibrate overnight in the incubator at 5% CO2. Actual pH in media was measured before and after each experiment.
Chemicals and antibodies
RPMI-1640 (SH30255.01), trypsin (SH40003.12), and phosphate-buffered saline (PBS, SH40003.12) were from HyClone. RPMI-1640 without NaHCO3 (51800) and Sodium Bicarbonate (25080) were from Gibco. BafA1 (B1793), CQ diphosphate salt (C6628), HCQ (H0915), protease cocktail tablets EDTA-free, phosphatase inhibitors (P5726, P0044) and bovine serum albumin (A7906) were from Sigma. The Lys-01 was synthesized by OncoTargeting AB (Sweden) and dissolved in DMSO. Protein assay standard I (5000-0007) and dry milk (170-6404) were from Bio-Rad. The following antibodies were used: LC3B (Cell Signaling Technology, 2775), cathepsin B (CTSB) (Calbiochem, PC41) and β-actin (ACTB) (Sigma, A5441). HRP-conjugated anti-rabbit (NA934V) and anti-mouse (NXA931) antibodies, ECL system (RPN2106) and PVDF membranes (RPN303F) were from GE Healthcare.
Treatment with acidic media and autophagy inhibitors
Cells were plated in standard RPMI buffered at pH 7.4. The next day the medium was replaced with media buffered at different pH (7.4, 6.8 and 6.5). After 20 h of exposure to medium at different pH, cells were treated with BafA1, CQ, HCQ and Lys-01 for 4 h and then collected for further analysis.
Western blotting
Cells were washed on ice and collected by scraping in cold PBS. The cell pellet was lysed in RIPA buffer (150 mM NaCl, 50 mM Tris pH 7.4, 1% Nonidet P-40 [Sigma, I3021], 0.1% SDS and 0.5% sodium deoxycholate) in presence of protease and phosphatase inhibitors. The protein concentration was determined by Biorad Protein Assay (Bio-Rad Laboratories, 500-0006) and an equal amount of proteins (20 μg) was loaded on precast acrylamide gels (4–12% SDS-PAGE, NuPage; Life Technologies, NP0335-NP0336). The proteins were transferred from the gel to PVDF membrane for 2 h at 4 °C. Red Ponceau staining of the membranes verified the proper loading and transfer. Membranes were blocked in 5% blotting grade dry milk in TBS with 0.1% Tween (TBS-T) for 1 h at room temperature and then incubated with primary antibodies diluted in 5% bovine serum albumin in TBS-T overnight at 4 °C. The next day membranes were washed and incubated for 1 h at room temperature with the appropriate HRP-conjugated secondary antibody and the binding was detected by the ECL system.
Cell viability assay
HCT116 and Me30966 cells (parental and low-pH adapted) were plated into 96-wells plates and the next day treated with different concentrations of CQ and Lys-01. Forty-eight h after treatment cell viability was evaluated by using the acid phosphatase assay as previously described.55
HPLC analysis of CQ and Lys-01 intracellular content
HCT116 cells were plated overnight and the next day medium was replaced with fresh medium at pH 7.4 and pH 6.8. Immediately after, CQ or Lys-01 (50 μM for both) were added to the cells for 4 h. Cells were then collected and whole cell lysate (5 μg/μl) was processed for HPLC analysis (OncoTargeting AB). Briefly, proteins were precipitated with acetonitrile containing the internal standard. Samples were subsequently shaken vigorously, centrifuged, and the resulting supernatant was transferred to HPLC-vials for analysis. Stock concentrations of 150 μM CQ and Lys-01 were used to prepare the calibration curves. All samples were analyzed by first separating them by reversed phase gradient LC and subsequently detecting them using positive electrospray ionization and multiple reaction monitoring (MRM) of the transitions m/z: 440.5 to 205.2 (Lys-01) and 320.5 to 247.2 (CQ).
Flow cytometry
HOS-GFP-LC3 cells (100,000 cells/dish) were plated into 20 cm2 dishes. The next day medium was replaced with medium at pH 7.4 or 6.8 for 24 h. BafA1 (50 nM), CQ (50 μM) or Lys-01 (50 μM) were added during the last 4 h incubation. Cells were collected by trypsinization and treated with 0.05% saponin (Biochemika, 47036) in PBS for 10 min at RT. After washing in PBS the cells were collected and immediately analyzed by a FACSCalibur instrument using Cellquest software (Becton Dickinson). GFP fluorescence was collected from at least 10,000 cells/sample.
Animal experiments
HCT116 and HT29 colon carcinoma cells (5 × 106) were injected subcutaneously in the right rear flank of female NMRI nu/nu mice. When tumors reached the size of 0.1 mL, mice were randomized into control or treatment groups and injected i.p. with CQ (20 mg/kg) every second day. Tumor volume was measured every second day and experiments were stopped 16 d after start of treatment. Animal studies were approved by the Stockholm North ethics committee and performed by Adlego AB (Stockholm), in accordance with Swedish governmental and European statutory regulations on animal welfare.
Immunohistochemistry
HCT116 tumors were fixed in 2% buffered formalin, dehydrated, embedded in paraffin, and sequential sections were obtained. The sections were deparaffinized with xylene, rehydrated, and microwaved. Sections were immunostained with rabbit anti-human LC3B (Cell Signaling Technology, 2775) and mouse anti-CA9 (clone M75, BioScience Slovakia) and visualized by the avidin–biotin–peroxidase complex technique (Vector Laboratories). Mayer hematoxylin was used for counterstaining. Histological slides were analyzed independently by 2 operators using the software Aperio ImageScope as previously described.12 For each vascularized tumor region, 5 inner regions and 5 outer regions containing 45000 pixels were randomly selected. Statistical analysis was performed on a total of 50 inner and 50 outer regions for each treatment group. Differences between groups were analyzed by the Mann-Whitney test.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by grants from the Association for International Cancer Research (grant #11-0522), the Swedish Cancer Society (grant #CAN 2012/415), and the Sigurd and Elsa Goljes Foundation. Angela Strambi was supported by the Boncompagni-Ludovisi Foundation. We are grateful to Patrice Codogno for helpful discussion and critical reading of the manuscript, and to Giuseppe Di Lernia for technical help.
Glossary
Abbreviations:
- BafA1
bafilomycin A1
- CQ
chloroquine
- HCQ
hydroxychloroquine
- HPLC
high-performance liquid chromatography
- CTSB
cathepsin B
- CA9
carbonic anhydrase IX
References
- 1.Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4:891–9. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- 2.Gatenby RA, Smallbone K, Maini PK, Rose F, Averill J, Nagle RB, Worrall L, Gillies RJ. Cellular adaptations to hypoxia and acidosis during somatic evolution of breast cancer. Br J Cancer. 2007;97:646–53. doi: 10.1038/sj.bjc.6603922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gillies RJ, Robey I, Gatenby RA. Causes and consequences of increased glucose metabolism of cancers. J Nucl Med. 2008;49(Suppl 2):24S–42S. doi: 10.2967/jnumed.107.047258. [DOI] [PubMed] [Google Scholar]
- 4.Fais S, De Milito A, You H, Qin W. Targeting vacuolar H+-ATPases as a new strategy against cancer. Cancer Res. 2007;67:10627–30. doi: 10.1158/0008-5472.CAN-07-1805. [DOI] [PubMed] [Google Scholar]
- 5.Gerweck LE. Tumor pH: implications for treatment and novel drug design. Semin Radiat Oncol. 1998;8:176–82. doi: 10.1016/S1053-4296(98)80043-X. [DOI] [PubMed] [Google Scholar]
- 6.Martínez-Zaguilán R, Raghunand N, Lynch RM, Bellamy W, Martinez GM, Rojas B, Smith D, Dalton WS, Gillies RJ. pH and drug resistance. I. Functional expression of plasmalemmal V-type H+-ATPase in drug-resistant human breast carcinoma cell lines. Biochem Pharmacol. 1999;57:1037–46. doi: 10.1016/S0006-2952(99)00022-2. [DOI] [PubMed] [Google Scholar]
- 7.Morita T, Nagaki T, Fukuda I, Okumura K. Clastogenicity of low pH to various cultured mammalian cells. Mutat Res. 1992;268:297–305. doi: 10.1016/0027-5107(92)90235-T. [DOI] [PubMed] [Google Scholar]
- 8.Calcinotto A, Filipazzi P, Grioni M, Iero M, De Milito A, Ricupito A, Cova A, Canese R, Jachetti E, Rossetti M, et al. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res. 2012;72:2746–56. doi: 10.1158/0008-5472.CAN-11-1272. [DOI] [PubMed] [Google Scholar]
- 9.Marino ML, Fais S, Djavaheri-Mergny M, Villa A, Meschini S, Lozupone F, Venturi G, Della Mina P, Pattingre S, Rivoltini L, et al. Proton pump inhibition induces autophagy as a survival mechanism following oxidative stress in human melanoma cells. Cell Death Dis. 2010;1:e87. doi: 10.1038/cddis.2010.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov. 2011;10:767–77. doi: 10.1038/nrd3554. [DOI] [PubMed] [Google Scholar]
- 11.Robey IF, Baggett BK, Kirkpatrick ND, Roe DJ, Dosescu J, Sloane BF, Hashim AI, Morse DL, Raghunand N, Gatenby RA, et al. Bicarbonate increases tumor pH and inhibits spontaneous metastases. Cancer Res. 2009;69:2260–8. doi: 10.1158/0008-5472.CAN-07-5575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wojtkowiak JW, Rothberg JM, Kumar V, Schramm KJ, Haller E, Proemsey JB, Lloyd MC, Sloane BF, Gillies RJ. Chronic autophagy is a cellular adaptation to tumor acidic pH microenvironments. Cancer Res. 2012;72:3938–47. doi: 10.1158/0008-5472.CAN-11-3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marino ML, Pellegrini P, Di Lernia G, Djavaheri-Mergny M, Brnjic S, Zhang X, Hägg M, Linder S, Fais S, Codogno P, et al. Autophagy is a protective mechanism for human melanoma cells under acidic stress. J Biol Chem. 2012;287:30664–76. doi: 10.1074/jbc.M112.339127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Amaravadi RK, Lippincott-Schwartz J, Yin XM, Weiss WA, Takebe N, Timmer W, DiPaola RS, Lotze MT, White E. Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res. 2011;17:654–66. doi: 10.1158/1078-0432.CCR-10-2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang ZJ, Chee CE, Huang S, Sinicrope FA. The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther. 2011;10:1533–41. doi: 10.1158/1535-7163.MCT-11-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, Thomas-Tikhonenko A, Thompson CB. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest. 2007;117:326–36. doi: 10.1172/JCI28833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mathew R, White E. Autophagy in tumorigenesis and energy metabolism: friend by day, foe by night. Curr Opin Genet Dev. 2011;21:113–9. doi: 10.1016/j.gde.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gong C, Bauvy C, Tonelli G, Yue W, Deloménie C, Nicolas V, Zhu Y, Domergue V, Marin-Esteban V, Tharinger H, et al. Beclin 1 and autophagy are required for the tumorigenicity of breast cancer stem-like/progenitor cells. Oncogene. 2013;32:2261–72, 1-11. doi: 10.1038/onc.2012.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rausch V, Liu L, Apel A, Rettig T, Gladkich J, Labsch S, Kallifatidis G, Kaczorowski A, Groth A, Gross W, et al. Autophagy mediates survival of pancreatic tumour-initiating cells in a hypoxic microenvironment. J Pathol. 2012;227:325–35. doi: 10.1002/path.3994. [DOI] [PubMed] [Google Scholar]
- 20.Yue W, Hamaï A, Tonelli G, Bauvy C, Nicolas V, Tharinger H, Codogno P, Mehrpour M. Inhibition of the autophagic flux by salinomycin in breast cancer stem-like/progenitor cells interferes with their maintenance. Autophagy. 2013;9:714–29. doi: 10.4161/auto.23997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Høyer-Hansen M, Jäättelä M. Autophagy: an emerging target for cancer therapy. Autophagy. 2008;4:574–80. doi: 10.4161/auto.5921. [DOI] [PubMed] [Google Scholar]
- 22.Kuwahara Y, Oikawa T, Ochiai Y, Roudkenar MH, Fukumoto M, Shimura T, Ohtake Y, Ohkubo Y, Mori S, Uchiyama Y, et al. Enhancement of autophagy is a potential modality for tumors refractory to radiotherapy. Cell Death Dis. 2011;2:e177. doi: 10.1038/cddis.2011.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lomonaco SL, Finniss S, Xiang C, Decarvalho A, Umansky F, Kalkanis SN, Mikkelsen T, Brodie C. The induction of autophagy by gamma-radiation contributes to the radioresistance of glioma stem cells. Int J Cancer. 2009;125:717–22. doi: 10.1002/ijc.24402. [DOI] [PubMed] [Google Scholar]
- 24.Zhuang W, Li B, Long L, Chen L, Huang Q, Liang Z. Induction of autophagy promotes differentiation of glioma-initiating cells and their radiosensitivity. Int J Cancer. 2011;129:2720–31. doi: 10.1002/ijc.25975. [DOI] [PubMed] [Google Scholar]
- 25.Solomon VR, Lee H. Chloroquine and its analogs: a new promise of an old drug for effective and safe cancer therapies. Eur J Pharmacol. 2009;625:220–33. doi: 10.1016/j.ejphar.2009.06.063. [DOI] [PubMed] [Google Scholar]
- 26.Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hurwitz SJ, Terashima M, Mizunuma N, Slapak CA. Vesicular anthracycline accumulation in doxorubicin-selected U-937 cells: participation of lysosomes. Blood. 1997;89:3745–54. [PubMed] [Google Scholar]
- 28.Lee CM, Tannock IF. Inhibition of endosomal sequestration of basic anticancer drugs: influence on cytotoxicity and tissue penetration. Br J Cancer. 2006;94:863–9. doi: 10.1038/sj.bjc.6603010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luciani F, Spada M, De Milito A, Molinari A, Rivoltini L, Montinaro A, Marra M, Lugini L, Logozzi M, Lozupone F, et al. Effect of proton pump inhibitor pretreatment on resistance of solid tumors to cytotoxic drugs. J Natl Cancer Inst. 2004;96:1702–13. doi: 10.1093/jnci/djh305. [DOI] [PubMed] [Google Scholar]
- 30.De Milito A, Marino ML, Fais S. A rationale for the use of proton pump inhibitors as antineoplastic agents. Curr Pharm Des. 2012;18:1395–406. doi: 10.2174/138161212799504911. [DOI] [PubMed] [Google Scholar]
- 31.Gatenby RA, Gillies RJ. A microenvironmental model of carcinogenesis. Nat Rev Cancer. 2008;8:56–61. doi: 10.1038/nrc2255. [DOI] [PubMed] [Google Scholar]
- 32.Trédan O, Galmarini CM, Patel K, Tannock IF. Drug resistance and the solid tumor microenvironment. J Natl Cancer Inst. 2007;99:1441–54. doi: 10.1093/jnci/djm135. [DOI] [PubMed] [Google Scholar]
- 33.Bristol ML, Emery SM, Maycotte P, Thorburn A, Chakradeo S, Gewirtz DA. Autophagy inhibition for chemosensitization and radiosensitization in cancer: do the preclinical data support this therapeutic strategy? J Pharmacol Exp Ther. 2013;344:544–52. doi: 10.1124/jpet.112.199802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McAfee Q, Zhang Z, Samanta A, Levi SM, Ma XH, Piao S, Lynch JP, Uehara T, Sepulveda AR, Davis LE, et al. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc Natl Acad Sci U S A. 2012;109:8253–8. doi: 10.1073/pnas.1118193109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.De Milito A, Fais S. Tumor acidity, chemoresistance and proton pump inhibitors. Future Oncol. 2005;1:779–86. doi: 10.2217/14796694.1.6.779. [DOI] [PubMed] [Google Scholar]
- 36.Raghunand N, Mahoney BP, Gillies RJ. Tumor acidity, ion trapping and chemotherapeutics. II. pH-dependent partition coefficients predict importance of ion trapping on pharmacokinetics of weakly basic chemotherapeutic agents. Biochem Pharmacol. 2003;66:1219–29. doi: 10.1016/S0006-2952(03)00468-4. [DOI] [PubMed] [Google Scholar]
- 37.Tannock IF, Rotin D. Acid pH in tumors and its potential for therapeutic exploitation. Cancer Res. 1989;49:4373–84. [PubMed] [Google Scholar]
- 38.Jensen PB, Sørensen BS, Sehested M, Grue P, Demant EJ, Hansen HH. Targeting the cytotoxicity of topoisomerase II-directed epipodophyllotoxins to tumor cells in acidic environments. Cancer Res. 1994;54:2959–63. [PubMed] [Google Scholar]
- 39.Wu H, Ding Z, Hu D, Sun F, Dai C, Xie J, Hu X. Central role of lactic acidosis in cancer cell resistance to glucose deprivation-induced cell death. J Pathol. 2012;227:189–99. doi: 10.1002/path.3978. [DOI] [PubMed] [Google Scholar]
- 40.Mort JS, Buttle DJ. Cathepsin B. Int J Biochem Cell Biol. 1997;29:715–20. doi: 10.1016/S1357-2725(96)00152-5. [DOI] [PubMed] [Google Scholar]
- 41.Eng KE, Panas MD, Karlsson Hedestam GB, McInerney GM. A novel quantitative flow cytometry-based assay for autophagy. Autophagy. 2010;6:634–41. doi: 10.4161/auto.6.5.12112. [DOI] [PubMed] [Google Scholar]
- 42.Kneen M, Farinas J, Li Y, Verkman AS. Green fluorescent protein as a noninvasive intracellular pH indicator. Biophys J. 1998;74:1591–9. doi: 10.1016/S0006-3495(98)77870-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.De Milito A, Fais S. Proton pump inhibitors may reduce tumour resistance. Expert Opin Pharmacother. 2005;6:1049–54. doi: 10.1517/14656566.6.7.1049. [DOI] [PubMed] [Google Scholar]
- 44.Sauvant C, Nowak M, Wirth C, Schneider B, Riemann A, Gekle M, Thews O. Acidosis induces multi-drug resistance in rat prostate cancer cells (AT1) in vitro and in vivo by increasing the activity of the p-glycoprotein via activation of p38. Int J Cancer. 2008;123:2532–42. doi: 10.1002/ijc.23818. [DOI] [PubMed] [Google Scholar]
- 45.Thews O, Gassner B, Kelleher DK, Schwerdt G, Gekle M. Impact of extracellular acidity on the activity of P-glycoprotein and the cytotoxicity of chemotherapeutic drugs. Neoplasia. 2006;8:143–52. doi: 10.1593/neo.05697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.De Milito A, Canese R, Marino ML, Borghi M, Iero M, Villa A, Venturi G, Lozupone F, Iessi E, Logozzi M, et al. pH-dependent antitumor activity of proton pump inhibitors against human melanoma is mediated by inhibition of tumor acidity. Int J Cancer. 2010;127:207–19. doi: 10.1002/ijc.25009. [DOI] [PubMed] [Google Scholar]
- 47.Raleigh JA, Calkins-Adams DP, Rinker LH, Ballenger CA, Weissler MC, Fowler WC, Jr., Novotny DB, Varia MA. Hypoxia and vascular endothelial growth factor expression in human squamous cell carcinomas using pimonidazole as a hypoxia marker. Cancer Res. 1998;58:3765–8. [PubMed] [Google Scholar]
- 48.Swietach P, Patiar S, Supuran CT, Harris AL, Vaughan-Jones RD. The role of carbonic anhydrase 9 in regulating extracellular and intracellular ph in three-dimensional tumor cell growths. J Biol Chem. 2009;284:20299–310. doi: 10.1074/jbc.M109.006478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ducharme J, Farinotti R. Clinical pharmacokinetics and metabolism of chloroquine. Focus on recent advancements. Clin Pharmacokinet. 1996;31:257–74. doi: 10.2165/00003088-199631040-00003. [DOI] [PubMed] [Google Scholar]
- 50.Raghunand N, He X, van Sluis R, Mahoney B, Baggett B, Taylor CW, Paine-Murrieta G, Roe D, Bhujwalla ZM, Gillies RJ. Enhancement of chemotherapy by manipulation of tumour pH. Br J Cancer. 1999;80:1005–11. doi: 10.1038/sj.bjc.6690455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maycotte P, Aryal S, Cummings CT, Thorburn J, Morgan MJ, Thorburn A. Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy. Autophagy. 2012;8:200–12. doi: 10.4161/auto.8.2.18554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sotelo J, Briceño E, López-González MA. Adding chloroquine to conventional treatment for glioblastoma multiforme: a randomized, double-blind, placebo-controlled trial. Ann Intern Med. 2006;144:337–43. doi: 10.7326/0003-4819-144-5-200603070-00008. [DOI] [PubMed] [Google Scholar]
- 53.Mancias JD, Kimmelman AC. Targeting autophagy addiction in cancer. Oncotarget. 2011;2:1302–6. doi: 10.18632/oncotarget.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Garber K. Inducing indigestion: companies embrace autophagy inhibitors. J Natl Cancer Inst. 2011;103:708–10. doi: 10.1093/jnci/djr168. [DOI] [PubMed] [Google Scholar]
- 55.Friedrich J, Eder W, Castaneda J, Doss M, Huber E, Ebner R, Kunz-Schughart LA. A reliable tool to determine cell viability in complex 3-d culture: the acid phosphatase assay. J Biomol Screen. 2007;12:925–37. doi: 10.1177/1087057107306839. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.