

Abstract
A group of disorders with disparate symptomatology, including congenital cerebellar ataxia, retinal blindness, liver fibrosis, polycystic kidney disease, and polydactyly, have recently been united under a single disease mechanism called ‘ciliopathies’. The ciliopathies are due to defects of the cellular antenna known as the primary cilium, a microtubule-based extension of cellular membranes found in nearly all cell types. Key among these ciliopathies is Joubert syndrome, displaying ataxia, oculomotor apraxia, and mental retardation* with a pathognomonic ‘molar tooth sign’ on brain magnetic resonance imaging. The importance of ciliary function in neuronal development has been appreciated only in the last decade with the classification of Joubert syndrome as a ciliopathy. This, together with the identification of many of the clinical features of ciliopathies in individuals with Joubert syndrome and the localization of Joubert syndrome’s causative gene products at or near the primary cilium, have defined a new class of neurological disease. Cilia are involved in diverse cellular processes including protein trafficking, photoreception, embryonic axis patterning, and cell cycle regulation. Ciliary dysfunction can affect a single tissue or manifest as multi-organ involvement. Ciliary defects have been described in retinopathies such as retinitis pigmentosa and Leber congenital amaurosis (defects in photoreceptor ciliary protein complexes), renal syndromes with nephronophthisis and cystic dysplastic kidneys, and liver conditions such as fibrosis and biliary cirrhosis. Recognizing the diverse presentations of the ciliopathies and screening strategies following diagnosis is an important part of the treatment plan of children with cilia-related disorders.
In 1969 Dr. Marie Joubert1 encountered four affected French Canadian siblings who were the product of consanguineous parents linked approximately 11 generations before. Joubert syndrome is an autosomal recessive disorder characterized by hypotonia, apnoea/hyperpnoea in infancy, oculomotor apraxia, and variable developmental delay/intellectual impairment with cerebellar vermal hypoplasia or aplasia. The unifying pathognomonic radiographic finding, known as the ‘molar tooth sign’ (MTS), which is visible on axial brain magnetic resonance imaging (MRI) at the level of the mid-brain–hindbrain junction (i.e. just above the pons),2 has helped to narrow the diagnosis to just those displaying this feature. Recent advancements have linked Joubert syndrome and other syndromic disorders to defects of cilia. Cilia are classically divided into motile and non-motile (primary) based on the organization of the microtubules in the ciliary axonemes. The axoneme is the microtubule backbone along which microtubule-mediated intraflagellar transport of various ciliary proteins occurs. The axoneme is anchored by the basal body in coordination with the action of centrosomal proteins and guanosine triphosphatases. Ciliogenesis refers to the process of the docking of the basal body at the plasma membrane, followed by recruitment of ciliary proteins and protrusion of the newly emerging axoneme into the plasma membrane. Subsequently, proteins are transported into and out of the cilia, further utilizing intraflagellar transport, which functions bidirectionally. A defect in the transport or arrangement of these cilia–centrosomal proteins adversely affects a variety of the critical developmental signalling pathways that are essential to cellular development such as Sonic hedgehog, Wnt signalling, planar cell polarity, and directional movement. However, the degree to which altered signalling leads to the eventual clinical presentation is not known, nor is it known whether modulation of these pathways might provide benefit to individuals.
Although primary cilia are located on nearly every animal cell, motile cilia are present only in highly specialized areas, including tracheal epithelium, sperm, oviducts, and brain ventricles. An important consequence of motile cilia disruption is ciliary dyskinesia (i.e. Kartagener syndrome), which is characterized by situs inversus totalis, amotile spermatozoa, and pulmonary insufficiency. On the other hand, individuals with primary cilia defects rarely show situs inversus and instead display a variety of multi-organ involvement syndromes known as genetic ciliopathy syndromes, such as Joubert syndrome. Recently, an association between Joubert syndrome and several other disorders, now known as Joubert syndrome-related disorders (JSRDs), was recognized, thus broadening the clinical spectrum of these disorders (Table I).3 Although Joubert syndrome was first defined based upon the presence of neurological features alone, JSRDs are characterized by extracentral nervous system involvement such as retinopathy, cystic dysplastic kidneys or nephronophthisis, hepatic fibrosis, and polydactyly and midline facial defects, each named for a particular constellation of signs and symptoms. JSRDs are now known to include cerebellar–ocular–renal syndrome, cerebellar vermis hypo-/aplasia, oligophrenia, congenital ataxia, ocular coloboma, and hepatic fibrosis (COACH) syndrome, and Varadi–Papp syndrome (also known as orofacial digital syndrome type VI) based upon the presence of the unifying MTS. The surprising aspect is that all of the proteins encoded by the responsible genes localize to the cellular primary cilium, suggesting common disease mechanisms, as well as future therapeutic strategies.
Table I.
Clinical features of each of the partially overlapping conditions within the Joubert syndrome and related disorders spectrum
| Clinical feature | JS | CORS | LCA | SLS | MKS | DKA | BBS | COACH | OFD6 | Cogan |
|---|---|---|---|---|---|---|---|---|---|---|
| Hypotonia | + | + | +/− | +/− | N/A | +/− | +/− | +/− | +/− | + |
| Intellectually impaired | +a | + | +/− | +/− | N/A | +/− | + | +/− | + | +/− |
| Hyperpnoea/apnoea | + | +/− | +/− | +/− | N/A | +/− | +/− | +/− | +/− | – |
| Oculomotor apraxia | + | + | +/− | − | N/A | − | − | − | − | + |
| Coloboma | − | − | − | − | +/− | − | − | + | − | − |
| Retinal dystrophy | +/− | + | + | + | − | + | + | − | − | − |
| Tongue hamartoma | − | − | − | − | − | − | − | − | + | − |
| Oral frenula | − | − | − | − | − | − | − | − | + | − |
| Orofacial clefting | − | − | − | − | +/− | − | − | − | + | − |
| Cystic renal dystrophy | − | + | − | − | + | + | + | − | − | − |
| Nephronophthisis | − | + | − | + | − | + | +/− | − | − | +/− |
| Hepatic fibrosis | − | − | − | − | − | − | +/− | + | − | − |
| Polymicrogyria | +/− | − | − | − | − | − | − | − | − | − |
| Encephalocele | − | − | − | − | + | − | − | − | − | − |
| MTS | + | + | +/− | +/− | +/− | +/− | +/− | +/− | +/− | +/− |
| Polydactyly | − | − | − | − | + | − | + | − | + | − |
| Obesity | − | − | − | − | N/A | − | + | − | − | − |
| Inheritance | ARb | AR | ARc | AR | AR | AR | AR | AR | AR | AR |
Most individuals are intellectually impaired but occasionally normal intelligence is seen with mild forms of disease.
Most cases of Joubert syndrome (JS) are inherited in a recessive fashion except for the OFD1, which can be X-linked.
Most cases of Leber congenital amaurosis (LCA) are inherited in a recessive fashion except forthe IMPDH1 gene mutation, inherited dominantly, and the CRX gene, inherited in an X-linked fashion. +, presence or −, absence of clinical sign and symptoms; +/−, a feature that can be present or absent within the various ciliopathies. CORS, cerebellar–ocular–renal syndrome; SLS, Senior–Løken syndrome; MKS, Meckel–Gruber syndrome; DKA, Dekaban–Arima syndrome; BBS, Bardet–Biedl syndrome; COACH, cerebellar vermis hypo-/aplasia, oligophrenia, congenital ataxia, ocular coloboma, and hepatic fibrosis; OFD, orofacial digital syndrome; Cogan, Cogan-type congenital oculomotor apraxia; MTS, molartooth sign; AR, autosomal recessive; N/A, not applicable.
Joubert syndrome
Joubert syndrome is an autosomal recessive disorder with a predicted incidence of one out of 100 000 in the United States.4 Classic Joubert syndrome can be clinically recognized in infancy by features of hypotonia, nystagmus, oculomotor apraxia, developmental delay, and variable intellectual impairment with episodes of apnoea and hyperpnoea that are typically notable within the first several months of life and undergo spontaneous improvement. Retinopathy may be present and can occur at a later stage depending on the age at diagnosis (Fig. 1). Certain dysmorphic features have been described, and include hypertelorism, arched eyebrows, broad forehead, and unilateral or bilateral ptosis with wide-shaped mouth that has been considered secondary to facial hypotonia. Significant phenotypic variability has been observed even among family members, making a clinical diagnosis difficult.
Figure 1.

Genotype–phenotype correlates in Joubert syndrome (JS) and Joubert syndrome and related disorders. CORS, cerebellar-ocular-renal syndrome; SLS, Senior-Løken syndrome; LCA, Leber congenital amaurosis; NPHP, nephronophthisis. Genes with major effect are in bold, other genes contributing to each phenotype are listed as well.
Diagnosis is based on the findings of radiological imaging findings of the head. Axial views on computed tomography or MRI reveal an MTS, comprising cerebellar vermis hypoplasia or aplasia, deepened interpeduncular fossa, and elongated superior cerebellar peduncles (Fig. 2). Joubert syndrome in its pure form does not involve other organs. Rarely, cases of Joubert syndrome with other cerebral malformations have been described, such as polymicrogyria. Individuals with such abnormalities have been known to develop epilepsy secondary to evidence of cortical dysplasia. However, the term ‘Dandy–Walker variant’, previously used to encompass all hindbrain anomalies, has been abandoned in favour of more precise terminology.5 While a diagnosis of ‘Joubert syndrome’ is applied to those individuals who specifically meet diagnostic criteria in the absence of additional non-neurological features, a diagnosis of the broader category of Joubert syndrome and related disorders (JSRD) is applied to any individual who displays MTS irrespective of the additional non-neurological features.
Figure 2.

Magnetic resonance imaging showing a normal cerebellar vermis and superior cerebellar peduncles on (a) axial and (b) sagittal images. (c) and (d) show cerebellar vermis hypoplasia and thickened superior cerebellar peduncles perpendicular to the brainstem (arrow), consistent with the molar tooth sign.
A number of genes have been identified as contributing to Joubert syndrome. Notably, individuals with Joubert syndrome most often display mutations in AHI1, RPGRIP1L, and CC2D2A, each accounting for about 10% of cases of the classic form of disease, as well as cases with additional retinal involvement.6–9 Individuals with more complex forms of Joubert syndrome including renal or hepatic disease typically display mutations in other genes.
Leber congenital amaurosis
Leber congenital amaurosis (LCA) was first described by The-odore Leber in 1869 as early infantile blindness due to retinal dysplasia. All but one form of LCA is inherited in a recessive fashion with findings of severe dystrophy of the retina. Visual impairment is noted from a history of poor visual response and lack of tracking that becomes evident in the first year of life, with visual acuity rarely better than 20/400 and limited to counting fingers and detecting hand motions and bright lights. Clinically, photophobia, nystagmus, sluggish or near-absent pupillary responses, keratoconus, and high hyperopia from impaired emmetropization can be seen. Children affected exhibit the oculodigital sign characterized by poking, rubbing, and pressing of the eyes, resulting in deep-set eyes. Although the oculodigital sign has been seen in other disorders associated with severe visual impairment, it has been claimed to be nearly pathognomonic for LCA. Fundoscopic examination typically reveals only subtle retinal pigment epithelial granularity, but impaired or flat responses on electroretinography are diagnostic at this age. Later in childhood, dystrophic changes worsen and a pigmentary retinopathy similar to retinitis pigmentosa is frequently observed. LCA is a disorder of photoreceptors, with failed transport of rhodopsin, loss of photoreceptor outer segments, and ultimately death of photoreceptors.
Although diagnosis is established by clinical findings, molecular genetic testing is currently available for 12 genes associated with LCA. Genetic mutations in RPGRIP1 and CEP290 have been found in LCA, and, as these are the same genes that are mutated in individuals with JSRD, it is clear that these conditions are allelic (i.e. resulting from the same genetic causes). Because of this, it is important to evaluate children diagnosed with LCA for neurological symptoms, and to consider brain MRI to determine if they meet the criteria for JSRD.
Cerebellar–ocular–renal syndrome
Cerebellar–ocular–renal syndrome describes an autosomal recessive disorder in which hypoplastic cerebellar vermis with MTS is accompanied by notable ocular and renal involvement. The first connection that was made was that individuals previously diagnosed with Senior–Løken syndrome (consisting of LCA and renal dysplasia) also displayed the MTS.10 The term ‘cerebellar–ocular–renal syndrome’ was also expanded to include individuals with a previous diagnosis of Dekaban–Arima syndrome (consisting of ocular abnormalities and cystic kidneys)10 once it was recognized that the renal pathology and MTS was the same in Senior–Løken syndrome (consisting of ocular abnormalities and nephronophthisis) and Dekaban–Arima syndrome.11 The renal pathology in conditions is manifested as nephronophthisis, a fibrocystic renal transformation that is most notable at the corticomedullary junction and is the most common recessive form of cystic kidney disease. The genetic causes of cerebellar–ocular–renal syndrome are largely mutations in the genes CEP290, TMEM216, and NPHP1.12–16
Cogan-type congenital oculomotor apraxia
Cogan-type congenital oculomotor apraxia is an autosomal recessive form of congenital oculomotor apraxia characterized by defective horizontal voluntary eye movements with jerkiness. Some individuals also have cerebellar vermis hypoplasia, with evidence of MTS,17 and occasionally develop nephronophthisis. The NPHP1 homozygous deletion has been identified in some individuals with JSRD,18 suggesting that this condition is allelic with NPHP1. It is uncertain whether Cogan-type oculomotor apraxia can exist as an isolated entity. Rather, it seems likely that the oculomotor apraxia observed in these individuals is linked to a cerebellar or brainstem disorder and JSRD.
Bardet–Biedl syndrome
Bardet–Biedl syndrome (BBS) is an autosomal recessive disorder characterized primarily by postaxial polydactyly, retinal dystrophy, truncal obesity, male hypogonadotropic hypogonadism, female genitourinary malformation, intellectual impairment, and renal abnormalities, with potential development of renal failure. It is diagnosed clinically with either four primary features or three primary and two secondary features. Postaxial polydactyly can be present on either the hands or feet or all four limbs, with the extra digits typically occurring on the ulnar side of the arm or the fibular side of the foot. Rod–cone dystrophy with pigmentary changes is described as lack of normal visual acuity resulting from loss of central cones, loss of dark adaptation, and poor peripheral visual field due to loss of rods. Abnormal visual findings can be delayed, leading to a late diagnosis. Male hypogonadism is characterized by small volume testes, cryptorchidism, or a small penile shaft. Female genitourinary abnormalities in BBS include hypoplastic fallopian tubes, uterus, or ovaries, vaginal atresia, or vesicovaginal fistula. Renal abnormalities, present in about 80% of individuals, typically include nephronophthisis, but other renal anomalies have been described19 and can eventually lead to end-stage renal disease. Secondary features of BBS include congenital cardiac defects (atrial septal defects, ventricular septal defects, aortic stenosis, patent ductus arteriosis), hepatic fibrosis or cirrhosis with biliary tract abnormalities, Hirsch-sprung disease, brachydactyly or syndactyly, strabismus, cataracts, ataxia, and a wide degree of behavioural and developmental problems including speech delay. The cause of the cognitive impairment is not well understood, but may relate to cerebellar hypoplasia or other subtle structural anomalies.20,21
Fifteen genes are known to be associated with BBS: BBS1, BBS2, ARL6/BBS3, BBS4, BBS5, MKKS/BBS6, BBS7, TTC8/BBS8, B1/BBS9, BBS10, TRIM32/BBS11, BBS12, MKS1/BBS13, CEP290/BBS14, and SDCCAG8. Mutations in the BBS1 and BBS10 gene account for over half of all cases, whereas the other genes make minor contributions.22–24 Like the other ciliopathies described here, nearly all of the encoded proteins localize at or near the primary cilium, and mutations are associated with alterations in structure or function of the primary cilium. BBS and Meckel–Gruber syndrome (MKS) have been described as eliciting a phenotypic overlap, and an underlying cause of BBS has been found with mutations in the MKS-related genes MKS1 and MKS3 as well as in CEP290.25
Meckel–Gruber syndrome
Meckel–Gruber syndrome is an autosomal recessive disorder that is usually lethal and is characterized by the presence of occipital encephalocele, polycystic kidney disease, and post- and (occasionally) preaxial polydactyly. Associated abnormalities include orofacial clefting, genitourinary anomalies, cerebral malformations such as a Dandy–Walker malformation, hydrocephalus, microcephaly, pulmonary hypoplasia, and fibrosis of the liver. Genitourinary problems reported include cryptorchidism, urethral atresia, and incomplete development of the internal and external genitalia resulting in ambiguous genitalia (requiring chromosomal analysis for sex determination). Cases can be identified by the presence of elevated alpha-fetoprotein levels or by prenatal ultrasonography, which can confirm a diagnosis based upon the unique presentation of occipital encephalocele/hydrocephalus and polydactytly. Prenatal MRI can document the constellation of findings as well as facial and skeletal deformities in addition to the renal malformations.26 Nephrogenic abnormalities causing oligohydramnios result in hypoplasia of the lungs, which is a common complication leading to death.
Five genes and six different loci have been identified in MKS: MKS1/MKS1, MKS2/TMEM216, MKS3/TMEM67, MKS4/CEP290, MKS5/RPGRIP1L, and MKS6/CC2D2A, demonstrating genetic heterogeneity. Individuals with JSRD have been reported to have similar phenotypic features as those with MKS in addition to the MTS, and in fact there are some families in which one child is diagnosed with JSRD and another with MKS. Malta syndrome, described in 2004 in Maltese siblings,3 is now believed to be a variant of MKS and is characterized by the presence of MTS, occipital encephalocele with hydrocephalus, cortical renal cysts, retinal abnormalities similar to LCA, and coloboma. Further, many of the same genes that cause JSRD when mutated can also lead to MKS, although the individuals with MKS typically have more severe forms of mutations such as stop codons. Thus, the data suggest that more severe forms of mutations can lead to MKS whereas less severe forms can lead to JSRD.
Orofacial digital syndrome type VI (Varadi–Papp syndrome)
Orofacial digital syndrome type VI (OFD6) is an autosomal recessive disorder that was described in 1980 by Varadi et al.27 They reported seven children from a consanguineous Gypsy group with reduplicated big toes, central polydactyly with Y-shaped metacarpal bones, orofacial clefting, lingual nodule or lingual hamartoma, and intellectual impairment. Affected children with OFD6 commonly have central nervous system abnormalities that include semilobar holoprosencephaly, cerebellar vermis dysgenesis, corpus callosum dysgenesis, and dysplastic hypothalamus and pituitary gland. Associated abnormalities include cryptorchidism, micropenis, buccoalveolar frenula, congenital heart anomalies, growth retardation, and strabismus in addition to the reduplicated big toes and orofacial clefting (Fig. 3). The lingual nodules are apparent typically along the inferior aspect or the sides of the tongue (Fig. 4) and are pathologically hamartomatous in nature. The collective deformities can be detected prenatally by ultrasonography in the second trimester. The presence of the MTS in several individuals with OFD6 suggests that it represents a ciliopathy,3 and mutations in the TMEM216 gene in two individuals conforming to OFD6, a gene implicated in JSRD and MKS, suggests that these are allelic ciliopathy disorders.28
Figure 3.
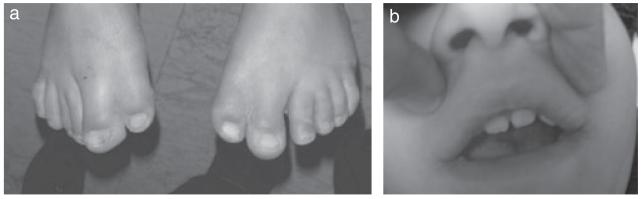
(a) Example of duplication of the great toes (preaxial polydactytly). (b) Notched upper lip seen in orofacial digital syndrome type VI. (Images courtesy of Dr. Maha Zaki, Clinical Genetics Department, Human Genetics and Genome Research Division, National Research Centre, Cairo, Egypt.)
Figure 4.

Lingual hamartoma and tongue tumors (arrows) seen in orofacial digital syndrome type VI.
Cerebellar vermis hypo-/aplasia, oligophrenia, congenital ataxia, ocular coloboma, and hepatic fibrosis (COACH) syndrome
The COACH acronym defines an autosomal recessive condition of cerebellar vermis hypoplasia or aplasia, oligophrenia, congenital ataxia, coloboma, and congenital hepatic fibrosis. The key feature of this syndrome is the malformation of the embryonic ductal plate, resulting in fibrosis of the liver. Phenotypic similarities between COACH and MKS led to a genetic evaluation of 14 individuals with COACH syndrome for the MKS3 mutation (TMEM67), which was found to be present in 57%.29 Doherty et al.30 also evaluated 23 individuals with COACH syndrome and found the MKS3 mutation in 83%. Another disorder, described as Gentile syndrome, which consists of cerebellar vermis dysplasia and MTS with hepatic fibrosis, is now considered to be a variant of COACH syndrome.
Clinical management of ciliopathies
The genotypic–phenotypic variability seen among individuals with Joubert syndrome and JSRD makes it very difficult to make an accurate diagnosis, especially when there may be a delay in the recognition of certain clinical findings and they may vary, even between siblings. Initial evaluation of any infant or child with hypotonia, ataxia, and abnormal eye movements should include a complete history and examination, with documentation of any dysmorphic features involving the face and skeletal deformities, any history of renal or liver abnormalities in the individual or family, and developmental delay. MRI of the brain and thorough review of sagittal and axial images of the brainstem for the MTS with cerebellar vermis hypoplasia should be considered. The presence of the MTS should initiate further clinical evaluation, with referral for ophthalmological examination, electroretinography, serum testing for liver and renal function, and ultrasound of the liver and kidney. If there is no abnormality on abdominal ultrasound, then it is recommended that testing be repeated every few years or so to detect delayed emergence of abnormalities that may show up as increased echogenicity or microcysts. It is important to note that, although the structural brain disorders are not progressive or degenerative, there can be progressive loss of renal, hepatic, or retinal function. Therefore, such proactive screening will aid earlier recognition of organ dysfunction, allowing for earlier treatment. Formal recommendations for screening can be found at the Joubert Syndrome Foundation website (http://www.joubertfoundation.com/).
Prenatal diagnosis as early as 12 weeks is reportedly feasible in the case of MKS and OFD6, with the ability to visualize the characteristic extracranial features such as polydactyly, renal cysts, and encephalocele.31 Genetic analysis can be undertaken prenatally in the case of families with a previously affected child and an identified genetic mutation. Fetal MRI is being used where available, and is more sensitive to detecting cerebral malformations. JSRDs are part of a spectrum of disorders of cilia with phenotypic–genotypic heterogeneity. There is a varying degree of intellectual impairment that ranges from mild to severe, but prognosis is largely dependent on the severity of involvement of the organ systems, in particular the retina, liver, and kidney. Unfortunately, there are currently no curative therapies for these genetic ciliopathic syndromes. Recent advances have resulted in the discovery of additional genetic mutations, which will allow us to study the outcome of these mutations and aid in the development of preventative and curative treatments.
ABBREVIATIONS
- BBS
Bardet–Biedl syndrome
- COACH
Cerebellar vermis hypo-/aplasia, oligophrenia, congenital ataxia, ocular coloboma, and hepatic fibrosis
- JSRD
Joubert syndrome-related disorders
- LCA
Leber congenital amaurosis
- MKS
Meckel–Gruber syndrome
- MTS
Molar tooth sign
- OFD6
Orofacial digital syndrome type VI
Footnotes
UK usage: learning disability.
REFERENCES
- 1.Joubert M, Eisenring JJ, Andermann F. Familial dysgenesis of the vermis: a syndrome of hyperventilation, abnormal eye movements and retardation. Neurology. 1968;18:302–3. [PubMed] [Google Scholar]
- 2.Maria BL, Quisling RG, Rosainz LC, et al. Molar tooth sign in Joubert syndrome: clinical, radiologic, and pathologic significance. J Child Neurol. 1999;14:368–76. doi: 10.1177/088307389901400605. [DOI] [PubMed] [Google Scholar]
- 3.Gleeson JG, Keeler LC, Parisi MA, et al. Molar tooth sign of the midbrain-hindbrain junction: occurrence in multiple distinct syndromes. Am J Med Genet A. 2004;125A:125–34. doi: 10.1002/ajmg.a.20437. discussion 117. [DOI] [PubMed] [Google Scholar]
- 4.Kroes HY, Fransen van de Putte DE, Ravesloot CJ, Lindhout D. The birth prevalence of Joubert syndrome: a population based study in the Netherlands. Eur J Hum Genet. 2007;15:68. [Google Scholar]
- 5.Barkovich AJ, Millen KJ, Dobyns WB. A developmental and genetic classification for midbrain-hindbrain malformations. Brain. 2009;132:3199–230. doi: 10.1093/brain/awp247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Valente EM, Brancati F, Silhavy JL, et al. AHI1 gene mutations cause specific forms of Joubert syndrome-related disorders. Ann Neurol. 2006;59:527–34. doi: 10.1002/ana.20749. [DOI] [PubMed] [Google Scholar]
- 7.Parisi MA, Doherty D, Eckert ML, et al. AHI1 mutations cause both retinal dystrophy and renal cystic disease in Joubert syndrome. J Med Genet. 2006;43:334–9. doi: 10.1136/jmg.2005.036608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brancati F, Travaglini L, Zablocka D, et al. RPGRIP1L mutations are mainly associated with the cerebello-renal phenotype of Joubert syndrome-related disorders. Clin Genet. 2008;74:164–70. doi: 10.1111/j.1399-0004.2008.01047.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gorden NT, Arts HH, Parisi MA, et al. CC2D2A is mutated in Joubert syndrome and interacts with the ciliopathy-associated basal body protein CEP290. Am J Hum Genet. 2008;83:559–71. doi: 10.1016/j.ajhg.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Satran D, Pierpont ME, Dobyns WB. Cerebello-oculo-renal syndromes including Arima, Senior-Loken and COACH syndromes: more than just variants of Joubert syndrome. Am J Med Genet. 1999;86:459–69. [PubMed] [Google Scholar]
- 11.Kumada S, Hayashi M, Arima K, et al. Renal disease in Arima syndrome is nephronophthisis as in other Joubert-related cerebello-oculo-renal syndromes. Am J Med Genet. 2004;131A:71–6. doi: 10.1002/ajmg.a.30294. [DOI] [PubMed] [Google Scholar]
- 12.Parisi MA, Bennett CL, Eckert ML, et al. The NPHP1 gene deletion associated with juvenile nephronophthisis is present in a subset of individuals with Joubert syndrome. Am J Hum Genet. 2004;75:82–91. doi: 10.1086/421846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brancati F, Barrano G, Silhavy JL, et al. CEP290 mutations are frequently identified in the oculo-renal form of Joubert syndrome-related disorders. Am J Hum Genet. 2007;81:104–13. doi: 10.1086/519026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Helou J, Otto EA, Attanasio M, et al. Mutation analysis of NPHP6/CEP290 in individuals with Joubert syndrome and Senior-Løken syndrome. J Med Genet. 2007;44:657–63. doi: 10.1136/jmg.2007.052027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sayer JA, Otto EA, O’Toole JF, et al. The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat Genet. 2006;38:674–81. doi: 10.1038/ng1786. [DOI] [PubMed] [Google Scholar]
- 16.Valente EM, Silhavy JL, Brancati F, et al. Mutations in CEP290, which encodes a centrosomal protein, cause pleiotropic forms of Joubert syndrome. Nat Genet. 2006;38:623–5. doi: 10.1038/ng1805. [DOI] [PubMed] [Google Scholar]
- 17.Whitsel EA, Castillo M, D’Cruz O. Cerebellar vermis and midbrain dysgenesis in oculomotor apraxia: MR findings. AJNR Am J Neuroradiol. 1995;16:831–4. [PMC free article] [PubMed] [Google Scholar]
- 18.Betz R, Rensing C, Otto E, et al. Children with ocular motor apraxia type Cogan carry deletions in the gene (NPHP1) for juvenile nephronophthisis. J Pediatr. 2000;136:828–31. [PubMed] [Google Scholar]
- 19.Imhoff O, Marion V, Stoetzel C, et al. Bardet-Biedl syndrome: a study of the renal and cardiovascular phenotypes in a French cohort. Clin J Am Soc Nephrol. 2010;6:22–9. doi: 10.2215/CJN.03320410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rooryck C, Pelras S, Chateil JF, et al. Bardet-Biedl syndrome and brain abnormalities. Neuropediatrics. 2007;38:5–9. doi: 10.1055/s-2007-981466. [DOI] [PubMed] [Google Scholar]
- 21.Baskin E, Kayiran SM, Oto S, Alehan F, Agildere AM, Saatçi U. Cerebellar vermis hypoplasia in a individual with Bardet-Biedl syndrome. J Child Neurol. 2002;17:385–7. doi: 10.1177/088307380201700514. [DOI] [PubMed] [Google Scholar]
- 22.Muller J, Stoetzel C, Vincent MC, et al. Identification of 28 novel mutations in the Bardet-Biedl syndrome genes: the burden of private mutations in an extensively heterogeneous disease. Hum Genet. 2010;127:583–93. doi: 10.1007/s00439-010-0804-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harville HM, Held S, Diaz-Font A, et al. Identification of 11 novel mutations in eight BBS genes by high-resolution homozygosity mapping. J Med Genet. 2010;47:262–7. doi: 10.1136/jmg.2009.071365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Otto EA, Ramaswami G, Janssen S, et al. Mutation analysis of 18 nephronophthisis associated ciliopathy disease genes using a DNA pooling and next generation sequencing strategy. J Med Genet. 2011;48:105–16. doi: 10.1136/jmg.2010.082552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leitch CC, Zaghloul NA, Davis EE, et al. Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat Genet. 2008;40:443–8. doi: 10.1038/ng.97. [DOI] [PubMed] [Google Scholar]
- 26.Hosny IA, Elghawabi HS. Ultrafast MRI of the fetus: an increasingly important tool in prenatal diagnosis of congenital anomalies. Magn Reson Imaging. 2010;28:1431–9. doi: 10.1016/j.mri.2010.06.024. [DOI] [PubMed] [Google Scholar]
- 27.Varadi V, Szabo L, Papp Z. Syndrome of polydactyly, cleft lip/palate or lingual lump, and psychomotor retardation in endogamic gypsies. J Med Genet. 1980;17:119–22. doi: 10.1136/jmg.17.2.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Valente EM, Logan CV, Mougou-Zerelli S, et al. Mutations in TMEM216 perturb ciliogenesis and cause Joubert, Meckel and related syndromes. Nat Genet. 2010;42:619–25. doi: 10.1038/ng.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brancati F, Dallapiccola B, Valente EM. Joubert Syndrome and related disorders. Orphanet J Rare Dis. 2010;5:20. doi: 10.1186/1750-1172-5-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Doherty D, Parisi MA, Finn LS, et al. Mutations in 3 genes (MKS3, CC2D2A and RPGRIP1L) cause COACH syndrome (Joubert syndrome with congenital hepatic fibrosis) J Med Genet. 2010;47:8–21. doi: 10.1136/jmg.2009.067249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saleem SN, Zaki MS. Role of MR imaging in prenatal diagnosis of pregnancies at risk for Joubert syndrome and related cerebellar disorders. AJNR Am J Neuroradiol. 2010;31:424–9. doi: 10.3174/ajnr.A1867. [DOI] [PMC free article] [PubMed] [Google Scholar]