

Abstract
Non alcoholic steatohepatitis (NASH) is the more severe form of nonalcoholic fatty liver disease. In NASH, fatty liver, hepatic inflammation, hepatocyte injury and fibrogenesis are associated, and thi condition may eventually lead to cirrhosis. Current treatment of NASH relies on the reduction of body weight and increase in physical activity, but there is no pharmacologic treatment approved as yet. Emerging data indicate that NASH progression results from parallel events originating from the liver as well as from the adipose tissue, the gut and the gastrointestinal tract. Thus, dysfunction of the adipose tissue through enhanced flow of free fatty acids and release of adipocytokines, and alterations in the gut microbiome generate proinflammatory signals that underly NASH progression. Additional ‘extrahepatic hits’ include dietary factors and gastrointestinal hormones. Within the liver, hepatocyte apoptosis, ER stress and oxidative stress are key contributors to hepatocellular injury. In addition, lipotoxic mediators and danger signals activate Kupffer cells which initiate and perpetuate the inflammatory response by releasing inflammatory mediators that contribute to inflammatory cell recruitment and development of fibrosis. Inflammatory and fibrogenic mediators include chemokines, the cannabinoid system, the inflammasome and activation of pattern-recognition receptors.
Here we review the major mechanisms leading to appearance and progression of NASH, focusing on both extrahepatic signals and local inflammatory mechanisms, in an effort to identify the most promising molecular targets for the treatment of this condition.
Keywords: Fatty Liver, physiopathology, therapy, Humans, Insulin Resistance
1. Introduction and dimension of the problem
Nonalcoholic fatty liver disease (NAFLD) is the most common hepatic alteration in the Western world, due to its well-established association with the metabolic syndrome, the prevalence of which is increasing in both affluent and developing countries. The more severe form of NAFLD, nonalcoholic steatohepatitis (NASH), is characterized by inflammatory infiltration and hepatocellular damage, with or without fibrosis. While simple (or ‘bland’) steatosis is considered a relatively benign condition due to the extremely low likelihood to progress to cirrhosis [1, 2], NASH is associated with the potential to progress to the entire clinical picture of advanced liver disease. Indeed, fat accumulation in the liver generates inflammatory signals and reactive oxygen species that can amplify liver injury and stimulates fibrosis. This is similar to the picture observed in different etiologies of chronic liver injury, such as chronic HCV or HBV infection, where the spectrum of alterations may range from a near normal liver to severe hepatitis and cirrhosis. The development of NASH is the result of a complex interaction between the environment, represented in the case of NAFLD by a sedentary lifestyle and excessive intake of calories, and individual predisposition, that affects both the development of excess adiposity and its consequences.
In Europe, NAFLD is the underlying cause of damage in 40–60% patients with mild fibrosis [3], and in the USA, where one third of the population is overweight, it is estimated that 9 million patients have different degrees of NASH [4]. The actual role of NASH as a primary cause of advanced liver disease is not well known. Some reports suggest that NASH is the underlying cause of cirrhosis in 1–3% of patients undergoing liver transplantation, but compared to other chronic liver diseases, NASH-related cirrhosis is less likely to undergo transplantation, and this may underestimate the relevance of this disease as a cause of end-stage liver disease. Nonetheless, NASH is considered the principal cause of most cases of cryptogenic cirrhosis, and therefore the significance of NAFLD as a cause of end-stage liver disease is probably not negligible [5]. More important, due to the worldwide epidemics in obesity, the prevalence of end-stage liver disease among NAFLD patients is expected to rise in the future.
Like for any other chronic liver disease, appearance and progression of fibrosis is a critical event in the course of NASH, although fibrosis does not represent a prerequisite for a diagnosis of NASH. Besides the role of NASH as a factor potentially leading to cirrhosis, decompensation, and hepatocellular carcinoma [6], several studies have highlighted the existence of an increased cardiovascular risk in these patients [1]. These data clearly indicate that treatment of NASH is an unmet need of outstanding relevance in Hepatology and Internal Medicine. This review will summarize the available evidence defining critical aspects of the pathophysiology of NASH. As no established treatment exists for this condition, discussion will be focused on the potential therapeutic targets that have been highlighted by experimental studies, and by recent advances in the field of adipose tissue biology and inflammation. In addition, reference will be made to clinical studies where approaches suggested by experimental investigation have been translated to the clinical practice. However, a detailed analysis of published clinical trials for the treatment of NASH is outside the scopes of this paper, and the reader may refer to several recent reviews [7–9].
2. Significance of NASH progression
Although the natural history of NAFLD is not well known, most studies suggest that the existence of NASH is a prerequisite to develop progressive fibrosis. On the other hand, the appearance and development of fibrosis represents a critical factor for the eventual progression to cirrhosis and end-stage liver disease. Like for other chronic liver diseases, identifying the patients that are more likely to progress is a clinical challenge. In the field of NASH, not only it is relevant to know the stage of fibrosis in the presence of established NASH, but it also often necessary to differentiate simple, non-evolutive steatosis from steatohepatitis. A number of clinical factors have been demonstrated to provide some prediction of the likelihood to observe advanced fibrosis or cirrhosis in a given patient. Age >45 years has been shown to predict fibrosis and cirrhosis in different studies [10, 11]. In contrast, race and female gender have been identified as predictive factors only in a minority of studies [11]. Components of the metabolic syndrome are also associated with disease progression, and the majority of patients presenting with NASH have metabolic abnormalities, including type 2 diabetes, obesity, dyslipidemia, and hypertension. In particular, diabetes, obesity, and hypertension have been independently associated with advanced disease [10–14]. Finally, more advanced fibrosis parallels the number of components of the metabolic syndrome present in the patients [15].
Genetic factors
Recent studies have demonstrated that polymorphisms of a number of candidate genes, including those encoding for immunoregulatory proteins, proinflammatory cytokines and fibrogenic factors, could influence the appearance of NASH and the eventual development of liver fibrosis in patients with NAFLD [16]. Genetic variations can explain susceptibility to develop NASH in patients with obesity and/or diabetes. Moreover, it has been hypothesized that genetic factors influence fibrosis progression. The role of HFE C282Y heterozygosity and iron accumulation are unclear, since studies have yielded contradictory results [17]. Combination of polymorphisms in profibrotic genes such as angiotensinogen and TGFβ1 are associated with advanced fibrosis, and polymorphisms in genes encoding for TNFα, microsomal triglyceride transfer protein, manganese superoxide dismutase, adrenergic receptors or interleukin 1α have been shown to influence fibrosis progression [16]. Similarly, variations of PNPLA3 are associated with histological severity in patients with NASH [18]. Finally, a recent report using a genome-wide association study identified several genes that regulate the susceptibility to develop NASH, including farnesyl diphosphate farnesyl transferase 1 [19], although other studies have not confirmed these findings. The results of these studies suggest that genetic variations could at least in part explain the broad variability in the natural history of NAFLD among patients with metabolic syndrome.
3. Fatty liver and insulin resistance: where it all begins?
Steatohepatitis develops only in a fraction of patients with NAFLD, while the majority presents with only ‘simple’ steatosis, but the two entities are considered part of the same disease spectrum. While fat accumulation is the common denominator of all forms of NAFLD, from a pathophysiological standpoint NASH develops when the excess lipid accumulation results in hepatic lipotoxicity. Toxic effects on hepatocytes, and possibly other liver cells, provokes injury, which in turn triggers an inflammatory and ‘would healing’ response that is the basis for the eventual development of fibrosis. Thus, a critical concept is that increased hepatic fat does not invariably result in hepatocellular damage, and understanding the mechanisms leading to lipotoxicity is of pivotal importance for the comprehension of this field. Anyhow, fat accumulation appears to be a prerequisite for the development of NASH. Therefore, it is also relevant to understand the events that lead to the initial accumulation of fat in the context of the metabolic syndrome.
Fatty liver
Fat accumulates in the liver mainly in the form of triglycerides, although several other lipid species are present. Human and animal studies have shown that accumulation of triglycerides in NAFLD is the result of the expansion of the intrahepatic pool of free fatty acids (FFA). FFA influx is dependent on: a) the amount of FFA released by the adipose tissue due to insulin resistance and excessive lipolysis (see below); b) dietary fat via chylomicron metabolism; c) de novo lipogenesis (Figure 1). On the other hand, the liver may dispose of FFA mainly via fatty acid oxidation, both in the mitochondria and at extramitochondrial sites, or through assembly and export of triglycerides with VLDL. Human and animals studies have shown that more than half of the FFA pool is derived from excess adipose tissue lipolysis [20]. Importantly, the pathways that lead to FFA disposition are usually activated at a higher level in patients with NAFLD, indicating that the critical factor leading to fat accumulation is related to excessive inflow of FFA rather than to limited oxidation. In fact, insulin is no longer capable to suppress VLDL secretion, and this results in atherogenic dyslipidemia, characterized by low HDL and high LDL-cholesterol [21]. A remarkable exception to this view is represented by patients with impairment of VLDL assembly, as occurs in familial hypobetalipoproteinemia, where high levels of hepatic TG are not associated with insulin resistance or steatohepatitis [22].
Figure 1. Central role of free fatty acids (FFA) in the pathogenesis of NAFLD.
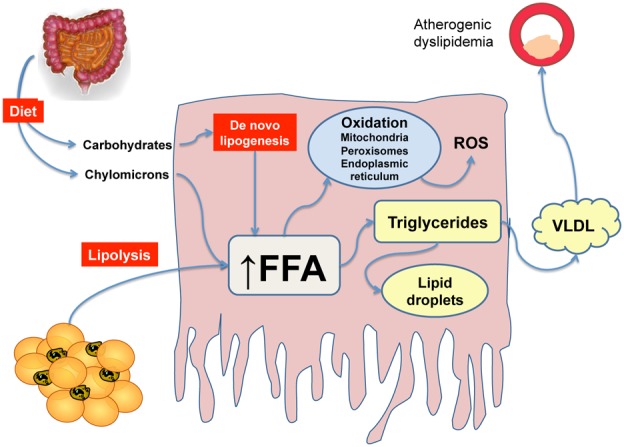
In the presence of insulin resistance, the majority of FFA reaching the liver derive from adipose tissue lipolysis. Lipids from the diet or de novo lipogenesis also contribute to expand the FFA pool. In the hepatocyte, FFA may be oxidized at mitochondrial and extramitochondrial sites, or incorporated in triglycerides. These in turn may be accumulated in lipid droplets, leading to steatosis or secreted in the circulation as VLDL, leading to a proatherogenic lipid pattern.
Although NASH occurs in the context of a fatty liver, it still remains to be established whether steatosis per se should be considered a primary target for the treatment of NASH. Some clinical studies would argue in this direction, as the amount of fat accumulating in the liver is a predictor of the presence of NASH. Nonetheless, experimental studies clearly demonstrate that interference with triglyceride accumulation and effective decrease of steatosis may not only be ineffective in ameliorating steatohepatitis, but could even worsen this condition. Diacylglycerol acyl transferases (DGAT) catalyze the final incorporation of a fatty acid in triglycerides. Inhibition of DGAT2 with antisense oligonucleotides was effective in reducing steatosis but caused more severe inflammation, oxidative stress and fibrosis [23]. In contrast, mice with hepatic overexpression of DGAT2 developed marked hepatic steatosis without liver injury or insulin resistance [24]. Nonetheless, the role of DGATs in NASH is more complex. Overexpression of DGAT1 in adipocytes and macrophages resulted in protection from adipose tissue and systemic inflammation, in spite of more severe obesity. On the other hand, DGAT1 knockout was found to be ineffective on steatosis, while it decreased fibrogenesis, increasing the amount of vitamin A stored in hepatic stellate cells (HSC) and inducing a less fibrogenic phenotype [25].
These results are of particular relevance, because they indicate that simply reducing the amount of triglycerides stored in the liver is unlikely to be an appealing target for the treatment of NASH. Dissociation between steatosis, insulin resistance and the features of NASH is also shown by data from the hepatic effects of bariatric surgery. While most of the available studies indicate that weight loss induced by this procedure is associated with reduced insulin resistance and an improvement of steatosis, the effects on necro-inflammation and fibrosis are more debatable [26]. Based on these observations, it appears that lipotoxicity that causes hepatocellular damage and subsequently NASH, should not be considered a synonym of steatosis (Figure 2). Moreover, these considerations introduce another unaddressed issue in the field of NASH, i.e. whether steatohepatitis develops in some patients after the appearance of a fatty liver or if the two entities originate as such in the individual patient. Although this uncertainty is difficult to be solved experimentally or with human studies, it is conceptually very important because it would allow the investigators to follow less strictly those patients without signs of inflammation and damage.
Figure 2. The fate of excess fatty acids is linked to the severity of nonalcoholic fatty liver disease.
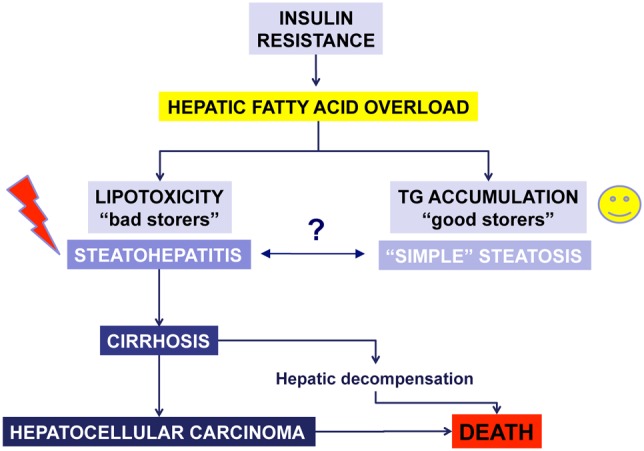
Increased hepatic inflow of fatty acids due to peripheral insulin resistance is associated in some patients with lipotoxic injury, while in other to limited damage. Experimental evidence indicates that accumulation of triglycerides may represent a protective mechanism to limit the toxic action of fatty acids and other lipotoxic products. In the presence of lipotoxicity, steatohepatitis develops possibly leading to end stage liver disease and its complications.
Insulin resistance
The degree of insulin resistance or the presence of diabetes are predictors of the existence of NASH or of a more advanced stage of fibrosis, suggesting a role for this disorder in the pathogenesis of steatohepatitis. Schematically, the current view suggests that insulin resistance originates at the level of the expanded and inflamed adipose tissue (see below), eventually involving the skeletal muscle, the pancreas, and the liver [7]. The presence of a fatty liver in turn contributes to the reduced response to insulin and aggravates alterations in glucose and lipid metabolism. It is well established that insulin resistance is associated with elevated levels of circulating insulin, and in hepatocytes, not all insulin-activated pathways are equally insensitive to the hormone. In fact, the ability of insulin to stimulate lipogenesis is maintained, via activation of the transcription factor sterol regulatory element binding protein-1c [20]. In addition, insulin preserves its mitogenic effects, that may contribute to promote carcinogenesis [27].
Along these lines, the possibility that other liver cells have an altered response to insulin has not been adequately addressed, but there are several experimental data suggest a possible direct role of insulin resistance in the pathogenesis of NASH and fibrosis. In particular, hepatic stellate cells (HSC) express receptors for insulin, insulin-like growth factor-I, and advanced glycation end-products, all of which generate profibrogenic signals, e.g. increasing the expression of type I collagen and connective tissue growth factor (CTGF) [28]. These data suggest that treatment of glucose dysmetabolism and/or of insulin resistance may contribute to ameliorate fibrosis occurring in the context of steatohepatitis. The possibility that insulin signaling directly affects the biology of other sinusoidal cells needs additional investigation [29].
In spite of the conceptual basis for the involvement of insulin resistance, the results of experimental studies and clinical trials did not provide definitive evidence indicating that modulation of this disturbance is effective for the treatment of NASH. Metformin, an activator of adenosine monophosphate-regulated kinase (AMPK), ameliorates glucose metabolism, has anti-inflammatory properties, and may interfere with profibrogenic pathways [30]. In spite of these actions, several published trials failed to show a consistent effect on NASH [31]. Thiazolidinediones (TZD) have been extensively evaluated for the treatment of NAFLD and NASH, and different studies in animal models and in patients have been published [7]. TZD mainly act activating the transcription factor peroxisome proliferator-activated receptor (PPAR)-gamma, which in turn targets pathways that may be beneficial in the treatment of NASH. PPAR-gamma activation improves insulin sensitivity in the adipose tissue and in the liver. In addition it induces adipose tissue differentiation towards a triglyceride-storing phenotype, limiting lipolysis. In the liver, TZD have been shown to be anti-inflammatory and anti-fibrogenic, acting on different targets, some of which are independent of PPAR-gamma, including NF-kappaB and AMPK. Specifically, PPAR-gamma has been shown to reduce Kupffer cell activation and cytokine secretion, and to polarize macrophages towards an anti-inflammatory phenotype [32].
PPAR-gamma-deficient mice fed a high-fat diet or administered a methionine- and choline-deficient diet are more sensitive to lipid accumulation or to the development of steatohepatitis [33]. PPAR-gamma is highly expressed in adipocytes, but is present also in different hepatic cells. In particular, PPAR-γ is expressed in quiescent HSCs and its expression decreases along the activation process that accompanies the acquisition of fibrogenic properties [34]. Moreover, exposure of HSCs to PPAR-γ ligands reverts most features of the activated phenotype of HSC and inhibits fibrosis in vivo [35]. However, it has been reported that starting the treatment with TZD once steatohepatitis is established results in limited effects on damage and fibrosis [36]. Making a parallel with studies in humans, treatment of patients with pioglitazone has been effective, as long as the treatment has been maintained, on steatosis and insulin resistance. In the recently published PIVENS trial the effects of pioglitazone on the NASH activity score were significantly higher than those of placebo, but similar to those of vitamin E. No treatment was superior to placebo in terms of fibrosis [37]. Based on these data it appears that solely treating insulin resistance is probably not sufficient to revert the complex effects of lipotoxicity that characterize NASH. Thus, treatments that specifically target the inflammatory and reparative process are more likely to interrupt the development and worsening of fibrosis and the eventual deterioration of liver function.
3. Signals from outside the liver
NAFLD is a typical systemic disease, where complex metabolic alterations affect the liver and many other tissues. While the critical step that leads to steatohepatitis is hepatocellular lipotoxic damage secondary to fat accumulation, the pathogenesis of NASH is far more complex and involves both a cross-talk among different resident and infiltrating liver cells, and the contribution of factors depending on extrahepatic systems.
3.1. Adipose tissue
Overweight and obesity are a major risk factor for NAFLD, and predict the eventual appearance of diabetes and other metabolic complications. Current knowledge points at the adipose tissue as a critical site for both the development of insulin resistance and the appearance of the changes that ultimately will cause NASH. While the initial site of alterations responsible for the reduction of insulin sensitivity remains debated, peripheral (i.e. adipose tissue) insulin resistance is considered a critical factor for the eventual development of NAFLD [38]. The first change is the expansion of the adipose tissue in the frame of obesity or overweight. Besides the contribution to insulin resistance, there are several mechanisms by which the expanded, inflamed, and overall dysfunctional adipose tissue leads to development of NASH (Figure 3).
Figure 3. Contribution of the adipose tissue to the development of nonalcoholic steatohepatitis.
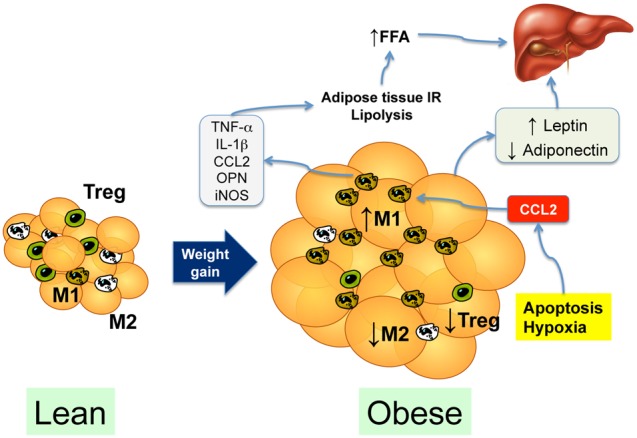
In the lean subject, M2-polarized macrophages and regulatory T cells (Treg) predominate in the vascular stromal fraction. Weight gain leads to expansion of the adipose tissue, resulting in tissue hypoxia and adipocyte apoptosis, leading to secretion of chemotactic cytokines such as CCL2 (MCP-1). CCL2 secretion recruits M1-polarized macrophages that secrete proinflammatory cytokines, contributing to insulin resistance, which increases lipolysis and the delivery of free fatty acids (FFA) to the liver. The expanded and inflamed adipose tissue also causes an imbalance in adipokine secretion, with an increase in leptin and a decrease in adiponectin circulating levels.
FFA release
A first consequence of the expanded and dysfunctional adipose tissue is the increased release of FFA in the circulation, due to the insufficient inhibition of lipolysis by insulin. Adipose tissue is the major energy storage site and triglyceride accumulation contributes to protect other tissues from the toxic effects of free fatty acids. The molecular mechanisms of lipid trafficking in adipocytes and in other cells have been recently reviewed [39]. The effects of insulin on the adipose tissue are mainly mediated by adipose tissue triglyceride lipase (ATGL) also known as PNPLA2, which belongs to the family of the patatin-like lipases. As flow of FFA to the liver is one of the initial and critical steps in the pathogenesis of NASH, regulation of ATGL activity may be a potential target in the effort to limit FFA flow to the liver. Recently, initial information on the phenotype of mice with genetic deletion of ATGL have been provided. Fuchs et al. [40] have demonstrated that lack of ATGL protects mice from tunicamycin-induced endoplasmic reticulum stress, in part via modification of the fatty acid profile. In mice with liver-specific inactivation of ATGL, severe progressive hepatic steatosis was found, although this was not associated with major metabolic abnormalities [41]. Moreover, while plasma ALT levels were elevated, inflammation and fibrosis were similar to or lower than those in controls.
The role of different lipases has also been evaluated in the light of the observation that mutations in PNPLA3 (adiponutrin) have an impact on the development of NAFLD and possibly on its progression. PNPLA3 has emerged from several genome-wide association studies as a potent factor predisposing to fatty liver [42]. Eventually, it has been reported that PNPLA3 mutations are also associated with a greater likelihood to have NASH and advanced fibrosis [18, 43]. The functional significance of PNPLA3 is still undetermined and only recently its major role as a nutritionally regulated lysophosphatidic acid acyltransferase has been clarified [44]. Interestingly, a sucrose-rich diet results in increased PNPLA3 activity, suggesting a mechanism for the increased susceptibility to steatosis in subjects with the I148M variant [44]. As expression of PNPLA3 is not unique to hepatocytes, it will be important to evaluate the possible role in other cells and the possible impact on steatohepatitis. Along these lines, the possibility to modulate the expression and/or function of this group of molecules represents a very interesting perspective for a targeted treatment in the field.
Adipose tissue inflammation
Obesity generates a state of chronic, low-grade inflammation, originating from the adipose tissue and the liver [45, 46]. Accumulation of immune cells in adipose tissue, particularly macrophages, is an important component of obesity-associated adipose tissue inflammation and contributes to the development of insulin resistance and fatty liver. Fat inflammatory response results from lipid accumulation into adipocytes, leading to chemokine production by adipocytes and recruitment of proinflammatory macrophages [45, 46].
The adipose tissue of lean subjects is characterized by a low number of macrophages that are maintained in an anti-inflammatory, M2 phenotype by regulatory T cells (Tregs) and eosinophil-derived cytokines (IL-4, IL-13) [47, 48]. In an obese environment, the recruitment of Tregs and the number of M2 macrophages decrease, whereas the enrichment of Th1 cells and endotoxemia drives recruitment of CCR2-expressing, proinflammatory M1 macrophages [45, 49–52]. The initial mechanism governing invasion of adipose tissue by CCR2-positive macrophages is most likely represented by production of cytokines and chemokines, including the CCR2 ligand CCL2 (MCP-1), by fat-laden adipocytes, although adipocyte death and hypoxia have also been incriminated [45, 46]. The relevance of inflammation appears evident considering the 30% of adipose tissue is constituted by the so-called ‘vascular-stromal’ fraction, that comprises macrophages, endothelial cells and immune cells [53].
Increased adipose tissue inflammation contributes to insulin resistance, disrupting insulin-dependent signaling pathways, and leading to enhanced delivery of FFA to the liver, and thereby to hepatic steatosis [54]. In keeping, ablation of Cd11c-positive adipose tissue macrophages is associated with reduction of steatosis and normalization of insulin sensitivity [55]. Moreover, invalidation of M1 genes, such as TNF alpha, IL-1 beta, iNOS, osteopontin, MCP-1 or its receptor CCR2, blunts insulin resistance, adipose tissue and hepatic inflammation, and steatosis (for reviews see [45, 46]). Interestingly, the decrease in M2-polarized adipose tissue macrophages that parallels the increase in the M1 counterpart may also be of importance, since exacerbation of liver fat accumulation is observed when disrupting transcription factors that govern M2 macrophage polarization, such as PPARg, PPARd, or KlF4 [32, 56, 57].
In this scenario, activation of JNK has been associated with increased serine phosphorylation of IRS, reducing the response to insulin [58]. In addition, NF-kappaB activation leads to increased expression of several proinflammatory cytokines and chemokines that amplify inflammation and critically contributes to insulin resistance [59]. Interestingly, activation of NF-kappaB appears to require ceramide biosynthesis and the following activation of toll-like receptor (TLR)-4 [60]. Thus, similar to what happens in the liver (see below), increased availability of free fatty acids, generation of oxidative stress-related products, and endoplasmic reticulum stress are major factors involved in the activation of proinflammatory signals in the adipose tissue. Enlargement of the subcutaneous adipose tissue compartment is associated with a variable proportion of abdominal (visceral) obesity, which has been found to correlate with hepatic inflammation and fibrosis in humans [61]. While the relative role of subcutaneous vs. abdominal obesity is still a matter of debate, several lines of evidence indicate that visceral fat is more ‘dysfunctional’, i.e. less sensitive to insulin and more prone to inflammation [46]. As the drainage of blood from the visceral district primarily encounters the liver, it is possible that visceral obesity has a greater impact on the development of NASH [62].
Adipokines
Another pivotal aspect of adipose tissue dysfunction is represented by an altered pattern of adipocytokine secretion. Several recent studies have highlighted a relation between adipokines and different aspects of NASH, including fibrosis [63, 64]. More consistent data have been obtained for leptin and adiponectin.
Leptin
Leptin levels are increased in obese patients, and leptin has been shown to mediate profibrogenic effects on the liver [64]. Hepatic stellate cells express functional leptin receptors and are directly responsive to leptin with a number of biological actions that collectively promote fibrogenesis, including activation of the hedgehog pathway [65]. Once activated, HSCs contribute to leptin expression, whereas low levels of leptin in quiescent HSCs are associated with higher expression of adiponectin [66], which in turn inhibits leptin signaling and limits the fibrogenic properties of HSC [67]. Interestingly, leptin has also been shown to mediate the angiogenic potential of HSC, which accompanies the development of fibrosis. This pathway requires activation of the mTOR pathway and generation of reactive oxygen species [68]. Moreover, hyperleptinemia during experimental steatohepatitis is associated with increased production of hepatic endocannabinoids, and enhanced responsiveness to the vasoconstrictor effects of endothelin-1, aggravating the microcirculatory dysfunction that characterizes chronic liver diseases [69].
Besides an action on HSC, leptin also targets Kupffer cells and sinusoidal endothelial cells, stimulating TGF-β expression [70]. More recently, an intriguing cross-talk between leptin’s action and endotoxin has been identified. Leptin was found to upregulate the expression of CD14 in Kupffer cells, resulting in enhanced response to low-dose endotoxin and accelerated progression of NASH [71]. When considering leptin as a target for therapy, it should be noted that this adipokine also reduces the deposition of ectopic fat in the liver, and has been used for the treatment of NAFLD in lipodystrophic patients [72]. However, in obese patients several tissues are resistant to the actions of leptin, and it is possible that, similarly to what is observed for insulin resistance, not all cells develop leptin resistance, and that fibrogenic and inflammatory cells remain sensitive to the actions of this adipokine, contributing to the progression of NASH. Thus, similar to other therapeutic approaches, it is likely that interference with the action of leptin is more promising if targeted to the cells that are more directly involved in inflammation and repair.
Adiponectin
Adiponectin increases insulin sensitivity and has in general a hepatoprotective and antifibrogenic effect in the liver wound healing process [63]. Although adiponectin and leptin share their effects counteracting ectopic fat deposition, they have divergent effects on inflammation. In fact, while leptin-deficient mice are protected from T cell-mediated hepatitis, lipodystrophic mice, that lack both adiponectin and leptin, are not, unless adiponectin is administered [73]. In general, adiponectin reduces inflammation, stimulating secretion of anti-inflammatory cytokines (e.g. IL-10), blocking NF-κB activation, and inhibiting release of TNF-α, IL-6, and chemokines [74]. Conversely, inflammation blocks adiponectin secretion, and specifically, adipose tissue inflammation contributes to reduce plasma adiponectin levels in obesity. Moreover, a direct antifibrogenic action of adiponectin has been demonstrated in models of toxic liver damage, and adiponectin ameliorates metabolic alterations and liver fibrosis in different models of steatohepatitis [75, 76]. Some of the anti-fibrogenic effects of adiponectin are dependent on activation of AMP-activated protein kinase (AMPK), that is activated in hepatic stellate cells upon interaction of adiponectin with its cognate ligands [77, 78].
Reduced generation of oxidative stress-related products in the adipose tissue and in the liver is another mechanism by which adiponectin may be beneficial in the context of NASH [79]. The antioxidant effects of adiponectin have been recently linked to activation of AdipoR1 and the resulting downstream release of intracellular calcium, together with increased activation of AMPK and sirtuin-1 [80]. In macrophages, adiponectin modulates the response to lipopolysaccaride dependent on TLR4, which leads to TNF-alpha secretion and generation of reactive oxygen species [81]. Interestingly, this effect, which is mediated by expression of interleukin-10, occurs after an initial phase during which adiponectin actually increases TNF-alpha expression, indicating a complex action of adiponectin on the pathways regulating inflammation. Along these lines, in fatty liver undergoing ischemia-reperfusion, adiponectin siRNA confer protection against oxidative stress and injury [82]. The anti-inflammatory effects of adiponectin are also mediated by its ability to favor an M2 antiinflammatory phenotype and to inhibit M1 polarization [83]. Thus adiponectin may maintain Kupffer cells in an M2 status in lean mice, while hypoadiponectinemia associated with obesity may be a key factor for Kupffer cell activation and steatohepatitis.
The adiponectin system represents therefore an appealing target for the treatment of NASH and the metabolic syndrome [84]. Some of the drugs that are currently used, including thiazolidinediones or metformin, exert at least part of their actions modulating the adiponectin system or its intracellular targets, such as AMPK. In addition, adiponectin receptors could be potentially targeted by the use of agonists. However, the relevance of these findings needs to be further validated in patients with NASH, because hypoadiponectinemia is a characteristic of patients with metabolic syndrome, but correlation to NASH has been inconsistently reported [7].
Other adipocytokines
Other adipokines could possibly participate in hepatic inflammation and fibrosis. These include resistin, that has been shown to modulate both inflammation and hepatic stellate cell biology [85, 86], and visfatin, which has been associated with propagation of lipotoxic inflammation [87]. Adipose tissue is a major site of expression of cytokines and chemokines that are also abundantly expressed within the liver. CCL2 (also known as monocyte chemoattractant protein-1, see above) has been implicated in the recruitment of inflammatory cells to the inflamed adipose tissue, possibly in concert with other chemokines [88]. Expression of TNF-alpha and IL-6 has been shown in the adipose tissue and it is considered to contribute to hepatic inflammation via an endocrine action, reaching the liver through the bloodstream. The possible role of these cytokines is discussed below (see 4.1).
The renin-angiotensin system
Accumulating data indicate that the renin-angiotensin system is a major modulator of insulin resistance and therefore may have a particular relevance in patients with NASH [28]. Local activation of this system has been shown to take place in adipose tissue, where it may contribute to the abnormalities associated with the metabolic syndrome [89], and in the liver, where it is critical for inflammation and fibrogenesis. Several experimental studies have demonstrated that interference with this system is accompanied by reduced damage, inflammation and fibrosis in models of steatohepatitis [90]. Based on the extensive clinical knowledge of drugs interfering with this system, renin-angiotensin system inhibition may be a promising strategy to prevent fibrosis progression in different chronic liver diseases, including NASH [91], and clinical trials are underway.
3.2. Intestine and the microbiome
In the last few years considerable attention has been directed to a possible direct role of the microbiome in the pathogenesis of NAFLD and NASH, with very intriguing results. Availability of novel sequencing techniques allows a comprehensive and detailed analysis of the microbiome, which has led to the identification of different ‘enterotypes’ in the general population [92]. Analysis of the role of different families of intestinal bacteria has been performed in several conditions, including obesity, and it has been proposed that the microbiome contributes to the susceptibility to become obese and to gain or lose weight more effectively why on a given dietary regimen [93]. These findings have a clear relevance for the pathophysiology of the metabolic syndrome and NAFLD [94, 95]. In fact, studies in mice and humans have demonstrated that gut bacteria contribute to determine the susceptibility to develop fatty liver, partly through modifications in choline metabolism [96, 97].
Epidemiological approaches have been recently associated with efforts to modify the microbiome. Henao-Mejia et al. have provided evidence that defects in the NALP3 inflammasome are associated with lower levels of IL-18 and other cytokines in the intestine, resulting in modulation of the gut microbiome [98]. This is associated with more severe development of NASH, through influx of TLR4 and TLR9 agonists into the portal circulation. Remarkably, co-housing of wild-type mice with mice defective in inflammasome, led to acquisition of the altered microbiome and transmission of a NASH phenotype [98]. These data indicate that experimentally, transfer of the microbiome even in the absence of previous destruction of the ‘normal’ flora, may be sufficient to transmit NASH. It still remains to be assessed at which level(s) the inflammasome is critical, in particular whether defects in the intestine are sufficient to transmit the phenotype. Moreover, these data should be reconciled with other studies implicating a deleterious effect of inflammasome activation in the pathogenesis of NASH [99, 100].
3.3. Dietary factors
While there is general agreement that a reduction in calorie intake is beneficial for NASH, as part of a complex lifestyle intervention [101], it is uncertain whether changes in the type of nutrients may be an additional approach to be pursued in the treatment of this condition. These aspects may be viewed as additional extrahepatic factors that ultimately modulate the response of the liver to the excess of calories and to lipid accumulation. Most of the available data are provided by studies in mice, and only initial evidence has been obtained in humans (Figure 4). Nonetheless, identification of the more ‘detrimental’ types of food could represent an important tool when counseling the patients, and it may ultimately contribute to the success of other therapies. Data from clinical studies suggest that patients with NASH have an imbalance in the type of fat consumed, including an excess in saturated fat and n-6 fatty acids, together with a relative deficiency in n-3 polyunsaturated fat [102, 103]. The different types of dietary lipid may have an impact on the development of hepatic lipotoxicity, although dietary fat contributes to approximately only 10% of the hepatic FFA pool [20]. In general, saturated fatty acids are more lipotoxic than mono- or polyunsaturated ones, mainly due to their lower ability to be incorporated in less toxic triglycerides and to higher induction of apoptosis [104–106].
Figure 4. Dietary factors influence the development of steatohepatitis.
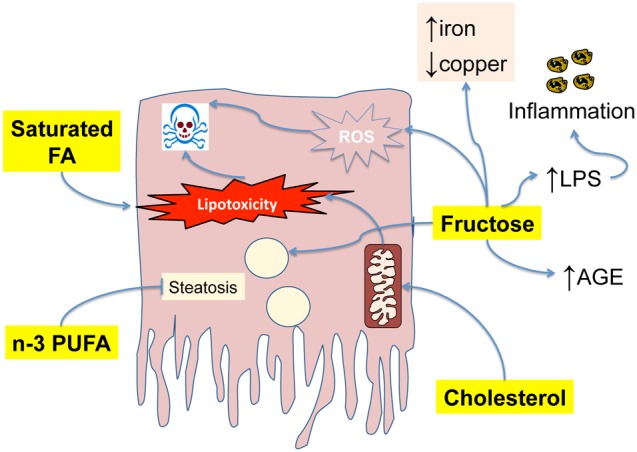
Saturated fatty acids, particularly palmitic acid, are highly lipotoxic and may lead to apoptotic cell death. In contrast, n-3 PUFA have a protective effect on steatosis. Free cholesterol targets the mitochondria, and contributes to lipotoxicity and apoptosis. Fructose favors steatosis and contributes to injury, inflammation and fibrogenesis increasing the availability of lipopolysaccharide (LPS), increasing the levels of advanced glycation end-products (AGE), increasing iron and depleting copper.
Monounsaturated fatty acids are part of the ‘Mediterranean diet’ which has been suggested to be beneficial in the metabolic syndrome [107]. In addition, several studied have addressed the potential beneficial effects of omega-3 PUFA in patients with NAFLD [108]. A recent metanalysis indicates that supplementation with omega-3 PUFA is associated with reduction of hepatic fat, whereas the effects on necroinflammation are more uncertain [109]. In addition, data obtained in population studies indicate that consumption of dietary n-3 PUFAs is inversely associated with the occurrence of hepatocellular carcinoma [110]. Omega-3 fatty acids have consistently been shown to reduce hepatic steatosis in murine models, and to lower plasma ALT in ob / ob mice [111, 112]. More recently, supplementation with eicosapentaenoic acid was investigated in mice with hepatocyte-specific deletion of PTEN, which develop steatohepatitis and cancer [113]. Supplementation was associated with reduced generation of ROS and lower inflammation, together with inhibition of HCC development. In this and other studies, part of the effects of omega-3 PUFA have been attributed to their ability to increase the expression of PPAR-alpha. Additionally, the anti-inflammatory effects of omega-3 fatty have been linked to modulation of eicosanoids [111], and of TNF or adiponectin [114].
Considerable recent interest has been placed on the role of cholesterol. Data from Mari et al. have indicated that upon administration of a cholesterol-rich diet, free cholesterol accumulates in mitochondria and sensitized hepatocytes to apoptosis and lipotoxic injury [115]. The relevance of hepatocellular free cholesterol is supported by recent findings obtained in foz/foz mice, which carry a spontaneous mutation associated with early obesity, insulin resistance, and development of NASH with fibrosis [116]. In these mice, increasing the amount of dietary cholesterol resulted in more severe inflammation, hepatocellular injury and fibrosis. These observations have a clear potential impact on the treatment of NASH, and several studies are underway. The excellent safety profile of cholesterol-lowering drugs and the well-established pharmacology of these compounds make them very attractive candidates for a pharmacological-based approach to NASH [117]. Solid data have also been obtained for the role of consumption of different types of carbohydrates, which increase insulin levels and contribute to elevate triglyceride levels, even in isocaloric conditions [118]. Moreover, histologic severity has been shown to be associated with higher carbohydrate intake, and a reduction in dietary carbohydrates was more effective in lowering ALT even with similar weight loss [119, 120]. These data have epidemiological and practical relevance, as the consumption of simple carbohydrates has dramatically increased in the past decades. In particular, fructose and sucrose, which contains 50% fructose, have been suggested to play a major role in metabolic disorders [121]. Among sugars, fructose has many peculiarities, as it poorly stimulates insulin or leptin secretion, and therefore it is less able to induce satiety [122]. Moreover, it is more efficient than glucose in inducing formation of advanced glycation end-products [121]. In a large clinical study performed by the NASH Clinical Research Network, dietary fructose consumption was found to be associated with major features of steatohepatitis, including higher fibrosis stage, hepatic inflammation and hepatocyte ballooning [123].
Experimental studies have unraveled at least in part the molecular mechanisms underlying the detrimental effects of fructose on the development of steatohepatitis. Fructose is directly proinflammatory, through elevation of endotoxin levels, induction of oxidative stress, and up-regulation of TNF-alpha expression [124]. Along these lines, chronic fructose intake in mice mutant for TLR4 caused lower hepatic triglyceride accumulation and reduced oxidative stress and expression of proinflammatory factors [125]. More recently, fructose was found to induce copper deficiency and to lead to iron overload in rats [126]. This resulted in worsened necroinflammation and suppression of antioxidant defenses, leading to lipid peroxidation. ER stress also contributes to the effects of fructose on the liver, as indicated by upregulated expression of GRP78 and increased phosphorylation of eIF2α and PERK [127]. These findings have a practical counterpart, and abstinence from beverages containing fructose should be suggested to patients with NAFLD. Accumulating evidence indicates that coffee has a relevant protective effects on the development of fibrosis in different chronic liver diseases [128]. In a recent study, patients with NASH reported a lower consumption of coffee compared to controls or subjects with simple steatosis [129]. Moreover, a negative relationship between coffee caffeine consumption and hepatic fibrosis was observed. Of note, other molecules besides caffeine may be protective in the setting of NASH. In a rodent model of steatohepatitis, addition of coffee to a high fat diet resulted in a reduction in hepatic fat accumulation, oxidative stress and liver inflammation [130]. These results were independent of caffeine, and were reproduced by administration of polyphenols or melanoidins. Nonetheless, caffeine appears to contribute to the actions of coffee, as demonstrated by data in patients [129, 131]. In addition, caffeine has been shown to modulate the effects of the profibrogenic cytokine TGF-beta on cultured hepatocytes. [132]. These data suggest that modification in the consumption of usual foods may be potentially helpful in preventing progression of NAFLD. Along these lines, even orange juice was recently shown to have anti-steatogenic effects in mice [133].
3.4. Gastrointestinal hormones
Hormones released in the gastrointestinal tract and targeting the liver and the pancreas have been recently implicated in the pathogenesis of metabolic disturbances. Glucagon-like peptide-1 (GLP-1) is an incretin secreted by the L-type cells of the intestine in response to food intake [134]. The half-life of GLP-1 is limited due to the rapid degradation by dipeptidyl dipeptidase-4 (DPP-4), and therefore long-acting agonists of the GLP-1 receptor such as exenatide or liraglutide have been generated. Similarly, DPP-4 inhibitors such as sitagliptin have been used in experimental and clinical studies. The main action of GLP-1 is the stimulation of glucose-dependent insulin secretion, but recent studies indicate that GLP-1 may be relevant in the pathogenesis of steatohepatitis. In ob/ob mice, exendin-4 was found to reduce hepatic steatosis and to improve insulin’s action [135]. Moreover, activation of the GLP-1 receptor led to induction of autophagy and to reduction of ER stress, two key events in the pathogenesis of steatohepatitis [136]. Ghrelin is another factor that has received recent attention in the pathogenesis of liver fibrosis [137]. Data in a model of steatohepatitis showed that necrosis and inflammation, oxidative stress, and lipid peroxidation were limited by treatment with ghrelin, acting through the LKB1/AMPK and PI3 K/Akt pathways [138].
4. Inside the liver: local inflammatory and fibrogenic mechanisms
Sustained inflammation in the liver is critical in the progression of NAFLD. Compelling evidences indicates that hepatic macrophages, comprising liver-resident macrophages (Kupffer cells) and monocytes infiltrating the injured liver, as well as specific lymphocyte subsets, play a pivotal role in the initiation and perpetuation of the inflammatory response, with a major deleterious impact on key steps of fatty liver progression to fibrosis.
4.1. Kupffer cell activation affects multiple steps of NAFLD
Kupffer cells are key players in the pathogenesis of NASH, as shown by the resistance to steatosis, inflammatory cell recruitment, hepatic injury and fibrosis, in mice where Kupffer cells are depleted with clodronate or gadolinium chloride [139–144]. Alike other macrophages, Kupffer cells and hepatic monocyte-derived macrophages are a highly heterogeneous and plastic populations that may adopt various phenotypes ranging between the extreme states known as M1 and M2. During NAFLD, Kupffer cells are exposed to a variety of exogenous and endogenous signals that shift their phenotype towards a proinflammatory M1 state, in particular danger signals released by steatotic hepatocytes and toxic lipids accumulated in Kupffer cells (Figure 5). In addition, exogenous inflammatory mediators originating from the adipose tissue and from bacterial products released from the gut, such as endotoxin (LPS), also activate Kupffer cells and trigger hepatic inflammation (see below). Inflammation driven by M1 Kupffer cells is counterbalanced by alternatively-polarized M2 macrophages, that produce anti-inflammatory cytokines such as IL-10, and promote resolution of inflammation [145, 146]. Macrophage polarization into an M2 phenotype is promoted by Th2-derived cytokines (IL-4, IL-13) [145, 146], that may also originate from hepatocytes [56, 147]. Factors promoting M2 Kupffer cell polarization are poorly characterized, but include the transcription factor PPARdelta [56, 147], adiponectin [83, 148] and the cannabinoid receptor CB2 [149] (see below).
Figure 5. Pivotal role of activated Kupffer cells in the pathogenesis of nonalcoholic steatohepatitis and fibrogenesis.
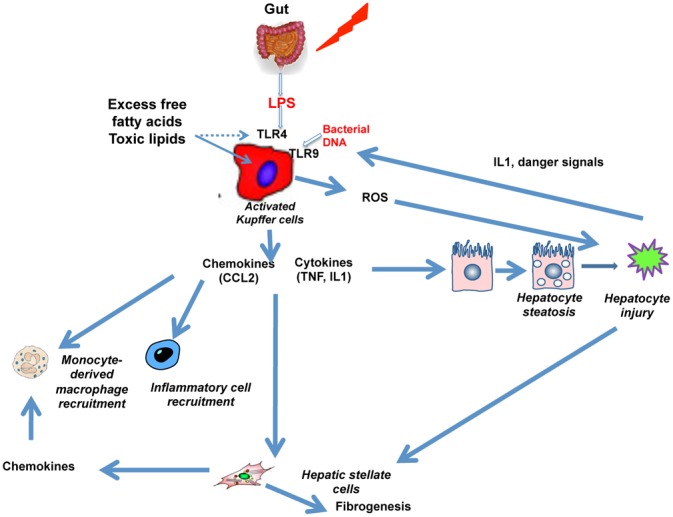
Increased levels of lipoipolysaccaride (LPS) and lipotoxic lipid products lead to activation of Kupffer cells, which release chemotactic factors (e.g. CCL2), and proinflammatory cytokines, and generate oxidative stress-related products including reactive oxygen species (ROS). These factors contribute to hepatocyte injury, which in turn, through danger signals, cause further activation of toll-like receptors (TLRs). In addition, inflammation and damage contribute to hepatic stellate cell activation and fibrosis.
Dysregulation of the hepatic M1/M2 phenotypic balance is emerging as a central mechanism governing NAFLD. Initially considered as a consequence of fat accumulation in hepatocytes, Kupffer cell activation is now viewed as a central mechanism triggering steatosis [56, 140, 150, 151]. In fact, recent studies have shown that IL-1beta released by M1-polarized Kupffer cells promotes triglyceride synthesis in hepatocytes following activation of diacylglycerol acyltransferase 2, and inhibits fatty acid oxidation by decreasing PPAR-alpha trans-activating activity [150, 151]. Moreover, invalidation of PPAR-delta in myeloid cells, which inhibits M2 and favors M1 Kupffer cell polarization, results in enhanced steatosis via inhibition of fatty acid oxidation in hepatocytes [56]. These studies demonstrate the causal relationship between the loss of alternative Kupffer cell polarization and/or acquisition of the M1 phenotype, and the development of steatosis.
Activation of Kupffer cell also governs the recruitment of blood–derived positive monocytes during NASH. Accumulating studies demonstrate that macrophage infiltration into the liver is primarily promoted by CCL2, a chemokine up-regulated in the serum and in the liver of patients with NASH [152], that drives the recruitment of C–C chemokine receptor 2 (CCR2) expressing monocytes. In particular, pharmacological or genetic invalidation of CCL2 or CCR2 reduces macrophage infiltration in mice fed CDAA or MCD, in addition to preventing steatosis [143, 153]. However, variable results have been obtained in mice with different genetic backgrounds [154]. Other molecules of the chemokine family have been shown to play a possible role in the pathogenesis of NASH, including CCL5 (RANTES) which s implicated in the recruitment of inflammatory cells and in the pathogenesis of fibrosis in different conditions. In principle, chemokine receptors represent an ideal target for the treatment of NASH. Inflammatory chemokines such as CCL2 or CCL5 are predominantly expressed in conditions of inflammation, and therefore inhibition of their action may possibly be achieved without major off-target effects. Moreover, chemokine receptors are G protein-coupled receptors that are generally easy to target with drugs. Therefore, it is likely that this group of molecules will represent an appealing target in the near future.
In addition to their key role in the initiation of the inflammatory reaction associated with early steps of NAFLD, Kupffer cells and recruited monocytes contribute to liver fibrosis progression, both by perpetuating inflammatory cell recruitment and triggering hepatic stellate cell activation. Emerging evidence also indicates that macrophages play a pivotal role in the resolution of liver fibrosis [155]. Although the characteristic profile of these pro- and anti-fibrotic macrophages is still unclear, recent data have demonstrated that M1 macrophages originating from resident Kupffer cells and Gr1+ monocytes promote progression of liver fibrosis by releasing inflammatory mediators such as IL-1beta, that activate hepatic stellate cells [150]. Conversely, M2-like Kupffer cells may display anti-fibrogenic functions [156–158]. Indeed, mice carrying a specific deletion of either the M2 marker arginase-1 or the chemokine receptor CX3CR1 in macrophages show reduced expression of M2 genes, and are prone to liver fibrosis [157, 158]. Along the same lines, the chemokine CX3CL1 suppresses hepatic stellate cell fibrogenic functions by triggering M2 Kupffer cell polarization [156]. However, M2 Kupffer cells have been also indicated as inducers of fibrosis, because of their capacity to produce TGF-beta and matrix metalloproteinases [145]. These studies highlight the fact that modulating Kupffer cell phenotype is an interesting strategy to protect against NAFLD progression, although better characterization/classification of pro and antifibrogenic macrophage populations is required.
4.2. Accumulation of lipids in Kupffer cells is proinflammatory
Recent studies have shown that alterations in lipid metabolism during NAFLD drive polarization of Kupffer cells into an M1 phenotype, via several mechanisms. First, uptake of oxidized lipoproteins by Kupffer cells, orchestrated by the scavenger receptors CD 36 and SR-A, leads to the formation of foamy macrophages with a proinflammatory phenotype [159]. In keeping with these data, immunization strategies against oxidized low-density lipoprotein or removal of cholesterol from the high fat diet in foz/foz or ldr−/− hyperlipidemic mice, ameliorates NASH-induced hepatic injury and inflammation [116, 160]. Second, excessive exposure to fatty acids, as induced by high fat diet, leads to accumulation of diacylglycerol and ceramide in Kupffer cells, that shift their phenotype towards an M1 state [161]. Noticeably, fat-laden M1 Kupffer cells from obese mice become hyperreactive to low levels of LPS, which may account for the vulnerability of these animals to develop liver inflammation and NASH [161]. Third, recognition of fatty acid moieties by TLR4 has been proposed as a mechanism by which lipids may activate Kupffer cells. However, free fatty acids most probably do not directly bind to TLR4 [162], but rather interact with TLR4 via the free fatty acid carrier fetuin A, a liver secretory glycoprotein recently identified as an endogenous TLR4 ligand [163].
4.3. Pattern recognition receptors (PRR)
Obesity is associated with increased gut permeability, leading to translocation of bacterial products which interact with pattern recognition receptors (PRR). Originally described as a defense mechanism against pathogens (bacteria, viruses and fungal components), PRR have now been shown to play an essential role in the pathogenesis of NAFLD. PRR comprise at least two sensing protein families, the NOD like receptors (NLR), which are associated with inflammasome complexes in the cytosol [164], and the Toll-like receptors (TLR), expressed at the cell surface or in the endosomal compartment [165]. Both NLR and TLRs detect danger signals, such as pathogens originating from gut-derived microorganisms (pathogen-associated molecular patterns, PAMPs), or molecules released endogenously from dying or stressed cells, in particular by hepatocytes (endogenous damage-associated molecular patterns, DAMPs) [164, 166]. Kupffer cells are the primary sensors of PAMPs and DAMPs, and TLR and inflammasomes have emerged as important components of Kupffer cell activation and NAFLD progression.
TLR are a family of receptors that recognize gut-derived bacterial products such as lipopolysaccharide (LPS), bacterial DNA, peptidoglycan, viral and fungal components originating from gut microbiota, but also denatured host DNA and indirectly free fatty acids [165]. Studies on the role of TLRs in the pathogenesis of NAFLD were initiated a decade ago, by the description of alterations in gut permeability and composition of gut microbiome in obesity, both in humans and experimental models [167–169]. As a result, Kupffer cells and other liver cells, in particular stellate cells and hepatocytes are exposed to a high load of gut-derived bacterial products that function as TLR ligands to promote NAFLD progression. Results from knock out animals indicate that TLR4 and TLR9 are deleterious in NAFLD, whereas TLR2 may display hepatoprotective effects [170, 171]. Although TLR5 is not expressed in the liver, Tlr5−/− mice fed a high fat diet are prone to obesity, insulin resistance and steatosis, related to alterations in the gut microbiome [172].
Among the TLRs, TLR4 has a central role in Kupffer cell activation by binding both LPS and danger signals such as HMGB1 or oxidized phospholipids. TLR4 acts in concert with its co-receptors CD14 or MD-2 to recognize LPS, leading to the recruitment of adaptor molecules MyD88 and TRIF. TLR4 triggers a cascade of signaling pathways that culminates in the activation of members of the NF-kappaB complex, and the resulting production of proinflammatory cytokines and type I interferons [166]. The role of TLR4 in NAFLD was demonstrated by the use of TLR4-deficient mice, which show impaired Kupffer cell activation, whereas LPS levels remain elevated [125, 144, 173]. Moreover, activation of TLR4 in Kupffer cells leads to generation of reactive oxygen species and the resulting activation of X-box binding protein-1, a transcription factor involved in the unfolded protein responses [174].
Mice deficient for TLR4 in hematopoietic cells are resistant to high fat diet-induced adipose tissue inflammation, insulin resistance and steatosis [175]. In addition, TLR4-deficient mice do not develop steatohepatitis and fibrosis when fed MCD, suggesting direct contribution of TLR4 expressed in liver cells to NASH [176]. On the other hand, when LPS release is reduced by administration of antibiotics or probiotics, obese mice are resistant to liver inflammation and liver injury [177]. Conversely, infusion of low-dose LPS to lean mice triggers hepatic steatosis, insulin resistance, and weight gain [178] and administration of LPS accelerates steatohepatitis in mice fed MCD [179]. Of note, activation of TLRs, including TLR4, has been shown to mediate the detrimental effects of fructose on the development of fatty liver [180]. These data demonstrate that activation of the LPS-TLR4 pathway in the liver and in the adipose tissue is a major component of NAFLD progression.
HSC also express several TLRs. Seki et al. have investigated the effects of TLR4 on the development of fibrosis, demonstrating that activation of this receptor in HSC potentiates the effects of TGF-beta downregulating the inhibitory co-receptor, Bambi [181]. Of note, fibrogenesis was dependent on the expression of TLR4 in HSC more than in other hepatic cell types, as demonstrated by experiments in chimeric mice. This view is supported by the observation that LPS and other bacterial products are also pro-inflammatory through a direct action on HSC [182]. More recently, the role of TLR9 in steatohepatitis has been addressed. Deletion of TLR9 resulted in a marked amelioration of experimental steatohepatitis, including the development of fibrosis [150]. Besides LPS, bacterial products such as bacterial DNA are increased during NASH, as shown in mice fed CDAA [150]. Bacterial DNA is a TLR9 ligand, and TLR9-deficient mice fed CDAA are resistant to steatosis, liver injury, inflammatory cell infiltration and fibrosis. In addition, these mice show a specific decrease in the production of IL-1 beta by Kupffer cells, a cytokine with apoptotic and steatogenic effects on in cultured hepatocytes and with fibrogenic properties towards isolated hepatic stellate cells [150]. Therefore, bacterial DNA binding toTLR9 Kupffer cells activates several features of NAFLD, via IL-1 release from Kupffer cells.
4.4. Inflammasome
Inflammasomes are cytoplasmic protein complexes that are key regulators of the innate immune response, by controlling the activation and secretion of IL-1 beta, IL-18 or IL33 in response to danger signals. Inflammasome structures comprise an intracellular NOD-, LRR- and pyrin domain-containing (NLRP) family member, an adaptor sensor molecule (ASC) and pro-caspase 1 [164]. Among NLRP, NLRP3 is the best characterized for its role in insulin resistance and NAFLD. NLRP3 is activated by PAMPs or DAMPs in intracellular compartments, resulting in the processing of pro-caspase 1 into activated caspase 1, and subsequent maturation of inactive pro IL-1-beta, IL-18 and IL33 into their active forms [164].
Recent data have highlighted the major but complex contribution of inflammasome components expressed in the adipose tissue, the gut and the liver at multiple steps of NAFLD progression. The expression of inflammasome components (NLRP3, ASC, caspase-1) and production of mature IL-1 beta are up-regulated in patients with NASH and in mice fed MCD or a high fat diet, but only after prolonged overfeeding and not just when simple steatosis develops [99]. Nevertheless, NLRP3-derived IL-1beta drives steatosis in HFD-fed mice, probably as a consequence of adipose tissue macrophage infiltration and enhanced insulin resistance [183, 184].
The contribution of inflammasome to NASH progression may involve the coordinated interplay between inflammatory cells, hepatocytes and hepatic stellate cells. Indeed, although inflammasome activation was initially thought to be restricted to inflammatory cells, accumulating data indicate that this complex is also expressed in hepatocytes and hepatic stellate cells. In hepatocytes, activation of inflammasome by lipotoxic lipids may represent an additional event governing activation of Kupffer cells during NAFLD. Thus, hepatocytes exposed to saturated fatty acids release IL-1beta and danger signals, that will activate inflammasome in Kupffer cells, and trigger subsequent release of proinflammatory signals [99]. The nature of the danger signal released by hepatocytes remains to be identified, but a good candidate could be HMGB1, a DAMP ligand of TLR4 released upon exposure to free fatty acids that results in inflammasome activation in macrophages [185, 186]. Direct activation of the inflammasome pathway in Kupffer cells could also be promoted by free fatty acids, free cholesterol and toxic lipids such as ceramides, as described in adipose tissue macrophages [164], thereby contributing to their polarization into an M1 state. Finally, activation of inflammasome in cultured hepatic stellate cells enhance their fibrogenic properties, suggesting that this pathway could also contribute to NASH-related fibrosis via direct effects on fibrogenic cells [187]. In keeping with these data, NLRP3−/−, ASC−/− or caspase 1−/− mice are protected against carbon tetrachloride or MCD-induced fibrosis [187, 188].
On the other hand, activation of inflammasome may also show beneficial effects against NASH, because mice invalidated for NLRP3, NLRP6, ASC or caspase-1 that are fed MCD show exacerbated steatosis [98] [142] and hepatic inflammation [98], in contrast to mice fed HFD, which are resistant to steatosis [183, 184]. Further studies are needed to reconcile these data, but the relative IL-18 to IL-1beta ratio in the two models (MCD, a NASH model without obesity and insulin resistance vs high fat diet, a model of metabolic syndrome with limited features of NASH) may be a key issue, because IL-18 but not IL-1 beta drives protection against MCD-induced NASH, in contrast to the major role of IL-1 beta in the high fat diet model [98].
4.5. NFκB and JNK as key signaling pathways linking inflammation to NAFLD
NFκB and the c-Jun-N terminal kinases (JNK) are strongly up-regulated in the liver and the adipose tissue of patients with NASH, and in obese mice or mice fed MCD. Both pathways are activated by a variety of danger signals, including endotoxin, oxidative stress, insulin, lipotoxic molecules or proinflammatory cytokines. Their activation in adipose tissue macrophages and in the liver, both in Kupffer cells and hepatocytes, contribute to steatosis, hepatic inflammation insulin resistance, and NASH-related liver injury.
Pharmacological inhibition of IKK beta, an upstream kinase which promotes NF-κB activation, prevents mice fed MCD from steatosis and NASH [189]. Selective deletion of IKK beta in myeloid cells results in suppression of inflammatory cytokine production, and protects from peripheral insulin resistance. In hepatocytes, manipulation of IKK-beta by specific gene deletion or overexpression in overfed mice has led to the demonstration of its contribution to Kupffer cell activation, steatosis and hepatic insulin resistance[190, 191]. Surprisingly, hepatocyte deletion of NEMO, a kinase which enhances IKK action, leads to increased inflammation, hepatocyte apoptosis and development of liver tumor in mice fed HFD [192]. These unexpected results probably reflect the consequences of a more severe inhibition of NF-κB when disrupting NEMO, with a resulting loss of the protective effects of NF-κB against hepatocyte apoptosis.
The two isoforms of JNK, JNK1 and JNK2, display distinct outcomes on NAFLD. In different models of obesity and NASH, pharmacological inhibition of JNK or global invalidation of JNK1, but not JNK2, protects against steatosis, insulin resistance, Kupffer cell activation, inflammatory cell recruitment and liver injury [193–196]. The requirement of myeloid JNK1 in Kupffer cell activation and steatosis was demonstrated by the use of chimeric mice generated by transplanting bone marrow cells lacking JNK1 into WT mice [197]. Although these results demonstrated the role of macrophage JNK1 in NASH pathogenesis, an additional study demonstrated that hepatocyte-specific deletion of JNK1 actually promotes steatosis and insulin resistance [198], suggesting that the deleterious effects of JNK1 in myeloid cells may predominate over the beneficial effects of JNK1 in hepatocytes.
Taken together, these data undoubtedly point out the major impact of the NF-κB and JNK pathways, in myeloid cells and hepatocytes, in insulin resistance and various aspects of NASH. They also further argue for a cross talk between hepatocytes and Kupffer cells in the pathogenesis of NASH. However, they underscore the complex cell specific roles of these signaling pathways in disease progression.
4.6. Lymphocytes
In addition to Kupffer cells, NAFLD progression is also governed by alterations in lymphocyte subsets. Indeed, fat-laden Kupffer cells promote recruitment of CD4+, CD8+ and B lymphocytes [161], and cause hyperresponsiveness to chemotactic agents in obese mice [199]. Moreover, alterations in the Treg/Th17 balance has also been hypothesized. Indeed, as described in the adipose tissue, the number of Tregs decreases in the liver of mice fed high fat diet, as a result of enhanced apoptosis promoted by oxidative stress [200]. Adoptive transfer of regulatory T cells reduces liver inflammation and injury upon exposure to low levels of LPS, suggesting that reduction in Tregs contributes to inflammation and hypersensitivity of obese mice to LPS-induced injury [200]. Whether, as described for adipose tissue M2 macrophages, Tregs also contribute to maintain the M2 Kupffer cell phenotype in lean mice is an intriguing possibility that deserves to be investigated. In parallel to the decrease in Tregs, an increase in hepatic Th17 lymphocytes has been observed in high fat diet fed mice senisitized to a low dose of LPS, and their neutralization reduces LPS-induced liver injury [201]. These data, together with the reported activation of Th17 responses in the adipose tissue of obese mice and in patients with type 2 diabetes [202], suggest that modifications of the Treg/Th17 balance could contribute to the inflammatory process associated with obesity and NAFLD, both in the adipose tissue and in the liver.
A ‘double-edged sword’ role has been demonstrated for natural killer T (NKT) cells. NKT cells are a subset of lymphocytes that are particularly abundant in healthy livers. They are activated by lipid antigens and can produce both proinflammatory Th1 and Th17 cytokines, and anti-inflammatory Th2 mediators. Liver NKT cells are decreased in steatotic human livers and in ob/ob mice or mice fed a high-fat high-sucrose diet [203, 204]. The loss of NKT cells in the steatotic liver is deleterious, since CD1d-deficient mice that lack NKT have exacerbated steatosis and glucose intolerance, whereas adoptive transfer of NKT cells in obese mice ameliorates steatosis and insulin resistance [203]. Depletion of NKT cells is also associated with increased Th1 and decreased Th2 cytokine production, and results from Kupffer cell-derived production of IL-12, that induces NKT cell death [204]. However, NKT cells accumulate in the liver of patients with advanced NASH and in mice fed MCD, via a Hedgehog-dependent pathway [205, 206]. In this context, NKT cells contribute to NAFLD progression, since CD1d-deficient mice fed MCD are protected from NASH-related fibrosis [207]. Therefore, NKT cells display beneficial effects at early steps of NAFLD (i.e steatosis), while promoting progression to steatohepatitis and fibrosis at later stages.
4.7. The endocannabinoid system
Dysregulation of the peripheral endocannabinoid (EC) system has been linked to the progression of NAFLD. Patients with NAFLD show increased serum levels of 2-arachydonoylglycerol, and endocannabinoid ligand, and enhanced hepatic expression of both cannabinoid receptors, CB1 and CB2 [208, 209]. In keeping with these data, convergent studies have highlighted the contribution of the endocannabinoid system in the pathogenesis of NAFLD [210].
CB1-deficient mice are resistant to high fat diet-induced obesity and show neither insulin resistance nor fatty liver [211]. Along the same lines, ob/ob mice treated with the CB1 receptor antagonist rimonabant show reversal of hepatic steatosis and improved insulin sensitivity [212]. Beneficial effects of CB1 receptor antagonism on steatosis and insulin resistance are not exclusively related to a decrease in body weight, but also results from interaction with peripheral receptors. Indeed, CB1 receptors are up-regulated in hepatocytes and adipocytes from mice fed a high fat diet. Moreover, mice bearing hepatocyte-specific deletion of CB1 receptors become obese in response to high fat diet, but are protected from steatosis, dyslipidemia and insulin resistance, demonstrating the direct contribution of hepatocyte CB1 to the steatogenic process [211]. Studies in cultured liver slices or in isolated hepatocytes have shown that increased fat accumulation directed by CB1 receptor activation results from the combination of an increase in hepatocyte lipogenesis following SREBP1c activation, a reduction of fatty acid oxidation via inhibition of AMP kinase, and a decrease in the hepatic production and release of TG-rich VLDL [211]. However, mice with liver-specific deletion of CB1 are only partially protected from steatosis, whereas mice with a global CB1 deletion are totally resistant, suggesting a role for adipose tissue-derived fat. Accordingly, activation of CB1 receptors in adipocytes enhance free fatty acid influx to the liver [213].
Data from epidemiological and therapeutic studies also support the causal role of the endocannabinoid pathway in the pathogenesis of steatosis. Daily cannabis use has been identified as an independent predictor of severe steatosis in patients with chronic hepatitis C [214]. In addition, hepatic CB1 expression correlates with the extent of steatosis in these patients, and is up-regulated in those with increased steatosis grade [215].
Activation of CB1 receptors may also participate to the inflammatory response and liver injury associated with NASH. CB1-dependent increase in fat TNFalpha is associated with reduced secretion of adiponectin, and administration of rimonabant to ob/ob mice reduces liver inflammation and liver injury, while restoring adiponectin levels [212]. Moreover, recent studies have shown that CB1 receptor activation in macrophages triggers the release of proinflammatory cytokines [216, 217], but whether CB1 is also expressed in Kupffer cells deserves future studies. Finally, CB1 receptors promote ER stress, via up-regulation of the Bip-PERK-eIF2alpha pathway and activates CREBH, a stress-associated liver-specific transcription factor inducible by LPS or proinflammatory cytokines [218–220]. Clinical data also support a role of CB1 in liver injury, since analysis of pooled 1-year data from four pivotal trials in overweight patients showed a significant decrease in alanine aminotransferase in patients under rimonabant [221]. Similar results were reported in the ADAGIO trial enrolling 800 patients with abdominal obesity and dyslipidemia [222].
CB1 receptors also play a major role in liver fibrosis. Enhanced hepatic CB1 receptor expression has been described in hepatic myofibroblasts from patients with NAFLD, and CB1 receptors were found to signal profibrogenic responses in experimental models of liver fibrosis [223]. Moreover, mice administered rimonabant or invalidated for CB1 are protected from fibrosis, by a mechanism involving reduced proliferation and increased apoptosis of hepatic myofibroblasts [223]. These results are corroborated by epidemiological data showing that daily cannabis smoking is an independent predictive factor of fibrosis severity and fibrosis stage in patient with chronic hepatitis C [224].
These data show that peripheral overactivation of CB1 receptors contributes to the progression of NAFLD, by acting at multiple steps of the disease. However, rimonabant has been withdrawn from the market, given an alarming rate of side effects on the central nervous system. This major drawback could soon be overcome with the emerging availability of CB1 antagonists unable to cross the blood-brain barrier. Along these lines, recent studies have validated the efficacy of two orally bioavailable CB1 antagonists with limited ability to penetrate the brain, AM6545 and JD 5037, on steatosis, liver injury, insulin resistance and blood glucose [220, 225, 226]. Interestingly, our recent unpublished findings indicate that AM6545 displays antifibrogenic properties similar to that of rimonabant (Lotersztajn, unpublished).
Enhanced hepatic expression of CB2 receptors has been documented in NAFLD patients and in fat stromal vascular fraction in obese mice [227]. Moreover, treatment of obese mice with the CB2 agonist JWH-133 enhance fat inflammation, insulin resistance and steatosis, while CB2-deficient animals are protected [227]. These data indicate that CB2 receptors contribute to metabolic steatosis, probably as a result of increased adipose tissue inflammation and insulin resistance. However, activation of CB2 receptors expressed in Kupffer cells protects hepatocytes from steatosis by promoting a shift of Kupffer cells towards an M2 phenotype and inhibiting the release of steatogenic cytokines (i.e TNF alpha and IL-1 beta, [149]. Moreover, CB2 receptors display potent hepatoprotective properties in a variety of models of acute and chronic liver injury [228]. Finally, CB2 receptors show antifibrogenic properties, via direct antiproliferative and apoptotic effects on hepatic myofibroblasts, and indirect anti-inflammatory effects on Kupffer cells and Th17 lymphocytes [149, 229, 230]. Therefore, CB2 receptors may display deleterious effects at early stages of NAFLD, but protective effects against NASH.
4.8. Apoptosis
NASH is characterized by hepatocyte apoptosis, and generation of apoptotic bodies has been recognized as a potent proinflammatory and profibrogenic stimulus [231]. Several pathways lead to lipotoxic apoptosis, including inflammation, ER stress, oxidative stress and fatty acids themselves, such as palmitic acid. After the appearance of apoptosis, apoptotic bodies are engulfed by Kupffer cells and HSC, resulting in release of proinflammatory and profibrogenic mediators. The direct link between apoptosis and NASH has been demonstrated by a number of experimental studies, which have shown that blockade of the apoptotic pathway results in a beneficial effect on inflammation, hepatocyte injury, and fibrosis. This mechanism also shows a cross-talk with the adipokine system, as leptin stimulates the phagocytotic process of apoptotic bodies by HSC, generating additional profibrogenic cues [232]. For these reasons, clinical trials have been performed to assess the possibility to translate these findings to the human setting [233]. Nonetheless, it should be kept in mind that strategies that chronically reduce apoptosis could potentially favor the appearance of hepatocellular carcinoma, the incidence of which has been found to be increased in patients with NAFLD even in the absence of cirrhosis.
4.9. ER stress
One of the consequences of lipotoxicity is the activation of the so-called ‘unfolded protein response’ (UPR) that is characterized by a series of intracellular events that counteract the excessive presence of misfolded proteins within cells, including hepatocytes [234]. The activation of the UPR includes increased activity of pathways that results in apoptosis and inflammation, primarily JNK. Of note, the UPR and the related ER stress have also been implicated in the determination of insulin resistance both at the level of adipose tissue and of the liver [235]. In particular, ER stress-mediated activation of JNK interferes with insulin receptor signaling, resulting in limited metabolic effects (see above). Like for many other pathways activated in conditions of hepatocyte injury or lipotoxicity, ER stress is to some extent protective, whereas its excessive activation may trigger cell death and injury. However, increasing understanding of the molecules implicated in its activation and fine tuning may eventually represent an important target to investigate for the treatment of chronic liver diseases.
4.10. Lipid droplets and autophagy
In hepatocytes, excess fat in form of triglycerides is stored in separate structures known as lipid droplets. Lipid droplets are separated by the cytosol by a protein layer, and have been considered relatively inert structures until recently. Recent studies have indicated that proteins lining the intracellular lipid droplets have a regulatory function which may have an impact on the dynamics of intracellular fat in both the adipose tissue and the liver, and in the development of inflammation [236]. In patients with NAFLD, expression of perilipin and adipophilin was found on the rim of lipid droplets in both NASH and simple steatosis. Interestingly, the frequency of adipophilin-positive ballooned hepatocytes correlated with inflammation and fibrosis [237].
The dynamics of intracellular lipid droplets are also associated with the role of autophagy. Autophagy is a metabolic process that degrades and recycles intracellular organelles and proteins. Autophagy delivers intracellular proteins and organelles sequestered in double-membrane vesicles (autophagosomes) to lysosomes for degradation and use as an energy source [238]. The Czaja’s group has demonstrated a connection between autophagy and steatosis, reporting that inhibition of autophagy in hepatocytes and in mouse liver increases triglyceride storage in lipid droplets [239]. While autophagy may be viewed as a protective mechanism against steatosis, its activation appears to favor the progression of fibrosis. In mouse stellate cells, an increase in autophagic flux was observed during activation in culture, and exposure of HSC to an inhibitor of autophagy decreased proliferation and expression of activation markers [240]. Moreover, the levels of autophagy were increased upon liver injury in mice and in human samples. Loss of autophagic function in mice following injury reduced fibrogenesis and matrix accumulation [241].
4.11. Mitochondria and oxidative stress
Oxidative stress has been traditionally considered one of the ‘second-hits’ responsible for the progression from steatosis to steatohepatitis [242] and demonstration of oxidative damage in the liver of patients with NAFLD has been provided [243]. One of the problems with this pathogenetic factor has been the observation that while the treatment with antioxidant was effective in animal models of steatohepatitis, it was rather ineffective in humans. This idea has been at least partially challenged by the results of the recent PIVENS study, where vitamin E was superior to placebo in reducing necroinflammation in patients with NASH [244]. In steatohepatitis, reactive oxygen species and reactive aldehydes may be generated by different sources [245]. An important contribution is the one provided by inflammatory cells, as shown by the observation that reduction of inflammation is associated with reduced oxidative stress [154]. The endoplasmic reticulum contributes to ROS generation mostly via induction of cytochrome P450, altered nutritional state, diabetes, and obesity [246]. The involvement of cytochrome P4502E1 in NASH has been elegantly shown in mice administered a methionine and choline-deficient (MCD) diet, where liver injury and fibrosis were associated with induction of CYP2E1, and Cyp2e1−/− mice were protected from damage [247]. Peroxisomes are sources of cytosolic hydrogen peroxide associated with fatty acid oxidation [245].
Mitochondria are a relevant source of oxidative stress-related products. [248]. Of note, mitochondrial alterations have been reported in NASH and in obese subjects, leading to uncoupling of oxidative phosphorylation and resulting in ROS generation. [248, 249]. Additionally, the role of iron in NAFLD and NASH has been recently be re-evaluated [250]. Iron overload has been recently shown to contribute to insulin resistance, altering post-receptor signaling. Moreover, mild hepatic iron overload promotes the progression of liver damage independently of IR, through oxidative stress [250]. Reactive aldehydes and ROS directly target hepatic stellate cells causing a fibrogenic stimulus [251], and this is favored by an increase in iron stores. Accordingly, iron depletion caused a decrease in oxidative stress and HSCs activation in experimental models of liver injury [252]. Modulation of iron stores may be done effectively and inexpensively with venesection, an approach which deserves consideration as a possible therapy for NASH [250].
Treatment with natural compounds with antioxidant activities may represent a new approach in this area, also considering the potential affordability of these molecules [253]. In this respect, curcumin is a very promising compound. Administration of curcumin in rodent models of steatohepatitis has been shown to prevent inflammation, injury and fibrosis [254, 255]. The beneficial actions of curcumin are based on modulation of different molecular effectors and inhibition of fibrogenic activities of hepatic stellate cells [256, 257]. Extracts of milk thistle, such as silybin, have demonstrated protective effects on fatty liver both in animals models and in patient studies [258]. These and other studies have highlighted to potential relevance of ‘nutraceuticals’ in this field.
5. Conclusions and perspectives
Steatohepatitis developing in patients with the metabolic syndrome is a relevant clinical problem, and a targeted therapy still an unmet need. While weight loss and physical exercise have provided clear benefits, a pharmacological therapy needs an in-depth knowledge of the pathogenetic aspects that are associated with the appearance of steatohepatitis. This entity appears to be separated by the simple steatosis observed in the majority of patients, and a wealth of information has accumulated in the past 10 years on several aspect of the pathogenesis of NASH. Application of translational research, which combines studies in humans with mechanistic information from preclinical investigation is the challenge for the discovery of new therapeutic targets. The data discussed above demonstrate not only that a wide number of potential therapeutic targets are available (Table 1), that for many of those a pharmacological targeting is feasible or even already available. Nonetheless, the active elucidation, through controlled clinical trials, of the therapies that could be of benefit for this condition, is still hampered by the need of a liver biopsy to differentiate simple steatosis from true NASH. Thus, the search for a targeted therapy is closely linked to research that aims to identify easy-to-use tests to at least narrow down the population of patients to subject to liver biopsy and to start on a therapeutic trial. The explosion of information on this condition witnessed in the past few years is likely to lead in reasonable time to new and targeted therapies.
Table 1.
Summary of the pathways representing possible targets for the treatment of NASH
| Pathway | Comments |
|---|---|
| Insulin resistance | Clinical studies show effectiveness on steatosis and inflammation, but little effects on fibrosis |
| Lipid accumulation | Evidence from experimental models suggests that NASH actually worsens if triglyceride accumulation is blocked. Lipases regulating intracellular lipid trafficking (e.g. ATGL) are an interesting target but little data are available. |
| Inflammation: macrophages | M1 macrophage accumulation is a key event in both the adipose tissue and the liver. Possible targets include chemokines (e.g. CCL2), their receptors, cytokines released by these cells (TNF, IL-6), and osteopontin. Increased M2 polarization of macrophages to generate anti-inflammatory signals. Consider possible detrimental effects on fibrosis? |
| Inflammation: lymphocytes | Increase in regulatory T cell function or number. |
| Inflammation: inflammasomes | IL-1beta is released by inflammasome activation and drives inflammation and fibrosis. However, Activation of inflammasome in the gut may be protective on NASH by modulating the microbiota. |
| Inflammation: signaling pathways | NF-kappaB has pleiotropic effects and prevents of cell death. Targeting selective downstream targets of NF-kappa B would be more realistic |
| Renin-angiotensin system | Relevant for insulin resistance, liver inflammation and fibrosis. Well known drugs in clinical practice for decades. |
| Microbiome | Prebiotics, probiotics, antibiotics. |
| TLRs | Strong evidence as mediators of inflammation and fibrosis. However, targeting TLR may increase susceptibility to infections. |
| Modulation of the GLP-1 system | Long-acting GLP-1 ligands or DPP4 inhibitors. |
| JNK | Involved in insulin resistance and cell death. However, possible risk of hepatocarcinogenesis upon prolonged inhibition |
| Oxidative stress | Clinical studies have shown efficacy of vitamin E on inflammation but not fibrosis. Need for new, more active molecules. |
| Autophagy | Antisteatogenic effects but drives fibrogenesis. |
| ER stress | Involved in both insulin resistance and liver damage |
| Omega-3 | Possible effects on steatosis. Beneficial action on inflammation and fibrosis still controversial. |
| Adipokines | Increase in adiponectin levels or signaling, e.g. AMPK, has potential to limit metabolic abnormalities, liver inflammation and fibrosis. |
| Cannabinoids | Upcoming availability of peripheral-restricted antagonists |
Acknowledgments
Work on nonalcoholic steatohepatitis in Dr. Marra’s laboratory is supported by grants from MIUR (Progetti PRIN), 7th Framework Programme (FLIP project) and the Fondazione CARIPLO. Work in Dr. Lotersztajn’s laboratory is supported by the INSERM, the Université Paris-Est, and by the Agence Nationale de la Recherche.
Footnotes
Conflict of Interest:
None
References
- 1.Ekstedt M, Franzen LE, Mathiesen UL, Thorelius L, Holmqvist M, Bodemar G, et al. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology. 2006;44:865–73. doi: 10.1002/hep.21327. [DOI] [PubMed] [Google Scholar]
- 2.Teli MR, James OF, Burt AD, Bennett MK, Day CP. The natural history of nonalcoholic fatty liver: a follow-up study. Hepatology. 1995;22:1714–9. [PubMed] [Google Scholar]
- 3.Roulot D, Costes J-L, Buyck J-Fo, Warzocha U, Gambier N, Czernichow Sb, et al. Transient elastography as a screening tool for liver fibrosis and cirrhosis in a community-based population aged over 45 years. Gut. 2011 doi: 10.1136/gut.2010.221382. in press. [DOI] [PubMed] [Google Scholar]
- 4.Angulo P. Nonalcoholic fatty liver disease. N Engl J Med. 2002;346:1221–31. doi: 10.1056/NEJMra011775. [DOI] [PubMed] [Google Scholar]
- 5.Ong J, Younossi ZM, Reddy V, Price LL, Gramlich T, Mayes J, et al. Cryptogenic cirrhosis and posttransplantation nonalcoholic fatty liver disease. Liver Transpl. 2001;7:797–801. doi: 10.1053/jlts.2001.24644. [DOI] [PubMed] [Google Scholar]
- 6.Bugianesi E, Leone N, Vanni E, Marchesini G, Brunello F, Carucci P, et al. Expanding the natural history of nonalcoholic steatohepatitis: From cryptogenic cirrhosis to hepatocellular carcinoma. Gastroenterology. 2002;123:134–40. doi: 10.1053/gast.2002.34168. [DOI] [PubMed] [Google Scholar]
- 7.Cusi K. Role of obesity and lipotoxicity in the development of nonalcoholic steatohepatitis: pathophysiology and clinical implications. Gastroenterology. 2012;142:711–725. e6. doi: 10.1053/j.gastro.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 8.Schattenberg JM, Schuppan D. Nonalcoholic steatohepatitis: the therapeutic challenge of a global epidemic. Curr Opin Lipidol. 2011;22:479–88. doi: 10.1097/MOL.0b013e32834c7cfc. [DOI] [PubMed] [Google Scholar]
- 9.Nguyen TAT, Sanyal AJ. Pathophysiology guided treatment of nonalcoholic steatohepatitis. Journal of Gastroenterology and Hepatology. 2012;27:58–64. doi: 10.1111/j.1440-1746.2011.07018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ratziu V, Giral P, Charlotte F, Bruckert E, Thibault V, Theodorou I, et al. Liver fibrosis in overweight patients. Gastroenterology. 2000;118:1117–23. doi: 10.1016/s0016-5085(00)70364-7. [DOI] [PubMed] [Google Scholar]
- 11.Harrison SA, Oliver D, Arnold HL, Gogia S, Neuschwander-Tetri BA. Development and validation of a simple NAFLD clinical scoring system for identifying patients without advanced disease. Gut. 2008;57:1441–1447. doi: 10.1136/gut.2007.146019. [DOI] [PubMed] [Google Scholar]
- 12.Dixon JB, Bhathal PS, O’Brien PE. Nonalcoholic fatty liver disease: predictors of nonalcoholic steatohepatitis and liver fibrosis in the severely obese. Gastroenterology. 2001;121:91–100. doi: 10.1053/gast.2001.25540. [DOI] [PubMed] [Google Scholar]
- 13.Angulo P, Keach JC, Batts KP, Lindor KD. Independent predictors of liver fibrosis in patients with nonalcoholic steatohepatitis. Hepatology. 1999;30:1356–62. doi: 10.1002/hep.510300604. [DOI] [PubMed] [Google Scholar]
- 14.Angulo P, Hui JM, Marchesini G, Bugianesi E, George J, Farrell GC, et al. The NAFLD fibrosis score: a noninvasive system that identifies liver fibrosis in patients with NAFLD. Hepatology. 2007;45:846–54. doi: 10.1002/hep.21496. [DOI] [PubMed] [Google Scholar]
- 15.Marchesini G, Bugianesi E, Forlani G, Cerrelli F, Lenzi M, Manini R, et al. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology. 2003;37:917–23. doi: 10.1053/jhep.2003.50161. [DOI] [PubMed] [Google Scholar]
- 16.Day CP. Genetic and Environmental Susceptibility to Non-Alcoholic Fatty Liver Disease. Digestive Diseases. 2010;28:255–260. doi: 10.1159/000282098. [DOI] [PubMed] [Google Scholar]
- 17.Wallace DF, Subramaniam VN. Co-factors in liver disease: The role of HFE-related hereditary hemochromatosis and iron. Biochimica et Biophysica Acta (BBA) - General Subjects. 2009;1790:663–670. doi: 10.1016/j.bbagen.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 18.Valenti L, Al-Serri A, Daly AK, Galmozzi E, Rametta R, Dongiovanni P, et al. Homozygosity for the patatin-like phospholipase-3/adiponutrin I148M polymorphism influences liver fibrosis in patients with nonalcoholic fatty liver disease. Hepatology. 2010;51:1209–17. doi: 10.1002/hep.23622. [DOI] [PubMed] [Google Scholar]
- 19.Chalasani N, Guo X, Loomba R, Goodarzi MO, Haritunians T, Kwon S, et al. Genome-Wide Association Study Identifies Variants Associated With Histologic Features of Nonalcoholic Fatty Liver Disease. Gastroenterology. 139:1567–1576.e6. doi: 10.1053/j.gastro.2010.07.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115:1343–51. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fabbrini E, Mohammed BS, Magkos F, Korenblat KM, Patterson BW, Klein S. Alterations in adipose tissue and hepatic lipid kinetics in obese men and women with nonalcoholic fatty liver disease. Gastroenterology. 2008;134:424–31. doi: 10.1053/j.gastro.2007.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Amaro A, Fabbrini E, Kars M, Yue P, Schechtman K, Schonfeld G, et al. Dissociation between intrahepatic triglyceride content and insulin resistance in familial hypobetalipoproteinemia. Gastroenterology. 2010;139:149–53. doi: 10.1053/j.gastro.2010.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamaguchi K, Yang L, McCall S, Huang J, Yu XX, Pandey SK, et al. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology. 2007;45:1366–74. doi: 10.1002/hep.21655. [DOI] [PubMed] [Google Scholar]
- 24.Monetti M, Levin MC, Watt MJ, Sajan MP, Marmor S, Hubbard BK, et al. Dissociation of Hepatic Steatosis and Insulin Resistance in Mice Overexpressing DGAT in the Liver. Cell Metab. 2007;6:69–78. doi: 10.1016/j.cmet.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 25.Yamaguchi K, Yang L, McCall S, Huang J, Yu XX, Pandey SK, et al. Diacylglycerol acyltranferase 1 anti-sense oligonucleotides reduce hepatic fibrosis in mice with nonalcoholic steatohepatitis. Hepatology. 2008;47:625–35. doi: 10.1002/hep.21988. [DOI] [PubMed] [Google Scholar]
- 26.Mathurin P, Hollebecque A, Arnalsteen L, Buob D, Leteurtre E, Caiazzo R, et al. Prospective study of the long-term effects of bariatric surgery on liver injury in patients without advanced disease. Gastroenterology. 2009;137:532–40. doi: 10.1053/j.gastro.2009.04.052. [DOI] [PubMed] [Google Scholar]
- 27.Godsland IF. Insulin resistance and hyperinsulinaemia in the development and progression of cancer. Clin Sci (Lond) 2010;118:315–32. doi: 10.1042/CS20090399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lanthier N, Horsmans Y, Leclercq IA. The metabolic syndrome: how it may influence hepatic stellate cell activation and hepatic fibrosis. Curr Opin Clin Nutr Metab Care. 2009;12:404–11. doi: 10.1097/MCO.0b013e32832c7819. [DOI] [PubMed] [Google Scholar]
- 29.Leclercq IA, Da Silva Morais A, Schroyen B, Van Hul N, Geerts A. Insulin resistance in hepatocytes and sinusoidal liver cells: mechanisms and consequences. J Hepatol. 2007;47:142–56. doi: 10.1016/j.jhep.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 30.Viollet B, Guigas B, Sanz Garcia N, Leclerc J, Foretz M, Andreelli F. Cellular and molecular mechanisms of metformin: an overview. Clin Sci (Lond) 2012;122:253–70. doi: 10.1042/CS20110386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ratziu V, Caldwell S, Neuschwander-Tetri BA. Therapeutic trials in nonalcoholic steatohepatitis: insulin sensitizers and related methodological issues. Hepatology. 2010;52:2206–15. doi: 10.1002/hep.24042. [DOI] [PubMed] [Google Scholar]
- 32.Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature. 2007;447:1116–20. doi: 10.1038/nature05894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu CW, Chu ES, Lam CN, Cheng AS, Lee CW, Wong VW, et al. PPARgamma is essential for protection against nonalcoholic steatohepatitis. Gene Ther. 2010;17:790–8. doi: 10.1038/gt.2010.41. [DOI] [PubMed] [Google Scholar]
- 34.Marra F, Efsen E, Romanelli RG, Caligiuri A, Pastacaldi S, Batignani G, et al. Ligands of peroxisome proliferator-activated receptor gamma modulate profibrogenic and proinflammatory actions in hepatic stellate cells. Gastroenterology. 2000;119:466–78. doi: 10.1053/gast.2000.9365. [DOI] [PubMed] [Google Scholar]
- 35.Galli A, Crabb DW, Ceni E, Salzano R, Mello T, Svegliati-Baroni G, et al. Antidiabetic thiazolidinediones inhibit collagen synthesis and hepatic stellate cell activation in vivo and in vitro. Gastroenterology. 2002;122:1924–40. doi: 10.1053/gast.2002.33666. [DOI] [PubMed] [Google Scholar]
- 36.Leclercq IA, Sempoux C, Starkel P, Horsmans Y. Limited therapeutic efficacy of pioglitazone on progression of hepatic fibrosis in rats. Gut. 2006;55:1020–9. doi: 10.1136/gut.2005.079194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chalasani NP, Sanyal AJ, Kowdley KV, Robuck PR, Hoofnagle J, Kleiner DE, et al. Pioglitazone versus vitamin E versus placebo for the treatment of non-diabetic patients with non-alcoholic steatohepatitis: PIVENS trial design. Contemp Clin Trials. 2009;30:88–96. doi: 10.1016/j.cct.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lomonaco R, Ortiz-Lopez C, Orsak B, Webb A, Hardies J, Darland C, et al. Effect of adipose tissue insulin resistance on metabolic parameters and liver histology in obese patients with nonalcoholic fatty liver disease. Hepatology. 2012;55:1389–97. doi: 10.1002/hep.25539. [DOI] [PubMed] [Google Scholar]
- 39.Watt MJ, Spriet LL. Triacylglycerol lipases and metabolic control: implications for health and disease. Am J Physiol Endocrinol Metab. 2010;299:E162–8. doi: 10.1152/ajpendo.00698.2009. [DOI] [PubMed] [Google Scholar]
- 40.Fuchs CD, Claudel T, Kumari P, Haemmerle G, Pollheimer MJ, Stojakovic T, et al. Absence of adipose triglyceride lipase protects from hepatic endoplasmic reticulum stress in mice. Hepatology. 2012;56:270–80. doi: 10.1002/hep.25601. [DOI] [PubMed] [Google Scholar]
- 41.Bazzichi L, Giannaccini G, Betti L, Fabbrini L, Schmid L, Palego L, et al. ATP, calcium and magnesium levels in platelets of patients with primary fibromyalgia. Clin Biochem. 2008;41:1084–90. doi: 10.1016/j.clinbiochem.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 42.Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40:1461–5. doi: 10.1038/ng.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Valenti L, Alisi A, Galmozzi E, Bartuli A, Del Menico B, Alterio A, et al. I148M patatin-like phospholipase domain-containing 3 gene variant and severity of pediatric nonalcoholic fatty liver disease. Hepatology. 2010;52:1274–80. doi: 10.1002/hep.23823. [DOI] [PubMed] [Google Scholar]
- 44.Kumari M, Schoiswohl G, Chitraju C, Paar M, Cornaciu I, Rangrez AY, et al. Adiponutrin functions as a nutritionally regulated lysophosphatidic acid acyltransferase. Cell Metab. 2012;15:691–702. doi: 10.1016/j.cmet.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kanneganti TD, Dixit VD. Immunological complications of obesity. Nat Immunol. 2012;13:707–12. doi: 10.1038/ni.2343. [DOI] [PubMed] [Google Scholar]
- 46.Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest. 2011;121:2111–7. doi: 10.1172/JCI57132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med. 2009;15:930–9. doi: 10.1038/nm.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu D, Molofsky AB, Liang HE, Ricardo-Gonzalez RR, Jouihan HA, Bando JK, et al. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science. 2011;332:243–7. doi: 10.1126/science.1201475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weisberg SP, Hunter D, Huber R, Lemieux J, Slaymaker S, Vaddi K, et al. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest. 2006;116:115–24. doi: 10.1172/JCI24335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112:1821–30. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bertola A, Deveaux V, Bonnafous S, Rousseau D, Anty R, Wakkach A, et al. Elevated expression of osteopontin may be related to adipose tissue macrophage accumulation and liver steatosis in morbid obesity. Diabetes. 2009;58:125–33. doi: 10.2337/db08-0400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gimble JM, Nuttall ME. Adipose-derived stromal/stem cells (ASC) in regenerative medicine: pharmaceutical applications. Curr Pharm Des. 2011;17:332–9. doi: 10.2174/138161211795164220. [DOI] [PubMed] [Google Scholar]
- 54.Postic C, Girard J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice. J Clin Invest. 2008;118:829–38. doi: 10.1172/JCI34275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Patsouris D, Li PP, Thapar D, Chapman J, Olefsky JM, Neels JG. Ablation of CD11c-positive cells normalizes insulin sensitivity in obese insulin resistant animals. Cell Metab. 2008;8:301–9. doi: 10.1016/j.cmet.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Odegaard JI, Ricardo-Gonzalez RR, Red Eagle A, Vats D, Morel CR, Goforth MH, et al. Alternative M2 activation of Kupffer cells by PPARdelta ameliorates obesity-induced insulin resistance. Cell Metab. 2008;7:496–507. doi: 10.1016/j.cmet.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liao X, Sharma N, Kapadia F, Zhou G, Lu Y, Hong H, et al. Kruppel-like factor 4 regulates macrophage polarization. J Clin Invest. 2011;121:2736–49. doi: 10.1172/JCI45444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang X, Xu A, Chung SK, Cresser JH, Sweeney G, Wong RL, et al. Selective inactivation of c-Jun NH2-terminal kinase in adipose tissue protects against diet-induced obesity and improves insulin sensitivity in both liver and skeletal muscle in mice. Diabetes. 2011;60:486–95. doi: 10.2337/db10-0650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yuan M, Konstantopoulos N, Lee J, Hansen L, Li ZW, Karin M, et al. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science. 2001;293:1673–7. doi: 10.1126/science.1061620. [DOI] [PubMed] [Google Scholar]
- 60.Fabbrini E, deHaseth D, Deivanayagam S, Mohammed BS, Vitola BE, Klein S. Alterations in fatty acid kinetics in obese adolescents with increased intrahepatic triglyceride content. Obesity (Silver Spring) 2009;17:25–9. doi: 10.1038/oby.2008.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.van der Poorten D, Milner KL, Hui J, Hodge A, Trenell MI, Kench JG, et al. Visceral fat: a key mediator of steatohepatitis in metabolic liver disease. Hepatology. 2008;48:449–57. doi: 10.1002/hep.22350. [DOI] [PubMed] [Google Scholar]
- 62.Petta S, Amato MC, Di Marco V, Camma C, Pizzolanti G, Barcellona MR, et al. Visceral adiposity index is associated with significant fibrosis in patients with non-alcoholic fatty liver disease. Aliment Pharmacol Ther. 2012;35:238–47. doi: 10.1111/j.1365-2036.2011.04929.x. [DOI] [PubMed] [Google Scholar]
- 63.Marra F, Bertolani C. Adipokines in liver diseases. Hepatology. 2009;50:957–69. doi: 10.1002/hep.23046. [DOI] [PubMed] [Google Scholar]
- 64.Bertolani C, Marra F. Role of adipocytokines in hepatic fibrosis. Curr Pharm Des. 2010;16:1929–40. doi: 10.2174/138161210791208857. [DOI] [PubMed] [Google Scholar]
- 65.Choi SS, Syn WK, Karaca GF, Omenetti A, Moylan CA, Witek RP, et al. Leptin promotes the myofibroblastic phenotype in hepatic stellate cells by activating the hedgehog pathway. J Biol Chem. 2010;285:36551–60. doi: 10.1074/jbc.M110.168542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ding X, Saxena NK, Lin S, Xu A, Srinivasan S, Anania FA. The roles of leptin and adiponectin: a novel paradigm in adipocytokine regulation of liver fibrosis and stellate cell biology. Am J Pathol. 2005;166:1655–69. doi: 10.1016/S0002-9440(10)62476-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Handy JA, Fu PP, Kumar P, Mells JE, Sharma S, Saxena NK, et al. Adiponectin inhibits leptin signalling via multiple mechanisms to exert protective effects against hepatic fibrosis. Biochem J. 2011;440:385–95. doi: 10.1042/BJ20102148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Aleffi S, Navari N, Delogu W, Galastri S, Novo E, Rombouts K, et al. Mammalian target of rapamycin mediates the angiogenic effects of leptin in human hepatic stellate cells. Am J Physiol Gastrointest Liver Physiol. 2011;301:G210–9. doi: 10.1152/ajpgi.00047.2010. [DOI] [PubMed] [Google Scholar]
- 69.Yang YY, Tsai TH, Huang YT, Lee TY, Chan CC, Lee KC, et al. Hepatic endothelin-1 and endocannabinoids-dependent effects of hyperleptinemia in nonalcoholic steatohepatitis-cirrhotic rats. Hepatology. 2012;55:1540–50. doi: 10.1002/hep.25534. [DOI] [PubMed] [Google Scholar]
- 70.Wang J, Leclercq I, Brymora JM, Xu N, Ramezani-Moghadam M, London RM, et al. Kupffer cells mediate leptin-induced liver fibrosis. Gastroenterology. 2009;137:713–23. doi: 10.1053/j.gastro.2009.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Imajo K, Fujita K, Yoneda M, Nozaki Y, Ogawa Y, Shinohara Y, et al. Hyperresponsivity to low-dose endotoxin during progression to nonalcoholic steatohepatitis is regulated by leptin-mediated signaling. Cell Metab. 2012;16:44–54. doi: 10.1016/j.cmet.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 72.Javor ED, Ghany MG, Cochran EK, Oral EA, Depaoli AM, Premkumar A, et al. Leptin reverses nonalcoholic steatohepatitis in patients with severe lipodystrophy. Hepatology. 2005;41:753–60. doi: 10.1002/hep.20672. [DOI] [PubMed] [Google Scholar]
- 73.Sennello JA, Fayad R, Morris AM, Eckel RH, Asilmaz E, Montez J, et al. Regulation of T cell-mediated hepatic inflammation by adiponectin and leptin. Endocrinology. 2005;146:2157–64. doi: 10.1210/en.2004-1572. [DOI] [PubMed] [Google Scholar]
- 74.Tilg H, Moschen AR. Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat Rev Immunol. 2006;6:772–83. doi: 10.1038/nri1937. [DOI] [PubMed] [Google Scholar]
- 75.Kamada Y, Tamura S, Kiso S, Matsumoto H, Saji Y, Yoshida Y, et al. Enhanced carbon tetrachloride-induced liver fibrosis in mice lacking adiponectin. Gastroenterology. 2003;125:1796–807. doi: 10.1053/j.gastro.2003.08.029. [DOI] [PubMed] [Google Scholar]
- 76.Xu A, Wang Y, Keshaw H, Xu LY, Lam KS, Cooper GJ. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J Clin Invest. 2003;112:91–100. doi: 10.1172/JCI17797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Caligiuri A, Bertolani C, Guerra CT, Aleffi S, Galastri S, Trappoliere M, et al. Adenosine monophosphate-activated protein kinase modulates the activated phenotype of hepatic stellate cells. Hepatology. 2008;47:668–76. doi: 10.1002/hep.21995. [DOI] [PubMed] [Google Scholar]
- 78.Adachi M, Brenner DA. High molecular weight adiponectin inhibits proliferation of hepatic stellate cells via activation of adenosine monophosphate-activated protein kinase. Hepatology. 2008;47:677–85. doi: 10.1002/hep.21991. [DOI] [PubMed] [Google Scholar]
- 79.Parola M, Marra F. Adipokines and Redox Signaling: Impact on Fatty Liver Disease. Antioxidant & Redox Signaling. 2011 doi: 10.1089/ars.2010.3848. in press. [DOI] [PubMed] [Google Scholar]
- 80.Iwabu M, Yamauchi T, Okada-Iwabu M, Sato K, Nakagawa T, Funata M, et al. Adiponectin and AdipoR1 regulate PGC-1alpha and mitochondria by Ca(2+) and AMPK/SIRT1. Nature. 2010;464:1313–9. doi: 10.1038/nature08991. [DOI] [PubMed] [Google Scholar]
- 81.Huang H, Park PH, McMullen MR, Nagy LE. Mechanisms for the anti-inflammatory effects of adiponectin in macrophages. J Gastroenterol Hepatol. 2008;23 (Suppl 1):S50–3. doi: 10.1111/j.1440-1746.2007.05284.x. [DOI] [PubMed] [Google Scholar]
- 82.Massip-Salcedo M, Zaouali MA, Padrissa-Altes S, Casillas-Ramirez A, Rodes J, Rosello-Catafau J, et al. Activation of peroxisome proliferator-activated receptor-alpha inhibits the injurious effects of adiponectin in rat steatotic liver undergoing ischemia-reperfusion. Hepatology. 2008;47:461–72. doi: 10.1002/hep.21935. [DOI] [PubMed] [Google Scholar]
- 83.Mandal P, Pratt BT, Barnes M, McMullen MR, Nagy LE. Molecular mechanism for adiponectin-dependent M2 macrophage polarization: link between the metabolic and innate immune activity of full-length adiponectin. J Biol Chem. 2011;286:13460–9. doi: 10.1074/jbc.M110.204644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Polyzos SA, Kountouras J, Zavos C, Tsiaousi E. The role of adiponectin in the pathogenesis and treatment of non-alcoholic fatty liver disease. Diabetes Obes Metab. 2010;12:365–83. doi: 10.1111/j.1463-1326.2009.01176.x. [DOI] [PubMed] [Google Scholar]
- 85.Filkova M, Haluzik M, Gay S, Senolt L. The role of resistin as a regulator of inflammation: Implications for various human pathologies. Clin Immunol. 2009;133:157–70. doi: 10.1016/j.clim.2009.07.013. [DOI] [PubMed] [Google Scholar]
- 86.Bertolani C, Sancho-Bru P, Failli P, Bataller R, Aleffi S, DeFranco R, et al. Resistin as an intrahepatic cytokine: overexpression during chronic injury and induction of proinflammatory actions in hepatic stellate cells. Am J Pathol. 2006;169:2042–53. doi: 10.2353/ajpath.2006.060081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Choi YJ, Choi SE, Ha ES, Kang Y, Han SJ, Kim DJ, et al. Involvement of visfatin in palmitate-induced upregulation of inflammatory cytokines in hepatocytes. Metabolism. 2011;60:1781–9. doi: 10.1016/j.metabol.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 88.Anderson EK, Gutierrez DA, Hasty AH. Adipose tissue recruitment of leukocytes. Curr Opin Lipidol. 2010;21:172–7. doi: 10.1097/MOL.0b013e3283393867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Thatcher S, Yiannikouris F, Gupte M, Cassis L. The adipose renin-angiotensin system: role in cardiovascular disease. Mol Cell Endocrinol. 2009;302:111–7. doi: 10.1016/j.mce.2009.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Matthew Morris E, Fletcher JA, Thyfault JP, Scott Rector R. The role of angiotensin II in nonalcoholic steatohepatitis. Mol Cell Endocrinol. 2012 doi: 10.1016/j.mce.2012.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Moreno M, Bataller R. Cytokines and renin-angiotensin system signaling in hepatic fibrosis. Clin Liver Dis. 2008;12:825–52. ix. doi: 10.1016/j.cld.2008.07.013. [DOI] [PubMed] [Google Scholar]
- 92.Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, et al. Enterotypes of the human gut microbiome. Nature. 2011;473:174–80. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–31. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 94.Compare D, Coccoli P, Rocco A, Nardone OM, De Maria S, Carteni M, et al. Gut--liver axis: the impact of gut microbiota on non alcoholic fatty liver disease. Nutr Metab Cardiovasc Dis. 2012;22:471–6. doi: 10.1016/j.numecd.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 95.Tilg H, Kaser A. Gut microbiome, obesity, and metabolic dysfunction. J Clin Invest. 2011;121:2126–32. doi: 10.1172/JCI58109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dumas ME, Barton RH, Toye A, Cloarec O, Blancher C, Rothwell A, et al. Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc Natl Acad Sci U S A. 2006;103:12511–6. doi: 10.1073/pnas.0601056103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Spencer MD, Hamp TJ, Reid RW, Fischer LM, Zeisel SH, Fodor AA. Association between composition of the human gastrointestinal microbiome and development of fatty liver with choline deficiency. Gastroenterology. 2011;140:976–86. doi: 10.1053/j.gastro.2010.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482:179–85. doi: 10.1038/nature10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Csak T, Ganz M, Pespisa J, Kodys K, Dolganiuc A, Szabo G. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology. 2011;54:133–44. doi: 10.1002/hep.24341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Stienstra R, van Diepen JA, Tack CJ, Zaki MH, van de Veerdonk FL, Perera D, et al. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc Natl Acad Sci U S A. 2011;108:15324–9. doi: 10.1073/pnas.1100255108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Thoma C, Day CP, Trenell MI. Lifestyle interventions for the treatment of non-alcoholic fatty liver disease in adults: a systematic review. J Hepatol. 2012;56:255–66. doi: 10.1016/j.jhep.2011.06.010. [DOI] [PubMed] [Google Scholar]
- 102.Musso G, Gambino R, De Michieli F, Cassader M, Rizzetto M, Durazzo M, et al. Dietary habits and their relations to insulin resistance and postprandial lipemia in nonalcoholic steatohepatitis. Hepatology. 2003;37:909–16. doi: 10.1053/jhep.2003.50132. [DOI] [PubMed] [Google Scholar]
- 103.McCarthy EM, Rinella ME. The role of diet and nutrient composition in nonalcoholic Fatty liver disease. J Acad Nutr Diet. 2012;112:401–9. doi: 10.1016/j.jada.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 104.Malhi H, Bronk SF, Werneburg NW, Gores GJ. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J Biol Chem. 2006;281:12093–101. doi: 10.1074/jbc.M510660200. [DOI] [PubMed] [Google Scholar]
- 105.Listenberger LL, Han X, Lewis SE, Cases S, Farese RV, Jr, Ory DS, et al. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci U S A. 2003;100:3077–82. doi: 10.1073/pnas.0630588100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ibrahim SH, Kohli R, Gores GJ. Mechanisms of lipotoxicity in NAFLD and clinical implications. J Pediatr Gastroenterol Nutr. 2011;53:131–40. doi: 10.1097/MPG.0b013e31822578db. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gillingham LG, Harris-Janz S, Jones PJ. Dietary monounsaturated fatty acids are protective against metabolic syndrome and cardiovascular disease risk factors. Lipids. 2011;46:209–28. doi: 10.1007/s11745-010-3524-y. [DOI] [PubMed] [Google Scholar]
- 108.Masterton GS, Plevris JN, Hayes PC. Review article: omega-3 fatty acids - a promising novel therapy for non-alcoholic fatty liver disease. Aliment Pharmacol Ther. 2010;31:679–92. doi: 10.1111/j.1365-2036.2010.04230.x. [DOI] [PubMed] [Google Scholar]
- 109.Parker HM, Johnson NA, Burdon CA, Cohn JS, O’Connor HT, George J. Omega-3 supplementation and non-alcoholic fatty liver disease: a systematic review and meta-analysis. J Hepatol. 2012;56:944–51. doi: 10.1016/j.jhep.2011.08.018. [DOI] [PubMed] [Google Scholar]
- 110.Sawada N, Inoue M, Iwasaki M, Sasazuki S, Shimazu T, Yamaji T, et al. Consumption of n-3 fatty acids and fish reduces risk of hepatocellular carcinoma. Gastroenterology. 2012;142:1468–75. doi: 10.1053/j.gastro.2012.02.018. [DOI] [PubMed] [Google Scholar]
- 111.Gonzalez-Periz A, Horrillo R, Ferre N, Gronert K, Dong B, Moran-Salvador E, et al. Obesity-induced insulin resistance and hepatic steatosis are alleviated by omega-3 fatty acids: a role for resolvins and protectins. Faseb J. 2009;23:1946–57. doi: 10.1096/fj.08-125674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sekiya M, Yahagi N, Matsuzaka T, Najima Y, Nakakuki M, Nagai R, et al. Polyunsaturated fatty acids ameliorate hepatic steatosis in obese mice by SREBP-1 suppression. Hepatology. 2003;38:1529–39. doi: 10.1016/j.hep.2003.09.028. [DOI] [PubMed] [Google Scholar]
- 113.Ishii H, Horie Y, Ohshima S, Anezaki Y, Kinoshita N, Dohmen T, et al. Eicosapentaenoic acid ameliorates steatohepatitis and hepatocellular carcinoma in hepatocyte-specific Pten-deficient mice. J Hepatol. 2009;50:562–71. doi: 10.1016/j.jhep.2008.10.031. [DOI] [PubMed] [Google Scholar]
- 114.Itoh M, Suganami T, Satoh N, Tanimoto-Koyama K, Yuan X, Tanaka M, et al. Increased adiponectin secretion by highly purified eicosapentaenoic acid in rodent models of obesity and human obese subjects. Arterioscler Thromb Vasc Biol. 2007;27:1918–25. doi: 10.1161/ATVBAHA.106.136853. [DOI] [PubMed] [Google Scholar]
- 115.Mari M, Caballero F, Colell A, Morales A, Caballeria J, Fernandez A, et al. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. 2006;4:185–98. doi: 10.1016/j.cmet.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 116.Van Rooyen DM, Larter CZ, Haigh WG, Yeh MM, Ioannou G, Kuver R, et al. Hepatic free cholesterol accumulates in obese, diabetic mice and causes nonalcoholic steatohepatitis. Gastroenterology. 2011;141:1393–403. 1403 e1–5. doi: 10.1053/j.gastro.2011.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Musso G, Cassader M, Gambino R. Cholesterol-lowering therapy for the treatment of nonalcoholic fatty liver disease: an update. Curr Opin Lipidol. 2011;22:489–96. doi: 10.1097/MOL.0b013e32834c37ee. [DOI] [PubMed] [Google Scholar]
- 118.McLaughlin T, Abbasi F, Lamendola C, Yeni-Komshian H, Reaven G. Carbohydrate-induced hypertriglyceridemia: an insight into the link between plasma insulin and triglyceride concentrations. J Clin Endocrinol Metab. 2000;85:3085–8. doi: 10.1210/jcem.85.9.6838. [DOI] [PubMed] [Google Scholar]
- 119.Kang H, Greenson JK, Omo JT, Chao C, Peterman D, Anderson L, et al. Metabolic syndrome is associated with greater histologic severity, higher carbohydrate, and lower fat diet in patients with NAFLD. Am J Gastroenterol. 2006;101:2247–53. doi: 10.1111/j.1572-0241.2006.00719.x. [DOI] [PubMed] [Google Scholar]
- 120.Ryan MC, Abbasi F, Lamendola C, Carter S, McLaughlin TL. Serum alanine aminotransferase levels decrease further with carbohydrate than fat restriction in insulin-resistant adults. Diabetes Care. 2007;30:1075–80. doi: 10.2337/dc06-2169. [DOI] [PubMed] [Google Scholar]
- 121.Yilmaz Y. Review article: fructose in non-alcoholic fatty liver disease. Aliment Pharmacol Ther. 2012;35:1135–44. doi: 10.1111/j.1365-2036.2012.05080.x. [DOI] [PubMed] [Google Scholar]
- 122.Lim JS, Mietus-Snyder M, Valente A, Schwarz JM, Lustig RH. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat Rev Gastroenterol Hepatol. 2010;7:251–64. doi: 10.1038/nrgastro.2010.41. [DOI] [PubMed] [Google Scholar]
- 123.Gregor MF, Yang L, Fabbrini E, Mohammed BS, Eagon JC, Hotamisligil GS, et al. Endoplasmic reticulum stress is reduced in tissues of obese subjects after weight loss. Diabetes. 2009;58:693–700. doi: 10.2337/db08-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bergheim I, Weber S, Vos M, Kramer S, Volynets V, Kaserouni S, et al. Antibiotics protect against fructose-induced hepatic lipid accumulation in mice: role of endotoxin. J Hepatol. 2008;48:983–92. doi: 10.1016/j.jhep.2008.01.035. [DOI] [PubMed] [Google Scholar]
- 125.Spruss A, Kanuri G, Wagnerberger S, Haub S, Bischoff SC, Bergheim I. Toll-like receptor 4 is involved in the development of fructose-induced hepatic steatosis in mice. Hepatology. 2009;50:1094–104. doi: 10.1002/hep.23122. [DOI] [PubMed] [Google Scholar]
- 126.Song M, Schuschke DA, Zhou Z, Chen T, Pierce WM, Jr, Wang R, et al. High fructose feeding induces copper deficiency in Sprague-Dawley rats: a novel mechanism for obesity related fatty liver. J Hepatol. 2012;56:433–40. doi: 10.1016/j.jhep.2011.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhang C, Chen X, Zhu RM, Zhang Y, Yu T, Wang H, et al. Endoplasmic reticulum stress is involved in hepatic SREBP-1c activation and lipid accumulation in fructose-fed mice. Toxicol Lett. 2012;212:229–40. doi: 10.1016/j.toxlet.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 128.Modi AA, Feld JJ, Park Y, Kleiner DE, Everhart JE, Liang TJ, et al. Increased caffeine consumption is associated with reduced hepatic fibrosis. Hepatology. 2010;51:201–9. doi: 10.1002/hep.23279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Molloy JW, Calcagno CJ, Williams CD, Jones FJ, Torres DM, Harrison SA. Association of coffee and caffeine consumption with fatty liver disease, nonalcoholic steatohepatitis, and degree of hepatic fibrosis. Hepatology. 2012;55:429–36. doi: 10.1002/hep.24731. [DOI] [PubMed] [Google Scholar]
- 130.Vitaglione P, Morisco F, Mazzone G, Amoruso DC, Ribecco MT, Romano A, et al. Coffee reduces liver damage in a rat model of steatohepatitis: the underlying mechanisms and the role of polyphenols and melanoidins. Hepatology. 2010;52:1652–61. doi: 10.1002/hep.23902. [DOI] [PubMed] [Google Scholar]
- 131.Birerdinc A, Stepanova M, Pawloski L, Younossi ZM. Caffeine is protective in patients with non-alcoholic fatty liver disease. Aliment Pharmacol Ther. 2012;35:76–82. doi: 10.1111/j.1365-2036.2011.04916.x. [DOI] [PubMed] [Google Scholar]
- 132.Gressner OA, Lahme B, Rehbein K, Siluschek M, Weiskirchen R, Gressner AM. Pharmacological application of caffeine inhibits TGF-beta-stimulated connective tissue growth factor expression in hepatocytes via PPARgamma and SMAD2/3-dependent pathways. J Hepatol. 2008;49:758–67. doi: 10.1016/j.jhep.2008.03.029. [DOI] [PubMed] [Google Scholar]
- 133.Salamone F, Li Volti G, Titta L, Puzzo L, Barbagallo I, La Delia F, et al. Moro orange juice prevents fatty liver in mice. World J Gastroenterol. 2012;18:3862–8. doi: 10.3748/wjg.v18.i29.3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kim W, Egan JM. The role of incretins in glucose homeostasis and diabetes treatment. Pharmacol Rev. 2008;60:470–512. doi: 10.1124/pr.108.000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ding X, Saxena NK, Lin S, Gupta NA, Anania FA. Exendin-4, a glucagon-like protein-1 (GLP-1) receptor agonist, reverses hepatic steatosis in ob/ob mice. Hepatology. 2006;43:173–81. doi: 10.1002/hep.21006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sharma S, Mells JE, Fu PP, Saxena NK, Anania FA. GLP-1 analogs reduce hepatocyte steatosis and improve survival by enhancing the unfolded protein response and promoting macroautophagy. PLoS ONE. 2011;6:e25269. doi: 10.1371/journal.pone.0025269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Marra F. Ghrelin and fibrogenesis: relief for a hungry liver. J Hepatol. 2011;55:221–3. doi: 10.1016/j.jhep.2011.01.027. [DOI] [PubMed] [Google Scholar]
- 138.Li Y, Hai J, Li L, Chen X, Peng H, Cao M, et al. Administration of ghrelin improves inflammation, oxidative stress, and apoptosis during and after non-alcoholic fatty liver disease development. Endocrine. 2012 doi: 10.1007/s12020-012-9761-5. [DOI] [PubMed] [Google Scholar]
- 139.Neyrinck AM, Cani PD, Dewulf EM, De Backer F, Bindels LB, Delzenne NM. Critical role of Kupffer cells in the management of diet-induced diabetes and obesity. Biochem Biophys Res Commun. 2009;385:351–6. doi: 10.1016/j.bbrc.2009.05.070. [DOI] [PubMed] [Google Scholar]
- 140.Huang W, Metlakunta A, Dedousis N, Zhang P, Sipula I, Dube JJ, et al. Depletion of liver Kupffer cells prevents the development of diet-induced hepatic steatosis and insulin resistance. Diabetes. 2010;59:347–57. doi: 10.2337/db09-0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lanthier N, Molendi-Coste O, Cani PD, van Rooijen N, Horsmans Y, Leclercq IA. Kupffer cell depletion prevents but has no therapeutic effect on metabolic and inflammatory changes induced by a high-fat diet. Faseb J. 2011;25:4301–11. doi: 10.1096/fj.11-189472. [DOI] [PubMed] [Google Scholar]
- 142.Dixon LJ, Berk M, Thapaliya S, Papouchado BG, Feldstein AE. Caspase-1-mediated regulation of fibrogenesis in diet-induced steatohepatitis. Lab Invest. 2012;92:713–23. doi: 10.1038/labinvest.2012.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Miura K, Yang L, van Rooijen N, Ohnishi H, Seki E. Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through CCR2. Am J Physiol Gastrointest Liver Physiol. 2012;302:G1310–21. doi: 10.1152/ajpgi.00365.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Rivera CA, Adegboyega P, van Rooijen N, Tagalicud A, Allman M, Wallace M. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J Hepatol. 2007;47:571–9. doi: 10.1016/j.jhep.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11:723–37. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122:787–95. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kang K, Reilly SM, Karabacak V, Gangl MR, Fitzgerald K, Hatano B, et al. Adipocyte-derived Th2 cytokines and myeloid PPARdelta regulate macrophage polarization and insulin sensitivity. Cell Metab. 2008;7:485–95. doi: 10.1016/j.cmet.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Fukushima J, Kamada Y, Matsumoto H, Yoshida Y, Ezaki H, Takemura T, et al. Adiponectin prevents progression of steatohepatitis in mice by regulating oxidative stress and Kupffer cell phenotype polarization. Hepatol Res. 2009;39:724–38. doi: 10.1111/j.1872-034X.2009.00509.x. [DOI] [PubMed] [Google Scholar]
- 149.Louvet A, Teixeira-Clerc F, Chobert MN, Deveaux V, Pavoine C, Zimmer A, et al. Cannabinoid CB2 receptors protect against alcoholic liver disease by regulating Kupffer cell polarization in mice. Hepatology. 2011;54:1217–26. doi: 10.1002/hep.24524. [DOI] [PubMed] [Google Scholar]
- 150.Miura K, Kodama Y, Inokuchi S, Schnabl B, Aoyama T, Ohnishi H, et al. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology. 2010;139:323–34. e7. doi: 10.1053/j.gastro.2010.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Stienstra R, Saudale F, Duval C, Keshtkar S, Groener JE, van Rooijen N, et al. Kupffer cells promote hepatic steatosis via interleukin-1beta-dependent suppression of peroxisome proliferator-activated receptor alpha activity. Hepatology. 2010;51:511–22. doi: 10.1002/hep.23337. [DOI] [PubMed] [Google Scholar]
- 152.Haukeland JW, Damas JK, Konopski Z, Loberg EM, Haaland T, Goverud I, et al. Systemic inflammation in nonalcoholic fatty liver disease is characterized by elevated levels of CCL2. J Hepatol. 2006;44:1167–74. doi: 10.1016/j.jhep.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 153.Baeck C, Wehr A, Karlmark KR, Heymann F, Vucur M, Gassler N, et al. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut. 2012;61:416–26. doi: 10.1136/gutjnl-2011-300304. [DOI] [PubMed] [Google Scholar]
- 154.Galastri S, Zamara E, Milani S, Novo E, Provenzano A, Delogu W, et al. Lack of CC chemokine ligand 2 differentially affects inflammation and fibrosis according to the genetic background in a murine model of steatohepatitis. Clin Sci (Lond) 2012;123:459–71. doi: 10.1042/CS20110515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115:56–65. doi: 10.1172/JCI22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Aoyama T, Inokuchi S, Brenner DA, Seki E. CX3CL1-CX3CR1 interaction prevents carbon tetrachloride-induced liver inflammation and fibrosis in mice. Hepatology. 2010;52:1390–400. doi: 10.1002/hep.23795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Pesce JT, Ramalingam TR, Mentink-Kane MM, Wilson MS, El Kasmi KC, Smith AM, et al. Arginase-1-expressing macrophages suppress Th2 cytokine-driven inflammation and fibrosis. PLoS Pathog. 2009;5:e1000371. doi: 10.1371/journal.ppat.1000371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Karlmark KR, Zimmermann HW, Roderburg C, Gassler N, Wasmuth HE, Luedde T, et al. The fractalkine receptor CXCR1 protects against liver fibrosis by controlling differentiation and survival of infiltrating hepatic monocytes. Hepatology. 2010;52:1769–82. doi: 10.1002/hep.23894. [DOI] [PubMed] [Google Scholar]
- 159.Wouters K, van Gorp PJ, Bieghs V, Gijbels MJ, Duimel H, Lutjohann D, et al. Dietary cholesterol, rather than liver steatosis, leads to hepatic inflammation in hyperlipidemic mouse models of nonalcoholic steatohepatitis. Hepatology. 2008;48:474–86. doi: 10.1002/hep.22363. [DOI] [PubMed] [Google Scholar]
- 160.Bieghs V, van Gorp PJ, Walenbergh SM, Gijbels MJ, Verheyen F, Buurman WA, et al. Specific immunization strategies against oxidized low-density lipoprotein: A novel way to reduce nonalcoholic steatohepatitis in mice. Hepatology. 2012;56:894–903. doi: 10.1002/hep.25660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Leroux A, Ferrere G, Godie V, Cailleux F, Renoud ML, Gaudin F, et al. Toxic lipids stored by Kupffer cells correlates with their pro-inflammatory phenotype at an early stage of steatohepatitis. J Hepatol. 2012;57:141–9. doi: 10.1016/j.jhep.2012.02.028. [DOI] [PubMed] [Google Scholar]
- 162.Erridge C, Samani NJ. Saturated fatty acids do not directly stimulate Toll-like receptor signaling. Arterioscler Thromb Vasc Biol. 2009;29:1944–9. doi: 10.1161/ATVBAHA.109.194050. [DOI] [PubMed] [Google Scholar]
- 163.Pal D, Dasgupta S, Kundu R, Maitra S, Das G, Mukhopadhyay S, et al. Fetuin-A acts as an endogenous ligand of TLR4 to promote lipid-induced insulin resistance. Nat Med. 2012 doi: 10.1038/nm.2851. [DOI] [PubMed] [Google Scholar]
- 164.Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature. 2012;481:278–86. doi: 10.1038/nature10759. [DOI] [PubMed] [Google Scholar]
- 165.Mencin A, Kluwe J, Schwabe RF. Toll-like receptors as targets in chronic liver diseases. Gut. 2009;58:704–20. doi: 10.1136/gut.2008.156307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Seki E, Brenner DA. Toll-like receptors and adaptor molecules in liver disease: update. Hepatology. 2008;48:322–35. doi: 10.1002/hep.22306. [DOI] [PubMed] [Google Scholar]
- 167.Vanni E, Bugianesi E. The gut-liver axis in nonalcoholic fatty liver disease: Another pathway to insulin resistance? Hepatology. 2009;49:1790–2. doi: 10.1002/hep.23036. [DOI] [PubMed] [Google Scholar]
- 168.Wigg AJ, Roberts-Thomson IC, Dymock RB, McCarthy PJ, Grose RH, Cummins AG. The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor alpha in the pathogenesis of non-alcoholic steatohepatitis. Gut. 2001;48:206–11. doi: 10.1136/gut.48.2.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Miele L, Valenza V, La Torre G, Montalto M, Cammarota G, Ricci R, et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology. 2009;49:1877–87. doi: 10.1002/hep.22848. [DOI] [PubMed] [Google Scholar]
- 170.Szabo G, Velayudham A, Romics L, Jr, Mandrekar P. Modulation of non-alcoholic steatohepatitis by pattern recognition receptors in mice: the role of toll-like receptors 2 and 4. Alcohol Clin Exp Res. 2005;29:140S–145S. doi: 10.1097/01.alc.0000189287.83544.33. [DOI] [PubMed] [Google Scholar]
- 171.Rivera CA, Gaskin L, Allman M, Pang J, Brady K, Adegboyega P, et al. Toll-like receptor-2 deficiency enhances non-alcoholic steatohepatitis. BMC Gastroenterol. 2010;10:52. doi: 10.1186/1471-230X-10-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, et al. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science. 328:228–31. doi: 10.1126/science.1179721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Tsukumo DM, Carvalho-Filho MA, Carvalheira JB, Prada PO, Hirabara SM, Schenka AA, et al. Loss-of-function mutation in Toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes. 2007;56:1986–98. doi: 10.2337/db06-1595. [DOI] [PubMed] [Google Scholar]
- 174.Ye D, Li FY, Lam KS, Li H, Jia W, Wang Y, et al. Toll-like receptor-4 mediates obesity-induced non-alcoholic steatohepatitis through activation of X-box binding protein-1 in mice. Gut. 2012;61:1058–67. doi: 10.1136/gutjnl-2011-300269. [DOI] [PubMed] [Google Scholar]
- 175.Saberi M, Woods NB, de Luca C, Schenk S, Lu JC, Bandyopadhyay G, et al. Hematopoietic cell-specific deletion of toll-like receptor 4 ameliorates hepatic and adipose tissue insulin resistance in high-fat-fed mice. Cell Metab. 2009;10:419–29. doi: 10.1016/j.cmet.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Csak T, Velayudham A, Hritz I, Petrasek J, Levin I, Lippai D, et al. Deficiency in myeloid differentiation factor-2 and toll-like receptor 4 expression attenuates nonalcoholic steatohepatitis and fibrosis in mice. Am J Physiol Gastrointest Liver Physiol. 2011;300:G433–41. doi: 10.1152/ajpgi.00163.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Li Z, Yang S, Lin H, Huang J, Watkins PA, Moser AB, et al. Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatology. 2003;37:343–50. doi: 10.1053/jhep.2003.50048. [DOI] [PubMed] [Google Scholar]
- 178.Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56:1761–72. doi: 10.2337/db06-1491. [DOI] [PubMed] [Google Scholar]
- 179.Kudo H, Takahara T, Yata Y, Kawai K, Zhang W, Sugiyama T. Lipopolysaccharide triggered TNF-alpha-induced hepatocyte apoptosis in a murine non-alcoholic steatohepatitis model. J Hepatol. 2009;51:168–75. doi: 10.1016/j.jhep.2009.02.032. [DOI] [PubMed] [Google Scholar]
- 180.Wagnerberger S, Spruss A, Kanuri G, Volynets V, Stahl C, Bischoff SC, et al. Toll-like receptors 1–9 are elevated in livers with fructose-induced hepatic steatosis. Br J Nutr. 2012;107:1727–38. doi: 10.1017/S0007114511004983. [DOI] [PubMed] [Google Scholar]
- 181.Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, et al. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med. 2007;13:1324–32. doi: 10.1038/nm1663. [DOI] [PubMed] [Google Scholar]
- 182.Brun P, Castagliuolo I, Pinzani M, Palu G, Martines D. Exposure to bacterial cell wall products triggers an inflammatory phenotype in hepatic stellate cells. Am J Physiol Gastrointest Liver Physiol. 2005;289:G571–8. doi: 10.1152/ajpgi.00537.2004. [DOI] [PubMed] [Google Scholar]
- 183.Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17:179–88. doi: 10.1038/nm.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Stienstra R, van Diepen JA, Tack CJ, Zaki MH, van de Veerdonk FL, Perera D, et al. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc Natl Acad Sci U S A. 108:15324–9. doi: 10.1073/pnas.1100255108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Lamkanfi M, Sarkar A, Vande Walle L, Vitari AC, Amer AO, Wewers MD, et al. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J Immunol. 2011;185:4385–92. doi: 10.4049/jimmunol.1000803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Li L, Chen L, Hu L, Liu Y, Sun HY, Tang J, et al. Nuclear factor high-mobility group box1 mediating the activation of Toll-like receptor 4 signaling in hepatocytes in the early stage of nonalcoholic fatty liver disease in mice. Hepatology. 2011;54:1620–30. doi: 10.1002/hep.24552. [DOI] [PubMed] [Google Scholar]
- 187.Watanabe A, Sohail MA, Gomes DA, Hashmi A, Nagata J, Sutterwala FS, et al. Inflammasome-mediated regulation of hepatic stellate cells. Am J Physiol Gastrointest Liver Physiol. 2009;296:G1248–57. doi: 10.1152/ajpgi.90223.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Dixon LJ, Berk M, Thapaliya S, Papouchado BG, Feldstein AE. Caspase-1-mediated regulation of fibrogenesis in diet-induced steatohepatitis. Lab Invest. 92:713–23. doi: 10.1038/labinvest.2012.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Beraza N, Malato Y, Vander Borght S, Liedtke C, Wasmuth HE, Dreano M, et al. Pharmacological IKK2 inhibition blocks liver steatosis and initiation of non-alcoholic steatohepatitis. Gut. 2008;57:655–63. doi: 10.1136/gut.2007.134288. [DOI] [PubMed] [Google Scholar]
- 190.Cai D, Yuan M, Frantz DF, Melendez PA, Hansen L, Lee J, et al. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med. 2005;11:183–90. doi: 10.1038/nm1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Arkan MC, Hevener AL, Greten FR, Maeda S, Li ZW, Long JM, et al. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med. 2005;11:191–8. doi: 10.1038/nm1185. [DOI] [PubMed] [Google Scholar]
- 192.Luedde T, Beraza N, Kotsikoris V, van Loo G, Nenci A, De Vos R, et al. Deletion of NEMO/IKKgamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell. 2007;11:119–32. doi: 10.1016/j.ccr.2006.12.016. [DOI] [PubMed] [Google Scholar]
- 193.Schattenberg JM, Singh R, Wang Y, Lefkowitch JH, Rigoli RM, Scherer PE, et al. JNK1 but not JNK2 promotes the development of steatohepatitis in mice. Hepatology. 2006;43:163–72. doi: 10.1002/hep.20999. [DOI] [PubMed] [Google Scholar]
- 194.Singh R, Wang Y, Xiang Y, Tanaka KE, Gaarde WA, Czaja MJ. Differential effects of JNK1 and JNK2 inhibition on murine steatohepatitis and insulin resistance. Hepatology. 2009;49:87–96. doi: 10.1002/hep.22578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Kodama Y, Brenner DA. c-Jun N-terminal kinase signaling in the pathogenesis of nonalcoholic fatty liver disease: Multiple roles in multiple steps. Hepatology. 2009;49:6–8. doi: 10.1002/hep.22710. [DOI] [PubMed] [Google Scholar]
- 196.Yang R, Wilcox DM, Haasch DL, Jung PM, Nguyen PT, Voorbach MJ, et al. Liver-specific knockdown of JNK1 up-regulates proliferator-activated receptor gamma coactivator 1 beta and increases plasma triglyceride despite reduced glucose and insulin levels in diet-induced obese mice. J Biol Chem. 2007;282:22765–74. doi: 10.1074/jbc.M700790200. [DOI] [PubMed] [Google Scholar]
- 197.Kodama Y, Kisseleva T, Iwaisako K, Miura K, Taura K, De Minicis S, et al. c-Jun N-terminal kinase-1 from hematopoietic cells mediates progression from hepatic steatosis to steatohepatitis and fibrosis in mice. Gastroenterology. 2009;137:1467–1477. e5. doi: 10.1053/j.gastro.2009.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Sabio G, Cavanagh-Kyros J, Ko HJ, Jung DY, Gray S, Jun JY, et al. Prevention of steatosis by hepatic JNK1. Cell Metab. 2009;10:491–8. doi: 10.1016/j.cmet.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Bigorgne AE, Bouchet-Delbos L, Naveau S, Dagher I, Prevot S, Durand-Gasselin I, et al. Obesity-induced lymphocyte hyperresponsiveness to chemokines: a new mechanism of Fatty liver inflammation in obese mice. Gastroenterology. 2008;134:1459–69. doi: 10.1053/j.gastro.2008.02.055. [DOI] [PubMed] [Google Scholar]
- 200.Ma X, Hua J, Mohamood AR, Hamad AR, Ravi R, Li Z. A high-fat diet and regulatory T cells influence susceptibility to endotoxin-induced liver injury. Hepatology. 2007;46:1519–29. doi: 10.1002/hep.21823. [DOI] [PubMed] [Google Scholar]
- 201.Tang Y, Bian Z, Zhao L, Liu Y, Liang S, Wang Q, et al. Interleukin-17 exacerbates hepatic steatosis and inflammation in non-alcoholic fatty liver disease. Clin Exp Immunol. 2011;166:281–90. doi: 10.1111/j.1365-2249.2011.04471.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Bertola A, Ciucci T, Rousseau D, Bourlier V, Duffaut C, Bonnafous S, et al. Identification of adipose tissue dendritic cells correlated with obesity-associated insulin-resistance and inducing th17 responses in mice and patients. Diabetes. 2012;61:2238–47. doi: 10.2337/db11-1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Elinav E, Pappo O, Sklair-Levy M, Margalit M, Shibolet O, Gomori M, et al. Adoptive transfer of regulatory NKT lymphocytes ameliorates non-alcoholic steatohepatitis and glucose intolerance in ob/ob mice and is associated with intrahepatic CD8 trapping. J Pathol. 2006;209:121–8. doi: 10.1002/path.1950. [DOI] [PubMed] [Google Scholar]
- 204.Kremer M, Thomas E, Milton RJ, Perry AW, van Rooijen N, Wheeler MD, et al. Kupffer cell and interleukin-12-dependent loss of natural killer T cells in hepatosteatosis. Hepatology. 2010;51:130–41. doi: 10.1002/hep.23292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Tajiri K, Shimizu Y, Tsuneyama K, Sugiyama T. Role of liver-infiltrating CD3+CD56+ natural killer T cells in the pathogenesis of nonalcoholic fatty liver disease. Eur J Gastroenterol Hepatol. 2009;21:673–80. doi: 10.1097/MEG.0b013e32831bc3d6. [DOI] [PubMed] [Google Scholar]
- 206.Syn WK, Oo YH, Pereira TA, Karaca GF, Jung Y, Omenetti A, et al. Accumulation of natural killer T cells in progressive nonalcoholic fatty liver disease. Hepatology. 2010;51:1998–2007. doi: 10.1002/hep.23599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Syn WK, Agboola KM, Swiderska M, Michelotti GA, Liaskou E, Pang H, et al. NKT-associated hedgehog and osteopontin drive fibrogenesis in non-alcoholic fatty liver disease. Gut. 2012;61:1323–9. doi: 10.1136/gutjnl-2011-301857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Liu J, Zhou L, Xiong K, Godlewski G, Mukhopadhyay B, Tam J, et al. Hepatic cannabinoid receptor-1 mediates diet-induced insulin resistance via inhibition of insulin signaling and clearance in mice. Gastroenterology. 2012;142:1218–1228. e1. doi: 10.1053/j.gastro.2012.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Mendez-Sanchez N, Zamora-Valdes D, Pichardo-Bahena R, Barredo-Prieto B, Ponciano-Rodriguez G, Bermejo-Martinez L, et al. Endocannabinoid receptor CB2 in nonalcoholic fatty liver disease. Liver Int. 2007;27:215–9. doi: 10.1111/j.1478-3231.2006.01401.x. [DOI] [PubMed] [Google Scholar]
- 210.Mallat A, Teixeira-Clerc F, Deveaux V, Manin S, Lotersztajn S. The endocannabinoid system as a key mediator during liver diseases: new insights and therapeutic openings. Br J Pharmacol. 2011;163:1432–40. doi: 10.1111/j.1476-5381.2011.01397.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Osei-Hyiaman D, Liu J, Zhou L, Godlewski G, Harvey-White J, Jeong WI, et al. Hepatic CB1 receptor is required for development of diet-induced steatosis, dyslipidemia, and insulin and leptin resistance in mice. J Clin Invest. 2008;118:3160–9. doi: 10.1172/JCI34827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Gary-Bobo M, Elachouri G, Gallas JF, Janiak P, Marini P, Ravinet-Trillou C, et al. Rimonabant reduces obesity-associated hepatic steatosis and features of metabolic syndrome in obese Zucker fa/fa rats. Hepatology. 2007;46:122–9. doi: 10.1002/hep.21641. [DOI] [PubMed] [Google Scholar]
- 213.Jourdan T, Djaouti L, Demizieux L, Gresti J, Verges B, Degrace P. CB1 antagonism exerts specific molecular effects on visceral and subcutaneous fat and reverses liver steatosis in diet-induced obese mice. Diabetes. 2010;59:926–34. doi: 10.2337/db09-1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Hezode C, Zafrani ES, Roudot-Thoraval F, Costentin C, Hessami A, Bouvier-Alias M, et al. Daily cannabis use: a novel risk factor of steatosis severity in patients with chronic hepatitis C. Gastroenterology. 2008;134:432–9. doi: 10.1053/j.gastro.2007.11.039. [DOI] [PubMed] [Google Scholar]
- 215.Van der Poorten D, Shahidi M, Tay E, Sesha J, Tran K, McLeod D, et al. Hepatitis C virus induces the cannabinoid receptor 1. PLoS One. 2010:5. doi: 10.1371/journal.pone.0012841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Dol-Gleizes F, Paumelle R, Visentin V, Mares AM, Desitter P, Hennuyer N, et al. Rimonabant, a selective cannabinoid CB1 receptor antagonist, inhibits atherosclerosis in LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2009;29:12–8. doi: 10.1161/ATVBAHA.108.168757. [DOI] [PubMed] [Google Scholar]
- 217.Miranville A, Herling AW, Biemer-Daub G, Voss MD. Reversal of inflammation-induced impairment of glucose uptake in adipocytes by direct effect of CB1 antagonism on adipose tissue macrophages. Obesity (Silver Spring) 2010;18:2247–54. doi: 10.1038/oby.2010.81. [DOI] [PubMed] [Google Scholar]
- 218.Chanda D, Kim DK, Li T, Kim YH, Koo SH, Lee CH, et al. Cannabinoid receptor type 1 (CB1R) signaling regulates hepatic gluconeogenesis via induction of endoplasmic reticulum-bound transcription factor cAMP-responsive element-binding protein H (CREBH) in primary hepatocytes. J Biol Chem. 2011;286:27971–9. doi: 10.1074/jbc.M111.224352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Mukhopadhyay B, Cinar R, Yin S, Liu J, Tam J, Godlewski G, et al. Hyperactivation of anandamide synthesis and regulation of cell-cycle progression via cannabinoid type 1 (CB1) receptors in the regenerating liver. Proc Natl Acad Sci U S A. 2011;108:6323–8. doi: 10.1073/pnas.1017689108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Tam J, Cinar R, Liu J, Godlewski G, Wesley D, Jourdan T, et al. Peripheral cannabinoid-1 receptor inverse agonism reduces obesity by reversing leptin resistance. Cell Metab. 2012;16:167–79. doi: 10.1016/j.cmet.2012.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Van Gaal LF, Scheen AJ, Rissanen AM, Rossner S, Hanotin C, Ziegler O. Long-term effect of CB1 blockade with rimonabant on cardiometabolic risk factors: two year results from the RIO-Europe Study. Eur Heart J. 2008;29:1761–71. doi: 10.1093/eurheartj/ehn076. [DOI] [PubMed] [Google Scholar]
- 222.Despres JP, Ross R, Boka G, Almeras N, Lemieux I. Effect of rimonabant on the high-triglyceride/ low-HDL-cholesterol dyslipidemia, intraabdominal adiposity, and liver fat: the ADAGIO-Lipids trial. Arterioscler Thromb Vasc Biol. 2009;29:416–23. doi: 10.1161/ATVBAHA.108.176362. [DOI] [PubMed] [Google Scholar]
- 223.Teixeira-Clerc F, Julien B, Grenard P, Tran-Van-Nhieu J, Deveaux V, Serriere-Lanneau, et al. CB1 cannabinoid receptor antagonism: a novel strategy for the treatment of liver fibrosis. Nature Medicine. 2006;12:671–676. doi: 10.1038/nm1421. [DOI] [PubMed] [Google Scholar]
- 224.Hezode C, Roudot-Thoraval F, Nguyen S, Grenard P, Julien B, Zafrani ES, et al. Daily cannabis smoking as a risk factor for fibrosis progression in chronic hepatitis C. Hepatology. 2005;42:63–71. doi: 10.1002/hep.20733. [DOI] [PubMed] [Google Scholar]
- 225.Cluny NL, Vemuri VK, Chambers AP, Limebeer CL, Bedard H, Wood JT, et al. A novel peripherally restricted cannabinoid receptor antagonist, AM6545, reduces food intake and body weight, but does not cause malaise, in rodents. Br J Pharmacol. 2010;161:629–42. doi: 10.1111/j.1476-5381.2010.00908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Tam J, Vemuri VK, Liu J, Batkai S, Mukhopadhyay B, Godlewski G, et al. Peripheral CB1 cannabinoid receptor blockade improves cardiometabolic risk in mouse models of obesity. J Clin Invest. 2010;120:2953–66. doi: 10.1172/JCI42551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Deveaux V, Cadoudal T, Ichigotani Y, Teixeira-Clerc F, Louvet A, Manin S, et al. Cannabinoid CB2 receptor potentiates obesity-associated inflammation, insulin resistance and hepatic steatosis. PLoS One. 2009;4:e5844. doi: 10.1371/journal.pone.0005844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Teixeira-Clerc F, Belot MP, Manin S, Deveaux V, Cadoudal T, Chobert MN, et al. Beneficial paracrine effects of cannabinoid receptor 2 on liver injury and regeneration. Hepatology. 2010;52:1046–59. doi: 10.1002/hep.23779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Julien B, Grenard P, Teixeira-Clerc F, Van Nhieu JT, Li L, Karsak M, et al. Antifibrogenic role of the cannabinoid receptor CB2 in the liver. Gastroenterology. 2005;128:742–55. doi: 10.1053/j.gastro.2004.12.050. [DOI] [PubMed] [Google Scholar]
- 230.Lafdil F, Guillot A, Bizy A, Zoltani K, Deveaux V, Zafrani S, et al. Inhibition of the pro-fibrogenic Th17 response by cannabinoid receptor 2 agonist decreases liver fibrosis. Journal of Hepatology. 2012;56:S152. [Google Scholar]
- 231.Guicciardi ME, Gores GJ. Apoptosis as a mechanism for liver disease progression. Semin Liver Dis. 2010;30:402–10. doi: 10.1055/s-0030-1267540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Jiang JX, Mikami K, Shah VH, Torok NJ. Leptin induces phagocytosis of apoptotic bodies by hepatic stellate cells via a Rho guanosine triphosphatase-dependent mechanism. Hepatology. 2008;48:1497–505. doi: 10.1002/hep.22515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Tiple D, Fabbrini G, Colosimo C, Ottaviani D, Camerota F, Defazio G, et al. Camptocormia in Parkinson disease: an epidemiological and clinical study. J Neurol Neurosurg Psychiatry. 2009;80:145–8. doi: 10.1136/jnnp.2008.150011. [DOI] [PubMed] [Google Scholar]
- 234.Colosimo C, Fabbrini G. Investigational drugs. Handb Clin Neurol. 2007;84:137–50. doi: 10.1016/S0072-9752(07)84037-1. [DOI] [PubMed] [Google Scholar]
- 235.Sellitto M, Genesio R, Conti A, Fabbrini F, Nitsch L, D’Armiento M, et al. Short 9q interstitial deletion in a neonate with lethal non-immune hydrops. Am J Med Genet A. 2008;146A:2566–9. doi: 10.1002/ajmg.a.32350. [DOI] [PubMed] [Google Scholar]
- 236.Defazio G, Berardelli A, Fabbrini G, Martino D, Fincati E, Fiaschi A, et al. Pain as a nonmotor symptom of Parkinson disease: evidence from a case-control study. Arch Neurol. 2008;65:1191–4. doi: 10.1001/archneurol.2008.2. [DOI] [PubMed] [Google Scholar]
- 237.Pini A, Brunetti J, Falciani C, Fabbrini M, Bracci L. Solubility improvement of an anthrax toxin peptide inhibitor by rational amino acid randomization. Protein Pept Lett. 2008;15:562–6. doi: 10.2174/092986608784966958. [DOI] [PubMed] [Google Scholar]
- 238.Strappaghetti G, Mastrini L, Lucacchini A, Giannaccini G, Betti L, Fabbrini L. Synthesis and biological affinity of new imidazo- and indol-arylpiperazine derivatives: further validation of a pharmacophore model for alpha(1)-adrenoceptor antagonists. Bioorg Med Chem Lett. 2008;18:5140–5. doi: 10.1016/j.bmcl.2008.07.084. [DOI] [PubMed] [Google Scholar]
- 239.Pasquini M, Fabbrini G, Berardelli I, Bonifati V, Biondi M, Berardelli A. Psychopathological features of obsessive-compulsive disorder in an Italian family with Gilles de la Tourette syndrome not linked to the SLITRK1 gene. Psychiatry Res. 2008;161:109–11. doi: 10.1016/j.psychres.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 240.Tiple D, Strano S, Colosimo C, Fabbrini G, Calcagnini G, Prencipe M, et al. Autonomic cardiovascular function and baroreflex sensitivity in patients with cervical dystonia receiving treatment with botulinum toxin type A. J Neurol. 2008;255:843–7. doi: 10.1007/s00415-008-0753-6. [DOI] [PubMed] [Google Scholar]
- 241.Korenblat KM, Fabbrini E, Mohammed BS, Klein S. Liver, muscle, and adipose tissue insulin action is directly related to intrahepatic triglyceride content in obese subjects. Gastroenterology. 2008;134:1369–75. doi: 10.1053/j.gastro.2008.01.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Day C, James O. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;114:842–845. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 243.Videla LA, Rodrigo R, Orellana M, Fernandez V, Tapia G, Quinones L, et al. Oxidative stress-related parameters in the liver of non-alcoholic fatty liver disease patients. Clin Sci (Lond) 2004;106:261–8. doi: 10.1042/CS20030285. [DOI] [PubMed] [Google Scholar]
- 244.Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med. 2010;362:1675–85. doi: 10.1056/NEJMoa0907929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Rolo AP, Teodoro JS, Palmeira CM. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic Biol Med. 2012;52:59–69. doi: 10.1016/j.freeradbiomed.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 246.Robertson G, Leclercq I, Farrell GC. Nonalcoholic steatosis and steatohepatitis. II. Cytochrome P-450 enzymes and oxidative stress. Am J Physiol Gastrointest Liver Physiol. 2001;281:G1135–9. doi: 10.1152/ajpgi.2001.281.5.G1135. [DOI] [PubMed] [Google Scholar]
- 247.Leclercq IA, Farrell GC, Field J, Bell DR, Gonzalez FJ, Robertson GR. CYP2E1 and CYP4A as microsomal catalysts of lipid peroxides in murine nonalcoholic steatohepatitis. J Clin Invest. 2000;105:1067–75. doi: 10.1172/JCI8814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Gambino R, Musso G, Cassader M. Redox balance in the pathogenesis of nonalcoholic fatty liver disease: mechanisms and therapeutic opportunities. Antioxid Redox Signal. 2011;15:1325–65. doi: 10.1089/ars.2009.3058. [DOI] [PubMed] [Google Scholar]
- 249.Sanyal AJ, Campbell-Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, et al. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology. 2001;120:1183–92. doi: 10.1053/gast.2001.23256. [DOI] [PubMed] [Google Scholar]
- 250.Dongiovanni P, Fracanzani AL, Fargion S, Valenti L. Iron in fatty liver and in the metabolic syndrome: a promising therapeutic target. J Hepatol. 2011;55:920–32. doi: 10.1016/j.jhep.2011.05.008. [DOI] [PubMed] [Google Scholar]
- 251.Parola M, Robino G. Oxidative stress-related molecules and liver fibrosis. J Hepatol. 2001;35:297–306. doi: 10.1016/s0168-8278(01)00142-8. [DOI] [PubMed] [Google Scholar]
- 252.Otogawa K, Ogawa T, Shiga R, Nakatani K, Ikeda K, Nakajima Y, et al. Attenuation of acute and chronic liver injury in rats by iron-deficient diet. Am J Physiol Regul Integr Comp Physiol. 2008;294:R311–20. doi: 10.1152/ajpregu.00735.2007. [DOI] [PubMed] [Google Scholar]
- 253.Singh S. From exotic spice to modern drug? Cell. 2007;130:765–8. doi: 10.1016/j.cell.2007.08.024. [DOI] [PubMed] [Google Scholar]
- 254.Leclercq IA, Farrell GC, Sempoux C, dela Pena A, Horsmans Y. Curcumin inhibits NF-kappaB activation and reduces the severity of experimental steatohepatitis in mice. J Hepatol. 2004;41:926–34. doi: 10.1016/j.jhep.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 255.Vizzutti F, Provenzano A, Galastri S, Milani S, Delogu W, Novo E, et al. Curcumin limits the fibrogenic evolution of experimental steatohepatitis. Lab Invest. 2010;90:104–15. doi: 10.1038/labinvest.2009.112. [DOI] [PubMed] [Google Scholar]
- 256.Rivera-Espinoza Y, Muriel P. Pharmacological actions of curcumin in liver diseases or damage. Liver Int. 2009;29:1457–66. doi: 10.1111/j.1478-3231.2009.02086.x. [DOI] [PubMed] [Google Scholar]
- 257.Tang Y, Zheng S, Chen A. Curcumin eliminates leptin’s effects on hepatic stellate cell activation via interrupting leptin signaling. Endocrinology. 2009;150:3011–20. doi: 10.1210/en.2008-1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Loguercio C, Andreone P, Brisc C, Brisc MC, Bugianesi E, Chiaramonte M, et al. Silybin combined with phosphatidylcholine and vitamin E in patients with nonalcoholic fatty liver disease: a randomized controlled trial. Free Radic Biol Med. 2012;52:1658–65. doi: 10.1016/j.freeradbiomed.2012.02.008. [DOI] [PubMed] [Google Scholar]