

Abstract
Aims
Peroxisome Proliferator-Activated Receptor (PPAR) α is a transcription factor controlling lipid metabolism in liver, heart, muscle and macrophages. PPARα-activation increases plasma HDL-cholesterol and exerts hypotriglyceridemic actions via the liver. However, the intestine expresses PPARα, produces HDL and chylomicrons and is exposed to diet-derived PPARα ligands. Therefore, we examined the effects of PPARα-activation on intestinal lipid and lipoprotein metabolism.
Methods and Results
The impact of PPARα-activation was evaluated in term of HDL-related gene expression in mice, ex-vivo in human jejunal biopsies and in Caco-2/TC7 cells. ApoAI/HDL secretion, cholesterol esterification and trafficking were also studied in-vitro.
In parallel to improving plasma lipid profiles and increasing liver and intestinal expression of fatty-acid-oxidation genes, treatment with the dual PPARα/δ-ligand GFT505 resulted in a more pronounced increase of plasma HDL compared to fenofibrate in mice. GFT505, but not fenofibrate, increased the expression of HDL-production genes such as apolipoprotein-AI and ATP-Binding-Cassette-A1 transporter in murine intestines. A similar increase was observed upon PPARα-activation of human biopsies and Caco-2/TC7 cells. Additionally, HDL secretion by Caco-2/TC7 cells increased. Moreover, PPARα-activation decreased the cholesterol-esterification capacity of Caco-2/TC7 cells, modified cholesterol trafficking and reduced apolipoprotein-B secretion.
Conclusions
PPARα-activation reduces cholesterol esterification, suppresses chylomicron- and increases HDL-secretion by enterocytes. These results identify the intestine as a target organ of PPARα-ligands with entero-hepatic tropism to reduce atherogenic dyslipidemia.
Keywords: Animals; Apolipoproteins B; metabolism; Butyrates; pharmacology; Caco-2 Cells; Cells, Cultured; Chalcones; pharmacology; Enterocytes; metabolism; Esterification; physiology; Fatty Acids; metabolism; Female; Humans; Jejunum; metabolism; Lipoproteins, HDL; metabolism; Mice; Mice, Knockout; PPAR alpha; antagonists & inhibitors; physiology; Phenylurea Compounds; pharmacology; Propionates; pharmacology
Introduction
High levels of low-density-lipoprotein (LDL)-cholesterol and low levels of high-density-lipoprotein (HDL)-cholesterol are risk factors for cardiovascular disease (1). In association with lifestyle intervention, LDL-cholesterol lowering therapy with statins is highly efficient to reduce cardiovascular risk, but has only modest effects on HDL-cholesterol (2). Furthermore, low HDL-cholesterol is an independent risk factor which contributes to the persistent residual cardiovascular risk in statin-treated patients (3; 4). Therefore, raising HDL may be a promising strategy to attenuate residual cardiovascular risk in statin-treated patients.
In enterocytes, dietary lipids are packaged with apolipoprotein-B48 (apoB48) into chylomicrons secreted into the lymph (5). Several studies demonstrated that post-prandial chylomicrons contribute to lipid accumulation in the arterial wall and to atherogenesis (6). Animal studies have shown that the intestine produces HDL (7). Indeed, intestinal-specific deletion of ATP-Binding-Cassette-A1 transporter (ABCA1) in mice results in a 30% decrease of HDL-cholesterol levels (8). Furthermore, exercise-training enhances HDL-cholesterol in rats in correlation with increased intestinal ABCA1 mRNA expression (9). Interestingly, human intestinal mucosa was shown to produce apolipoprotein-AI (apoAI) (10) and HDL (11).
Peroxisome Proliferator-Activated Receptor (PPAR)α is a transcription factor belonging to the nuclear receptor superfamily and activated by fibrates, eicosanoids and fatty-acids (12; 13). PPARα controls lipid and glucose metabolism in several tissues and cell types including liver, heart, kidney, adipose tissue and macrophages (14–16). PPARα-activation enhances hepatic fatty-acid-oxidation and peripheral triglyceride clearance, thus reducing hypertriglyceridemia (13; 17). PPARα increases hepatic apoAI synthesis and ABCA1 expression, resulting in increased plasma HDL-cholesterol (18; 19). In addition, PPARα-activation controls cholesterol trafficking in human macrophages and raises cholesterol efflux via ABCA1 contributing to increased reverse cholesterol transport (15; 20). Since diet-derived compounds (fatty-acids and phospholipids) can activate PPARα and since the intestine is the site of dietary lipid absorption, it is highly exposed to PPARα-ligands (21; 22). The role of PPARα in the intestine has been addressed in only a few studies. Previous studies in rodents did not evidence pronounced effects of the PPARα-ligand fenofibrate (FF) in the intestine contrary to the liver, possibly because of its rapid urinary excretion and hence relatively low intestinal exposure (24). However, a genome wide analysis of intestinal RNA of mice treated with Wy14643 revealed that PPARα controls numerous metabolic pathways and in particular lipid metabolism (25), pointing to a role for PPARα in this organ.
We thus investigated whether intestinal PPARα-activation regulates HDL production. Treatment of mice with two different PPAR-agonists, FF, a pure PPARα-agonist and GFT505 (26), a PPARα/δ-modulator, which, unlike FF, undergoes extensive enterohepatic cycling, resulted in comparable hypotriglyceridemic effects. However, only GFT505 treatment increased apoAI and ABCA1 mRNA levels in the murine intestine, an effect which was associated with a more pronounced increase of plasma HDL levels. These effects were not observed in PPARα-deficient mice. Using the human Caco-2/TC7 model cultured on porous filters, we found that PPARα-activation reduces secretion of chylomicrons while enhancing HDL production probably by increasing ABCA1 expression, apoAI secretion and by reducing cholesterol esterification. The physiological relevance of these regulatory processes was verified in human jejunal biopsies. Collectively, our data suggest that the intestine is a target organ for entero-hepato-tropic PPARα-ligands to increase HDL, an effect which may result in a reduction of residual cardiovascular risk.
Materials and methods
For details, see supplementary materials and methods (supp.M&M).
Animal study
Wild-type (+/+) and homozygous (−/−) PPARα-deficient female mice in the apoE2-KI background (12 week-old) fed a western-diet were treated for 14d with GFT505 (10 or 30mpk) or FF (200mpk) or carboxy-methyl-cellulose (0.5%) (27).
Human intestine culture
Intestinal biopsies were recovered during gastric bypass surgery from obese patients enrolled in the ABOS study (ClinicalTrials.gov; NCT01129297).
Caco-2/TC7 cell culture and PPARα knock-down
Caco-2/TC7 were grown as described (28). For stable PPARα invalidation (29) and culture details, see supp.M&M.
HDL preparation
HDL (d=1.12–1.21g/ml) from human plasma and basolateral media of Caco-2/TC7 were prepared by sequential ultracentrifugations. For details in preparation, electron microscopy and apolipoprotein quantification, see supp.M&M.
Post-prandial micelle preparation
Synthetic micelles prepared as described (28).
Confocal microscopy
Cholesterol esterification assay
Caco-2/TC7 were incubated with [3H]-cholesterol-micelles (7.5μCi/well), lipids were extracted and separated by TLC.
Macrophage cell culture and cholesterol efflux
Mononuclear cells were isolated from blood of healthy donors, cholesterol-loaded with [3H]-cholesterol-AcLDL and efflux assays performed as described (15).
Statistics
For details, see supp.M&M.
Results
PPARα activation in mice and in human jejunal biopsies increases the expression of genes involved in intestinal HDL production
To determine whether PPARα-activation regulates HDL production in the small intestine, apoE2-KI mice, which display a similar lipid-response to PPARα-agonists as humans (36), were fed a western-diet with daily oral administration of FF or the dual PPARα/δ-agonist GFT505 for 14d. As reported (36), FF decreased plasma triglycerides (TG) and total-cholesterol (tab1). Similarly, GFT505 lowered plasma TG and total-cholesterol. Whereas plasma HDL-cholesterol increased approximately 2.5-fold upon treatment with FF, the increase of HDL-cholesterol was significantly more pronounced upon GFT505 (tab1). FF and GFT505 induced the hepatic expression of PPARα target genes, such as Acyl-CoenzymeA-oxidase-1 (ACOX1) at a similar extent (supp.fig 1) indicating equipotent dosing. The effects of GFT505 on TG and HDL-cholesterol were abolished in PPARα-deficient apoE2-KI mice, demonstrating that the effects are PPARα-dependent (supp.tab1). Interestingly, a decrease, albeit less pronounced, of total and nonHDL-cholesterol was maintained in GFT505-treated PPARα-deficient apoE2-KI mice, suggesting a contribution of its PPARδ activity on these lipid parameters (supp.tab1). GFT505-treated mice displayed a PPARα dependent higher ABCA1 and apoAI gene expression in the small intestine compared to untreated mice (fig 1A, fig 1B), whereas FF did not regulate ABCA1 nor apoAI mRNA expression (fig 1C, fig 1D). Treatment of mice with GW0742, a pure PPARδ-agonist, did not modify ABCA1 nor ApoAI gene expression (not shown).
Figure 1. PPARα-activation with GFT505 in-vivo in mice and GFT505 and GW7647 ex-vivo in human jejunal biopsies increases the expression of genes involved in HDL production.
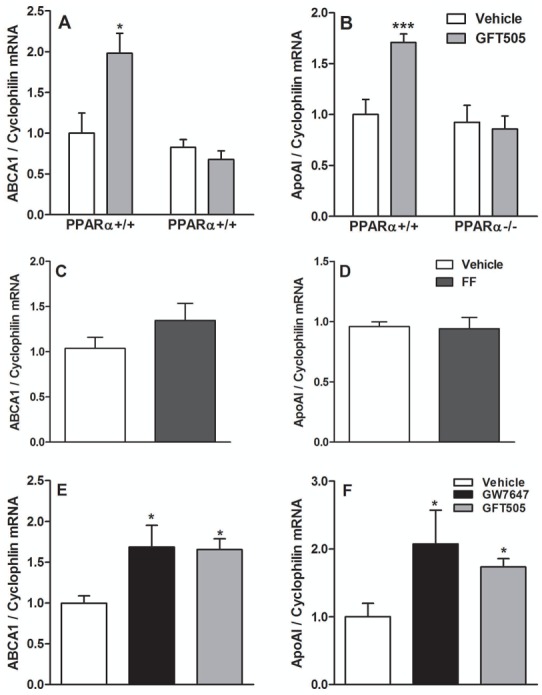
Q-PCR analysis of ABCA1 (A, C, E) and apoAI (B, D, F) was performed on RNA isolated from scrapped intestinal mucosa (A, B, C, D) of GFT505 (30mpk) or FF (200mpk)-treated apoE2-KI mice for 14d and from human jejunal biopsies (E, F) treated with GW7647 (600nM) and GFT505 (1μM) for 18h. In vivo values are expressed as means ± SEM and ex vivo values as means ± SD. t test; *, P<0.05; ***,P<0.001 Vehicle vs treatment).
To determine whether PPARα-activation also regulates these genes in human intestine, human jejunal biopsies were treated with GFT505 and a synthetic PPARα-ligand, GW7647, for 18h. Both compounds significantly increased ABCA1 and apoAI mRNA expression (fig 1E, fig 1F).
PPARα-activation increases the production of HDL in Caco-2/TC7
Based on observations in mice (8), we hypothesized that the up-regulation of ABCA1 and apoAI gene expression observed upon PPARα-activation would translate in increased intestinal HDL production. To test this, we used an in-vitro model of the human intestinal barrier, polarized Caco-2/TC7 cells grown on porous filters (28; 32). PPARα expression was knocked-down (ShPPARα) or not (ShControl) by lentiviral infection and cells were subsequently incubated with GW7647. HDL secreted by Caco-2/TC7 were isolated by ultracentrifugation and analyzed by electron microscopy. PPARα-activation increased the number of HDL secreted at the basolateral side of Caco-2/TC7 (fig 2A). ApoAI secreted in the HDL fraction increased when non-infected Caco-2/TC7 and ShControl-infected cells were treated with GW7647, whereas apoAI secretion was not modified upon treatment of ShPPARα-infected cells by GW7647 (fig 2B), demonstrating that this effect is PPARα-dependent. Interestingly, the quantity of apoAI in the chylomicron fraction was not modified (not shown). To evaluate the HDL functionality, we investigated their capacity to promote cholesterol efflux from macrophages. Human monocyte-derived macrophages were cholesterol-loaded with [3H]-cholesterol-AcLDL and subsequently incubated with HDL secreted by Caco-2/TC7 treated with GW7647 for 24h. Equivalent volumes of isolated HDL from non-infected or ShControl-infected Caco-2/TC7 treated with GW7647 enhanced macrophage cholesterol efflux compared to HDL isolated from vehicle-treated cells (fig 2C). HDL from ShPPARα-infected cells treated with GW7647 did not show enhanced efflux capacity compared to HDL from untreated cells. ABCA1 gene expression increased upon GW7647 treatment in a PPARα-dependent manner (fig 2D). A similar up-regulation was observed with GFT505 (supp.fig 2). In contrast to the in-vivo and ex-vivo response, apoAI mRNA was not induced by PPARα-agonists in Caco-2/TC7 (not shown).
Figure 2. GW7647 increases the production of HDL by Caco-2/TC7.
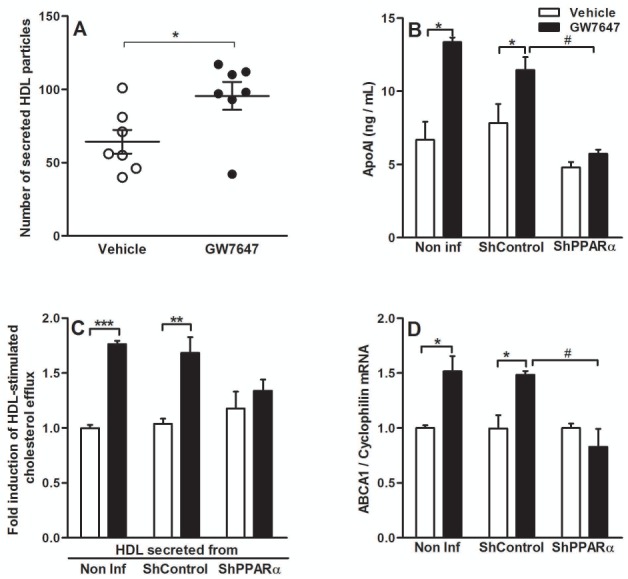
Caco-2/TC7 (non-infected, ShControl- or ShPPARα-infected) were treated with GW7647 (600nM) for 48h (Q-PCR and ELISA analysis) or 72h (cholesterol efflux assay).
(A) Number of HDL secreted by non-infected Caco-2/TC7 per equivalent volume. (B) ELISA analysis of apoAI in the HDL fraction (representative of 3 independent experiments, values are expressed as means ± SEM). (C) Cholesterol-loaded primary human macrophages were incubated with HDL secreted by Caco-2/TC7 (representative of 2 independent experiments, values are expressed as means ± SEM). (D) Q-PCR analysis of ABCA1. Values are expressed as means ± SD. t-test; *P<.05; **P<.01; ***P<.001 Vehicle vs treatment; #P<.05 treated ShControl- vs treated-ShPPARα-infected cells.
PPARα-activation modulates cholesterol distribution, increases cytoplasmic lipid droplet formation and reduces apoB secretion in Caco-2/TC7
To test the effect of PPARα-activation on intestinal cholesterol absorption known to occur in sequential steps, from uptake at the brush border membrane and transport into the cell to packaging and secretion of lipoproteins at the basolateral side of enterocytes (33), Caco-2/TC7 were incubated for 24h with [3H]-cholesterol-micelles and activated with GW7647 24h before and 24h after cholesterol-loading. Cholesterol content in the apical medium, the cellular compartment and the basolateral medium of Caco-2/TC7 was not modulated by GW7647 (not shown), suggesting that PPARα regulates neither total cholesterol uptake nor efflux by enterocytes. Next, GW7647-treated Caco-2/TC7 were incubated with fluorescent-NBD-cholesterol-containing micelles for 4h, followed by non-fluorescent micelles for an additional 4h period. Quantitative analysis of fluorescence intensity at each optical slice showed that, without affecting the total amount of absorbed cholesterol (assessed by radioactive assay), GW7647 treatment modifies cholesterol distribution inducing retention of cholesterol in the subapical compartments (fig 3A). Interestingly, as shown by confocal microscopy (fig 3B), GW7647 also increased the size of cholesterol-containing lipid droplets in the apical and basolateral compartments.
Figure 3. PPARα-activation modulates intracellular cholesterol trafficking, increases cytoplasmic lipid droplet formation and reduces apoB secretion by Caco-2/TC7.
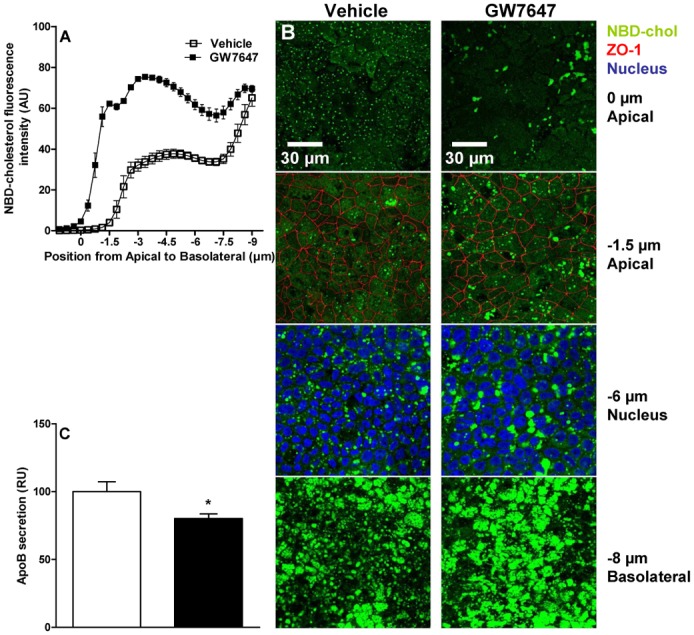
Caco-2/TC7 were incubated with NBD-cholesterol-micelles (50μg/well) for 4h and then for 4h with non-fluorescent micelles. GW7647 (600nM) was added 24h before and during the NBD-cholesterol-loading period (representative of 3 independent experiments). (A) NBD-fluorescence was quantified using Image J at each optical slice. (B) NBD-cholesterol (green), ZO-1 (red) and Hoechst (blue) were visualized by confocal microscopy. (C) ApoB in the basolateral medium was quantified by ELISA. Values are expressed as means ± SEM. t-test; *P<.05 Vehicle vs treatment.
Since PPARα-activation in Caco-2/TC7 does not affect the total amount of cholesterol uptake and efflux, we hypothesized that PPARα-activation may decrease apoB-containing lipoprotein secretion to counterbalance the increase of secreted HDL. Indeed, GW7647 decreased apoB secretion in the basolateral compartment of Caco-2/TC7 (fig 3C) as previously shown (34).
PPARα-activation decreases cholesterol esterification capacity of Caco-2/TC7
Cholesterol trafficking from the plasma membrane to the endoplasmic reticulum is a limiting step for its esterification and subsequent secretion in chylomicrons (35). To address whether, by interfering with cholesterol trafficking, PPARα reduces cholesterol esterification, Caco-2/TC7 were incubated for 24h with [3H]-cholesterol-micelles and activated with GW7647 24h before and 24h after cholesterol loading. PPARα-activation decreased the quantity of [3H]-cholesteryl esters (CE) assayed by TLC both in the cells (fig 4A) and secreted in the basolateral compartment (fig 4B). The decreased CE production does not result only from a perturbation of cholesterol trafficking since ACAT-2 (acyl-coenzymeA:cholesterol-acyltransferase-2) gene expression was also decreased (fig 4C).
Figure 4. PPARα-activation decreases cholesterol esterification in Caco-2/TC7.
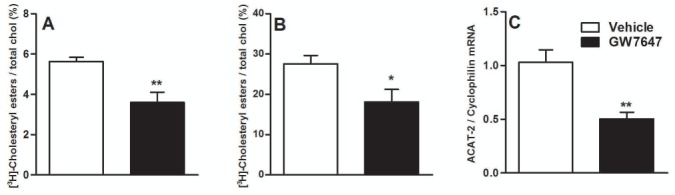
Caco-2/TC7 were incubated with [3H]-cholesterol-micelles (7.5μCi/well) for 48h. GW7647 (600nM) was added 24h before and during the [3H]-cholesterol-loading. Intracellular lipids (A) and lipids secreted in the basolateral compartment (B) were extracted and separated by TLC. Results are expressed as means of CE percent of total cholesterol (±SEM). Q-PCR analysis of ACAT-2 (C) was performed on GW7647 (600nM)-treated Caco-2/TC7 for 24h. Values are expressed as means ± SD. t-test; *P<.05; **P<.01 Vehicle vs treatment.
PPARα-activation increases the expression of lipid droplet formation-genes in Caco-2/TC7 cells
To investigate the mechanism behind the increase of cytoplasmic lipid droplet size upon PPARα-activation (fig 3B), we analyzed the expression of lipid droplet-associated proteins. PPARα-activation increased mRNA levels of perilipin-2 (PLIN2), which coats lipid droplets with its partner ABHD5 (AB-hydrolase-domain-containing-5) (fig 5A) (36). In line, ABHD5 gene expression was also increased (fig 5B). Furthermore, the increase of PLIN2 was strongly reduced and the increase of ABHD5 was totally abolished in ShPPARα-infected cells treated with GW7647 (supp.fig 3A, supp.fig 3B). In addition, PLIN2 and ABHD5 mRNA levels increased only in intestines of GFT505-treated PPARα+/+, but not PPARα−/− apoE2-KI mice (supp.fig 3C, suppl.fig 3D). The increased PLIN2 and ABHD5 gene expression was also observed, albeit less pronounced, in human jejunal biopsies treated with GW7647 or GFT505 (fig 5C, fig 5D).
Figure 5. PPARα-activation increases cytoplasmic lipid droplet formation-associated genes in Caco-2/TC7 and in human jejunal biopsies.
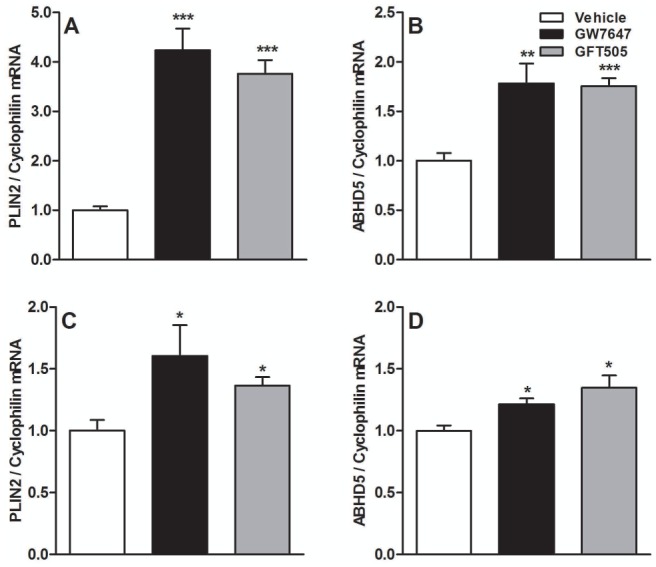
Q-PCR analysis of PLIN2 (A, C) and ABHD5 (B, D) was performed on RNA isolated from Caco-2/TC7 (A, B) and human jejunal biopsies (C, D) treated with GW7647 (600nM) or GFT505 (1μM) for 24h or 18h respectively. Values are expressed as means ± SD. t test; *P<.05; **P<.01; ***P<.001 Vehicle vs treatment.
PPARα-activation increases fatty-acid-oxidation genes in human enterocytes and murine intestines
In addition to the decreased apoB secretion (fig 3C) and in accordance with previous data showing that intestinal PPARα-activation suppresses postprandial lipidemia by increasing fatty-acid-oxidation (25; 34), PPARα ligands increased mRNA levels of fatty-acid-oxidation genes including Carnitine-Palmitoyl-Transferase-1 (CPT-1A), ACOX1 and long-chain Acyl-CoA synthetase family member 5 (ACSL5) (fig 6A, fig 6B, fig 6C) in Caco-2/TC7. CPT-1A and ACOX1 mRNA levels were increased in human biopsies treated with PPARα-ligands (fig 6D, fig 6E). The effect of GW7647 on CPT-1A and ACSL5 was strongly reduced in treated ShPPARα-infected cells and totally abolished for ACOX1 (supp.fig 4A, supp.fig 4B, supp.fig 4C). GFT505 treatment increased the intestinal expression of fatty-acid-oxidation genes in a PPARα-dependent manner in apoE2-KI mice (supp.fig 4D, supp.fig 4E). The expression of fatty-acid-oxidation genes was induced in intestines of FF-treated mice, in contrast to the lack of regulation of HDL-related genes, suggesting that FF acts as a selective PPAR modulator in the intestine (supp.fig 4F, supp.fig 4G, supp.fig 4H).
Figure 6. PPARα-activation increases fatty-acid oxidation genes in Caco-2/TC7 and in human jejunal biopsies.
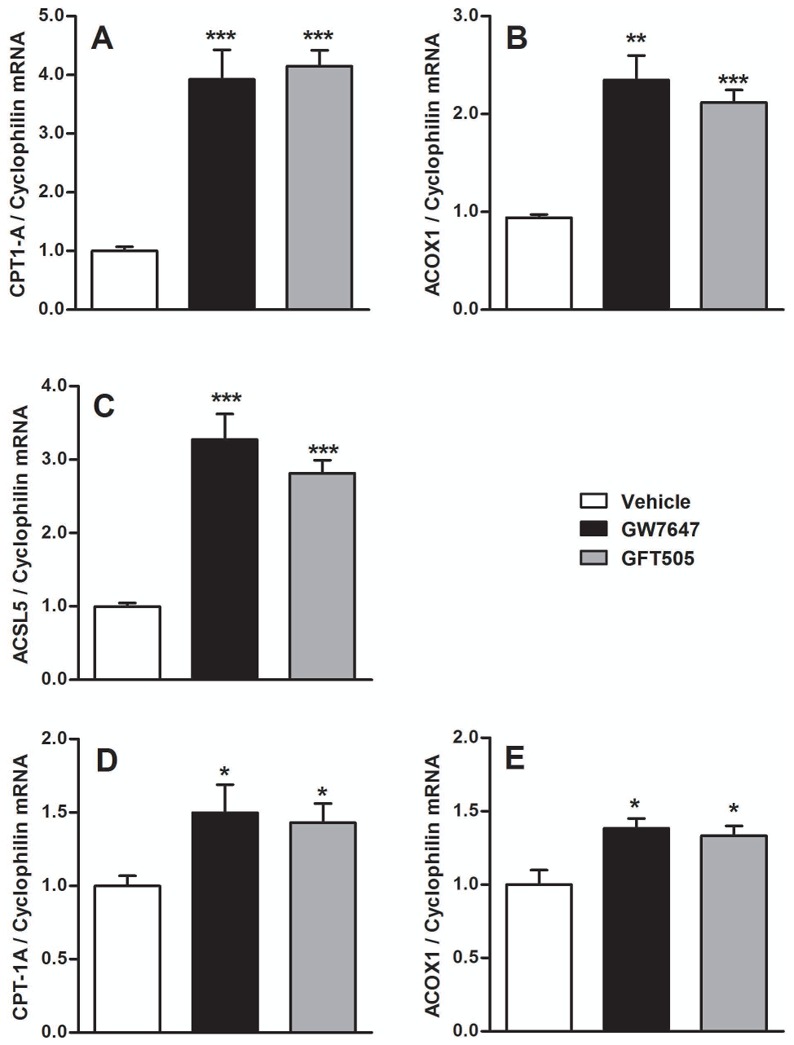
Q-PCR analysis of CPT-1 (A, D), ACOX1 (B, E) and ACSL5 (C) was performed on RNA isolated from Caco-2/TC7 (A, B, C) or human jejunal biopsies (D, E) treated with GW7647 (600nM) or GFT505 (1μM) for 24h or 18h respectively. Values are expressed as means ± SD. t-test; *P<.05; **P<.01; ***P<.001 Vehicle vs treatment.
Interestingly, PPARα-ligands increased cAMP-responsive-element-binding protein-3-like-3 (CREB3L3) mRNA levels in Caco-2/TC7 (supp.fig 5A), a transcription factor involved in the regulation of TG metabolism for which mutations were recently associated with hypertriglyceridemia in humans (37). The induction of CREB3L3 was observed in human biopsies treated with GW7647 and GFT505 (supp.fig 5B). Furthermore, PPARα-ligands did not regulate this gene in ShPPARα-infected cells nor in PPARα-deficient mice (supp.fig 5C, supp.fig 5D). Collectively, these results indicate that PPARα-activation increases the fatty-acid-oxidation pathway, the transcription factor CREB3L3 and increases lipid droplet size.
Discussion
In addition to the liver, the intestine has emerged more recently as a central organ participating in lipid metabolism by regulating lipid absorption, trafficking and lipoprotein metabolism through its involvement in the production of chylomicrons (5; 32), HDL (8) and in trans-intestinal cholesterol excretion (38). Thus, both the liver and intestine are involved in the regulation of whole body lipoprotein metabolism and play a role in the pathophysiology of atherogenic dyslipidemia (39; 40). Fibrates, PPARα-agonists, are often used in the treatment of dyslipidemia characterized by high TG and low HDL-cholesterol levels. Until now, the effects of PPARα-agonists on the improvement of lipid profiles were essentially attributed to their action on the liver and the arterial wall (41; 42). However, PPARα is also expressed in the intestine. In a genome wide analysis of intestinal RNA isolated from PPARα-ligand-treated mice, intestinal PPARα was found to regulate fatty-acid metabolism (25). Moreover, PPARα-activation decreased the secretion of apoB by enterocytes (34). Therefore, we studied whether intestinal PPARα-activation could contribute to the control of HDL metabolism.
Using a humanized mouse model of mixed dyslipidemia, the apoE2-KI mouse, we show in line with previous studies that FF and GFT505-treatments improve the lipid profile by decreasing TG, total cholesterol and by increasing HDL-cholesterol (27; 31). We show that PPARα mediates the effects of GFT505 on TG and HDL-cholesterol levels. Although all clinical studies illustrate the hypotriglyceridemic efficiency of PPARα-agonists, the effects on HDL levels are variable. Such discrepancies could be explained by differences in population selection and heterogeneity or in the molecule used (43–45). In our study, the HDL-increasing effect of GFT505 was more pronounced than FF at concentrations (10mpk vs 100mpk) inducing similar expression levels of ACOX1 in the liver. Although FF did induce intestinal fatty-acid-oxidation-related genes, suggesting that FF can activate intestinal PPARα, only GFT505 induced expression of intestinal HDL production-related genes such as apoAI and ABCA1. Thus, GFT505 and FF differ in the spectrum of regulated target genes, giving the rational to design new selective PPAR modulators. Furthermore, the different activity of fenofibrate vs GFT505 on intestinal gene expression is due to the extensive enterohepatic recycling and fecal excretion of GTF505, whereas fenofibrate is excreted in the urine (Rubenstrunk A and al., manuscript in preparation).
Using the Caco-2/TC7 model (28), we confirmed that PPARα-ligands decrease apoB secretion (34), suggesting a decrease of TG-rich lipoprotein secretion. Moreover, we showed that PPARα-activation increased ABCA1 gene expression and apoAI secretion. Although the previously reported absence of apoAI mRNA regulation (46) demonstrates the limits of the Caco-2/TC7 model (apoAI mRNA was increased in PPARα-agonist-treated human jejunal biopsies), PPARα-activation increased the number of HDL secreted, which in turn enhanced cholesterol efflux from human monocyte-derived macrophages.
As we did not observe any difference in the levels of apical cholesterol uptake nor in the levels of cholesterol secreted at the basal side of treated Caco-2/TC7, we hypothesized that PPARα-activation induces a shift of the absorbed cholesterol from the chylomicron to the HDL production pathway. This hypothesis is strengthened by the fact that less cholesterol was esterified in treated cells. The decrease of esterification may be explained by a decrease of ACAT-2 gene expression which results, as shown in human hepatic cells and macrophages (47; 48), in a decrease of ACAT-2 activity which is also under the control of substrate availability at its site of action, the endoplasmic reticulum (35). By investigating cholesterol trafficking using a fluorescent cholesterol derivative, we observed cholesterol retention in the sub-apical compartment of GW7647-treated Caco-2/TC7 and an increase of lipid droplet size concomitantly with an increase of PLIN2expression, which is necessary for lipid droplet stabilization, and ABHD5, sequestered by PLIN2 at the lipid surface (49). We did not observe any regulation of Rab proteins which are required for vesicular trafficking, nor NPC1 and NPC2 (not shown) which contrasts to macrophages (47). Thus, the mechanism responsible for the effects of PPARα-activation on intracellular cholesterol trafficking remains to be clarified. However, the decrease of ACAT-2 gene expression was concomitant to an increase of CPT-1A expression, which could decrease the availability of fatty-acid for cholesterol esterification, as shown in macrophages (47). In line, PPARα-activation increased the esterase-arylacetamide-deacetylase (AADAC) (not shown) and ACSL5 gene expression, both associated with a reduction of TG accumulation and an increase of fatty-acid-oxidation in rodent hepatocytes (50). Different mechanisms could thus act in concert to divert cholesterol from esterification: decrease of ACAT-2 gene expression, reduction of fatty-acid availability through an increase of CPT-1A expression and fatty-acid-oxidation enzymes and/or retention of cholesterol in lipid droplets.
It has been recently shown that a mutation of CREB3L3 in humans is associated with hypertriglyceridemia. CREB3L3-deficient mice are hypertriglyceridemic and display defective TG clearance (37). Interestingly, we observed a PPARα-dependent up-regulation of CREB3L3 expression in GW7647-treated Caco-2/TC7 and in GFT505-treated mice. The importance of the regulation of this gene expression and its implication in the modulation of lipid homeostasis upon PPARα-activation still remain to be established.
The results of our study collectively show that the increase of ABCA1 expression, the modification in cholesterol trafficking and the decreased cholesterol esterification capacity induced by PPARα-activation potentially contribute to the overall effects on intestinal secretion of apoAI-containing HDL by Caco-2/TC7. The transcriptional regulation of HDL-related genes was observed in mice and in human jejunal biopsies. Therefore, the decrease of TG and the increase of HDL levels observed in PPARα-agonist-treated patients could be due, in addition to hepatic actions, to effects in the intestine. Our work strengthens the idea that the intestine is an important target organ for strategies to increase HDL production via activation of intestinal PPARα. Moreover, our data demonstrate the relevance of designing new specific PPARα modulators with appropriate tissue and gene selective profiles.
Supplementary Material
Table 1. Lipid profiles of apoE2-KI mice treated with FF or GFT505.
ApoE2-KI mice on a western-diet were treated by oral gavage with FF (200mpk) or GFT505 (10mpk) or vehicle for 14d (n=6 mice/group).
| Vehicle | FF | GFT505 | |
|---|---|---|---|
|
| |||
| TG (mg/dL) | 278 ± 76 | 156 ±62* | 110 ±50* |
| Total cholesterol (mg/dL) | 1006 ± 190 | 459 ± 201** | 384 ± 142*** |
| HDL-cholesterol (mg/dL) | 26 ± 7 | 62 ± 11*** | 71 ± 10***,# |
| non HDL-cholesterol (mg/dL) | 980 ± 196 | 397 ± 203** | 312 ± 142*** |
Values are expressed as means ± SEM. ANOVA;
P<.05;
P<.01;
P<.001 vs untreated mice;
P<.05 vs FF-treated mice.
Acknowledgments
Funding:
This work was supported by grants from Université Lille 2, Région Nord/Pas-de-Calais, the FEDER and the “Fondation Leducq”.
We thank M. Tardivel, C. Allet and A. Loyens of the BICeL (IMPRT-IFR114) for access to confocal microscopy and electron microscopy platforms and technical advices.
Abbreviations
- ABCA1
ATP-Binding Cassette A1 transporter
- ABHD5
AB-Hydrolase Domain containing 5
- ACAT-2
Acyl-coenzyme A: Cholesterol Acyltransferase-2
- ACOX1
Acyl-Coenzyme A oxidase 1
- ACSL5
Acyl-CoA Synthetase Long-chain family member 5
- apo
apolipoprotein
- CE
Cholesteryl Ester
- CPT-1
Carnitine Palmitoyl Transferase I
- CREB3L3
cAMP responsive element binding protein 3 like 3
- FC
Free Cholesterol
- FF
Fenofibrate
- HDL
High Density Lipoproteins
- LDL
Low Density Lipoproteins
- PLIN2
Perilipin 2
- PPAR
Peroxisome proliferator-activated receptor
Footnotes
Disclosures:
No conflicts of interest exist
Bibliography
- 1.Castelli W, Garrison R, Wilson P, Abbott R, Kalousdian S, Kannel W. Incidence of coronary heart disease and lipoprotein cholesterol levels. the Framingham study. JAMA. 1986;256:2835–2838. [PubMed] [Google Scholar]
- 2.McTaggart F, Jones P. Effects of statins on HDL: a potential contribution to cardiovascular benefit. Cardiovasc Drugs Ther. 2008;22:321–338. doi: 10.1007/s10557-008-6113-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baigent C, Keech A, Kearney P, Blackwell L, Buck G, Pollicino C, Kirby A, Sourjina T, Peto R, Collins R, Simes R, CTTC Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;366:1267–1278. doi: 10.1016/S0140-6736(05)67394-1. [DOI] [PubMed] [Google Scholar]
- 4.Barter P, Gotto A, LaRosa J, Maroni J, Szarek M, Grundy S, Kastelein J, Bittner V, Fruchart J Treating to New Targets Investigators. HDL-cholesterol, very low levels of LDL-cholesterol, and cardiovascular events. N Engl J Med. 2007;357:1301–1310. doi: 10.1056/NEJMoa064278. [DOI] [PubMed] [Google Scholar]
- 5.Black D. Development and physiological regulation of intestinal lipid absorption. I. Development of intestinal lipid absorption: cellular events in chylomicron assembly and secretion. Am J Physiol Gastrointest Liver Physiol. 2007;293:G519–24. doi: 10.1152/ajpgi.00189.2007. [DOI] [PubMed] [Google Scholar]
- 6.Su J, Nzekwu M, Cabezas M, Redgrave T, Proctor S. Methods to assess impaired post-prandial metabolism and the impact for early detection of cardiovascular disease risk. Eur J Clin Invest. 2009;39(9):741–754. doi: 10.1111/j.1365-2362.2009.02179.x. [DOI] [PubMed] [Google Scholar]
- 7.Green P, Tall A, Glickman R. Rat intestine secretes discoid HDL. J Clin Invest. 1978;61:528–534. doi: 10.1172/JCI108963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brunham L, Kruit J, Pape T, Parks J, Kuipers F, Hayden M. Intestinal ABCA1 directly contributes to HDL biogenesis in-vivo. J Clin Invest. 2006;116:1052–1062. doi: 10.1172/JCI27352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khabazian B, Ghanbari-Niaki A, Safarzadeh-Golpordesari A, Ebrahimi M, Rahbarizadeh F, Abednazari H. Endurance training enhances ABCA1 expression in rat small intestine. Eur J Appl Physiol. 2009;107:351–358. doi: 10.1007/s00421-009-1133-3. [DOI] [PubMed] [Google Scholar]
- 10.Rachmilewitz D, Fainaru M. ApolipoproteinA-I synthesis and secretion by cultured human intestinal mucosa. Metab Clin Exp. 1979;28:739–743. doi: 10.1016/0026-0495(79)90179-3. [DOI] [PubMed] [Google Scholar]
- 11.Johansson C, Rössner S, Walldius G. HDL secretion from human intestinal tract. Lancet. 1978;8059:324–325. doi: 10.1016/s0140-6736(78)90091-0. [DOI] [PubMed] [Google Scholar]
- 12.Forman B, Chen J, Evans R. Hypolipidemic drugs, polyunsaturated fatty-acids, and eicosanoids are ligands for PPAR-alpha and delta. Proc Natl Acad Sci US A. 1997;94:4312–4317. doi: 10.1073/pnas.94.9.4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lefebvre P, Chinetti G, Fruchart J, Staels B. Sorting out the roles of PPAR-alpha in energy metabolism and vascular homeostasis. J Clin Invest. 2006;116:571–580. doi: 10.1172/JCI27989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Braissant O, Foufelle F, Scotto C, Dauça M, Wahli W. Differential expression of peroxisome proliferator-activated receptors: tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology. 1996;137:354–366. doi: 10.1210/endo.137.1.8536636. [DOI] [PubMed] [Google Scholar]
- 15.Chinetti G, Lestavel S, Bocher V, Remaley A, Neve B, Pineda Torra I, Teissier E, Minnich A, Jaye M, Duverger N, Brewer H, Fruchart J, Clavey V, Staels B. PPAR-alpha and PPAR-gamma activators induce cholesterol removal from human macrophage foam cells through stimulation of the ABCA1 pathway. Nat Med. 2001;7:53–58. doi: 10.1038/83348. [DOI] [PubMed] [Google Scholar]
- 16.Colin S, Bourguignon E, Boullay A, Tousaint J, Huet S, Caira F, Staels B, Lestavel S, Lobaccaro J, Delerive P. Intestine-specific regulation of PPAR-alpha gene transcription by LXR. Endocrinology. 2008;149:5128–5135. doi: 10.1210/en.2008-0637. [DOI] [PubMed] [Google Scholar]
- 17.Watts G, Barrett P, Ji J, Serone A, Chan D, Croft K, Loehrer F, Johnson A. Differential regulation of lipoprotein kinetics by atorvastatin and fenofibrate in subjects with the metabolic syndrome. Diabetes. 2003;52:803–811. doi: 10.2337/diabetes.52.3.803. [DOI] [PubMed] [Google Scholar]
- 18.Berthou L, Duverger N, Emmanuel F, Langouët S, Auwerx J, Guillouzo A, Fruchart J, Rubin E, Denèfle P, Staels B, Branellec D. Opposite regulation of human versus mouse apolipoproteinA-I by fibrates in human apolipoproteinA-I transgenic mice. J Clin Invest. 1996;97:2408–2416. doi: 10.1172/JCI118687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hossain M, Tsujita M, Gonzalez F, Yokoyama S. Effects of fibrate drugs on expression of ABCA1 and HDL biogenesis in hepatocytes. J Cardiovasc Pharmacol. 2008;51:258–266. doi: 10.1097/FJC.0b013e3181624b22. [DOI] [PubMed] [Google Scholar]
- 20.Chinetti G, Lestavel S, Fruchart J, Clavey V, Staels B. PPAR-alpha reduces cholesterol esterification in macrophages. Circ Res. 2003;92:212–217. doi: 10.1161/01.res.0000053386.46813.e9. [DOI] [PubMed] [Google Scholar]
- 21.Chakravarthy M, Lodhi I, Yin L, Malapaka R, Xu H, Turk J, Semenkovich C. Identification of a physiologically relevant endogenous ligand for PPAR-alpha in liver. Cell. 2009;138(3):476–488. doi: 10.1016/j.cell.2009.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Delerive P, Furman C, Teissier E, Fruchart J, Duriez P, Staels B. Oxidized phospholipids activate PPAR-alpha in a phospholipase A2-dependent manner. FEBS Lett. 2000;471:34–38. doi: 10.1016/s0014-5793(00)01364-8. [DOI] [PubMed] [Google Scholar]
- 23.Staels B, van Tol A, Andreu T, Auwerx J. Fibrates influence the expression of genes involved in lipoprotein metabolism in a tissue-selective manner in the rat. Arterioscler Thromb. 1992;12:286–294. doi: 10.1161/01.atv.12.3.286. [DOI] [PubMed] [Google Scholar]
- 24.Bünger M, van den Bosch H, van der Meijde J, Kersten S, Hooiveld G, Müller M. Genome-wide analysis of PPAR-alpha activation in murine small intestine. Physiol Genomics. 2007;30(2):192–204. doi: 10.1152/physiolgenomics.00198.2006. [DOI] [PubMed] [Google Scholar]
- 25.Cariou B, Zaïr Y, Staels B, Bruckert E. Effects of the new dual PPAR-alpha/delta agonist GFT505 on lipid and glucose homeostasis in abdominally obese patients with combined dyslipidemia or impaired glucose metabolism. Diabetes Care. 2011;34:2008–2014. doi: 10.2337/dc11-0093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lalloyer F, Wouters K, Baron M, Caron S, Vallez E, Vanhoutte J, Baugé E, Shiri-Sverdlov R, Hofker M, Staels B, Tailleux A. PPAR-alpha gene level differently affects lipid metabolism and inflammation in apolipoprotein-E2 knock-in mice. Arterioscler Thromb Vasc Biol. 2011;31:1573–1579. doi: 10.1161/ATVBAHA.110.220525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chateau D, Pauquai T, Delers F, Rousset M, Chambaz J, Demignot S. Lipid micelles stimulate the secretion of triglyceride-enriched apolipoprotein-B48-containing lipoproteins by Caco-2 cells. J Cell Physiol. 2005;202:767–776. doi: 10.1002/jcp.20173. [DOI] [PubMed] [Google Scholar]
- 28.Béaslas O, Cueille C, Delers F, Chateau D, Chambaz J, Rousset M, Carrière V. Sensing of dietary lipids by enterocytes: a new role for SR-BI/cla-1. PLoS ONE. 2009;4:e4271–4278. doi: 10.1371/journal.pone.0004278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rasband W. Imagej. US National Institutes of Health; Bethesda, Maryland, USA: 1997–2009. http://rsb.info.nih.gov/ij/ [Google Scholar]
- 30.Hennuyer N, Tailleux A, Torpier G, Mezdour H, Fruchart J, Staels B, Fiévet C. PPAR-alpha, but not PPAR-gamma, activators decrease macrophage-laden atherosclerotic lesions in a nondiabetic mouse model of mixed dyslipidemia. Arterioscler Thromb Vasc Biol. 2005;25:1897–1902. doi: 10.1161/01.ATV.0000175756.56818.ee. [DOI] [PubMed] [Google Scholar]
- 31.Duval C, Touche V, Tailleux A, Fruchart J, Fievet C, Clavey V, Staels B, Lestavel S. NPC1L1 gene expression is down regulated by LXR activators in the intestine. BBRC. 2006;340:1259–1263. doi: 10.1016/j.bbrc.2005.12.137. [DOI] [PubMed] [Google Scholar]
- 32.Iqbal J, Hussain M. Intestinal lipid absorption. Am J Physiol Endocrinol Metab. 2009;296:1183–1194. doi: 10.1152/ajpendo.90899.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kimura R, Takahashi N, Murota K, Yamada Y, Niiya S, Kanzaki N, Murakami Y, Moriyama T, Goto T, Kawada T. Activation of PPAR-α suppresses postprandial lipidemia through fatty-acid-oxidation in enterocytes. Biochem Biophys Res Commun. 2011;410:1–6. doi: 10.1016/j.bbrc.2011.05.057. [DOI] [PubMed] [Google Scholar]
- 34.Field F, Watt K, Mathur S. Ezetimibe interferes with cholesterol trafficking from the plasma membrane to the endoplasmic reticulum in Caco-2 cells. J Lipid Res. 2007;48:1735–1745. doi: 10.1194/jlr.M700029-JLR200. [DOI] [PubMed] [Google Scholar]
- 35.Lee B, Zhu J, Wolins N, Cheng J, Buhman K. Differential association of adipophilin and TIP47 proteins with cytoplasmic lipid droplets in mouse enterocytes during dietary fat absorption. Biochim Biophys Acta. 2009;1791(12):1173–1180. doi: 10.1016/j.bbalip.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 36.Lee J, Giannikopoulos P, Duncan S, Wang J, Johansen C, Brown J, Plutzky J, Hegele R, Glimcher L, Lee A. The transcription factor cyclic amp-responsive element-binding protein H regulates triglyceride metabolism. Nat Med. 2011;17(7):812–815. doi: 10.1038/nm.2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van der Velde A, Brufau G, Groen A. Transintestinal cholesterol efflux. Curr Opin Lipidol. 2010;21:167–171. doi: 10.1097/MOL.0b013e3283395e45. [DOI] [PubMed] [Google Scholar]
- 38.Weingärtner O, Lütjohann D, Böhm M, Laufs U. Relationship between cholesterol synthesis and intestinal absorption is associated with cardiovascular risk. Atherosclerosis. 2010;210:362–365. doi: 10.1016/j.atherosclerosis.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 39.Xiao C, Lewis G. Regulation of chylomicron production in humans. Biochim Biophys Acta. 2011 doi: 10.1016/j.bbalip.2011.09.019. [DOI] [PubMed] [Google Scholar]
- 40.Fruchart J, Duriez P. Mode of action of fibrates in the regulation of triglyceride and HDL-cholesterol metabolism. Drugs Today. 2006;42:39–64. doi: 10.1358/dot.2006.42.1.963528. [DOI] [PubMed] [Google Scholar]
- 41.Rigamonti E, Chinetti-Gbaguidi G, Staels B. Regulation of macrophage functions by PPAR-alpha, PPAR-gamma, and LXRs in mice and men. Arterioscler Thromb Vasc Biol. 2008;28:1050–1059. doi: 10.1161/ATVBAHA.107.158998. [DOI] [PubMed] [Google Scholar]
- 42.Keech A, Simes R, Barter P, Best J, Scott R, Taskinen M, Forder P, Pillai A, Davis T, Glasziou P, Drury P, Kesäniemi Y, Sullivan D, Hunt D, Colman P, d’Emden M, Whiting M, Ehnholm C, Laakso M, FIELD SI. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the field study): randomised controlled trial. Lancet. 2005;366:1849–1861. doi: 10.1016/S0140-6736(05)67667-2. [DOI] [PubMed] [Google Scholar]
- 43.Tenenbaum A, Motro M, Fisman E, Tanne D, Boyko V, Behar S. Bezafibrate for the secondary prevention of myocardial infarction in patients with metabolic syndrome. Arch Intern Med. 2005;165:1154–1160. doi: 10.1001/archinte.165.10.1154. [DOI] [PubMed] [Google Scholar]
- 44.The BIP Study Group. Secondary prevention by raising HDL-cholesterol and reducing triglycerides in patients with coronary artery disease: the bezafibrate infarction prevention study. Circulation. 2000;102:21–27. doi: 10.1161/01.cir.102.1.21. [DOI] [PubMed] [Google Scholar]
- 45.Dullens S, Mensink R, Mariman E, Plat J. Differentiated Caco-2 cells as an in-vitro model to evaluate de-novo apolipoproteinA-I production in the small intestine. Eur J Gastroenterol Hepatol. 2009;21:642–649. doi: 10.1097/meg.0b013e328321b0c8. [DOI] [PubMed] [Google Scholar]
- 46.Chinetti-Gbaguidi G, Rigamonti E, Helin L, Mutka A, Lepore M, Fruchart J, Clavey V, Ikonen E, Lestavel S, Staels B. PPAR-alpha controls cellular cholesterol trafficking in macrophages. J Lipid Res. 2005;46:2717–2725. doi: 10.1194/jlr.M500326-JLR200. [DOI] [PubMed] [Google Scholar]
- 47.Pramfalk C, Angelin B, Eriksson M, Parini P. Cholesterol regulates ACAT2 gene expression and enzyme activity in human hepatoma cells. Biochem Biophys Res Commun. 2007;364:402–409. doi: 10.1016/j.bbrc.2007.10.028. [DOI] [PubMed] [Google Scholar]
- 48.Granneman J, Moore H, Krishnamoorthy R, Rathod M. Perilipin controls lipolysis by regulating the interactions of AB-hydrolase containing 5 and adipose triglyceride lipase. J Biol Chem. 2009;284:34538–34544. doi: 10.1074/jbc.M109.068478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lo V, Erickson B, Thomason-Hughes M, Ko K, Dolinsky V, Nelson R, Lehner R. Arylacetamide deacetylase attenuates fatty-acid-induced triacylglycerol accumulation in rat hepatoma cells. J Lipid Res. 2010;51:368–377. doi: 10.1194/jlr.M000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhou Y, Abidi P, Kim A, Chen W, Huang T, Kraemer F, Liu J. Transcriptional activation of hepatic ACSL3 and ACSL5 by oncostatin m reduces hypertriglyceridemia through enhanced beta-oxidation. Arterioscler Thromb Vasc Biol. 2007;27:2198–2205. doi: 10.1161/ATVBAHA.107.148429. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.