

Abstract
Objective
In the wake of intracerebral hemorrhage (ICH), a devastating stroke with no effective treatment, hemoglobin/iron-induced oxidative injury leads to neuronal loss and poor neurologic outcomes. (-)-Epicatechin (EC), a brain-permeable flavanol that modulates redox/oxidative stress via the NF-E2–related factor (Nrf) 2 pathway, has been shown to be beneficial for vascular and cognitive function in humans. Here, we examined whether EC can reduce early brain injury in ICH mouse models and investigated the underlying mechanisms.
Methods
ICH was induced by injecting collagenase, autologous blood, or thrombin into mouse striatum. EC was administered orally at 3 h after ICH and then every 24 h. Lesion volume, neurologic deficits, brain edema, reactive oxygen species, and protein expression and activity were evaluated.
Results
EC significantly reduced lesion volume and ameliorated neurologic deficits in both male and female ICH mice. Cell death and neuronal degeneration were decreased in the perihematomal area and were associated with reductions in caspase-3 activity and high-mobility group protein B1 (HMGB-1) level. These changes were accompanied by attenuation of oxidative insults, increased phase II enzyme expression, and increased Nrf2 nuclear accumulation. Interestingly, in addition to providing neuroprotection via Nrf2 signaling, EC diminished heme oxygenase-1 induction and brain iron deposition via an Nrf2-independent pathway that downregulated ICH-induced activating protein-1 activation and decreased matrix metalloproteinase 9 activity, lipocalin-2 levels, iron-dependent cell death, and ferroptosis-related gene expression.
Interpretation
Collectively, our data show that EC protects against ICH by activation of Nrf2-dependent and -independent pathways and may serve as a potential intervention for patients with ICH.
Keywords: (-)-Epicatechin, ferroptosis, heme oxygenase-1, iron, NF-E2-related factor 2
Introduction
Intracerebral hemorrhage (ICH) is the stroke subtype with the highest mortality and morbidity. Unfortunately, this condition has received far less research attention than has ischemic stroke.1 In the aftermath of ICH, in addition to the hematoma causing primary physical compression, blood components, hemoglobin, and nonheme iron elevate the production of reactive oxygen species (ROS), which contribute to secondary brain injury.2,3 Evidence suggests that excessive hemoglobin and iron released from the hematoma accumulate in the brain parenchyma, causing neurotoxicity and accelerating neurodegeneration.4,5 Recently, iron-dependent, nonapoptotic cell death known as ferroptosis has been identified as a potential therapeutic target to reduce iron-related cell death.6 Strategies to limit iron toxicity and ROS production could be used to reduce brain injury7 and improve quality of life in patients with ICH.3
The NF-E2–related factor (Nrf) 2/antioxidant response element (ARE) pathway plays a pivotal role in orchestrating expression of the phase II detoxification and antioxidant genes that combat the pathogenic events associated with ICH.8 Although most products of the Nrf2/ARE pathway are protective, heme oxygenase 1 (HO-1), which is responsible for iron metabolism, appears to act as a prooxidant in the ICH brain.9 No clear link between detoxification of iron overload and Nrf2 activation has been reported. Nevertheless, our previous studies in Nrf2- and HO-1-deficient mice confirmed the neuroprotective function of Nrf2 and deleterious effects of HO-1 in the pathologic process of ICH.10,11 Although free radical scavenger interventions hold promise for treating ICH, their lack of success in clinical trials argues for additional investigation.12,13
Another potential target for manipulating ICH insults is activator protein (AP)-1, a dimeric transcription factor composed of α-fetoprotein, Jun, or Fos protein subunits that bind to a common DNA site.14 AP-1 activation leads to the expression of many proinflammatory mediators, including matrix metalloproteinases (MMPs), which contribute to blood-brain barrier (BBB) breakdown and exacerbate brain edema in both hemorrhagic and ischemic stroke models.15,16 Although inhibition or deletion of MMPs was shown to be neuroprotective in a mouse model of ICH,17,18 the role of AP-1 in ICH remains to be elucidated.
The cerebrovascular benefits of fruits, vegetables, and green tea have been ascribed in part to the polyphenols present, particularly flavanols.19,20 Cocoa and green tea are enriched with flavanols such as epicatechins, catechins, and procyanidins. Oral consumption of (-)-epicatechin (EC) elicits positive effects on vascular function, cognition, and ischemic brain injury in animals and humans.21–23 Additionally, pharmacokinetic studies of EC have shown that it can penetrate the BBB after intravenous or oral administration in rats.24–26 This simple molecule scavenges prooxidants and free radicals through activation of the Nrf2 signaling pathway.23,27,28 However, whether EC can activate Nrf2 in the brain after ICH is still unknown.
In this study, we examined whether oral EC administration after ICH provides neuroprotection in mice and investigated potential underlying molecular mechanisms. We specifically tested the therapeutic efficacy of EC on early ICH-induced brain injury because half of human deaths occur within the first 2 days.13,29,30 We conclude that the flavanol EC could potentially be a therapeutic agent for treating ICH. Moreover, the results indicate that EC achieves this protective effect through Nrf2-dependent and alternative Nrf2-independent pathways.
Materials and Methods
Animals
This study was conducted in accordance with the National Institutes of Health guidelines for the use of experimental animals. Experimental protocols were approved by the Johns Hopkins University Animal Care and Use Committee. Twelve-month-old (30–36 g) C57BL/6 male and female mice (Charles River Laboratories, Germantown, MD), specific gene-deficient mice (Nrf2 knockout [KO], HO-1 KO), and wild-type (WT) littermates were bred in a pathogen-free environment. Nrf2 KO mice and HO-1 KO mice were originally generated by Masayuki Yamamoto31 and by Drs. Poss and Tonegawa,32 respectively. Mice were genotyped for Nrf2 and HO-1 status by polymerase chain reaction (PCR) amplification of genomic DNA extracted from tail snips.10,11
Experimental groups
Mice were randomly assigned to receive vehicle (sterilized water; Hospira, Chicago, IL) or 5, 15, or 45 mg/kg EC (Sigma, St. Louis, MO) by gavage at 3 h after ICH and then every 24 h thereafter for 72 h. Studies have shown that one oral dose of EC (30 mg/kg) can cross the BBB24–26,33 and provide marked neuroprotection in β-amyloid-, N-methyl-D-aspartate (NMDA)-, and ischemia-induced brain injury.23,34 We chose the delivery route, dosing, and treatment regimens for EC based on previous work22–24,26,34,35 and our preliminary tests. By characterizing the purity and stability of EC (Fig. S1), we determined that the compound remained stable in water for 24 h, which was established as a suitable window for sample usage. An experimenter blind to treatment group tested the mice on a 24-point neurologic scoring system11,36 before EC administration. Mice that had neurologic deficit scores greater than 20 or less than 3 were excluded from the study. A total of 172 WT mice were allocated into the vehicle group and 216 into the EC groups. Fifty-two mice were excluded because of severe neurologic deficit (vehicle: 5/172, EC: 7/216) or death (vehicle: 18/172; EC: 22/216). Five Nrf2 KO mice were excluded because of neurologic deficit standard or death. Sixty-six additional sham-operated WT, Nrf2 KO, and HO-1 KO and WT control mice were used for histology, biochemical assays, and reverse transcriptase PCR (n = 5–10/group). No animals were excluded from analysis once they proceeded into the tests. All behavioral, anatomical, and biochemical measures and analyses were carried out with a blinded experimental design according to published guidelines.37–39
For complete details of all the experimental procedures, see Data S1.
Statistical analyses
Data are presented as mean ± SD. We evaluated all behavioral tests by two-way repeated measure analysis of variance (ANOVA) to detect significant differences between and among treatment groups. In anatomical and biochemical studies, one-way or two-way ANOVA was used for comparisons among multiple groups. Bonferroni post hoc analysis was used to determine where those differences occurred. Differences between two groups were tested with the Student's t-test (SigmaStat 3.5; Systat Software Inc., San Jose, CA). The criterion for statistical significance was P < 0.05.
Results
EC reduces ICH-induced early brain injury and neurologic deficits
To define the optimal dose of EC in ICH, we first administered different doses of EC to 12-month-old male mice subjected to collagenase injection. Injury volume was assessed 72 h post-ICH, as collagenase-induced brain parenchyma lesion reaches a maximum by this time.40 Brain sections were stained with Luxol fast blue/Cresyl violet, and lesions were identified by lack of color and destruction of myelin architecture (Fig. 1A). The lesions were smaller in mice that received 72 h of 15 mg/kg EC (EC 15; 5.5 ± 1.5 mm3) or 45 mg/kg EC (EC 45; 4.5 ± 0.9 mm3) than in those that received vehicle (8.9 ± 1.5 mm3; both P < 0.01). Lesion size did not differ between mice that received vehicle and those that received 5 mg/kg EC (EC 5; 7.8 ± 0.8 mm3; P = 0.328) or between EC 15 and EC 45 groups (P = 0.318; Fig. 1B). Also, no obvious lesion was observed in the sham-operated group.
Figure 1.

Effects of EC on brain injury volume and neurologic function in mice subjected to ICH. (A) Representative Luxol fast blue/Cresyl violet-stained brain sections at 72 h post-ICH; injured areas lack staining and are circled in white. Scale bar: 1 mm. (B) Quantification analysis shows significantly smaller brain injury volumes in EC-treated groups than in the vehicle-treated group at 72 h post-ICH. (C) EC posttreatment improved neurologic deficit score (top), forelimb placing (middle), and corner turn test performance (bottom) of mice subjected to ICH. Values are mean ± SD; n = 10 mice per group. *P < 0.05 versus vehicle.
Several sets of behavioral experiments were performed at 24 and 72 h post-ICH to determine the effect of EC on neurologic deficits. Compared with vehicle, EC 15 showed a beneficial effect on neurologic score (9.9 ± 1.7 vs. 6.7 ± 1.1; P < 0.01), forelimb placing (37.0 ± 11.5% vs. 72.0 ± 11.3%; P < 0.01), and corner turns (74.0 ± 12.6% vs. 60.0 ± 11.5%; P < 0.05) 72 h post-ICH. Although EC 45 improved performance in the forelimb placing (P < 0.01) and corner turn (P < 0.01) tests, it failed to improve neurologic score (P = 0.054) because mice in this treatment group had deficiency in front limb symmetry and compulsory circling tests.
Sex is one determinant of ICH outcome, and evidence has shown that some drugs lack therapeutic efficacy in female animals.41,42 To this end, we tested the efficacy of EC for injury reduction after ICH in age-matched female mice. Compared to vehicle-treated mice, mice that were administrated EC 15 had smaller injury volume (6.2 ± 0.8 mm3 vs. 4.3 ± 1.2 mm3; P < 0.05) and better neurologic function (10.4 ± 0.9 vs. 7.5 ± 1.4; P < 0.01) 72 h post-ICH (Fig. S2A and B). Because every rodent model of ICH has drawbacks that limit its clinical relevance,3 we further characterized the efficacy of EC for ICH in the blood model, another common strategy to recapitulate ICH. EC 15 reduced neurologic deficit scores in both male (8.7 ± 1.0 vs. 6.8 ± 1.0; P < 0.05) and female (9.3 ± 0.8 vs. 7.4 ± 0.8; P < 0.01) mice 72 h post-ICH (Fig. S2C and D).
To assess whether the reduction in early brain injury and the improvement of neurologic deficits in EC-treated mice correlated with reduced brain edema, we examined brain water content 72 h after ICH. EC 15 reduced brain water content in ipsilateral striatum in collagenase-injected male (P < 0.05) and female (P = 0.004) mice, blood-injected male (P = 0.021) and female (P < 0.01) mice, and thrombin-injected male mice (P < 0.05; Fig. S2E–I). These results imply that efficacy of oral EC administration is not affected by sex. Clinical study has suggested an association between brain edema and subsequent neurologic deficits.43 Therefore, we examined long-term neurobehavioral function by neurologic score and the corner turn test on day 28 post-ICH. Compared with the vehicle group, EC 15 mice had better neurologic scores (6.8 ± 1.0 vs. 4.5 ± 1.0; P < 0.01) and better performance in the corner turn test (79.1 ± 3.8% vs. 70.0 ± 4.5%; P < 0.01) on day 28 post-ICH (Fig. S3A and B). Because lesion size did not differ between EC 15 and EC 45 groups, we used EC 15 in the following mechanistic studies.
EC decreases perihematoma cell death and neuronal degeneration
Because EC 15 significantly alleviated brain injury and improved neurologic deficits, we investigated whether this treatment paradigm ameliorates perihematomal cell death. Cells positive for propidium iodide (PI), terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL), and Fluoro-Jade B (FJB) were evident 72 h after ICH in and around the hematoma. Compared to vehicle treatment, EC 15 reduced the number of PI-positive cells (from 221.5 ±13.7 cells/mm2 to 170.4 ± 26.1 cells/mm2; P < 0.05), the percentage of TUNEL-positive cells (from 41.4 ± 11.4% to 22.8 ± 7.1%; P < 0.01), and the number of FJB-positive cells (from 121.4 ± 23.5 cells/mm2 to 78.8 ± 19.3 cells/mm2; P < 0.01) in the perihematoma area 72 h post-ICH (Fig. 2A, B, D, and F). Few PI-, TUNEL-, and FJB-positive cells were observed in the contralateral hemisphere of either group. EC 15 also reduced ICH-induced upregulation of high-mobility group protein B1 (HMGB-1) at 72 h post-ICH (P < 0.05; Fig. 2C) and the increase in caspase-3 activity at 48 h (P < 0.05) and 72 h (P < 0.01) post-ICH (Fig. 2E). The neuroprotective effect of EC was further confirmed by using primary neurons. Hemoglobin-induced lactate dehydrogenase release was decreased in primary neurons co-treated with EC for 24 h (160.2 ± 4.8% vs. 135.8 ± 2.6%; P < 0.05; Fig. S4). These results further confirm the anatomical observations.
Figure 2.
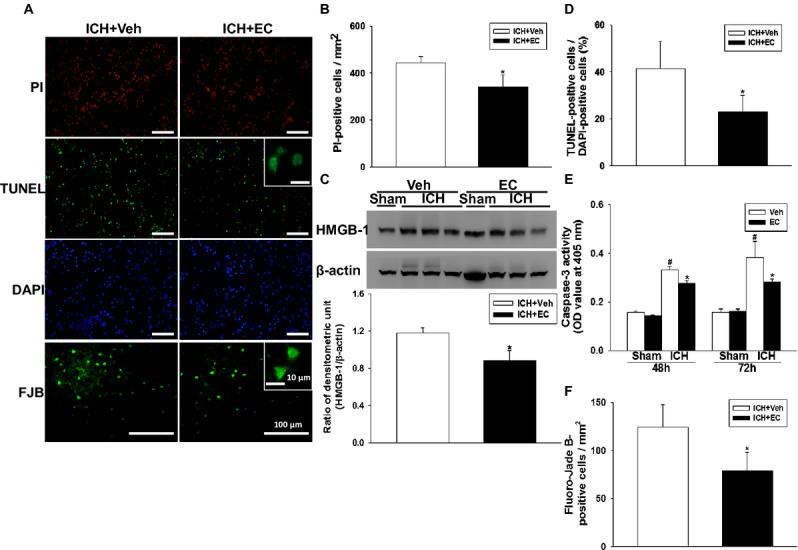
Identification of cell death, apoptosis, and neuronal degeneration in mice subjected to ICH. (A) Representative propidium iodide (PI)-, TUNEL-, DAPI-, and Fluoro-Jade B (FJB)-stained brain sections. The insets are representative TUNEL- and FJB-positive cells at higher magnification. Scale bars: 100 μm; 10 μm (inset). (B) Quantification analysis indicates that 15 mg/kg EC (EC 15) significantly reduced PI-positive cells at 72 h post-ICH. (C) Top: Representative immunoblots of the HMGB-1 protein in ipsilateral hemispheres from sham-operated and ICH mice treated with or without EC 15. Bottom: Densitometric analysis shows that HMGB-1 expression in the ipsilateral hemispheres was significantly lower in EC 15 mice than in vehicle-treated mice 72 h after ICH. (D) Quantification analysis shows that the percentage of TUNEL-positive cells was significantly lower in the EC 15 mice than in the vehicle-treated mice 72 h after ICH. (E) Bar graphs showing caspase-3 enzyme activity in EC and vehicle groups at 48 and 72 h after sham surgery or ICH induction. ICH significantly increased caspase-3 enzyme activity in both EC- and vehicle-treated mice. The hemorrhagic hemispheres in the EC-treated group exhibited significantly less caspase-3 activity than did those in the vehicle-treated group. (F) Quantification analysis of FJB staining indicates that EC-treated mice had significantly fewer degenerating neurons than did vehicle-treated mice 72 h after ICH. Values are mean ± SD; n = 6–9 mice per group. *P < 0.05 versus vehicle; #P < 0.05 versus sham.
EC alleviates ICH-induced brain oxidative injury and increases Nrf2 nuclear accumulation
We used the hydroethidine technique to study the effect of EC on posthemorrhagic ROS production. The presence of ROS was observed as red particles in perihematomal cells 24 h post-ICH (Fig. 3A). On the contralateral side, ROS signals were present but relatively weak (data not shown). EC 15 mice exhibited less ROS production in the perihematomal area than did vehicle-treated mice (fluorescence intensity: 0.056 ± 0.007 vs. 0.038 ± 0.007; P < 0.01; Fig. 3A). The final product of lipid peroxidation, malondialdehyde (MDA), was markedly elevated in hemorrhagic brain tissue compared to that in sham groups at 48 h (P < 0.01) and 72 h (P < 0.01) post-ICH. Compared with that in the vehicle group, EC 15 reduced MDA level in hemorrhagic tissues at 48 h (9.2 ± 0.7 nmol/mg vs. 7.3 ± 0.6 nmol/mg; P < 0.01) and 72 h (10.0 ± 1.0 nmol/mg vs. 8.1 ± 1.0 nmol/mg; P < 0.01) post-ICH (Fig. 3B). Protein carbonyls, another marker of oxidative stress, were also reduced in the EC 15 group at 72 h post-ICH (Fig. 3C).
Figure 3.

EC alleviates ICH-induced reactive oxygen species (ROS) production. (A) Top: Ethidium fluorescent signal was evident in brain sections from mice 24 h after ICH. Scale bar: 50 μm. Bottom: Quantification analysis of fluorescence intensity indicated that EC significantly reduced ROS production at 1 day post-ICH. (B) Bar graphs showing malondialdehyde (MDA) level at 48 and 72 h after sham surgery or ICH induction. ICH significantly increased MDA level in hemorrhagic brain tissue of both EC- and vehicle-treated mice. EC-treated mice had less MDA than did vehicle-treated mice. (C) Representative immunoblot of hemorrhagic brain tissue shows that EC-treated mice had less protein carbonylation than did vehicle-treated mice. β-actin served as a loading control. The arrows show the carbonylated proteins at different molecular weights. (D) Top: Representative immunoblot of ipsilateral hemispheres from mice treated with or without 15 mg/kg EC; brains were collected 6 h after sham surgery or ICH. Bottom: Bar graph of densitometric analysis shows Nrf2 protein level in nuclear extracts from ipsilateral hemispheres of EC- and vehicle-treated mice. (E and F) Line graphs show SOD-1 and NQO-1 mRNA expression in EC- and vehicle-treated mice at 6, 24, and 72 h after ICH. Compared with vehicle treatment, EC significantly increased SOD-1 mRNA expression at 24 and 72 h and NQO-1 mRNA expression at 6 and 72 h in the ipsilateral hemisphere. Values are mean ± SD; n = 5–6 mice per group. *P < 0.05 versus vehicle; #P < 0.05 versus sham.
To test whether EC induces Nrf2 nuclear translocation, we assayed Nrf2 protein in cytosolic and nuclear brain extracts by Western blot. EC administration did not alter cytosolic Nrf2 levels; however, nuclear Nrf2 accumulation was increased ∼1.8-fold by EC administration at 6 h post-ICH (P < 0.05; Fig. 3D). We then examined the effect of Nrf2 nuclear accumulation on its downstream transcripts, SOD1 and NQO1, by RT-qPCR. In EC 15 mice, SOD1 mRNA expression was increased by ∼13%, 33%, and 70% of that in the vehicle group at 6 h (P < 0.05), 24 h (P < 0.05), and 72 h (P < 0.01) post-ICH (Fig. 3E). Similarly, NQO1 mRNA expression was increased by ∼20% and 42% of that in the vehicle group at 6 h (P < 0.001) and 72 h (P < 0.05) post-ICH (Fig. 3F).
EC decreases hemorrhage-induced iron deposition and HO-1 expression through an Nrf2-independent mechanism
HO-1 is a key downstream effector of Nrf2. It is upregulated after ICH and exacerbates neuronal injury.11 Furthermore, genetic overexpression of HO-1 contributes to pathologic iron deposition in astrocytes.44 Here, ICH induced an increase in HO-1 protein level in the hemorrhagic hemisphere at 72 h that was reduced approximately twofold by EC 15 (P < 0.05; Fig. 4A). The relationship between HO-1 and iron deposition in ICH brain was further confirmed by using HO-1 KO mice. Few iron-positive cells were observed in hemorrhagic brain sections from HO-1 KO mice (Fig. 4B). Additionally, at 72 h post-ICH, iron deposition was lower in EC-treated WT mice (15.6 ± 4.6 cells/0.12 mm2) than in vehicle-treated WT mice (38.3 ± 3.3 cells/0.12 mm2; P < 0.05; Fig. 4C). To identify whether the lower HO-1 expression and iron deposition observed in the EC-treated mice can be attributed to a difference in collagenase-induced bleeding volume, we measured hemoglobin content at 24 h post-ICH when hematoma reaches its maximum in the collagenase model.11,45 No significant difference was observed between vehicle- and EC-treated mice (P > 0.05; Fig. 4D).
Figure 4.

Effects of EC on HO-1 protein expression, iron deposition, and hemorrhagic injury volume in WT, Nrf2 KO, and HO-1 KO mice subjected to ICH. (A) Top: Representative immunoblot of HO-1 protein from sham-operated and ICH mice treated with or without EC. Bottom: Densitometric analysis shows that HO-1 protein level in the ipsilateral hemisphere was significantly less in EC-treated mice than in vehicle-treated mice at 72 h after ICH. (B) Representative Perls-stained brain sections from WT and HO-1 KO mice 72 h post-ICH. In the perihematomal region, many iron-positive cells can be seen in the WT mice; based on cell morphology, most of them are likely activated microglia/macrophages with cytoplasmic iron deposits (arrows). The iron-positive cells with similar morphology are rarely detectable in the HO-1 KO mice. (C) Ferric iron accumulation in the perihematomal region of mice at 72 h post-ICH was significantly less in EC-treated mice than in vehicle-treated mice. (D) Hemoglobin levels were not significantly different in brains of vehicle- and EC-treated mice at 24 h post-ICH. (E) Representative brain sections from Nrf2 KO mice 72 h post-ICH were stained with Luxol fast blue/Cresyl violet. Areas of injury lack staining and are circled with a black line. Bottom: Quantification analysis shows significantly smaller brain injury volumes in EC-treated than in vehicle-treated mice. (F) Representative Perls-stained brain sections from Nrf2 KO mice with or without EC administration 72 h post-ICH. Fewer iron-positive cells are present in the EC-treated mice. The inset images show neuron-like and microglia/macrophage-like morphology (Left) and iron-positive (P) and -negative (N) cells (Right). (G) Top: Representative immunoblot of HO-1 protein from WT and Nrf2 KO mice subjected to sham operation or ICH induction. Bottom: Bar graph of densitometric analysis shows a significant increase in HO-1 expression in the ipsilateral hemispheres of Nrf2 KO mice at 72 h after ICH compared with that in WT mice. (H) Nrf2 KO mice had significantly more Perls-positive cells than did WT mice in the perihematomal region 72 h post-ICH. Scale bars: 50 μm (B); 1 mm (E); 20 μm (F). Values are mean ± SD; n = 5–10 mice per group. *P < 0.05 versus vehicle or WT.
To determine whether the effects on ICH injury were due to activation of the Nrf2 pathway, we administered EC (15 mg/kg per day) to Nrf2 KO mice with or without ICH. Interestingly, deletion of Nrf2 did not totally abolish the therapeutic effect of EC in ICH brain. Compared with vehicle treatment, EC was still able to reduce brain injury volume (11.0 ± 1.2 mm3 vs. 7.9 ± 1.9 mm3; P < 0.05 at 72 h post-ICH; Fig. 4E). Accordingly, EC 15 per se decreased iron-positive cells in the perihematomal region at 72 h post-ICH (50.8 ± 6.1 cells/0.12 mm2 vs. 33.6 ± 5.6 cells/0.12 mm2; P < 0.01; Fig. 4F). Furthermore, ICH-induced HO-1 protein expression (Fig. 4G) and iron deposition were significantly higher in Nrf2 KO brain (57.1 ± 6.4 cells/0.12 mm2) than in WT brain (38.3 ± 3.3 cells/0.12 mm2; P < 0.01) 72 h post-ICH (Fig. 4H).
EC reduces AP-1 and MMP-9 enzyme activity
To elucidate the possible alternative pathway by which EC provides neuroprotection, we screened 56 transcription factors in brains of mice 72 h post-ICH using a protein/DNA array. We observed increases in activity of AP-1, nuclear factor of activated T cells (NFAT-1), and growth hormone factor 1 (Pit-1) in brain tissue nuclear extracts that were diminished by EC treatment (Fig. S5A). Because stress–response elements that contain an AP-1 (Fos/Jun)-binding consensus sequence are major cis-elements for HO-1 induction,46 we further examined transcription factor AP-1 activity at 48 and 72 h post-ICH. AP-1 activity was increased after ICH in the vehicle-treated group (P < 0.01; Fig. 5A). However, EC 15 treatment reduced AP-1 activity compared with vehicle treatment at 48 h (P = 0.013) and 72 h (P < 0.01) post-ICH. We also determined the activity of MMP-2 and MMP-9, the downstream transcripts regulated by AP-1,47 by gelatin in situ zymography. EC 15 decreased gelatinolytic activity in cells (positive cells, 9.8 ± 1.8 vs. 7.4 ± 0.9 per 0.01 mm2; P < 0.05) in the perihematomal area 72 h post-ICH (Fig. 5B). Gelatinolytic activity was weak in the striatum of sham-operated brain sections (data not shown). Gelatin gel zymography showed that EC 15 reduced MMP-9 activity by ∼66% compared with that in the vehicle-treated group. MMP-2 gelatinolytic activity was relatively weak (P < 0.01; Fig. 5C).
Figure 5.
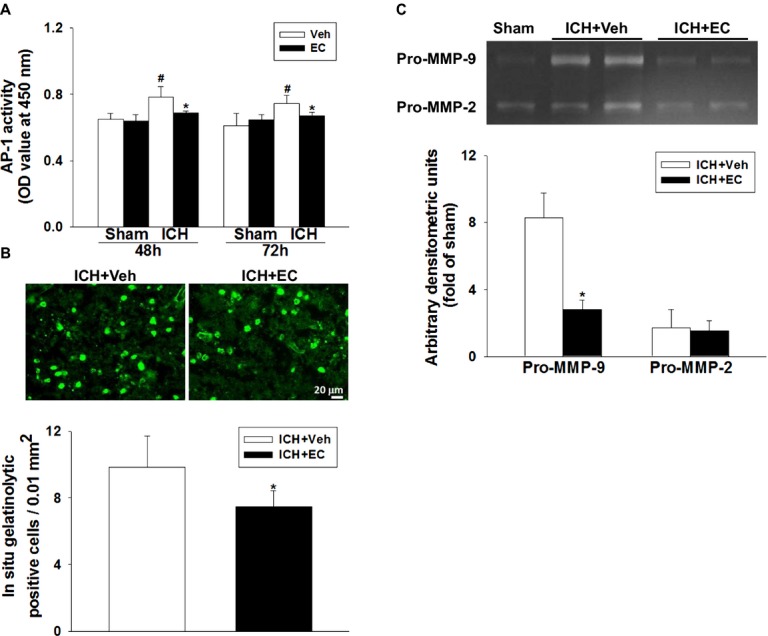
Effects of EC on AP-1 and MMP activity in mice subjected to ICH. (A) AP-1 activity was significantly greater in the ipsilateral hemisphere of ICH mice than in that of sham-operated mice. EC administration significantly reduced AP-1 activity at 48 and 72 h post-ICH. (B) Top: Representative gelatin in situ zymography fluorescent images from ICH mice treated with vehicle or EC. Bottom: Quantification shows that gelatinolytic activity was lower in EC-treated mice than in vehicle-treated mice at 72 h post-ICH. (C) Top: Representative gel zymography of MMP-9 and MMP-2 activity from vehicle- and EC-treated mice at 72 h post-ICH. The gelatinase activity of pro-MMP-9 was increased in brains from both vehicle- and EC-treated mice. Bottom: Quantification analysis indicated that pro-MMP-9 activity was decreased by EC administration. Pro-MMP-2 gelatinase activity was weak in all groups. Values are mean ± SD; n = 5–6 mice per group. *P < 0.05 versus vehicle; #P < 0.05 versus sham.
EC modulates iron-related chemokine LCN2 and ferroptosis gene expression
Ferroptosis is a newly recognized, iron-dependent form of cell death.6 A previous study reported that lipocalin-2 (LCN2) participates in iron homeostasis and enhances brain iron clearance after ICH.48 We sought to determine whether EC might act via an iron-related mechanism. Compared with that in the sham group, LCN2 protein expression increased in the brains of both vehicle- and EC-treated mice 72 h after ICH. However, LCN2 expression was ∼1.6-fold higher in the EC-treated mice (P < 0.01; Fig. 6A). The enzyme-linked immunosorbent assay (ELISA) further confirmed that LCN2 protein level was higher in brain tissue of EC 15 mice than in brain tissue of vehicle-treated mice at 6 h (6976.1 ± 232.3 pg/mL vs. 6430.9 ± 76.7 pg/mL; P < 0.05) and 24 h (7329.5 ± 30.5 pg/mL vs. 6913.2 ± 225.1 pg/mL; P < 0.05) post-ICH (Fig. 6B). To determine if EC alters the expression of ferroptosis-related genes, we measured the expression of iron response element binding protein 2 (Ireb2), ATP synthase F0 complex subunit C3 (ATP5G3), citrate synthase (Cs), and ATP ribosomal protein L8 (Rpl8) at 6, 24, and 72 h after ICH induction. Compared with levels in the vehicle-treated group, EC 15 reduced Ireb2 mRNA by ∼20% at 6 h (P < 0.01) post-ICH and increased ATP5G3 mRNA by ∼25% at 6 h (P < 0.05), 24 h (P < 0.01), and 72 h (P < 0.01) post-ICH. However, expression of Cs mRNA and Rpl8 mRNA did not differ significantly between the two groups.
Figure 6.
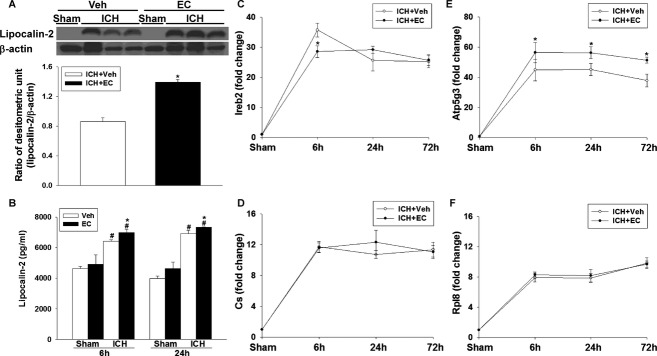
Effects of EC on lipocalin-2 (LCN2) and ferroptosis-related genes in mice subjected to ICH. (A) Top: Representative immunoblot of LCN2 protein expression in hemorrhagic tissue from sham-operated and ICH mice treated with or without EC. Bottom: Densitometric analysis shows a significant increase in LCN2 protein expression in the ipsilateral hemisphere of EC-treated mice 72 h after ICH compared with that in vehicle-treated mice. (B) Bar graph shows LCN2 protein concentration in the ipsilateral hemispheres of vehicle- and EC-treated mice at 6 and 24 h post-ICH. ICH significantly increased LCN2 protein levels in both groups, but LCN2 protein level was significantly higher in the EC-treated group than in the vehicle-treated group. (C–F) Line graphs show mRNA expression of Ireb2, Cs, Atp5g3, and Rpl8 in the hemorrhagic hemispheres of vehicle- and EC-treated mice at 6, 24, and 72 h post-ICH. EC treatment significantly decreased Ireb2 mRNA expression at 6 h and increased Atp5g3 mRNA expression at 6, 24, and 72 h after ICH compared with vehicle treatment. Cs and Rpl8 mRNA expression did not differ significantly between the two groups. Values are mean ± SD; n = 5–6 mice per group. *P < 0.05 versus vehicle; #P < 0.05 versus sham.
Discussion
Natural flavanols have the potential to improve an individual's oxidant defense system by activating endogenous protective signaling pathways. When flavanol-rich food is consumed, EC is absorbed into circulation22 and crosses the BBB.24–26 In animal models, EC has been shown to have neuroprotective effects, such as improving cognitive performance,21 enhancing memory retention,26 reducing β-amyloid toxicity,34 preventing tau aggregation,49 attenuating ischemic brain injury, and inhibiting excitotoxicity.23 In this study, using a collagenase model of ICH, we showed that EC reduces early brain injury in 12-month-old male and female mice and that the improved neurologic function is sustained for at least 4 weeks after ICH. We also confirmed the efficacy of EC in the blood and thrombin models of ICH. Furthermore, we found that EC reduces ICH-induced cell death in the perihematomal region, promotes Nrf2 nuclear accumulation, and reduces oxidative brain damage. Interestingly, we showed that EC decreases HO-1 protein expression and iron deposition through an Nrf2-independent mechanism, decreases AP-1 and MMP-9 activity, increases LCN2 protein level, and modulates ferroptosis-related gene expression. Taken together, these findings provide clear evidence that oral administration of EC protects brain against early hemorrhagic injury through Nrf2-dependent and -independent pathways.
Most animal studies of ICH have been carried out in 2- or 3-month-old animals, limiting the direct translation of animal studies into clinical trials. We used 12-month-old mice in this study to enhance the clinical relevance, as stroke is most common in older adults. Additionally, because the current, commonly used blood- and collagenase-induced ICH models cannot fully and adequately mirror human ICH events, we applied multiple models in our study to reduce possible translational pitfalls.3,38,50 We followed the published guidelines for translational studies37–39 and confirmed the efficacy of EC in three ICH-related models. We found that posttreatment of mice for 72 h with 15 mg/kg oral EC decreased injury volume and brain edema formation in the early stage of ICH and improved neurologic deficits for as long as 28 days after ICH onset. The dose of 15 mg/kg per day in mice is approximately equivalent to a human dose of 1.22 mg/kg per day, based on calculations with a body surface area conversion factor.51 Previous studies also provide supportive information on the feasibility of using oral EC to treat central nervous system disorders.22,23,26
Sex differences may influence stroke outcomes and response to pharmacologic therapy.52 Although sex differences and possible molecular mechanisms have been studied in ischemic stroke,53 few animal studies of ICH have been conducted in female mice.42 In this study, although post hoc analysis showed no significant difference between males and females owing to limited sample size, female mice tended to have smaller lesion volumes and less brain water content than did male mice after ICH, supporting the idea that sex affects brain injury after ICH. It is noteworthy, however, that the efficacy of EC did not differ in males and females.
Cell death from acute ICH in central and perihematomal areas is a sophisticated process that involves several different pathways. Necrosis and apoptosis are the major forms of pathologic cell death after ICH in animals and humans.54–57 It was reported that the number of necrotic and apoptotic cells in the perihematomal area peaks at 72 h post-ICH.57,58 This peak correlates with an increase in HMGB-1, which is released from necrotic cells and correlates positively with stroke severity.59,60 The EC-induced attenuation of neuronal death, as evidenced by a reduction in FJB staining, is consistent with in vitro studies showing that EC increases the viability of neurons exposed to hemoglobin and suggests a direct protective effect of EC on neurons.
Iron-dependent ferroptosis, a newly recognized form of programmed cell death, occurs in hippocampal slice cultures exposed to glutamate.6 This process is characterized by iron-dependent accumulation of ROS, similar to what is observed after ICH. To date, however, the role of ferroptosis after ICH remains unknown. Research in this area has been hindered by a lack of specific markers that can be used to characterize this phenomenon in vivo and in vitro. On the basis of available information, we tested ferroptosis-related gene expression in mice subjected to ICH with or without EC posttreatment. The reduction in Ireb2 gene expression and iron deposition in the hemorrhagic territory demonstrates the ability of EC to ameliorate iron-induced cell death after ICH. Ireb2 encodes a master regulator of iron metabolism. Neurons lacking Ireb2 are highly resistant to hemoglobin toxicity, and Ireb2 KO increases cell viability after ICH.61 Furthermore, extensive evidence has shown that iron chelation reduces hemoglobin/iron-induced toxicity and could be a promising therapy for ICH.7,62
Although flavanols have been speculated to act as iron chelators in an in vitro model of Alzheimer's disease,63 to our knowledge, this is the first report to provide evidence that EC has iron elimination properties after ICH, when the brain is exposed to iron overload. Iron, one of the hemoglobin/heme degradation products of HO-1, damages neurons by catalyzing reactions with hydrogen peroxide to generate hydroxyl radicals, believed to be the most reactive and dangerous form of ROS. ICH-induced HO-1 upregulation and overexpression of HO-1 in mice each result in iron overload in the brain.44,64 We showed previously that HO-1 deletion reduces early brain injury by decreasing oxidative stress in ICH brain.11 Here, we provide direct evidence of a relationship between HO-1 expression and iron deposition in ICH brain using HO-1 KO mice. Notably, EC administration reduced HO-1 protein expression in hemorrhagic tissue and thereby reduced ROS-induced DNA damage (hydroethidine), lipid peroxidation (MDA), and protein oxidation (dinitrophenyl hydrazone). These antioxidant effects were accompanied by increases in Nrf2 nuclear accumulation and phase II enzyme (SOD1 and NQO1) expression.
It has been shown in the middle cerebral artery occlusion model of ischemic stroke that the neuroprotection conferred by EC is abolished in mice that lack Nrf2. Similarly, EC-induced neuroprotection in neurons exposed to tert-butyl hydroperoxide is absent in neurons isolated from Nrf2 KO mice.23 We and others have shown that Nrf2 protects brain against ICH injury.8,10 Here, we confirmed that Nrf2 KO mice have larger ICH-induced lesion volume and more iron-loaded cells than do WT mice at 72 h post-ICH. Interestingly, EC 15 was still able to decrease ICH-induced lesion by 28.1% in Nrf2 KO mice (compared to 38.2% in WT mice). The fact that Nrf2 KO mice still showed some protection from posttreatment with EC, suggests that an Nrf2-independent pathway might act synergistically under ICH conditions. Although no residual neuroprotection from EC was apparent in Nrf2 KO mice in a previously published study of ischemic stroke,23 many differences between the studies could account for the discrepancy. Most importantly, hemorrhagic and ischemic strokes produce different pathological events. Additionally, the previous study used younger animals, a single-dose intervention, and an earlier endpoint. Moreover, our findings are in line with those of recently published studies which showed that the gene regulation of HO-1 can occur through an Nrf2-independent pathway in other organs.65,66
Nrf2/small Maf heterodimers and AP-1 complexes are the two major transcription factors that participate in HO-1 activation. In Nrf2 KO fibroblasts treated with arsenite, HO-1 gene expression levels increased gradually, probably via AP-1 activation.65 The transactivation of AP-1, a stress-activated transcription factor, has been defined as an inflammatory signaling molecule in ischemic stroke and traumatic brain injury.67 However, the role of AP-1 in ICH and iron-induced cell death has not been explored. One study in an in vitro mouse epidermal model showed that AP-1 activation increases after UVB stimulation and can be inhibited by deferoxamine, an iron chelator that can reduce iron deposition and neuronal death after ICH.68–70 Furthermore, EC suppresses TNFα-mediated AP-1–DNA binding in adipocytes.71 These insights imply that although EC increases antioxidant gene expression through Nrf2 activation, its ability to chelate iron and suppress AP-1 activity may downregulate HO-1 and subsequently reduce iron-induced brain damage after ICH. In addition, MMPs and LCN2 are coeffectors of AP-1 activation and iron overload after brain injury.48,72 ROS can activate MMP-9, which degrades the neurovascular matrix, leading to brain edema and tissue injury.17 LCN2, an acute phase protein that is upregulated under various stress conditions, plays a central role in iron transport. LCN2 captures iron particles, reduces cell death, and acts as an antioxidant in vivo by regulating iron homeostasis after ICH, sepsis, and renal ischemia.48,73–75 In this study, EC significantly reduced ICH-induced brain injury in WT and Nrf2 KO mice, suppressed AP-1 activation, reduced MMP-9 activity, and upregulated LCN2. Taken together, these findings suggest that an alternative, Nrf2-independent pathway is involved.
Although we provide proof of concept and elucidate the mechanisms of action of EC in our ICH models, additional efforts should be made before clinical trials can be considered in the future. (1) The effects of a delayed treatment regimen and preventive use of EC need to be tested in different ICH models and in different animal species. We speculate that pretreatment with EC should be protective in ICH; however, in this study, EC 45 had unfavorable effects on neurologic function, though it did not increase brain lesion volume. Additionally, green tea catechins were reported to have antiplatelet activity.76 Therefore, the pretreatment dosage of EC needs to be titrated carefully in animals to prevent bleeding risk. (2) Our selection of dose and delivery route in this study was based on available pharmacologic studies as described in the Methods. Nonoral systemic administration of EC such as by intravenous infusion should be tested as an emergency treatment option for ICH patients. Pharmacokinetic parameters of EC in humans and rabbits support this option.35,77 (3) Although our data show that EC has a direct neuroprotective effect on neurons in our in vitro ICH model, several lines of evidence indicate that EC can regulate blood pressure and improve vascular function by targeting endothelial cells.22,78 Because hypertension is one of the risk factors for ICH,2,38 multiple cellular therapeutic targets of EC should be validated. Moreover, green tea catechins have the ability to modify the activity of multiple intracellular molecular targets, including mitogen-activated protein kinase (MAPK), protein kinase C, and PI3K/Akt pathways.79 Studies are needed to determine whether EC targets multiple signaling pathways that trigger signaling crosstalk.
In conclusion, we found that oral posttreatment of mice with EC protects against early hemorrhagic brain injury. We confirmed that EC has both antioxidant and iron-chelating properties and may act through Nrf2-dependent and -independent pathways. EC's ability to act on multiple targets may provide an opportunity for application in other neurologic disorders, such as head and spinal cord injury. Although more work is needed, targeting AP-1 could provide new mechanistic insight in the search for drugs to treat ICH. Furthermore, our evidence may help the general public and health care providers make informed decisions on whether EC could be accepted as an adjunct for ICH treatment.
Acknowledgments
This study was supported by AHA 13GRNT15730001, NIH K01AG031926, R01AT007317, and R01NS078026 (J. W.). We thank Lingshu Liu, Joy Ziyi Zhou, and Weizhu Tang for blind analysis of histology and immunofluorescence; Herman Kwansa for HPLC analysis of (-)-epicatechin purity; and Paul Talalay, PhD, for providing Nrf2−/− mice. We thank Claire Levine, MS, ELS, for assistance with manuscript preparation; Adam Sapirstein, MD, and Zeng-Jin Yang, MD, PhD, for comments on the manuscript; Raymond Koehler, PhD, and the Wang lab team members for insightful input.
Author Contributions
C. C. and J. W. designed the research, C. C., J. W., and S. C. performed the research, C. C. and J. W. analyzed the data and wrote the paper.
Conflict of Interest
None declared.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Data S1. Materials and methods.
Figure S1. HPLC assessment of the stability of EC. The stability of an aqueous solution of EC at 4°C after 24 h was assessed by HPLC using fluorescence detection. The analyte of EC (Sigma, E1753) was 100% pure as can be seen from the chromatogram. EC in water remained stable for more than 24 h.
Figure S2. EC administration improves early ICH outcomes in male and female mice subjected to three hemorrhagic models. (A and B) At 72 h after female mice were subjected to collagenase-induced ICH, brain injury volumes were significantly smaller (A) and neurologic deficit scores were significantly lower (B) in the EC-treated group than in the vehicle-treated group. (C and D) At 72 h after induction of ICH by the blood model, male (C) and female (D) mice treated with EC had significantly lower neurologic deficit scores than did those treated with vehicle. (E and F) At 72 h after collagenase-induced ICH, brain water content was significantly lower in male (E) and female (F) mice treated with EC than in those treated with vehicle. (G and H) At 72 h after blood-induced ICH, brain water content was significantly lower in male (G) and female (H) mice treated with EC than in those treated with vehicle. (I) At 72 h after male mice were subjected to the thrombin ICH model, brain water content in the EC-treated group was significantly lower than that in the vehicle-treated group. Values are mean ± SD (n = 6–10 animals per group). *P < 0.05 versus vehicle. Ips, ipsilateral; Cont, contralateral; Stri, striatum; Cerebel, cerebelum.
Figure S3. EC administration improves long-term functional outcomes in mice subjected to ICH. (A) Neurologic deficit scores of male mice were significantly lower in the EC-treated group than in the vehicle-treated group 28 days after collagenase-induced ICH. (B) Compared with vehicle treatment, EC administration improved corner turn test performance of male mice on day 28 after collagenase injection. Values are mean ± SD; n = 10 mice per group. *P < 0.05 versus vehicle; #P < 0.05 versus 1 day.
Figure S4. EC administration decreases neuronal vulnerability to hemoglobin-induced toxicity. Exposure of primary neuronal cultures to hemoglobin (Hb; 6 µmol/L) for 24 h caused a significant increase in lactate dehydrogenase (LDH) release. Co-treatment of neurons with EC (20 µmol/L) significantly reduced Hb-induced LDH release. Values are mean ± SD; n = 3 per group. *P < 0.05 versus vehicle; #P < 0.05 versus Hb alone.
Figure S5. EC administration modulates transcription factor activity profile. (A) Representative chemiluminescence images of protein/DNA array from sham-operated mice and ICH mice treated with or without EC. At 72 h after ICH, activity of AP-1, NFAT-1, and Pit-1 was decreased in the ipsilateral hemisphere of EC-treated mice compared with that of vehicle-treated mice. (B) The DNA probe sequences for detecting specific transcription factors. AP-1, activator protein-1; CREB, cAMP response element-binding protein 1; GATA, globin transcription factor; NFAT-1, nuclear factor of activated T cells; Pit-1, growth hormone factor 1; SP-1, Sp1 transcription factor; TR, thyroid hormone receptor; PC, positive control.
References
- 1.Donnan GA, Hankey GJ, Davis SM. Intracerebral haemorrhage: a need for more data and new research directions. Lancet Neurol. 2010;9:133–134. doi: 10.1016/S1474-4422(10)70001-6. [DOI] [PubMed] [Google Scholar]
- 2.Xi G, Keep RF, Hoff JT. Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurol. 2006;5:53–63. doi: 10.1016/S1474-4422(05)70283-0. [DOI] [PubMed] [Google Scholar]
- 3.Wang J. Preclinical and clinical research on inflammation after intracerebral hemorrhage. Prog Neurobiol. 2010;92:463–477. doi: 10.1016/j.pneurobio.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zecca L, Youdim MB, Riederer P, et al. Iron, brain ageing and neurodegenerative disorders. Nat Rev Neurosci. 2004;5:863–873. doi: 10.1038/nrn1537. [DOI] [PubMed] [Google Scholar]
- 5.Wang X, Mori T, Sumii T, et al. Hemoglobin-induced cytotoxicity in rat cerebral cortical neurons: caspase activation and oxidative stress. Stroke. 2002;33:1882–1888. doi: 10.1161/01.str.0000020121.41527.5d. [DOI] [PubMed] [Google Scholar]
- 6.Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu H, Wu T, Li M, et al. Efficacy of the lipid-soluble iron chelator 2,2′-dipyridyl against hemorrhagic brain injury. Neurobiol Dis. 2012;45:388–394. doi: 10.1016/j.nbd.2011.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao X, Sun G, Zhang J, et al. Transcription factor Nrf2 protects the brain from damage produced by intracerebral hemorrhage. Stroke. 2007;38:3280–3286. doi: 10.1161/STROKEAHA.107.486506. [DOI] [PubMed] [Google Scholar]
- 9.Wang J, Dore S. Inflammation after intracerebral hemorrhage. J Cereb Blood Flow Metab. 2007;27:894–908. doi: 10.1038/sj.jcbfm.9600403. [DOI] [PubMed] [Google Scholar]
- 10.Wang J, Fields J, Zhao C, et al. Role of Nrf2 in protection against intracerebral hemorrhage injury in mice. Free Radic Biol Med. 2007;43:408–414. doi: 10.1016/j.freeradbiomed.2007.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang J, Dore S. Heme oxygenase-1 exacerbates early brain injury after intracerebral haemorrhage. Brain. 2007;130:1643–1652. doi: 10.1093/brain/awm095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ziai WC. Hematology and inflammatory signaling of intracerebral hemorrhage. Stroke. 2013;44:S74–S78. doi: 10.1161/STROKEAHA.111.000662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gomes JA, Manno E. New developments in the treatment of intracerebral hemorrhage. Neurol Clin. 2013;31:721–735. doi: 10.1016/j.ncl.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 14.Shaulian E, Karin M. AP-1 as a regulator of cell life and death. Nat Cell Biol. 2002;4:E131–E136. doi: 10.1038/ncb0502-e131. [DOI] [PubMed] [Google Scholar]
- 15.Zhao BQ, Wang S, Kim HY, et al. Role of matrix metalloproteinases in delayed cortical responses after stroke. Nat Med. 2006;12:441–445. doi: 10.1038/nm1387. [DOI] [PubMed] [Google Scholar]
- 16.Ma Q, Huang B, Khatibi N, et al. PDGFR-alpha inhibition preserves blood-brain barrier after intracerebral hemorrhage. Ann Neurol. 2011;70:920–931. doi: 10.1002/ana.22549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang J, Tsirka SE. Neuroprotection by inhibition of matrix metalloproteinases in a mouse model of intracerebral haemorrhage. Brain. 2005;128:1622–1633. doi: 10.1093/brain/awh489. [DOI] [PubMed] [Google Scholar]
- 18.Xue M, Fan Y, Liu S, et al. Contributions of multiple proteases to neurotoxicity in a mouse model of intracerebral haemorrhage. Brain. 2009;132:26–36. doi: 10.1093/brain/awn215. [DOI] [PubMed] [Google Scholar]
- 19.Kokubo Y, Iso H, Saito I, et al. The impact of green tea and coffee consumption on the reduced risk of stroke incidence in Japanese population: the Japan public health center-based study cohort. Stroke. 2013;44:1369–1374. doi: 10.1161/STROKEAHA.111.677500. [DOI] [PubMed] [Google Scholar]
- 20.Knekt P, Jarvinen R, Reunanen A, et al. Flavonoid intake and coronary mortality in Finland: a cohort study. BMJ. 1996;312:478–481. doi: 10.1136/bmj.312.7029.478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nehlig A. The neuroprotective effects of cocoa flavanol and its influence on cognitive performance. Br J Clin Pharmacol. 2013;75:716–727. doi: 10.1111/j.1365-2125.2012.04378.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schroeter H, Heiss C, Balzer J, et al. (-)-Epicatechin mediates beneficial effects of flavanol-rich cocoa on vascular function in humans. Proc Natl Acad Sci USA. 2006;103:1024–1029. doi: 10.1073/pnas.0510168103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shah ZA, Li RC, Ahmad AS, et al. The flavanol (-)-epicatechin prevents stroke damage through the Nrf2/HO1 pathway. J Cereb Blood Flow Metab. 2010;30:1951–1961. doi: 10.1038/jcbfm.2010.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abd El Mohsen MM, Kuhnle G, Rechner AR, et al. Uptake and metabolism of epicatechin and its access to the brain after oral ingestion. Free Radic Biol Med. 2002;33:1693–1702. doi: 10.1016/s0891-5849(02)01137-1. [DOI] [PubMed] [Google Scholar]
- 25.Wu L, Zhang QL, Zhang XY, et al. Pharmacokinetics and blood-brain barrier penetration of (+)-catechin and (-)-epicatechin in rats by microdialysis sampling coupled to high-performance liquid chromatography with chemiluminescence detection. J Agric Food Chem. 2012;60:9377–9383. doi: 10.1021/jf301787f. [DOI] [PubMed] [Google Scholar]
- 26.van Praag H, Lucero MJ, Yeo GW, et al. Plant-derived flavanol (-)epicatechin enhances angiogenesis and retention of spatial memory in mice. J Neurosci. 2007;27:5869–5878. doi: 10.1523/JNEUROSCI.0914-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ruijters EJ, Weseler AR, Kicken C, et al. The flavanol (-)-epicatechin and its metabolites protect against oxidative stress in primary endothelial cells via a direct antioxidant effect. Eur J Pharmacol. 2013;715:147–153. doi: 10.1016/j.ejphar.2013.05.029. [DOI] [PubMed] [Google Scholar]
- 28.Litterio MC, Jaggers G, Sagdicoglu Celep G, et al. Blood pressure-lowering effect of dietary (-)-epicatechin administration in L-NAME-treated rats is associated with restored nitric oxide levels. Free Radic Biol Med. 2012;53:1894–1902. doi: 10.1016/j.freeradbiomed.2012.08.585. [DOI] [PubMed] [Google Scholar]
- 29.Zurasky JA, Aiyagari V, Zazulia AR, et al. Early mortality following spontaneous intracerebral hemorrhage. Neurology. 2005;64:725–727. doi: 10.1212/01.WNL.0000152045.56837.58. [DOI] [PubMed] [Google Scholar]
- 30.Feigin VL, Lawes CM, Bennett DA, et al. Worldwide stroke incidence and early case fatality reported in 56 population-based studies: a systematic review. Lancet Neurol. 2009;8:355–369. doi: 10.1016/S1474-4422(09)70025-0. [DOI] [PubMed] [Google Scholar]
- 31.Enomoto A, Itoh K, Nagayoshi E, et al. High sensitivity of Nrf2 knockout mice to acetaminophen hepatotoxicity associated with decreased expression of ARE-regulated drug metabolizing enzymes and antioxidant genes. Toxicol Sci. 2001;59:169–177. doi: 10.1093/toxsci/59.1.169. [DOI] [PubMed] [Google Scholar]
- 32.Poss KD, Tonegawa S. Heme oxygenase 1 is required for mammalian iron reutilization. Proc Natl Acad Sci USA. 1997;94:10919–10924. doi: 10.1073/pnas.94.20.10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Faria A, Pestana D, Teixeira D, et al. Insights into the putative catechin and epicatechin transport across blood-brain barrier. Food Funct. 2011;2:39–44. doi: 10.1039/c0fo00100g. [DOI] [PubMed] [Google Scholar]
- 34.Cuevas E, Limon D, Perez-Severiano F, et al. Antioxidant effects of epicatechin on the hippocampal toxicity caused by amyloid-beta 25-35 in rats. Eur J Pharmacol. 2009;616:122–127. doi: 10.1016/j.ejphar.2009.06.013. [DOI] [PubMed] [Google Scholar]
- 35.Chen YA, Hsu KY. Pharmacokinetics of (-)-epicatechin in rabbits. Arch Pharm Res. 2009;32:149–154. doi: 10.1007/s12272-009-1129-x. [DOI] [PubMed] [Google Scholar]
- 36.Clark W, Gunion-Rinker L, Lessov N, et al. Citicoline treatment for experimental intracerebral hemorrhage in mice. Stroke. 1998;29:2136–2140. doi: 10.1161/01.str.29.10.2136. [DOI] [PubMed] [Google Scholar]
- 37.Landis SC, Amara SG, Asadullah K, et al. A call for transparent reporting to optimize the predictive value of preclinical research. Nature. 2012;490:187–191. doi: 10.1038/nature11556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.MacLellan CL, Paquette R, Colbourne F. A critical appraisal of experimental intracerebral hemorrhage research. J Cereb Blood Flow Metab. 2012;32:612–627. doi: 10.1038/jcbfm.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fisher M, Feuerstein G, Howells DW, et al. Update of the stroke therapy academic industry roundtable preclinical recommendations. Stroke. 2009;40:2244–2250. doi: 10.1161/STROKEAHA.108.541128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Del Bigio MR, Yan HJ, Buist R, et al. Experimental intracerebral hemorrhage in rats. Magnetic resonance imaging and histopathological correlates. Stroke. 1996;27:2312–2319. doi: 10.1161/01.str.27.12.2312. discussion 2319–2320. [DOI] [PubMed] [Google Scholar]
- 41.Nakamura T, Hua Y, Keep RF, et al. Estrogen therapy for experimental intracerebral hemorrhage in rats. J Neurosurg. 2005;103:97–103. doi: 10.3171/jns.2005.103.1.0097. [DOI] [PubMed] [Google Scholar]
- 42.Nakamura T, Xi G, Hua Y, et al. Intracerebral hemorrhage in mice: model characterization and application for genetically modified mice. J Cereb Blood Flow Metab. 2004;24:487–494. doi: 10.1097/00004647-200405000-00002. [DOI] [PubMed] [Google Scholar]
- 43.Gebel JM, Jr, Jauch EC, Brott TG, et al. Relative edema volume is a predictor of outcome in patients with hyperacute spontaneous intracerebral hemorrhage. Stroke. 2002;33:2636–2641. doi: 10.1161/01.str.0000035283.34109.ea. [DOI] [PubMed] [Google Scholar]
- 44.Song W, Zukor H, Lin SH, et al. Unregulated brain iron deposition in transgenic mice over-expressing HMOX1 in the astrocytic compartment. J Neurochem. 2012;123:325–336. doi: 10.1111/j.1471-4159.2012.07914.x. [DOI] [PubMed] [Google Scholar]
- 45.Chang CF, Chen SF, Lee TS, et al. Caveolin-1 deletion reduces early brain injury after experimental intracerebral hemorrhage. Am J Pathol. 2011;178:1749–1761. doi: 10.1016/j.ajpath.2010.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alam J, Camhi S, Choi AM. Identification of a second region upstream of the mouse heme oxygenase-1 gene that functions as a basal level and inducer-dependent transcription enhancer. J Biol Chem. 1995;270:11977–11984. doi: 10.1074/jbc.270.20.11977. [DOI] [PubMed] [Google Scholar]
- 47.Brinckerhoff CE, Matrisian LM. Matrix metalloproteinases: a tail of a frog that became a prince. Nat Rev Mol Cell Biol. 2002;3:207–214. doi: 10.1038/nrm763. [DOI] [PubMed] [Google Scholar]
- 48.Dong M, Xi G, Keep RF, et al. Role of iron in brain lipocalin 2 upregulation after intracerebral hemorrhage in rats. Brain Res. 2013;1505:86–92. doi: 10.1016/j.brainres.2013.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.George RC, Lew J, Graves DJ. Interaction of cinnamaldehyde and epicatechin with tau: implications of beneficial effects in modulating Alzheimer's disease pathogenesis. J Alzheimers Dis. 2013;36:21–40. doi: 10.3233/JAD-122113. [DOI] [PubMed] [Google Scholar]
- 50.Kirkman MA, Allan SM, Parry-Jones AR. Experimental intracerebral hemorrhage: avoiding pitfalls in translational research. J Cereb Blood Flow Metab. 2011;31:2135–2151. doi: 10.1038/jcbfm.2011.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J. 2008;22:659–661. doi: 10.1096/fj.07-9574LSF. [DOI] [PubMed] [Google Scholar]
- 52.Hurn PD, Vannucci SJ, Hagberg H. Adult or perinatal brain injury: does sex matter? Stroke. 2005;36:193–195. doi: 10.1161/01.STR.0000153064.41332.f6. [DOI] [PubMed] [Google Scholar]
- 53.Liu F, Li Z, Li J, et al. Sex differences in caspase activation after stroke. Stroke. 2009;40:1842–1848. doi: 10.1161/STROKEAHA.108.538686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang YX, Yan A, Ma ZH, et al. Nuclear factor-kappaB and apoptosis in patients with intracerebral hemorrhage. J Clin Neurosci. 2011;18:1392–1395. doi: 10.1016/j.jocn.2010.11.039. [DOI] [PubMed] [Google Scholar]
- 55.Rodrigues CM, Sola S, Nan Z, et al. Tauroursodeoxycholic acid reduces apoptosis and protects against neurological injury after acute hemorrhagic stroke in rats. Proc Natl Acad Sci USA. 2003;100:6087–6092. doi: 10.1073/pnas.1031632100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brunswick AS, Hwang BY, Appelboom G, et al. Serum biomarkers of spontaneous intracerebral hemorrhage induced secondary brain injury. J Neurol Sci. 2012;321:1–10. doi: 10.1016/j.jns.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 57.Zhu X, Tao L, Tejima-Mandeville E, et al. Plasmalemma permeability and necrotic cell death phenotypes after intracerebral hemorrhage in mice. Stroke. 2012;43:524–531. doi: 10.1161/STROKEAHA.111.635672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Matsushita K, Meng W, Wang X, et al. Evidence for apoptosis after intercerebral hemorrhage in rat striatum. J Cereb Blood Flow Metab. 2000;20:396–404. doi: 10.1097/00004647-200002000-00022. [DOI] [PubMed] [Google Scholar]
- 59.Zhou Y, Xiong KL, Lin S, et al. Elevation of high-mobility group protein box-1 in serum correlates with severity of acute intracerebral hemorrhage. Mediators Inflamm. 2010;2010:142458. doi: 10.1155/2010/142458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ohnishi M, Katsuki H, Fukutomi C, et al. HMGB1 inhibitor glycyrrhizin attenuates intracerebral hemorrhage-induced injury in rats. Neuropharmacology. 2011;61:975–980. doi: 10.1016/j.neuropharm.2011.06.026. [DOI] [PubMed] [Google Scholar]
- 61.Regan RF, Chen M, Li Z, et al. Neurons lacking iron regulatory protein-2 are highly resistant to the toxicity of hemoglobin. Neurobiol Dis. 2008;31:242–249. doi: 10.1016/j.nbd.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Selim M. Deferoxamine mesylate: a new hope for intracerebral hemorrhage: from bench to clinical trials. Stroke. 2009;40:S90–S91. doi: 10.1161/STROKEAHA.108.533125. [DOI] [PubMed] [Google Scholar]
- 63.Reznichenko L, Amit T, Zheng H, et al. Reduction of iron-regulated amyloid precursor protein and beta-amyloid peptide by (-)-epigallocatechin-3-gallate in cell cultures: implications for iron chelation in Alzheimer's disease. J Neurochem. 2006;97:527–536. doi: 10.1111/j.1471-4159.2006.03770.x. [DOI] [PubMed] [Google Scholar]
- 64.Wu J, Hua Y, Keep RF, et al. Iron and iron-handling proteins in the brain after intracerebral hemorrhage. Stroke. 2003;34:2964–2969. doi: 10.1161/01.STR.0000103140.52838.45. [DOI] [PubMed] [Google Scholar]
- 65.Harada H, Sugimoto R, Watanabe A, et al. Differential roles for Nrf2 and AP-1 in upregulation of HO-1 expression by arsenite in murine embryonic fibroblasts. Free Radic Res. 2008;42:297–304. doi: 10.1080/10715760801975735. [DOI] [PubMed] [Google Scholar]
- 66.Kang J, Jeong MG, Oh S, et al. A FoxO1-dependent, but NRF2-independent induction of heme oxygenase-1 during muscle atrophy. FEBS Lett. 2014;588:79–85. doi: 10.1016/j.febslet.2013.11.009. [DOI] [PubMed] [Google Scholar]
- 67.Raivich G, Behrens A. Role of the AP-1 transcription factor c-Jun in developing, adult and injured brain. Prog Neurobiol. 2006;78:347–363. doi: 10.1016/j.pneurobio.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 68.Kramer-Stickland K, Edmonds A, Bair WB, III, et al. Inhibitory effects of deferoxamine on UVB-induced AP-1 transactivation. Carcinogenesis. 1999;20:2137–2142. doi: 10.1093/carcin/20.11.2137. [DOI] [PubMed] [Google Scholar]
- 69.Wu H, Wu T, Xu X, et al. Iron toxicity in mice with collagenase-induced intracerebral hemorrhage. J Cereb Blood Flow Metab. 2011;31:1243–1250. doi: 10.1038/jcbfm.2010.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hua Y, Keep RF, Hoff JT, et al. Brain injury after intracerebral hemorrhage: the role of thrombin and iron. Stroke. 2007;38:759–762. doi: 10.1161/01.STR.0000247868.97078.10. [DOI] [PubMed] [Google Scholar]
- 71.Vazquez-Prieto MA, Bettaieb A, Haj FG, et al. (-)-Epicatechin prevents TNFalpha-induced activation of signaling cascades involved in inflammation and insulin sensitivity in 3T3-L1 adipocytes. Arch Biochem Biophys. 2012;527:113–118. doi: 10.1016/j.abb.2012.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rosenberg GA. Matrix metalloproteinases in brain injury. J Neurotrauma. 1995;12:833–842. doi: 10.1089/neu.1995.12.833. [DOI] [PubMed] [Google Scholar]
- 73.Jung M, Sola A, Hughes J, et al. Infusion of IL-10-expressing cells protects against renal ischemia through induction of lipocalin-2. Kidney Int. 2012;81:969–982. doi: 10.1038/ki.2011.446. [DOI] [PubMed] [Google Scholar]
- 74.Srinivasan G, Aitken JD, Zhang B, et al. Lipocalin 2 deficiency dysregulates iron homeostasis and exacerbates endotoxin-induced sepsis. J Immunol. 2012;189:1911–1919. doi: 10.4049/jimmunol.1200892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Goetz DH, Willie ST, Armen RS, et al. Ligand preference inferred from the structure of neutrophil gelatinase associated lipocalin. Biochemistry. 2000;39:1935–1941. doi: 10.1021/bi992215v. [DOI] [PubMed] [Google Scholar]
- 76.Kang WS, Chung KH, Chung JH, et al. Antiplatelet activity of green tea catechins is mediated by inhibition of cytoplasmic calcium increase. J Cardiovasc Pharmacol. 2001;38:875–884. doi: 10.1097/00005344-200112000-00009. [DOI] [PubMed] [Google Scholar]
- 77.Lee MJ, Maliakal P, Chen L, et al. Pharmacokinetics of tea catechins after ingestion of green tea and (-)-epigallocatechin-3-gallate by humans: formation of different metabolites and individual variability. Cancer Epidemiol Biomarkers Prev. 2002;11:1025–1032. [PubMed] [Google Scholar]
- 78.Jimenez R, Duarte J, Perez-Vizcaino F. Epicatechin: endothelial function and blood pressure. J Agric Food Chem. 2012;60:8823–8830. doi: 10.1021/jf205370q. [DOI] [PubMed] [Google Scholar]
- 79.Mak JC. Potential role of green tea catechins in various disease therapies: progress and promise. Clin Exp Pharmacol Physiol. 2012;39:265–273. doi: 10.1111/j.1440-1681.2012.05673.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Materials and methods.
Figure S1. HPLC assessment of the stability of EC. The stability of an aqueous solution of EC at 4°C after 24 h was assessed by HPLC using fluorescence detection. The analyte of EC (Sigma, E1753) was 100% pure as can be seen from the chromatogram. EC in water remained stable for more than 24 h.
Figure S2. EC administration improves early ICH outcomes in male and female mice subjected to three hemorrhagic models. (A and B) At 72 h after female mice were subjected to collagenase-induced ICH, brain injury volumes were significantly smaller (A) and neurologic deficit scores were significantly lower (B) in the EC-treated group than in the vehicle-treated group. (C and D) At 72 h after induction of ICH by the blood model, male (C) and female (D) mice treated with EC had significantly lower neurologic deficit scores than did those treated with vehicle. (E and F) At 72 h after collagenase-induced ICH, brain water content was significantly lower in male (E) and female (F) mice treated with EC than in those treated with vehicle. (G and H) At 72 h after blood-induced ICH, brain water content was significantly lower in male (G) and female (H) mice treated with EC than in those treated with vehicle. (I) At 72 h after male mice were subjected to the thrombin ICH model, brain water content in the EC-treated group was significantly lower than that in the vehicle-treated group. Values are mean ± SD (n = 6–10 animals per group). *P < 0.05 versus vehicle. Ips, ipsilateral; Cont, contralateral; Stri, striatum; Cerebel, cerebelum.
Figure S3. EC administration improves long-term functional outcomes in mice subjected to ICH. (A) Neurologic deficit scores of male mice were significantly lower in the EC-treated group than in the vehicle-treated group 28 days after collagenase-induced ICH. (B) Compared with vehicle treatment, EC administration improved corner turn test performance of male mice on day 28 after collagenase injection. Values are mean ± SD; n = 10 mice per group. *P < 0.05 versus vehicle; #P < 0.05 versus 1 day.
Figure S4. EC administration decreases neuronal vulnerability to hemoglobin-induced toxicity. Exposure of primary neuronal cultures to hemoglobin (Hb; 6 µmol/L) for 24 h caused a significant increase in lactate dehydrogenase (LDH) release. Co-treatment of neurons with EC (20 µmol/L) significantly reduced Hb-induced LDH release. Values are mean ± SD; n = 3 per group. *P < 0.05 versus vehicle; #P < 0.05 versus Hb alone.
Figure S5. EC administration modulates transcription factor activity profile. (A) Representative chemiluminescence images of protein/DNA array from sham-operated mice and ICH mice treated with or without EC. At 72 h after ICH, activity of AP-1, NFAT-1, and Pit-1 was decreased in the ipsilateral hemisphere of EC-treated mice compared with that of vehicle-treated mice. (B) The DNA probe sequences for detecting specific transcription factors. AP-1, activator protein-1; CREB, cAMP response element-binding protein 1; GATA, globin transcription factor; NFAT-1, nuclear factor of activated T cells; Pit-1, growth hormone factor 1; SP-1, Sp1 transcription factor; TR, thyroid hormone receptor; PC, positive control.