

Abstract
Enantioenriched potassium β-trifluoroboratoamides have been synthesized via an asymmetric, copper-catalyzed 1,4-addition of tetrahydroxydiboron (BBA) and tetrakis(dimethylamino)diboron to α,β-unsaturated amides. These dibora reagents provide access to the desired organotrifluoroborates using effective and atom economical sources of boron. The copper-catalyzed β-boration is extended to α,β-unsaturated ketones and esters. Desired potassium organotrifluoroborates are synthesized with yields up to 92% and enantiomeric ratios up to 98:2. The enantioenriched potassium β-trifluoroboratoamides are successfully cross-coupled with an array of aryl and heteroaryl chlorides in high yield with complete stereochemical fidelity as the transmetalation proceeds through an SE2 mechanism via an open transition state.
Keywords: Catalytic, Asymmetric Borylation, Potassium Organotrifluoroborate, Stereospecific Suzuki-Miyaura Cross-Coupling, Tetrakis(dimethylamino)diboron, Tetrahydroxydiboron
Introduction
The synthesis of β-borylated carbonyl compounds via metal-catalyzed conjugate addition of a dibora species to an α,β-unsaturated carbonyl compound represents an important transformation in organic synthesis, as it provides access to a useful class of synthetic intermediates. Yun reported two examples of the asymmetric addition of bis(pinacolato)diboron (B2pin2) to α,β-unsaturated amides with ees up to 99% after oxidation of the pinacol boronate to the corresponding alcohol.[1] Additionally, two enantioenriched potassium β-trifluoroboratoamides[2] have been synthesized by a method based on the pioneering work of Yun,[1] utilizing a copper-catalyzed 1,4-borylation of α,β-unsaturated amides with B2pin2 (eq 1). Although extremely effective in providing the enantioenriched boronates, these methods suffer from the use of the atom inefficient B2pin2 as the borylating agent.
Several other transition metal complexes, such as those based on rhodium,[3] palladium,[4] platinum,[5] and nickel,[6] have been shown to mediate the conjugate addition of bis(pinacolato)diboron across α,β-unsaturated carbonyl compounds. Additionally Fernandez[7] and Hoveyda[8] have published asymmetric, metal-free additions of B2pin2, catalyzed by chiral phosphines or chiral N-heterocyclic carbenes, respectively. However, copper has been shown to be quite effective and is also the cheapest and least toxic of the transition metals[9] shown to facilitate this transformation.
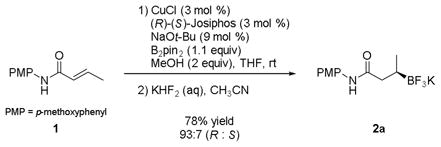 |
(1) |
Recently, tetrahydroxydiboron[10] (bisboronic acid or BBA) and tetrakis(dimethylamino)diboron[11] have been demonstrated to be more sustainable and more atom economical dibora species as compared to bis(pinacolato)diboron (Figure 1). Tetrakis(dimethylamino)diboron is synthesized in a three-step sequence starting from boron tribromide.[12] Addition of pinacol and HCl provides B2pin2, and reaction with aq. HCl affords BBA. Because B2(NMe2)4 is the direct synthetic precursor to B2pin2, the use of the latter is inherently wasteful because the addition of pinacol is completely unnecessary. If the desired product of a borylation reaction is a boronic acid (or an organotrifluoroborate), less than 10% of the mass of B2pin2 is incorporated into the final product because of the two equivalents of pinacol generated upon hydrolysis of the boronate ester. Aside from generating an exorbitant amount of waste in the form of pinacol, this diol is not an easy byproduct to remove from reaction mixtures.[13] Replacing the dibora reagent with BBA (3) or tetrakis(dimethylamino)diboron (4) allows the direct installation of a boronic acid, thus avoiding these problems. In the case of the conversion to the trifluoroborate, the byproducts are water or dimethylamine in place of pinacol, thus greatly facilitating isolation of the final product.
Figure 1.
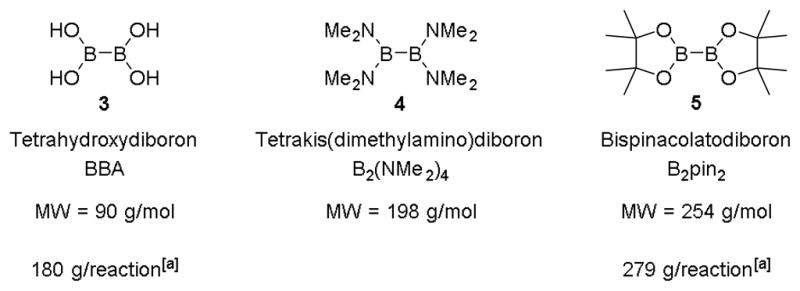
Structures and Atom Economy of Dibora Sources. [a]Calculations based on optimized conditions for a 1 mole reaction.
Even accounting for the two equivalents of BBA required for the optimized conjugate borylation using BBA, the latter is a much more atom economical dibora species than B2pin2, because on this basis a >50% reduction in the mass of the starting material can be achieved (Figure 1). Both BBA and tetrakis(dimethylamino)diboron are commercially available.
BBA and tetrakis(dimethylamino)diboron are capable of undergoing many of the same types of transformations as B2pin2, including Miyaura borylation.[10a],[10b],[11] BBA can also undergo conjugate additions to α,β-unsaturated carbonyl compounds in a racemic manner.[10c] The goal of the current project was to employ BBA and/or tetrakis(dimethylamino)diboron in the catalytic, asymmetric synthesis of potassium β-trifluoroboratoamides.
The synthesis of enantioenriched, air-stable secondary organometallic reagents has gained much attention over the past few years.[14] The synthesis of chiral, nonracemic organotrifluoroborates is particularly appealing owing to their favorable properties.[15] Because of their tetracoordinate nature, trifluoroborates are stable to both air and moisture, and thus they are capable of being stored on the bench indefinitely without decomposition. Trifluoroborates are less prone to protodeboronation during cross-coupling reactions than their boronic acid or boronate ester counterparts, which can require an excess of up to 1 equivalent.[16] Consequently, trifluoroborates can be used in near stoichiometric amounts in these important transformations.[15]
Recently, Matteson homologation chemistry was utilized to synthesize enantioenriched potassium 1-(benzyloxy)-3-phenylpropyltrifluoroborate in an enantiopure form.[17] The use of this material was demonstrated in its cross-coupling with aryl- and heteroaryl chlorides, with the reactions proceeding in high yield with complete stereochemical fidelity. The synthesis of related materials in an enantioenriched form is therefore appealing for the rapid installation of a stereogenic center via a cross-coupling.
Reported herein is the asymmetric synthesis of potassium β-trifluoroborato carbonyl compounds via a copper-catalyzed 1,4-addition of bisboronic acid and tetrakis(dimethylamino)diboron to α,β-unsaturated amides, esters, and ketones. To the best of our knowledge, this method represents the first asymmetric addition of bisboronic acid and tetrakis(dimethylamino)diboron. The potassium β-trifluoroboratoamides generated therefrom are successfully cross-coupled stereospecifically with aryl and heteroaryl chlorides. The paradigm of employing a hemilabile ligand to avoid β-hydride elimination is further expanded to include a wide array of amides, which provides an effective route to the direct installation of a stereocenter through cross-coupling.
Results and Discussion
Seeking to develop a synthesis of enantioenriched potassium β-trifluoroboratoamides utilizing BBA and/or tetrakis(dimethylamino)diboron, we hoped to utilize high throughput experimentation (HTE) to determine optimal reaction conditions.[18] (E)-N-(4-Methoxyphenyl)but-2-enamide was chosen as the model substrate (1) as it was previously shown to undergo the copper-catalyzed conjugate addition with B2pin2 to provide the corresponding organotrifluoroborate with an enantiomeric ratio of 93:7 (eq 1).[2]
One of the benefits in utilizing HTE in the development of synthetic methods is the minimal use of expensive chiral ligands. Performing the reaction on a 10 μmol scale allows the use of minute amounts of ligand over a range of reaction conditions.
The purpose of the initial screening was to determine if there was an ideal ligand for the desired transformation. Yun reported bidentate phosphine ligands that coordinate to a single copper as being the most effective for the conjugate borylation with B2pin2. For the asymmetric addition, chiral ligands whose backbone or chain length are similar to those effective in the racemic borylation (dppbz or dppp) provided the desired product in high yield. Specifically, (R,S)-Josiphos provided two enantioenriched products in high yield (81%) and enantiomeric ratio (up to 99:1).[1]
Therefore, 50 different chiral ligands, based largely on Yun’s platform, but also including several structurally diverse ligands, were screened under various reaction conditions with BBA as the borylating agent. Several copper sources were screened in this reaction, including CuCl, which was shown to be effective in the racemic synthesis of potassium β-trifluoroboratoamides.[10c] As a protic solvent is required to promote catalytic turnover as well as for esterification of BBA and tetrakis(dimethylamino)diboron, which are converted to the tetra(alkoxy)dibora species in situ,[10a] several alcohol solvents were tested. The final variable screened was the source and amount of alkoxide base. Base is required to form a [Cu]-OR complex, which then undergoes transmetalation with the dibora species to form the active [Cu]-B(OR)2 catalyst. In the case of the racemic addition of BBA, excess NaOt-Bu served to sequester the produced B(OR)3 in the form of NaB(OR)4.[10c]
Because the reaction generates an alkylboron species, this intermediate was converted to the corresponding alcohol via oxidation with NaBO3•4H2O to facilitate analysis (eq 2).
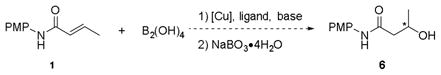 |
(2) |
The relative amounts of product and unreacted starting material were compared by addition of an internal standard followed by HPLC analysis of the microscale reactions. The ratios of product to internal standard (P/IS) and starting material to internal standard (SM/IS) can be directly compared for microscale reactions in one screen. If the same internal standard and HPLC conditions are used for future screens, the results can be correlated to determine the ideal reaction conditions.
Some of the results of the initial screen are displayed in Figure 2. The enantiomeric ratios were determined for those reactions with high P/IS ratios by subjecting those reactions to Supercritical Fluid Chromatographic (SFC) analysis. The P/IS ratios were normalized to the highest hit for ease of comparison. Only the top twelve ligands that provided an appreciable amount of desired product are shown; see Supporting Information for the screening results with all 50 ligands.
Figure 2.
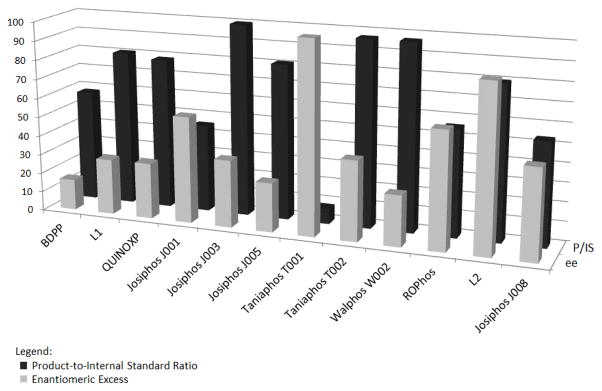
Graphical summary of Screen 1. Reaction conditions: 1.0 equiv of 1, 2.0 equiv of BBA, 0.05 equiv of CuCl, 0.05 equiv of ligand, 0.30 equiv of NaOt-Bu or KOMe, EtOH, rt, 24 h. L1 = 1,2-bis[(2S,5S)-2,5-diisopropylphospholano]ethane. L2 = (2R,5R)-1-{[(2R,5R)-2,5-diphenylpyrrolidin-1-yl]methylene}-2,5-diphenylpyrrolidinium tetrafluoroborate.
As shown in Figure 2, out of the 50 ligands tested in the reaction, the three ligands providing the highest enantiomeric excess were ROPhos,[19] Tania-phos T001,[20] and (2R,5R)-1-{[(2R,5R)-2,5-diphenylpyrrolidin-1-yl]methylene}-2,5-diphenylpyrrolidinium tetrafluoroborate (L2)[21] (Figure 3). With BBA as the borylating agent, (R,S)-Josiphos, which proved successful for the addition of B2pin2, was not effective in the borylation of α,β-unsaturated amides, and led to a trifluoroboratoamide with modest yield and enantiomeric ratio.
Figure 3.
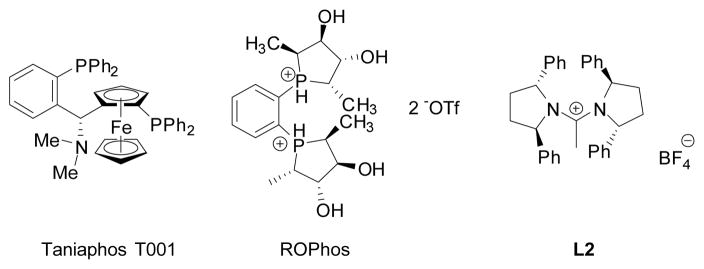
Structures of Taniaphos T001, ROPhos, and L2.
With ROPhos as the ligand, an enantiomeric ratio of 86:14 was obtained, whereas Taniaphos T001 provided an enantiomeric excess of >99:1. Further optimization with ROPhos did not lead to an enantiomeric excess higher than that shown here, and attempts at optimizing the conjugate addition with Taniaphos T001 as the ligand resulted in only 50% yield of the oxidized product 6.[22]
Figure 2 shows L2 is the only ligand that provided both high levels of enantiomeric induction as well as a high P/IS ratio. L2 is a carbene precursor, forming a carbene upon deprotonation, which then coordinates copper. Two structurally similar ligands provided low levels of enantioselectivity, and therefore L2 was chosen for further study.
Optimization with this ligand was completed with the aid of HTE and required only one additional screen, which employed CuCl as the copper source with L2 as the ligand in the presence of three different bases (KOMe, NaOt-Bu, LiOMe) and four different solvents (EtOH, MeOH, THF/EtOH, toluene/EtOH).
Based on the results outlined in Figure 4, it is evident that both KOMe and NaOt-Bu are superior bases to LiOMe. Also apparent is that the solvent system of 1:1 THF/EtOH provides the highest P/IS ratio, slightly greater than that of 1:1 toluene/EtOH.
Figure 4.

Graphical summary of Screen 2. Reaction conditions: 1.0 equiv of 1, 2.0 equiv of BBA, 0.05 equiv of CuCl, 0.05 equiv of L2, 1.0 or 2.0 equiv of base, rt, 24 h.
Although it seems as if several different systems provide a high P/IS ratio, the base and solvent system only marginally affect the enantioselectivity of the reaction.[23] SFC analysis of the top reactions yielded one set of conditions that provided both high P/IS ratio and a high enantiomeric ratio, with the (R) alcohol predominating. The conditions of CuCl as the copper source, NaOt-Bu as the base, and L2 as the ligand in a solvent system of 1:1 THF/EtOH resulted in complete conversion of the starting material with an enantiomeric ratio of 93:7. This enantiomeric ratio is the same as that reported using Yun’s conditions with bis(pinacolato)diboron as the borylation reagent (eq 1).
Having conditions in which there was complete conversion of the starting material, final optimization was conducted on a 0.5 mmol scale at the bench in order to synthesize the desired organotrifluoroborate product (Table 1). Despite screening with CuCl, the copper source was changed to the air-stable Cu(MeCN)4PF6 so that a glovebox would not be required to set up these reactions. It is important to note that changing the copper source did not have an effect on the yield or enantiomeric excess. Varying the concentration of the reaction also had little effect on the yield or the enantiomeric excess of this reaction (entries 1–3). Decreasing the equivalents of BBA in the reaction resulted in a modestly lower yields of the desired trifluoroborate (entries 4–5). Lowering the temperature to 0 °C did not improve the enantioselectivity of the reaction and provided the desired product in lower yield (entry 6). Therefore, the optimized conditions were established as Cu(MeCN)4PF6 as the copper source (5 mol %), L2 as the ligand (5 mol %), NaOt-Bu as the base (1 equiv) in a solvent system of THF/EtOH (1:1). These conditions provided the desired product in 90% yield with an enantiomeric ratio of 93:7 (R:S). The enantiomeric excess of these reactions was determined by oxidation of 2a to 6, followed by SFC analysis.
Table 1.
Final Optimization of the Asymmetric, Conjugate Addition of BBA.

| |||||
|---|---|---|---|---|---|
| entry | concentration | temperature | equiv of BBA | yield (%) | enantiomeric ratio[a] |
| 1 | 0.1 M | rt | 2 | 87 | 91:9 |
| 2 | 0.25 M | rt | 2 | 82 | 94:6 |
| 3 | 0.5 M | rt | 2 | 90 | 93:7 |
| 4 | 0.5 M | rt | 1.5 | 80 | 92:8 |
| 5 | 0.5 M | rt | 1.2 | 67 | 92:8 |
| 6 | 0.5 M | 0 °C | 2 | 77 | 93:7 |
Enantiomeric excess determined by oxidation of 2a to 6 followed by SFC analysis of 6
As the carbene-catalyzed, asymmetric conjugate addition of B2pin2 to α,β-unsaturated carbonyl compounds is known,[8] it was important to confirm that the current borylation was copper-catalyzed. Preforming the active complex by adding the copper salt, ligand, and base in THF before adding BBA and the amide in EtOH provided the same yield and enantiomeric ratio of product as when the complex was not preformed. Additionally, Cu(MeCN)4PF6 was determined to be necessary, as the reaction conducted in the absence of the copper salt did not result in product formation.
The optimized conditions were applied to an array of α,β-unsaturated amides, providing the desired enantioenriched β-trifluoroboratoamides in high yields and enantiomeric ratios (Table 2). Enantiomeric ratios were determined by oxidation and SFC analysis of the corresponding alcohol. Installation of a phenyl group on the terminal olefin resulted in a decrease in the enantiomeric excess in most cases (entries 13, 14); however, changing the group to a longer carbon chain provided high levels of enantiomeric excess (entries 2, 6). Secondary and tertiary amides subjected to the reaction conditions afforded the desired products. An N-cyclopropylamide provided the desired trifluoroborate with a yield of 51% and an enantiomeric ratio of 93:7 (entry 5). Heteroatoms, as appearing in the heterocyclic amide derived from morpholine, do not interfere with the catalysis, as the corresponding trifluoroborate can be synthesized in a yield of 84% (entry 8). Unsymmetrical tertiary amides can also be subjected to the borylation, obtaining the desired product in 71% yield (entry 10). The borylation was scaled to 5.5 mmol, with the catalyst loading cut in half to 2.5 mol %. The desired product was isolated in 85% yield with an enantiomeric ratio of 93:7 (entry 1). Lowering the equivalents of BBA on a 3 mmol scale resulted in a decreased yield of the β-trifluoroboratoamide (entry 4).
Table 2.
Scope of the Catalytic, Asymmetric Borylation of α,β-Unsaturated Amides with BBA.

| |
|---|---|
| entry | product[a] |
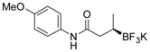
| 1 |
 2a 90%, 93:7 er (85%, 93:7 er)[b] |
| 2 |
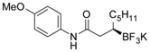 2b 82%, 97:3 er |
| 3 |
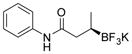 2c 80%, 92:8 er |
| 4 |
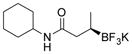 2d 82%, 95:5 er (71%, 1.5 equiv BBA)[c] |
| 5 |
2e 51%, 93:7 er |
| 6 |
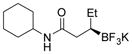 2f 86%, 98:2 er |
| 7 |
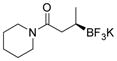 2g 82%, 92:8 er |
| 8 |
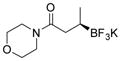 2h 84%, 97:3 er |
| 9 |
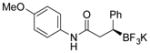 2i 92%, 96:4 er |
| 10 |
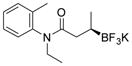 2j 71%, 76:24 er |
| 11 |
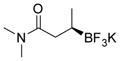 2k 86%, 70:30 er |
| 12 |
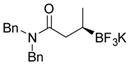 2l 80%, 66:34 er |
| 13 |
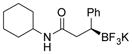 2m 77%, 72:28 er |
| 14 |
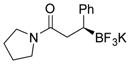 2n 85%, 78:22 er |
Enantiomeric excess was determined by oxidation of 2 followed by SFC analysis of the corresponding alcohol.
Reaction performed on a 5.5 mmol scale with 2.5 mol % Cu(MeCN)4PF6, 2.5 mol % L2.
Reaction performed on a 3 mmol scale.
The reaction conditions were extended to the β-boration of other α,β-unsaturated carbonyl compounds, including esters and ketones (Table 3). Different substitution patterns on the substrates, including cyclic and acyclic partners, proved to work well in the reaction, providing the corresponding enantioenriched trifluoroborates in high yield and enantiomeric ratios (as judged by oxidation and SFC analysis of the resulting alcohols). The presence of a phenyl ring on the terminal carbon of the olefin led to a drop of enantiomeric excess in these reactions (entry 4).
Table 3.
Scope of the Catalytic, Asymmetric Borylation of α,β-Unsaturated Ketones and Esters.
| |||
|---|---|---|---|
| entry | product | yield (%) | enantiomeric ratio[a] |
| 1 |
|
91 | 95:5 |
| 2 |
|
55 | 90:10 |
| 3 |
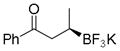 7c
7c
|
75 | 86:14 |
| 4 |
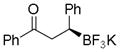 7d
7d
|
52 | 79:21 |
| 5 |
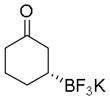 7e
7e
|
90 | 74:26 |
Enantiomeric excess was determined by oxidation of 7 followed by SFC analysis of the corresponding alcohol.
Tetrakis(dimethylamino)diboron was tested as the borylating agent under the reaction conditions developed for BBA. The desired β-trifluoroboratoamide was synthesized in 19% yield. However, simply changing the co-solvent from EtOH to MeOH resulted in an increased yield of 91% with an enantiomer ratio of 93:7. A similar solvent trend has been observed in the Miyaura borylation with BBA and tetrakis(dimethylamino)diboron, where improved yields were obtained with MeOH as the solvent.[10a],[10b],[11] Using these conditions, the yield obtained with BBA as the borylating agent is practically the same as that using B2(NMe2)4, whereas the enantiomeric ratio obtained is the same for both dibora sources. Therefore, these modified conditions were applied to an array of α,β-unsaturated amides (Table 4). For the most part, the use of tetrakis(dimethylamino)diboron led to similar or improved enantiomeric ratios but a lower yield of the isolated product as compared to analogous reactions with BBA.
Table 4.
Scope of the Catalytic, Asymmetric Borylation of α,β-Unsaturated Amides with Tetrakis(dimethylamino)diboron.
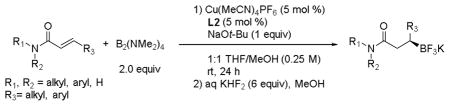
| |||
|---|---|---|---|
| entry | product | yield (%) | enantiomeric ratio[a] |
| 1 |
 2a
2a
|
91 | 93:7 |
| 2 |
 2d
2d
|
79 | 95:5 |
| 3 |
 2h
2h
|
68 | 92:8 |
| 4 |
 2j
2j
|
71 | 84:16 |
| 5 |
 2k
2k
|
72 | 70:30 |
| 6 |
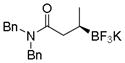 2l
2l
|
73 | 90:10 |
| 7 |
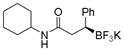 2m
2m
|
62 | 76:24 |
Enantiomeric excess was determined by oxidation of 2 followed by SFC analysis of the corresponding alcohol.
Because tetrakis(dimethylamino)diboron is converted to a tetra(alkoxy)diboron in situ during the reaction, up to four equivalents of dimethylamine are released per equivalent of the dibora species. In an attempt to determine whether an amine could be used as an additive to improve the enantiomeric ratio of some borylations, one equivalent of dimethylamine or N-methylaniline was added to the reaction with BBA as the borylating agent. In the case of dimethylamine, only starting amide was recovered after 24. Although the desired product was observed when N-methylaniline was added to the reaction with BBA, the same enantiomeric ratio was observed as when the amine was absent.
After synthesizing enantioenriched potassium β-trifluoroboratoamides, research focused on their Suzuki–Miyaura cross-coupling with aryl and heteroaryl chlorides. The Suzuki–Miyaura cross-coupling reaction[24] has become one of the most utilized reactions for the construction of C-C bonds in organic synthesis. The cross-coupling of potassium β-trifluoroboratoamides has increased importance in the synthesis of enantioenriched β-arylated amides owing to the complementarity of its implementation relative to existing methods. Thus, the main disconnections to synthesize β-arylated amides are summarized in Scheme 1.
Scheme 1.
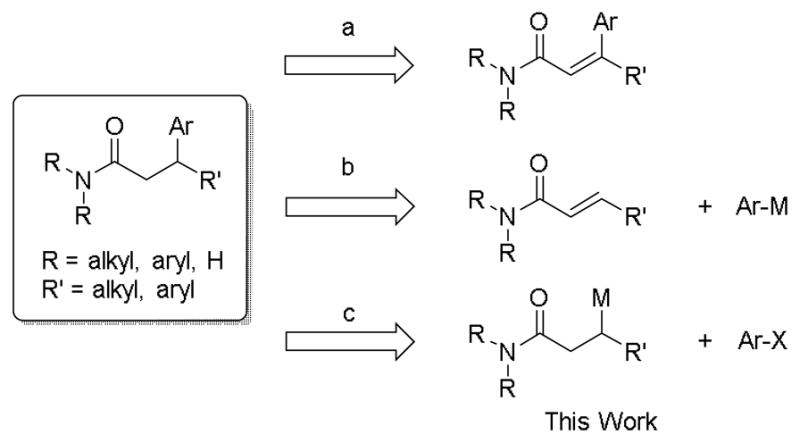
Disconnections To Access β-Arylated Amides
One common method to synthesize these types of substrates is a transition metal-catalyzed hydrogenation (route a).[25] In the case of β,β-diaryl substituted amides, an asymmetric reduction provides the desired product with an enantiomeric excess up to 74%.[26] Perhaps the most common method for the generation of β-arylated amides is a transition metal-catalyzed conjugate addition of an arylmetallic species to an α,β-unsaturated amide (route b).[27] A rhodium-catalyzed addition of arylboronic acids to lactams provides the desired β-arylated products in yields and enantiomeric excess up to 99%.[27b] However, the rhodium-catalyzed addition of phenylboronic acid to an acyclic amide provided the desired product in only 40% yield with an enantiomeric ratio of 90:10.[27c] As noted by this analysis, the most challenging aspect of asymmetric synthesis is the late stage conversion of a prochiral center to a stereocenter with high enantiomeric excess and yield. Utilizing a stereospecific cross-coupling as outlined herein moves the asymmetric step to a more reliable stage through the preparation of enantioenriched starting materials (route C). The assembly of enantioenriched nucleophilic reagents thus allows the direct installation of a stereogenic center into a molecule with complete stereochemical integrity. Furthermore, the polarity-inverted mode of coupling increases the diversity of products that can be accessed owing to the tremendous availability of readily accessible and highly functionalized aryl and heteroaryl halide partners.
The cross-coupling of potassium β- trifluoroboratoamides has been demonstrated to proceed in high yield with complete inversion of stereochemistry.[2] This stereochemical outcome, first demonstrated by Suginome,[28] is surmised to occur from the presence of a strongly coordinating carbonyl group, which can interact with the boron center to disfavor a four-centered transmetalation pathway. We sought to extend the scope of this type of cross-coupling by demonstrating that diverse, enantioenriched potassium β-trifluoroboratoamides, with various substitution patterns, cross-couple without loss of stereochemical integrity through inversion of configuration. Furthermore, there was a desire to extend the coupling to heteroaryl chlorides, which had not been examined previously.
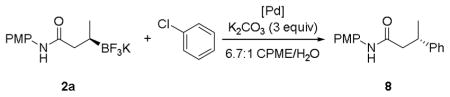
Reported conditions for the coupling of 2a with chlorobenzene incorporated 10 mol % of Pd(OAc)2 and 20 mol % of XPhos with 3 equiv of Cs2CO3 in a solvent system of CPME/H2O, resulting in an 82% yield of the desired product (eq 3).[2] With the report of Buchwald’s second and third generation precatalysts,[29] which rapidly form the active L1Pd(0) species by reductively eliminating carbazole after loss of HX (X = Cl or OMs), an attempt was made to lower the catalyst loading by changing the palladium source to one of these complexes.
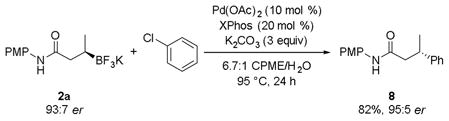 |
(3) |
Optimization with the Buchwald precatalysts is outlined in Table 5. Side products include the sum of materials resulting from protodeboronation of the potassium β-trifluoroborato-amide, homocoupling of the aryl chloride, and unidentifiable side products. The original conditions for this cross-coupling are shown in entry 7. Decreasing the catalyst load of Pd(OAc)2 resulted in incomplete consumption of the aryl halide (entry 6). Switching from Pd(OAc)2/XPhos to XPhos-Pd-G2, the XPhos derivative of Buchwald’s 2nd generation precatalyst, provided a superior ratio of product to side-product at the same temperature (entry 2). Cutting the catalyst load in half to 5 mol % resulted in a similar ratio (entry 1). No advantage was gained in utilizing the 3rd generation XPhos precatalyst (entry 4), and lowering the temperature to 80 °C resulted in incomplete conversion of the chlorobenzene after 24 h (entry 5).
Table 5.
Optimization of the Cross-Coupling of 2a with Chlorobenzene.
| |||
|---|---|---|---|
| entry | palladium source | temperature | SM : P : SPa |
| 1 | XPhos-Pd-G2 (5 mol %) | 100 °C | 0.0 : 1.0 : 0.4 |
| 2 | XPhos-Pd-G2 (10 mol %) | 100 °C | 0.0 : 1.0 : 0.3 |
| 3 | XPhos-Pd-G2 (5 mol %) | 80 °C | 0.3 : 1.0 : 1.1 |
| 4 | XPhos-Pd-G3 (5 mol %) | 100 °C | 0.6 : 1.0 : 0.7 |
| 5 | XPhos-Pd-G3 (5 mol %) | 80 °C | 2.9 : 1.0 : 1.9 |
| 6 | Pd(OAc)2/XPhos (5/10 mol %) | 100 °C | 2.0 : 1.0 : 0.0 |
| 7 | Pd(OAc)2/XPhos (10/20 mol %) | 100 °C | 0.0 : 1.0 : 0.2 |
Starting Material : Product : Side Products
Therefore, based on this optimization, the palladium loading was decreased to 5 mol % with XPhos-Pd-G2 as the palladium source. XPhos-Pd-G2 is easily synthesized from the aminobiphenyl palladium μ-Cl dimer in 90% yield (eq 4).[30]
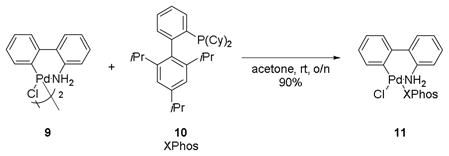 |
(4) |
With this new palladium source, the desired cross-coupled product was obtained in 85% yield without loss of stereochemical integrity (eq 5). These results are comparable to those reported previously (eq 3).[2]
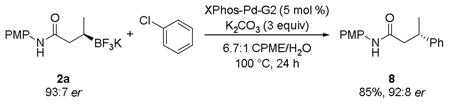 |
(5) |
The general reaction conditions were applied to a representative set of heteroaryl chlorides with potassium N-(4-methoxyphenyl)-3-(trifluoroborato)-butanamide 2a as the nucleophilic partner in the cross-coupling reaction (Table 6).
Table 6.
Cross-Coupling of 2a with Heteroaryl Chlorides and Diverse Aryl Electrophiles.
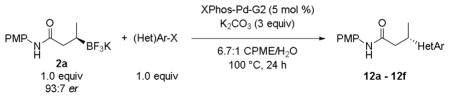
| ||||
|---|---|---|---|---|
| entry | product | yield (%) | enantiomeric ratio | |
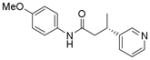
| 1 |
 12a
12a
|
X = Cl | 56 (52)[a] | 91:9 (95:5)[a] |
| 2 |
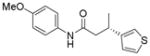 12b
12b
|
X = Cl | 60 | 94:6 |
| 3 |
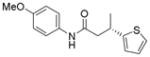 12c
12c
|
X = Cl | 53 | 95:5 |
| 4 |
 12d
12d
|
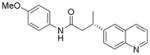
X = Cl | 58 | 92:8 |
| 5 |
 12e
12e
|
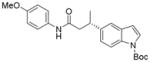
X = Cl | 43 | 92:8 |
| 6 |
 12f
12f
|
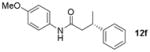
X = Cl | 85 | 92:8 |
| X = Br | 68 | 95:5 | ||
| X = l | (−)[b] | (−) | ||
| X = OMs | 73 | 92:8 | ||
| X = OTf | 78 | 90:10 | ||
| X = OTs | (−)[b] | (−) | ||
| X = OSO2NMe2 | (−)[b] | (−) | ||
Reaction performed on a 3.33 mmol scale with 2.5 mol % XPhos-Pd-G2.
No product observed.
When the electrophile was 3-chloropyridine, the coupled product was obtained in 56% yield (entry 1). Isomeric thiophenes provide the desired products with yields up to 60% (entries 2 and 3). A chloroquinoline was also an efficient electrophile for this reaction, affording the desired product in 56% yield (entry 4). An unprotected chloroindole did not result in product formation; however, an N-Boc protected chloroindole was successfully cross-coupled in 43% yield (entry 5). The cross-coupling was performed on a 1 g scale of 2a using 3-chloropyridine as a coupling partner with a catalyst loading of 2.5 mol %, and the desired product was obtained in 52% yield. Most importantly, however, is that all of the cross-couplings with heteroaryl chlorides proceed with virtually complete stereochemical integrity.
The compatibility of the cross-coupling was next examined for diverse electrophilic partners (Table 6, entry 6). When iodobenzene was employed as the electrophilic partner in the reaction, none of the desired product was obtained after 24 h. However, several other electrophiles tested provided the desired product in moderate to high yield. Chlorobenzene was previously shown to work efficiently (eq 4). When changing the electrophile to bromobenzene, a lower yield of 68% was obtained. Aryl bromides have been previously shown to be effective coupling partners with potassium β-trifluoroboratoamides.[2] These coupling conditions are also efficient for C-O activation as both phenyl mesylate and -triflate provided the cross-coupled product in good yield. Most importantly, these couplings proceed with virtually complete stereochemical fidelity regardless of the type of electrophile.
To expand the range of nucleophiles, the general reaction conditions were then applied to several different enantioenriched potassium β-trifluoroboratoamides with 1-chloro-4-fluorobenzene as the electrophile (Table 7). Secondary and tertiary amides both provide the desired product with yields between 81–89%. The unsymmetrical amide 2j was efficient in this reaction, as the cross-coupled product was obtained in 81% yield. As with the heteroaryl chlorides, these couplings proceed with little to no loss of enantioselectivity and are presumed to proceed with inversion of configuration based on previously established precedents.
Table 7.
Cross-Coupling of Various Potassium β-Trifluoroboratoamides.
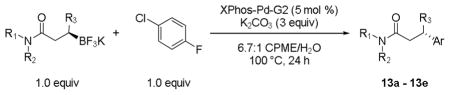
| ||||
|---|---|---|---|---|
| entry | SM er | product | yield (%) | enantiomeric ratio |
| 1 | 93:7 |
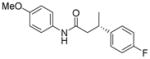 13a
13a
|
89 | 93:7 |
| 2 | 92:8 |
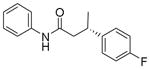 13b
13b
|
88 | 92:8 |
| 3 | 74:26 |
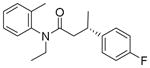 13c
13c
|
81 | 73:27 |
| 4 | 93:7 |
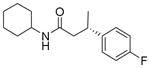 13d
13d
|
83 | 95:5 |
| 5 | 95:5 |
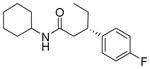 13e
13e
|
85 | 96:4 |
The inversion of configuration for the cross-coupling reaction has been attributed to the presence of the electron-donating group on the nucleophilic species. Chiral benzylstannanes,[31] silanes,[32] and α-(acylamino)benzyl boronates[28] have all been shown to couple with inversion of configuration. However, enantioenriched secondary benzylic pinacol boronates proceed through retention of configuration.[33] As reported by Suginome[28] and then by our group,[2] the stereochemical–determining step in this cross-coupling reaction is the transmetalation, which does not occur through the traditional four centered transition structure.[34] Because the carbonyl of the amide can coordinate to the intermediate boronate species after hydrolysis of the trifluoroborate, the transmetalation proceeds through an SE2 mechanism via an open transition structure to provide the product with inversion of configuration (Scheme 2). Borate substrates that proceed with inversion of configuration through SE2-type reaction pathways have been previously revealed in the literature.[35]
Scheme 2.
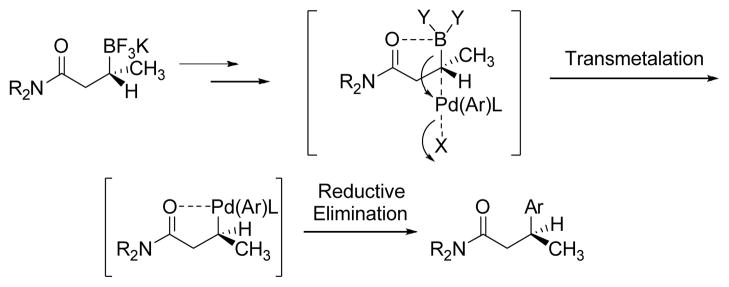
Rationale for the Sense of Asymmetry Observed in the Cross-Coupling Reaction to Access β-Arylated Amides.
Suginome reported that the addition of a Lewis acid, such as Zr(OiPr)4•iPrOH, changes the stereochemistry of the reaction. Thus, with enantioenriched α-(acylamino)benzyl boronate esters the carbonyl selectively binds to the Lewis acid, resulting in coupling that proceeds with retention of configuration.[28] To determine if Zr(OiPr)4•iPrOH was a suitable Lewis acid to invert the stereochemistry in the present study, the cross-coupling of a potassium β-trifluoroboratoamide with chlorobenzene was performed with Zr(OiPr)4•iPrOH as an additive (eq 6). Unfortunately, the desired crosscoupled product was not formed under these conditions. The reaction showed consumption of the aryl halide; however, an array of side products formed, including some β-hydride elimination product.
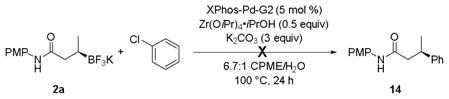 |
(6) |
Conclusion
The synthesis of enantioenriched β-trifluoroborato carbonyl compounds was developed utilizing an asymmetric copper-catalyzed conjugate addition of bisboronic acid and tetrakis(dimethylamino)diboron. The enantioenriched β-trifluoroboratoamides were successfully cross-coupled with 1-chloro-4-fluorobenzene in high yield, and potassium N-(4-methoxyphenyl)-3-(trifluoroborato)butanamide was successfully coupled to an array of heteroaryl chlorides in good yield. The carbonyl group on the amide contributes to the stereochemical outcome of the coupling reaction. The cross-coupling proceeds with complete inversion of stereochemistry, as the carbonyl group on the nucleophilic species coordinates the boronate intermediate, allowing the transmetalation to proceed through an SE2 mechanism.
Experimental Section
General Considerations
All starting materials, reagents, and catalysts were used as received. THF, EtOH, MeOH and CPME were distilled from sodium/benzophenone prior to use. Both solvents and H2O was degassed prior to use. Tetrakis(dimethylamino)diboron was distilled prior to use and stored under Ar. Standard benchtop techniques were employed for handling air–sensitive reagents. Melting points (°C) are uncorrected. 19F NMR chemical shifts were referenced to external CFCl3 (0.0 ppm). 11B NMR spectra at 128.4 MHz were obtained on a spectrometer equipped with the appropriate decoupling accessories. All 11B NMR chemical shifts were referenced to external BF3·OEt2 (0.0 ppm) with a negative sign indicating an upfield shift. Determination of the absolute configurations was based on our previous work.[2]
General Procedure for the Preparation of Enantioenriched Potassium β-Trifluoroboratoamides with BBA
An oven dried microwave vial with stir bar was charged with Cu(MeCN)4PF6 (0.0093 g, 0.025 mmol, 5 mol %), NaOt-Bu (0.0480 g, 0.5 mmol, 1 equiv), (2R, 5R)-1{[(2R, 5R)-2,5-diphenylpyrrolidin-1-yl]methylene}2,5-diphenylpyrrolidiniumtetrafluoroborate (0.0136 g, 0.025 mmol, 5 mol %), (HO)2B-B(OH)2 (0.0896 g, 1 mmol, 2 equiv), and amide (0.5 mmol, 1 equiv). The vial was sealed with a cap lined with a disposable Teflon septum, evacuated under vacuum and purged with Ar (three times). Anhyd THF (0.5 mL) was added under Ar followed by anhyd EtOH (0.5 mL), and the reaction mixture was stirred at rt for 24 h. After cooling to 0 °C, MeOH (1 mL) and sat. aq KHF2 (3 mmol, 6 equiv) was added dropwise. The reaction mixture was stirred at rt until deemed complete by 11B NMR (about 10 min), concentrated, and placed on the high vacuum overnight. The crude product was isolated by filtration with hot acetone (3 × 10 mL), then concentrated to a minimal amount (~2 mL), and precipitating with Et2O (20 mL).
Potassium (R)-N-(4-Methoxyphenyl)-3-(trifluoroborato)butanamide (2a).[2]
According to the general procedure using (E)-N-(4-methoxyphenyl)but-2-enamide (0.0955 g, 0.5 mmol), the product was obtained as a white crystalline solid in 90% yield (135 mg). mp =168–170 °C. 1H NMR (360 MHz, acetone-d6) δ 8.82 (br s, 1H), 7.56 (d, J = 8.7 Hz, 2H), 6.80 (d, J = 8.8 Hz, 2H), 3.73 (s, 3H), 2.26–2.42 (m, 1H), 1.99–2.03 (m, 1H), 0.82 (m, 4H). 13C NMR (125.8 MHz, acetone-d6): δ 174.2, 155.2, 133.2, 120.6, 113.4, 54.6, 41.8, 15.5. 19F NMR (338.8 MHz, acetone-d6): δ −147.17. 11B NMR (128.4 MHz, acetone-d6) δ 5.25. [α]D20= +10.8 (c = 0.34, MeOH).
Potassium (R)-N-(4-Methoxyphenyl)-3-(trifluoroborato)methyloctanamide (2b)
According to the general procedure using (E)-N-(4-methoxyphenyl)oct-2-enamide (0.1235 g, 0.5 mmol), the product was obtained as a white crystalline solid in 82% yield (146 mg). mp = 224–226 °C. 1H NMR (360 MHz, DMSO-d6) δ 9.31 (s, 1H), 7.47 (d, J = 8.6 Hz, 2H), 6.80 (d, J = 8.5 Hz, 2H), 3.68 (s, 3H), 2.19 (dd, J = 14.0, 4.8 Hz, 1H), 1.90 (dd, J = 14.0, 9.8 Hz, 1H), 1.00–1.31 (m, 8H), 0.79 (t, J = 7.0 Hz, 3H), 0.62 (brs, 1H). 13C NMR (90.5 MHz, DMSO-d6) δ 174.8, 154.9, 133.7, 120.8, 114.0, 55.5, 32.9, 31.8, 28.83, 22.7, 14.5. 19F NMR (338.8 MHz, DMSO-d6): δ −141.73. 11B NMR (128.4 MHz, DMSO-d6) δ 4.92. IR (neat) 3290, 2953, 2920, 2851, 1650, 1523, 1409, 1247, 1080, 980, 825, 657, 636 cm−1. HRMS (ESI) calcd. for C16H24BFNO3 [M-(KF2)+(CH2O)]− 308.1833, found 308.1833. [α]D20= −3.5 (c = 0.2, MeOH).
Potassium (R)-N-Phenyl-3-(trifluoroborato)butanamide (2c)
According to the general procedure using (E)-N-phenylbut-2-enamide (0.0805 g, 0.5 mmol), the product was obtained as a white crystalline solid in 80% yield (108 mg). mp = 135–138 °C. 1H NMR (300 MHz, acetone-d6) δ 8.99 (s, 1H), 7.76-7.64 (m, 2H), 7.24 (d, J = 8.5 Hz, 2H), 6.90–7.05 (m, 1H), 2.46 (dd, J = 13.8, 5.1 Hz, 1H), 2.09–2.17 (m, 1H), 0.83–0.95 (m, 4H). 13C NMR (75.4 MHz, acetone-d6) δ 174.6, 139.9, 128.2, 122.4, 119.1, 41.9, 15.43. 19F NMR (282.4 MHz, acetone-d6): δ −147.18 11B NMR (128.4 MHz, acetone-d6) δ 5.74. IR (Neat) 3290, 2949, 2876, 1639, 1599, 1513, 1441, 1308, 1244, 1059, 1010, 907, 748, 690, 656 cm−1. HRMS (ESI) calcd. for C11H14BFNO2 [M-(KF2)+(CH2O)]− 222.1102, found 222.1102. [α]D20= +18.8 (c = 0.26, MeOH).
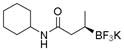
Potassium (R)-N-Cyclohexyl-3-(trifluoroborato)butanamide (2d).[2]
According to the general procedure using (E)-N-cyclohexylbut-2-enamide (0.0836 g, 0.5 mmol), the product was obtained as a white crystalline solid in 82% yield (113 mg). mp = 213–215 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.19 (d, J = 8.0 Hz, 1H), 3.37–3.58 (m, 1H), 1.99 (dd, J = 13.7, 3.8 Hz, 1H), 1.47–1.72 (m, 6H), 1.16 (m, 5H), 0.43–0.65 (m, 4H). 13C NMR (75.4 MHz, DMSO-d6) δ 174.5, 47.3, 40.7, 33.0, 25.6, 25.0, 15.9. 19F NMR (282.4 MHz, DMSO-d6) δ −128.4. 11B NMR (128.4 MHz, DMSO-d6) δ 5.25. [α]D20= −8.0 (c = 0.1, MeOH).
Potassium (R)-N-Cyclopropyl-3-(trifluoroborato)butanamide (2e).[10c]
According to the general procedure using (E)-N-cyclopropylbut-2-enamide (0.0.625 g, 0.5 mmol), the product was obtained as a white crystalline solid in 51% yield (60 mg). mp =165–167 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.42 (d, J = 5.0 Hz, 1H), 2.53–2.56 (m, 1H), 1.95–2.07 (m, 1H), 1.53–1.66 (m, 1H), 0.55–0.65 (m, 4H), 0.50–0.56 (m, 2H), 0.33 (s, 2H). 13C NMR (125.8 MHz, DMSO-d6) δ 176.9, 22.4, 16.0, 6.1, 5.9. 19F NMR (470.8 MHz, DMSO-d6): δ −144.97. 11B NMR (128.4 MHz, DMSO-d6) δ 5.76. [α]D20= +19.0 (c = 0.2, MeOH).
Potassium (R)-N-Cyclohexyl-3-(trifluoroborato)pentanamide (2f).[10c]
According to the general procedure using (E)-N-cyclohexylpent-3-enamide (0.0906 g, 0.5 mmol), the product was obtained as a white crystalline solid in 86% yield (124 mg). mp = 237–239 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.13 (d, J = 7.9 Hz, 1H), 3.47 (m, 1H), 1.96 (dd, J = 14.1, 4.6 Hz, 1H), 1.46–1.74 (m, 6H), 0.97–1.28 (m, 7H), 0.75 (t, J = 7.4 Hz, 3H), 0.45 (br s, 1H). 13C NMR (75.4 MHz, DMSO-d6) δ 174.8, 47.3, 33.1, 32.9, 25.8, 25.1, 24.0, 14.3. 19F NMR (338.8 MHz, DMSOd6): δ −141.10. 11B NMR (128.4 MHz, DMSO-d6) δ 4.94. [α]D20= −14.6 (c = 0.13, MeOH).
Potassium 1-(Piperidin-1-yl)-3-(trifluoroborato)butan-1-one (2g).[2]
According to the general procedure for the racemic preparation using (E)-1-(piperidin-1-yl)but-2-en-1-one (0.0765 g, 0.5 mmol), the product was obtained as a light yellow amorphous solid in 82% yield (107 mg). 1H NMR (500 MHz, acetone-d6) δ 3.33–3.58 (m, 4H), 2.22–2.29 (m, 1H), 2.24–210 (m, 1H), 1.43–1.63 (m, 6H), 0.76–0.79 (m, 3H), 0.71 (br s, 1H). 13C NMR (125.8 MHz, acetone-d6) δ 173.7, 46.5, 41.7, 38.4, 26.3, 25.4, 24.4, 16.2. 19F NMR (470.8 MHz, acetone-d6) δ −147.68. 11B NMR (128.4 MHz, acetone-d6) δ 5.46. [α]D20= −13.0 (c = 0.1, MeOH).
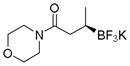
Potassium (R)-1-Morpholino-3-(trifluoroborato)butan-1-one (2h).[10c]
According to the general procedure for the racemic preparation using (E)-1-morpholinobut-2-en-1-one (0.0775 g, 0.5 mmol), the product was obtained as a white crystalline solid in 84% yield (111 mg). mp = 175–177 °C. 1H NMR (500 MHz, DMSO-d6) δ 3.46–4.00 (m, 4H), 3.27–3.46 (m, 4H), 2.29 (d, J = 13.7 Hz, 1H), 1.66–1.86 (m, 1H), 0.66 (d, J = 6.9 Hz, 3H), 0.45 (brs, 1H). 13C NMR (125.8 MHz, DMSO-d6) δ 174.2, 66.6, 46.3, 41.5, 37.3, 16.3. 19F NMR (470.8 MHz, DMSO-d6) δ − 145.78. 11B NMR (128.4 MHz, acetone-d6) δ 4.25. [α]D20= +2.9 (c = 0.34, MeOH).
Potassium (S)-N-(4-Methoxyphenyl)-3-phenyl-3-(trifluoroborato)propanamide (2i)
According to the general procedure using N-(4-methoxyphenyl)cinnamide (0.1265 g, 0.5 mmol), the product was obtained as a white crystalline solid in 92% yield (166 mg). mp = 176–179 °C. 1H NMR (300 MHz, acetone-d6) δ8.66 (s, 1H), 7.45 (d, J = 8.6 Hz, 2H), 7.26 (d, J = 7.6 Hz, 2H), 7.10 (t, J = 7.5 Hz, 2H), 6.89–6.98 (m, 1H), 6.77 (d, J = 8.5 Hz, 2H), 3.73 (s, 3H), 2.61–2.84 (m, 2H), 2.36–2.39 (m, 1H). 13C NMR (75.4 MHz, acetone-d6) δ 173.2, 155.3, 148.9, 133.0, 128.1, 127.0, 122.9, 120.6, 113.3, 54.6, 40.2. 19F NMR (282.4 MHz, DMSO-d6) δ − 144.72. 11B NMR (128.4 MHz, DMSO-d6) δ 5.46. IR (neat) 3367, 3342, 1658, 1641, 1515, 1241, 980, 824, 703, 659, 640 cm−1. HRMS (ESI) calcd. for C16H16BF3NO2 [M]− 322.1222, found 322.1223. [α]D20= +21.0 (c = 0.2, MeOH).
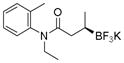
Potassium N-Ethyl-N-(o-tolyl)-3-(trifluoroborato)butanamide (2j).[2]
According to the general procedure for the racemic preparation using (E)-N-ethyl-N-(o-tolyl)but-2-enamide (0.1015 g, 0.5 mmol), the product was obtained as a white crystalline solid in 71% yield (110 mg). mp = 228–230 °C. 1H NMR (300 MHz, asterisk denotes minor rotamer peaks, DMSO-d6) δ 7.31–7.33 (m, 1H), 7.16–7.29 (m, 2H), 7.01–7.07 (m, 1H), 3.88–3.99 (m, 1H), 3.11–3.23* (m, 1H), 2.96–3.01 (m, 1H), 2.08–2.14 (m, 3H), 1.72–1.91 (m, 1H), 1.48* (dd, J = 14.6, 9.8 Hz, 1H), 1.32 (dd, J = 15.7, 10.3 Hz, 1H), 0.96–1.08 (m, 3H), 0.56–0.77 (m, 4H). 13C NMR (75.4 MHz, asterisk denotes rotamer peaks, DMSO-d6) δ 174.6, 174.2*, 142.1, 141.9*, 136.1, 135.6*, 131.4, 131.3*, 129.9, 129.8*, 127.9, 127.8*, 127.1, 42.6, 42.2*, 38.4, 37.1*, 17.5, 16.5, 16.3*, 13.4, 13.2*. 19F NMR (470.8 MHz, DMSO-d6) δ − 147.65. 11B NMR (128.4 MHz, DMSO-d6) δ 4.97. [α]D20= −8.1 (c = 0.16, MeOH).
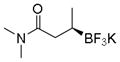
Potassium (R)-N,N-Dimethyl-3-(trifluoroborato)butanamide (2k).[10c]
According to the general procedure for the racemic preparation using (E)-N,N-dimethylbut-2-enamide (0.0565 g, 0.5 mmol), the product was obtained as a yellow amorphous solid in 86% yield (95 mg). 1H NMR (500 MHz, acetone-d6) δ 3.00 (s, 3H), 2.83 (s, 3H), 2.24–2.31 (m, 1H), 2.05 (s, 1H), 0.79 (m, 4H). 13C NMR (125.8 MHz, acetone-d6) δ 175.9, 37.6, 37.0, 34.4, 15.9. 19F NMR (470.8 MHz, acetone-d6) δ − 147.34. 11B NMR (128.4 MHz, DMSO-d6) δ 5.42. [α]D20= −5.6 (c = 0.34, MeOH).
Potassium (R)-N,N-Dibenzyl-3-(trifluoroborato)butanamide (2l).[2]
According to the general procedure for the racemic preparation using (E)-N,N-dibenzylbut-2-enamide (0.1865 g, 0.5 mmol), the product was obtained as a white crystalline solid in 80% yield (149 mg). mp = 236–238 °C. 1H NMR (360 MHz, DMSO-d6) δ 7.09–7.44 (m, 10H), 4.67 (d, J = 15.2 Hz, 2H), 4.59 (d, J = 17.1 Hz, 2H), 4.31 (d, J = 17.0 Hz, 1H), 4.18 (d, J = 15.1 Hz, 1H), 2.40 (dd, J = 13.9, 3.1 Hz, 1H), 1.80 (dd, J = 13.9, 11.1 Hz, 1H), 0.72 (d, J = 6.9 Hz, 3H), 0.59 (br s, 1H). 13C NMR (90.5 MHz, DMSO-d6) δ 176.1, 138.2, 129.0, 128.7, 127.8, 127.5, 127.2, 126.8, 126.4, 50.1, 47.5, 37.6, 16.5. 19F NMR (338.8 MHz, acetone-d6) δ − 148.19. 11B NMR (128.4 MHz, acetone-d6) δ 5.46. [α]D20= −7.0 (c = 0.1, MeOH).
Potassium (S)-N-Cyclohexyl-3-phenyl-3-(trifluoroborato)propanamide (2m).[10c]
According to the general procedure for the racemic preparation using N-cyclohexylcinnamide (0.1145 g, 0.5 mmol), the product was obtained as a white crystalline solid in 77% yield (130 mg). mp = 168–170 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.10–7.00 (m, 4H), 6.96 (d, J = 8.2 Hz, 1H), 6.87 (t, J = 6.9 Hz, 1H), 3.28–3.36 (m, 1H), 2.27–2.36 (m, 2H), 1.99–2.02 (m, 1H), 1.41–1.62 (m, 4H), 1.31–1.41 (m, 1H), 0.85–1.18 (m, 5H). 13C NMR (125.8 MHz, DMSO-d6) δ 173.5, 149.4, 128.5, 126.9, 122.7, 47.1, 39.3, 38.7, 32.7, 25.6, 24.8, 24.8. 19F NMR (470.8 MHz, DMSO-d6) δ − 142.19. 11B NMR (128.4 MHz, DMSO-d6) δ 5.22. [α]D20= +5.0 (c = 0.2, MeOH).
Potassium (S)-3-Phenyl-1-(pyrrolidin-1-yl)-3-(trifluoroborato)propan-1-one (2n).[2]
According to the general procedure for the racemic preparation using (E)-3-phenyl-1-(pyrrolidin-1-yl)prop-2-en-1-one (0.1545 g, 0.5 mmol), the product was obtained as a white amorphous solid in 85% yield (131 mg). 1H NMR (500 MHz, acetoned6) δ 7.20 (d, J = 7.0 Hz, 2H), 7.08 (t, J = 6.7 Hz, 2H), 6.93 (t, J = 8.1 Hz, 1H), 3.39 (s, 1H), 3.23–3.34 (m, 3H), 2.63 (dd, J = 13.2, 7.2 Hz, 1H), 2.45 (dd, J = 14.0, 7.5 Hz, 1H), 2.21 (s, 1H), 1.70–1.82 (m, 4H). 13C NMR (125.8 MHz, acetone-d6) δ 173.3, 149.6, 128.3, 126.9, 122.9, 46.1, 45.0, 38.2, 25.7, 23.9. 19F NMR (470.8 MHz, acetone-d6) δ − 144.31. 11B NMR (128.4 MHz, acetone-d6) δ 5.14. [α]D20= +37.1 (c = 0.1, MeOH).
Potassium Ethyl (R)-3-(Trifluoroborato)butanoate (7a).[10c]
According to the general procedure using ethyl (E)-but-2-enoate (0.0720 g, 0.5 mmol), the product was obtained as a white crystalline solid in 91% yield (100 mg). mp = 128–130 °C. 1H NMR (500 MHz, DMSO-d6) δ 4.48 (q, J = 7.1 Hz, 2H), 2.71–2.80 (m, 1H), 2.31–2.42 (m, 1H), 1.63 (t, J = 7.1 Hz, 3H), 1.17–1.32 (m, 4H). 13C NMR (125.8 MHz, acetone-d6) δ 176.2, 59.1, 38.7, 15.57, 14.1. 19F NMR (470.8 MHz, acetone-d6) δ −148.24. 11B NMR (128.4 MHz, acetone-d6) δ 5.96. [α]D20 = +4.0 (c = 0.25, MeOH).
Potassium Ethyl (R)-3-(Trifluoroborato)octanoate (7b)
According to the general procedure using ethyl (E)-oct-2-enoate (0.0851 g, 0.5 mmol), the product was obtained as a white crystalline solid in 55% yield (77 mg). mp = 220–222 °C. 1H NMR (500 MHz, acetone-d6) δ 4.01 (q, J = 7.0 Hz, 2H), 2.23 (dd, J = 13.9, 6.2 Hz, 1H), 2.00 (dd, J = 13.9, 8.9 Hz, 1H), 1.07–1.45 (m, 11H), 0.85 (t, J = 7.1 Hz, 3H), 0.75 (br s, 1H). 13C NMR (125.8 MHz, acetone-d6) δ 176.3, 58.5, 36.7, 32.6, 31.4, 22.5, 13.6, 13.5. 19F NMR (470.8 MHz, acetone-d6) δ −145.99. 11B NMR (128.4 MHz, acetone-d6) δ 5.79. IR (neat) 2949, 2924, 2851, 2356, 2332, 1717, 1467, 1445, 1368, 1249, 1217, 1126, 1073, 996, 937, 643 cm−1. HRMS (ESI) calcd. For C11H22BF2O3 [M-(KF)+(MeO)]− 251.1630, found 251.1631. [α]D20= +5.7 (c = 0.2, MeOH).
Potassium (R)-3-Trifluoroborato-1-phenylbutan-1-one (7c)
According to the general procedure using (E)-1-phenylbut-2-en-1-one (0.0730 g, 0.5 mmol), the product was obtained as a white crystalline solid in 75% yield (95 mg). mp =110–112 °C. 1H NMR (300 MHz, acetone-d6) δ 8.00 (d, J = 7.5 Hz, 2H), 7.38–7.3861 (m, 3H), 3.13 (m, 1H), 2.56 (m, 1H), 1.07–0.92 (br s, 1H), 0.83 (d, J = 6.9 Hz, 3H). 13C NMR (75.4 MHz, acetone-d6) δ 203.3, 137.8, 131.9, 128.1, 128.1, 42.9, 15.2. 19F NMR (282.4 MHz, acetone-d6) δ −148.08. 11B NMR (128.4 MHz, acetone-d6) δ 5.03. IR (neat) 2937, 2868, 1667, 1597, 1446, 1306, 1278, 1073, 1014, 924, 893, 690, 645 cm−1. HRMS (ESI) calcd. For C10H11BF3O [M-K]− 251.0855, found 251.0934. [α]D20= +61.0 (c = 0.1, MeOH).
Potassium (S)-1,3-Diphenyl-3-(trifluoroborato)butan-1-one (7d).[10c]
According to the general procedure using (E)-chalcone (0.104 g, 0.5 mmol), the product was obtained as a white crystalline solid in 52% yield (82 mg). mp = 147–150 °C. 1H NMR (500 MHz, acetone-d6) δ 7.94 (d, J = 8.0 Hz, 2H), 7.50 (t, J = 8.1 Hz, 1H), 7.41 (t, J = 7.8 Hz, 2H), 7.21 (d, J = 9.5 Hz, 2H), 7.00–7.14 (m, 2H), 6.83–6.99 (m, 1H), 3.39 (d, J = 7.7 Hz, 2H), 2.44–2.59 (m, 1H). 13C NMR (125.8 MHz, acetone-d6) δ 202.0, 148.5, 137.8, 131.9, 128.1, 128.1, 128.0, 127.1, 123.0, 41.3. 19F NMR (470.8 MHz, acetone-d6) δ −144.96. 11B NMR (128.4 MHz, acetone-d6) δ 4.57. [α]D20= +27.3 (c = 0.23, MeOH).
Potassium (R)-3-(Trifluoroborato)cyclohexanone (7e).[10c]
According to the general procedure using cyclohex-2-enone (0.0480 g, 0.5 mmol), the product was obtained as a white crystalline solid in 90% yield (92 mg). mp = 235–237 °C. 1H NMR (360 MHz, DMSO-d6) δ 2.13–2.22 (m, 1H), 2.11-1.87 (m, 4H), 1.56–1.59 (m, 1H), 1.36–1.53 (m, 1H), 1.21–1.32 (m, 1H), 0.47 (br s, 1H). 13C NMR (90.5 MHz, DMSO-d6) δ 215.4, 45.21, 42.3, 30.2, 27.7. 19F NMR (338.8 MHz, DMSO-d6) δ −145.3. 11B NMR (128.4 MHz, acetone-d6) δ 5.25. [α]D20= +55.0 (c = 0.34, MeOH).
General Procedure for the Preparation of Enantioenriched Potassium β-Trifluoroboratoamides with Tetrakis(dimethylamino)diboron
An oven dried microwave vial with stir bar was charged with Cu(MeCN)4PF6 (0.0093 g, 0.025 mmol, 5 mol %), NaOt-Bu (0.0480 g, 0.5 mmol, 1 equiv), and (2R, 5R)-1{[(2R,5R)-2,5-diphenylpyrrolidin-1-yl]methylene}2,5-diphenylpyrrolidinium tetrafluoroborate (0.0136 g, 0.025 mmol, 5 mol %). The vial was sealed with a cap lined with a disposable Teflon septum, evacuated under vacuum and purged with Ar (three times). Anhyd THF (0.5 mL) was added under Ar followed by tetrakis(dimethylamino)diboron (1 mmol, 2 equiv) in anhyd MeOH (0.5 mL). The reaction was stirred at rt for 5 min before adding the amide (0.5 mmol, 1 equiv). The reaction mixture was stirred at rt for 24 h. After cooling to 0 °C, MeOH (1 mL) and sat. aq KHF2 (3 mmol, 6 equiv) was added dropwise. The reaction mixture was stirred at rt until complete by 11B NMR (about 10 min), concentrated, and placed on the high vacuum overnight. The crude product was isolated by hot filtration with acetone (3 × 10 mL), concentrating to a minimal amount (~2 mL), and precipitating with Et2O (20 mL).
Spectral data for the enantioenriched cross-coupled products is the same as the racemic products and can be seen in “General Procedure for the Preparation of Enantioenriched Potassium β-Trifluoroboratoamides with BBA.”
Determination of Absolute Configuration and Enantiomeric Ratio of Enantioenriched Potassium Trifluoroboratoamides
The purpose of the oxidation of the enantioenriched potassium trifluoroboratoamides was to determine the enantiomeric ratio of the corresponding alcohol. Therefore a small sample of the potassium trifluoroboratoamide was oxidized with oxone. After confirming the product by NMR, the sample was subjected to SFC analysis.
General Procedure for the Oxidation of Enantioenriched Potassium β-Trifluoroboratoamides (Method A)
To a flask containing a mixture of potassium β-trifluoroboratoamides, esters, or ketones (1 mmol, 1 equiv) and acetone (2.5 mL) (MeOH was added dropwise in the case of insoluble trifluoroborates), was added Oxone (2.5 mL of a 0.2 M solution in H2O, 1 equiv) in one portion. The reaction mixture was stirred at rt until 11B NMR indicated completion of the reaction (~10 min). To the crude reaction mixture was added H2O (2.5 mL) and aq HCl (0.3 M, 3 mL), and the aqueous layer was extracted with CH2Cl2 (3×15 mL). The combination organic layers were dried (MgSO4), filtered, concentrated in vacuo.
General Procedure for the Preparation of Both (R)- and (S)-MTPA Ester Enantiomers (Method B)
To a solution of β-hydroxy amides or ketones (which was prepared by method A) (0.027 mmol) in CH2Cl2 (1.8 mL) in a round bottom flask under Ar was added Et3N (19 mL, 0.137 mmol), (R)-MTPA chloride (25.5 mL, 0.137 mmol), and DMAP (3.6 mg, 0.030 mmol). The resulting mixture was stirred for 16 h at rt and then the solvent was concentrated in vacuo. Enantiomeric ratios were determined by 19F NMR of the unpurified material.
3-Hydroxy-N-(4-methoxyphenyl)butanamide.[2]
According to the general procedure (Method A) using potassium (R)-N-(4-methoxyphenyl)-3-(trifluoroborato)butanamide (0.299 g, 1 mmol) the title compound was obtained as a white crystalline solid (90%, 188 mg). mp = 134–136 °C. 1H NMR (300 MHz, CDCl3) δ 7.68 (s, 1H), 7.33 (d, J = 8.9 Hz, 3H), 6.79 (d, J = 8.9 Hz, 2H), 4.17–4.27 (m, 1H), 3.72 (s, 3H), 2.90 (s, 1H), 2.50-2.32 (m, 2H), 1.22 (d, J = 6.3 Hz, 3H). 13C NMR (90.5 MHz, CDCl3) δ 170.2, 156.5, 130.5, 122.0, 114.1, 64.9, 55.4, 44.9, 29.6, 22.9. A method was developed to separate the enantiomers using SFC analysis (Column OD-H, 10% i-PrOH, 2 mL, 10 MPa); (S)-isomer retention time = 8.76 min and (R)-isomer retention time = 9.80 min.
(R)-3-Hydroxy-N-(4-methoxyphenyl)butanamide (2a-OH)
Following the procedure described above for oxidation using the enantioselective borylated intermediate, the title compound was obtained as a white crystalline solid with spectra in accordance with that described above for the racemate (0.25 mmol scale, 88%, 46 mg). mp = 135–136 °C. [α]D20 = −31.5 (c = 1.0, MeOH). A method was developed to separate the enantiomers using SFC analysis (Column OD-H, 10% i-PrOH, 2 mL, 10 MPa); the enantiomeric ratio was measured to be 5:95. The major enantiomer having an absolute configuration of R. (S)-isomer retention time = 8.64 min and (R)-isomer retention time = 9.55 min.
3-Hydroxy-N-(4-methoxyphenyl)octanamide
According to the general procedure (Method A) using potassium N-(4-methoxyphenyl)-3-(trifluoroborato)methyloctanamide (0.355 g, 1 mmol) the title compound was obtained as a white solid. mp = 120–122 °C. 1H NMR (300 MHz, CDCl3) δ 7.67 (s, 1H), 7.39 (d, J = 9.0 Hz, 2H), 6.85 (d, J = 9.0 Hz, 2H), 4.07 (d, J = 11.3 Hz, 1H), 3.79 (s, 3H), 3.19 (s, 1H), 2.37–2.60 (m, 2H), 1.70-1.17 (m, 8H), 0.90 (t, J = 6.9 Hz, 3H). 13C NMR (125.8 MHz, CDCl3) δ 170.3, 1566.4, 130.5, 123.4, 121.8, 114.0, 113.0, 68.6, 55.3, 43.4, 36.8, 31.6, 25.0, 22.4, 13.9.IR (neat) 3314, 2957, 2924, 2855, 1655, 1548, 1513, 1250, 1031, 825, 665, 648 cm−1. HRMS (ESI) calcd. For C15H24NO3 [M+H]+ 266.1756, found 266.1751. A method was developed to separate the enantiomers using SFC analysis (Column OD-H, 10% i-PrOH, 2 mL, 10 MPa); (S)-isomer retention time = 9.47 min and (R)-isomer retention time = 11.55 min.
(R)-3-Hydroxy-N-(4-methoxyphenyl)octanamide (2b-OH)
Following the procedure described above for oxidation using the enantioselective borylated intermediate, the title compound was obtained as a white solid with spectra in accordance with that described above for the racemate. mp = 119–122 °C. [α]D20 = −20.8 (c = 0.25, MeOH). Using SFC analysis (Column AS-H, 10% i-PrOH, 2 mL, 10 MPa); the enantiomeric ratio was measured to be 3:97. The major enantiomer having an absolute configuration of R. (S)-isomer retention time = 9.67 min and (R)-isomer retention time = 11.71 min.
3-Hydroxy-N-phenylbutanamide.[ 36 ]
According to the general procedure (Method A) using potassium (R)-N-phenyl-3-(trifluoroborato)butanamide (0.269 g, 1 mmol) the title compound was obtained as a white crystalline solid. mp = 92–94 °C. 1H NMR (500 MHz, acetone-d6) δ 9.19 (s, 1H), 7.64 (d, J = 8.7 Hz, 2H), 7.22–7.34 (m, 2H), 7.09-6.98 (m, 1H), 4.19–4.30 (m, 1H), 4.10 (br s, 1H), 2.39–2.53 (m, 2H), 1.19 (d, J = 6.4 Hz, 3H). 13C NMR (125.8 MHz, acetone–d6) δ 170.1, 139.1, 128.48, 123.14, 119.17, 64.33, 45.70, 22.60. A method was developed to separate the enantiomers using SFC analysis (Column OD-H, 10% i-PrOH, 2 mL, 10 MPa); (S)-isomer retention time = 8.51 min and (R)-isomer retention time = 10.49 min.
(R)-3-Hydroxy-N-phenylbutanamide (2c-OH)
Following the procedure described above for oxidation using the enantioselective borylated intermediate, the title compound was obtained as a white crystalline solid with spectra in accordance with that described above for the racemate. mp = 93–95 °C [α]D20 = −14.8 (c = 0.25, MeOH). A method was developed to separate the enantiomers using SFC analysis (Column OD-H, 10% i-PrOH, 2 mL, 10 MPa); the enantiomeric ratio was measured to be 8:92. The major enantiomer having an absolute configuration of R. (S)-isomer retention time = 8.51 min and (R)-isomer retention time = 10.11 min.
N-Cyclohexyl-3-hydroxybutanamide.[2]
According to the general procedure (Method A) using potassium N-cyclohexyl-3-(trifluoroborato)butanamide (0.274 g, 1 mmol) the title compound was obtained as a white crystalline solid. mp = 134–136 °C. 1H NMR (300 MHz, CDCl3) δ 5.74 (s, 1H), 4.16 (m, 1H), 3.95 (s, 1H), 3.78 (m, 1H), 2.16–2.40 (m, 2H), 1.84–1.99 (m, 2H), 1.54–1.80 (m, 3H), 1.04–1.48 (m, 8H). 13C NMR (75.4 MHz, CDCl3) δ 171.3, 64.7, 48.0, 43.8, 32.9, 25.3, 24.6, 22.6. A method was developed to separate the enantiomers using SFC analysis (Column OD-H, 10% i-PrOH, 2 mL, 10 MPa); (S)-isomer retention time = 5.27 min and (R)-isomer retention time = 5.57 min.
(R)-N-Cyclohexyl-3-hydroxybutanamide (2d-OH).[2]
Following the procedure described above for oxidation using the enantioselective borylated intermediate, the title compound was obtained as a white crystalline solid with spectra in accordance with that described above for the racemate. mp = 134–136 °C [α]D20 = −17.0 (c = 0.2, MeOH). Using SFC analysis (Column AS-H, 10% i-PrOH, 2 mL, 10 MPa); the enantiomeric ratio was measured to be 5:95. The major enantiomer having an absolute configuration of R. (S)-isomer retention time = 6.19 min and (R)-isomer retention time = 6.93 min.
N-Cyclopropyl-3-hydroxyoctanamide
According to the general procedure (Method A) using potassium N-(4-methoxyphenyl)-3-(trifluoroborato)methyloctanamide (0.233 g, 1 mmol) the title compound was obtained as a yellow oil. 1H NMR (500 MHz, CDCl3) δ 6.20 (s, 1H), 4.03–4.22 (m, 1H), 3.56 (br s, 1H), 2.64 (s, 1H), 2.12–2.29 (m, 2H), 1.15 (t, J = 5.5 Hz, 3H), 0.71 (t, J = 6.6 Hz, 2H), 0.44 (t, J = 6.3 Hz, 2H). 13C NMR (125.8 MHz, CDCl3) δ 173.9, 64.7, 43.6, 22.7, 22.3, 8.0, 6.4. IR (neat) 3283, 2965, 2920, 1641, 1542, 1455, 1376, 1302, 1196, 1123, 852, 664, 638 cm−1. HRMS (ESI) calcd. For C7H13NO2 [M]+ 143.0946, found 143.0948. A method was developed to separate the enantiomers using SFC analysis (Column AS-H, 10% i-PrOH, 2 mL, 10 MPa); (S)-isomer retention time = 6.87 min and (R)-isomer retention time = 7.81 min.
(R)-N-Cyclopropyl-3-hydroxyoctanamide (2e-OH)
Following the procedure described above for oxidation using the enantioselective borylated intermediate, the title compound was obtained as a yellow oil with spectra in accordance with that described above for the racemate. [α]D20 = −25.0 (c = 0.1, MeOH). Using SFC analysis (Column AS-H, 10% i-PrOH, 2 mL, 10 MPa); the enantiomeric ratio was measured to be 7:93. The major enantiomer having an absolute configuration of R. (S)-isomer retention time = 6.07 min and (R)-isomer retention time = 7.44 min.
N-Cyclohexyl-3-hydroxypentanamide
According to the general procedure (Method A) using potassium N-cyclohexyl-3-(trifluoroborato)pentanamide (0.289 g, 1 mmol) the title compound was obtained as a white crystalline solid (76%, 151 mg). mp = 106–108 °C. 1H NMR (300 MHz, CDCl3) δ 5.98 (br s, 1H), 3.79–3.88 (m, 2H), 3.6174–3.74 (m, 1H), 2.05–2.32 (m, 2H), 1.78–1.86 (m, 2H), 1.70-0.96 (m, 10H), 0.86 (t, J = 7.4 Hz, 3H). 13C NMR (75.4 MHz, CDCl3) δ 171.6, 69.9, 48.0, 41.8, 32.9, 29.6, 25.3, 24.6, 9.7.IR (neat) 3292, 2932, 2852, 1639, 1556, 1445, 1386, 1194, 1125, 984, 870, 699, 630 cm−1. HRMS (ESI) calcd. For C11H22NO2 [M+H]− 200.1651, found 200.1651. A method was developed to separate the enantiomers using SFC analysis (Column AS-H, 10% i-PrOH, 2 mL, 10 MPa); (S)-isomer retention time = 8.77 min and (R)-isomer retention time = 10.67 min.
(R)-N-Cyclohexyl-3-hydroxypentanamide (2f-OH)
Following the procedure described above for oxidation using the enantioselective borylated intermediate, the title compound was obtained as a white crystalline solid with spectra in accordance with that described above for the racemate. mp = 105–106 °C. [α]D20 = −17.0 (c = 0.2, MeOH). Using SFC analysis (Column AS-H, 10% i-PrOH, 2 mL, 10 MPa); the enantiomeric ratio was measured to be 2:98. The major enantiomer having an absolute configuration of R. (S)-isomer retention time = 8.75 min and (R)-isomer retention time = 9.21 min.
3-Hydroxy-1-(piperidin-1-yl)butan-1-one.[37]
According to the general procedure (Method A) using potassium 1-(piperidin-1-yl)-3-(trifluoroborato)butan-1-one (0.261 g, 1 mmol) the title compound was obtained as a yellow oil. 1H NMR (500 MHz, CDCl3) δ 4.20 (s, 1H), 3.98–4.2 (m, 1H), 3.49–3.61 (m, 2H), 3.27–3.44 (m, 2H), 2.49 (d, J = 16.0 Hz, 1H), 2.31 (dd, J = 16.5, 9.4 Hz, 1H), 1.64–1.72 (m, 2H), 1.51–1.60 (m, 4H), 1.10–1.28 (m, 3H). 13C NMR (125.8 MHz, CDCl3) δ 170.6, 64.1, 46.2, 42.3, 40.5, 26.1, 25.3, 24.2, 22.0. A method was developed to separate the enantiomers using SFC analysis (Column AD-H, 10% i-PrOH, 2 mL, 10 MPa); (S)-isomer retention time= 6.04 min and (R)-isomer retention time = 5.51 min.
(R)-3-Hydroxy-1-(piperidin-1-yl)butan-1-one (2g-OH)
Following the procedure described above for oxidation using the enantioselective borylated intermediate, the title compound was obtained as a yellow oil with spectra in accordance with that described above for the racemate. [α]D20 = −24.4 (c = 0.25, MeOH). A method was developed to separate the enantiomers using SFC analysis (Column AD-H, 10% i-PrOH, 2 mL, 10 MPa); the enantiomeric ratio was measured to be 8:92. The major enantiomer having an absolute configuration of R. (S)-isomer retention time = 6.10 min and (R)-isomer retention time = 5.49 min.
3-Hydroxy-1-morpholinobutan-1-one.[ 38 ]
According to the general procedure (Method A) using potassium morpholino-3-(trifluoroborato)butan-1-one (0.263 g, 1 mmol) the title compound was obtained as a colorless oil (35%, 60 mg). 1H NMR (360 MHz, CDCl3) δ 4.19–4.23 (m, 1H), 4.10 (s, 1H), 3.51–3.75 (m, 6H), 3.44 (dd, J = 5.9, 3.8 Hz, 2H), 2.45 (dd, J = 16.4, 2.6 Hz, 1H), 2.30 (dd, J = 16.4, 9.4 Hz, 1H), 1.22 (d, J = 5.3 Hz 3H). 13C NMR (90.5 MHz, CDCl3) δ 171.1, 66.7, 66.4, 64.1, 45.6, 41.7, 40.7, 22.2. A method was developed to separate the enantiomers using SFC analysis (Column AD-H, 10% i-PrOH, 2 mL, 10 MPa); (S)-isomer retention time = 4.96 min and (R)-isomer retention time = 5.61 min.
(R)-3-Hydroxy-1-morpholinobutan-1-one (2h-OH)
Following the procedure described above for oxidation using the enantioselective borylated intermediate, the title compound was obtained as a colorless oil with spectra in accordance with that described above for the racemate. [α]D20 = −10.7 (c = 1.0, MeOH). A method was developed to separate the enantiomers using SFC analysis (Column AD-H, 10% i-PrOH, 2 mL, 10 MPa); the enantiomeric ratio was measured to be 28:72. The major enantiomer having an absolute configuration of R. (S)-isomer retention time = 4.85 min and (R)-isomer retention time = 5.11 min.
3-Hydroxy-N-(4-methoxyphenyl)-3-phenylpropanamide
According to the general procedure (Method A) using potassium (S)-N-(4-methoxyphenyl)-3-phenyl-3-(trifluoroborato)propanamide (0.361 g, 1 mmol) the title compound was obtained as a white crystalline solid. mp = 144–146 °C. 1H NMR (500 MHz, DMSO-d6) δ 9.73 (s, 1H), 7.50 (d, J = 8.8 Hz, 2H), 7.30–7.47 (m, 4H), 7.24 (m, 1H), 6.86 (d, J = 8.5 Hz, 2H), 5.06 (dd, J = 8.9, 4.7 Hz, 1H), 3.71 (s, 3H), 3.40 (br s, 1H), 2.62–2.66 (m, 1H), 2.46–2.59 (m, 1H). 13C NMR (125.8 MHz, DMSO-d6) δ 168.9, 155.4, 145.7, 132.7, 128.4, 127.2, 126.0, 121.0, 114.1, 70.1, 55.4, 47.2. IR (neat) 3326, 2953, 2916, 2851, 1653, 1536, 1511, 1469, 1409, 1244, 1030, 1018, 822, 756, 701, 689, 656 cm− 1. HRMS (ESI) calcd. For C16H18NO3 [M+H]+ 272.1287, found 272.1296. Both (R)- and (S)-MTPA esters were prepared according to general procedure B, and the enantiomeric ratio was determined by 19F NMR (338.8 MHz, CDCl3): (R)-MTPA derivative of (S) enantiomer δ = −71.73, (R) enantiomer δ = −71.50.
(S)-3-Hydroxy-N-(4-methoxyphenyl)-3-phenylpropanamide (2i-OH)
Following the procedure described above for oxidation using the enantioselective borylated intermediate, the title compound was obtained as a white crystalline solid with spectra in accordance with that described above for the racemate. mp = 144–147 °C. [α]D20= −18.0 (c = 0.2, MeOH). Both (R)- and (S)-MTPA esters were prepared according to general procedure B, and the enantiomeric ratio was determined by 19F NMR (338.8 MHz, CDCl3): (R)-MTPA derivative of (S) enantiomer δ = −71.78, (R) enantiomer δ = −71.52. The enantiomeric ratio was measured to be 4:96, the major enantiomer having an absolute configuration of S.
N-Ethyl-3-hydroxy-N-(o-tolyl)butanamide
According to the general procedure (Method A) using potassium N-ethyl-N-(o-tolyl)-3-(trifluoroborato)butanamide (0.311 g, 1 mmol) the title compound was obtained as a yellow oil (90%, 199 mg). 1H NMR (500 MHz, asterisk denotes minor rotamer peaks, CDCl3) δ 7.19–7.31 (m, 3H), 7.03 (t, J = 8.9 Hz, 1H), 4.21 (s, 1H), 4.00–4.17 (m, 2H), 3.21 (m, 1H), 2.19 (d, J = 7.9 Hz, 3H), 1.77–2.16 (m, 2H, 1H*), 1.09–1.13 (m, 3H), 1.03 (dd, J = 6.4, 2.9 Hz, 3H). 13C NMR (125.8 MHz, asterisk denotes minor rotamer peaks, CDCl3) δ 172.5, 172.3*, 140.1, 140.0*, 135.6, 135.3*, 131.5, 131.4*, 129.1, 128.8*, 128.4, 127.1, 127.1*, 64.4, 64.2*, 42.7, 42.7*, 41.6, 22.1, 22.1*, 17.3, 12.7, 12.6*. IR (neat) 3436, 2977, 2929, 2876, 1712, 1631, 1493, 1447, 1409, 1293, 1255, 1138, 1091, 958, 77, 728, 648 cm−1. HRMS (ESI) calcd. For C13H20NO2 [M+H]− 222.1494, found 222.1493. A method was developed to separate the enantiomers using SFC analysis (Column AD-H, 10% i-PrOH:CH3CN (85:15), 2 mL, 10 MPa); (S)-isomer retention time = 4.63 min and (R)-isomer retention time = 5.22 min.
(R)-N-Ethyl-3-hydroxy-N-(o-tolyl)butanamide (2j-OH)
Following the procedure described above for oxidation using the enantioselective borylated intermediate, the title compound was obtained as a yellow oil with spectra in accordance with that described above for the racemate. [α]D20 = −18.0 (c = 0.1, MeOH). A method was developed to separate the enantiomers using SFC analysis (Column AD-H, 10% i-PrOH/CH3CN (85:15), 2 mL, 10 MPa); the enantiomeric ratio was measured to be 28:72. The major enantiomer having an absolute configuration of R. (S)-isomer retention time = 4.85 min and (R)-isomer retention time = 5.38 min.
3-Hydroxy-N,N-dimethylbutanamide.[ 39 ]
According to the general procedure (Method A) using potassium N-(4-methoxyphenyl)-3-(trifluoroborato)methyloctanamide (0.221 g, 1 mmol) the title compound was obtained as a yellow oil. 1H NMR (500 MHz, CDCl3) δ 4.39 (br s, 1H), 4.13–4.23 (m, 1H), 2.98 (s, 3H), 2.95 (s, 3H), 2.47 (dd, J = 16.5, 2.4 Hz, 1H), 2.29 (dd, J = 16.5, 9.6 Hz, 1H), 1.21 (d, J = 6.4 Hz, 3H). 13C NMR (125.8 MHz, CDCl3) δ 172.6, 64.0, 40.7, 36.9, 35.0, 22.0. A method was developed to separate the enantiomers using SFC analysis (Column R,R-Whelk, 10% i-PrOH, 2 mL, 10 MPa); (S)-isomer retention time = 12.41 min and (R)-isomer retention time = 11.87 min.
(R)-3-Hydroxy-N,N-dimethylbutanamide (2k-OH)
Following the procedure described above for oxidation using the enantioselective borylated intermediate, the title compound was obtained as a yellow oil with spectra in accordance with that described above for the racemate. [α]D20 = +11.7 (c = 1.0, MeOH). Using SFC analysis (Column R,R-Whelk, 10% i-PrOH, 2 mL, 10 MPa); the enantiomeric ratio was measured to be 30:70. The major enantiomer having an absolute configuration of R. (S)-isomer retention time = 12.41 min and (R)-isomer retention time = 11.47 min.
N,N-Dibenzyl-3-hydroxybutanamide
According to the general procedure (Method A) using potassium (R)-N,N-dibenzyl-3-(trifluoroborato)butanamide (0.373 g, 1 mmol) the title compound was obtained as a white crystalline solid (98%, 277 mg). mp = 64–66 °C. 1H NMR (500 MHz, CDCl3) δ 7.27–7.42 (m, 6H), 7.23 (d, J = 5.0 Hz, 2H), 7.16 (d, J = 5.0 Hz, 2H), 4.71 (d, J = 14.8 Hz, 1H), 4.55 (d, J = 14.8 Hz, 1H), 4.40–4.52 (m, 1H), 4.27–4.31 (m, 2H), 4.22 (br s, 1H), 2.60 (dd, J = 16.4, 2.5 Hz, 1H), 2.42–2.54 (m, 1H), 1.23 (dd, J = 6.6, 3.0 Hz, 3H). 13C NMR (125.8 MHz, CDCl3) δ 173.3, 136.7, 135.8, 128.9, 128.6, 128.1, 127.7, 127.4, 126.2, 64.3, 49.7, 47.9, 40.8, 22.1. A method was developed to separate the enantiomers using SFC analysis (Column OD-H, 10% i-PrOH, 2 mL, 10 MPa); (S)-isomer retention time = 12.11 min and (R)-isomer retention time = 11.15 min.
(R)-N,N-Dibenzyl-3-hydroxybutanamide (2l-OH)
Following the procedure described above for oxidation using the enantioselective borylated intermediate, the title compound was obtained as a white crystalline solid with spectra in accordance with that described above for the racemate. mp = 65–66 °C. [α]D20 = −5.0 (c = 0.3, MeOH). A method was developed to separate the enantiomers using SFC analysis (Column OD-H, 10% i-PrOH, 2 mL, 10 MPa); the enantiomeric ratio was measured to be 34:66. The major enantiomer having an absolute configuration of R. (S)-isomer retention time = 12.20 min and (R)-isomer retention time = 11.09 min.
N-Cyclohexyl-3-hydroxy-3-phenylpropanamide
According to the general procedure (Method A) using potassium (S)-N-cyclohexyl-3-phenyl-3-(trifluoroborato)propanamide (0.337 g, 1 mmol) the title compound was obtained as a white crystalline solid (89%, 219 mg). mp = 132–134 °C. 1H NMR (300 MHz, CDCl3) δ 7.17–7.41 (m, 5H), 6.30 (s, 1H), 4.97 (dd, J = 7.4, 4.6 Hz, 1H), 4.74 (s, 1H), 3.66–3.69 (m, 1H), 2.36–2.54 (m, 2H), 1.30–1.82 (m, 2H), 1.49–1.74 (m, 3H), 1.27–1.31 (m, 2H), 1.05–1.17 (m, 3H). 13C NMR (75.4 MHz, CDCl3) δ 170.9, 143.1, 128.2, 127.4, 125.5, 70.7, 48.1, 44.5, 32.74, 25.3, 24.6. IR (neat) 3395, 3299, 2932, 2847, 1635, 1552, 1445, 1359, 1206, 1052, 1016, 984, 891, 755, 696, 646 cm−1. HRMS (ESI) calcd. For C15H22NO2 [M+H]− 248.1651, found 248.1650. A method was developed to separate the enantiomers using SFC analysis (Column OD-H, 10% i-PrOH, 2 mL, 10 MPa); (S)-isomer retention time = 11.16 min and (R)-isomer retention time = 10.69 min.
(S)-N-Cyclohexyl-3-hydroxy-3-phenylpropanamide (2m-OH)
Following the procedure described above for oxidation using the enantioselective borylated intermediate, the title compound was obtained as a white crystalline solid with spectra in accordance with that described above for the racemate. mp = 131–134 °C. [α]D20 = +140.5 (c = 0.2, MeOH). A method was developed to separate the enantiomers using SFC analysis (Column OD-H, 10% i-PrOH, 2 mL, 10 MPa); the enantiomeric ratio was measured to be 28:72. The major enantiomer having an absolute configuration of S. (S)-isomer retention time = 10.95 min and (R)-isomer retention time = 10.50 min.
3-Hydroxy-3-phenyl-1-(pyrrolidin-1-yl)propan-1-one.[38]
According to the general procedure (Method A) using potassium (S)-3-phenyl-1-(pyrrolidin-1-yl)-3-(trifluoroborato)propan-1-one (0.309 g, 1 mmol) the title compound was obtained as a white crystalline solid. mp = 48–50 °C. 1H NMR (500 MHz, CDCl3) δ 7.32–7.53 (m, 4H), 7.25–7.32 (m, 1H), 5.18 (s, 1H), 4.93 (s, 1H), 3.49–3.51 (m, 2H), 3.27–3.43 (m, 2H), 2.51–2.76 (m, 2H), 1.91 (m, 4H). 13C NMR (75.4 MHz, CDCl3) δ170.0, 143.0, 128.3, 127.3, 125.6, 70.2, 46.5, 45.4, 42.9, 25.8, 24.2. A method was developed to separate the enantiomers using SFC analysis (Column OD-H, 10% i-PrOH, 2 mL, 10 MPa); (S)-isomer retention time = 13.12 min and (R)-isomer retention time = 14.10 min.
(S)-3-Hydroxy-3-phenyl-1-(pyrrolidin-1-yl)propan-1- one (2n-OH)
Following the procedure described above for oxidation using the enantioselective borylated intermediate, the title compound was obtained as a white crystalline solid with spectra in accordance with that described above for the racemate. mp = 48–51 °C. [α]D20= −12.0 (c = 0.1, MeOH). A method was developed to separate the enantiomers using SFC analysis (Column OD-H, 10% i-PrOH, 2 mL, 10 MPa); the enantiomeric ratio was measured to be 22:78. The major enantiomer having an absolute configuration of S. (S)-isomer re= 14.41 min and (R)-isomer retention time = 15.11 min.
Ethyl 3-Hydroxybutanoate.[40]
According to the general procedure using potassium (R)-ethyl 3-(trifluoroborato)butanoate (0.222 g, 1 mmol) the title compound was obtained as a yellow oil (98%, 129 mg). 1H NMR (300 MHz, CDCl3) δ 4.11–4.26 (m, 3H), 3.07 (s, 1H), 2.35–2.53 (m, 2H), 1.27 (t, J = 7.1 Hz, 3H), 1.22 (d, J = 6.3 Hz, 3H). 13C NMR (75.4 MHz, CDCl3) δ 172.7, 64.1, 60.5, 42.6, 22.3, 14.0. A method was developed to separate the enantiomers using SFC analysis (Column OD-H, 10% i-PrOH, 2 mL, 10 MPa); (S)-isomer retention time = 10.3 min and (R)-isomer retention time = 11.29 min.
Ethyl (R)-3-Hydroxybutanoate (7a-OH)
Following the procedure described above for oxidation using the enantioselective borylated intermediate, the title compound was obtained as a yellow oil with spectra in accordance with that described above for the racemate. [α]D20= −6.7 (c = 0.55, MeOH). A method was developed to separate the enantiomers using SFC analysis (Column OD-H, 10% i-PrOH, 2 mL, 10 MPa); the enantiomeric ratio was measured to be 5:95, the major enantiomer having an absolute configuration of R. (S)-isomer retention time = 10.71 min and (R)-isomer retention time = 11.64 min.
Ethyl 3-Hydroxyoctanoate.[41]
According to the general procedure using potassium ethyl (R)-3-(trifluoroborato)octanoate (0.278 g, 1 mmol) the title compound was obtained as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 4.36-4.10 (m, 2H), 4.10-3.93 (m, 1H), 3.01 (br s, 1H), 2.45 (m, 2H), 1.65-1.16 (m, 11H), 1.02-0.80 (m, 3H). 13C NMR (125.8 MHz, CDCl3) δ 172.9, 67.9, 60.5, 41.2, 36.4, 31.6, 25.0, 22.4, 14.0, 13.8. A method was developed to separate the enantiomers using SFC analysis (Column AD-H, 10% i-PrOH, 2 mL, 10 MPa); (S)-isomer retention time = 3.97 min and (R)-isomer retention time = 3.09 min.
Ethyl (R)-3-Hydroxyoctanoate (7b-OH)
Following the procedure described above for oxidation using the enantioselective borylated intermediate, the title compound was obtained as a colorless oil with spectra in accordance with that described above for the racemate. [α]D20= −4.61 (c = 0.26, MeOH). A method was developed to separate the enantiomers using SFC analysis (Column AD-H, 10% i-PrOH, 2 mL, 10 MPa); the enantiomeric ratio was measured to be 12:88, the major enantiomer having an absolute configuration of R. (S)-isomer retention time = 3.93 min and (R)-isomer retention time = 2.99 min.
3-Hydroxy-1-phenylbutan-1-one.[ 42 ]
According to the general procedure using potassium (R)-3-trifluoroborato-1-phenylbutan-1-one (0.254 g, 1 mmol) the title compound was obtained as a yellow oil (65%, 106 mg). 1H NMR (300 MHz, CDCl3) δ 7.96 (d, J = 7.1 Hz, 2H), 7.53–7.64 (m, 1H), 7.42–7.53 (m, 2H), 4.41 (m, 1H), 3.40 (br s, 1H), 3.23-2.98 (m, 2H), 1.31 (d, J = 6.3 Hz, 3H). 13C NMR (75.4 MHz, CDCl3) δ 200.6, 136.6, 133.4, 128.5, 127.9, 76.5, 63.9, 46.4, 22.3. A method was developed to separate the enantiomers using SFC analysis (Column AS-H, 10% i-PrOH, 2 mL, 10 MPa); (S)-isomer retention time = 3.13 min and (R)-isomer retention time = 3.47 min.
(R)-3-Hydroxy-1-phenylbutan-1-one (7c-OH)
Following the procedure described above for oxidation using the enantioselective borylated intermediate, the title compound was obtained as a yellow oil with spectra in accordance with that described above for the racemate. [α]D20 = −1.53 (c = 0.26, MeOH). A method was developed to separate the enantiomers using SFC analysis (Column AS-H, 10% i-PrOH, 2 mL, 10 MPa); the enantiomeric ratio was measured to be 14:86, the major enantiomer having an absolute configuration of R. (S)-isomer retention time = 3.17 min and (R)-isomer retention time = 3.47 min.
3-Hydroxy-1,3-diphenylpropan-1-one.[ 43 ]
According to the general procedure using potassium (S)-1,3-diphenyl-3-(trifluoroborato)butan-1-one (0.316 g, 1 mmol) the title compound was obtained as a yellow oil. 1H NMR (360 MHz, CDCl3) δ 7.97 (d, J = 7.1 Hz, 2H), 7.55–7.65 (m, 1H), 7.28–7.53 (m, 7H), 5.34–5.41 (m, 1H), 3.58 (s, 1H), 3.39 (d, J = 6.0 Hz, 2H). 13C NMR (90.5 MHz, CDCl3) δ 200.1, 142.9, 136.6, 133.6, 128.7, 128.5, 128.1, 127.6, 125.7, 70.0, 47.4. A method was developed to separate the enantiomers using SFC analysis (Column R,R-Whelk, 10% i-PrOH, 2 mL, 10 MPa); (R)-isomer retention time = 8.79 min and (S)-isomer retention time = 11.35 min.
(S)-3-Hydroxy-1,3-diphenylpropan-1-one (7d-OH)
Following the procedure described above for oxidation using the enantioselective borylated intermediate, the title compound was obtained as a yellow oil with spectra in accordance with that described above for the racemate. [α]D20 = +8.0 (c = 0.25, MeOH). A method was developed to separate the enantiomers using SFC analysis (Column R,R-Whelk, 10% i-PrOH, 2 mL, 10 MPa); the enantiomeric ratio was measured to be 21:79, the major enantiomer having an absolute configuration of S. (R)-isomer retention time = 8.82 min and (S)-isomer retention time = 11.47 min.
3-Hydroxycyclohexan-1-one
According to the general procedure using potassium (R)-3-(trifluoroborato)cyclohexanone (0.204 g, 1 mmol) the title compound was obtained as a yellow oil (87%, 99 mg). 1H NMR (500 MHz, CDCl3) δ 4.18–4.22 (m, 1H), 2.67 (dd, J = 14.0, 4.4 Hz, 1H), 2.40–2.45 (m, 1H), 2.30–2.40 (m, 2H), 2.18-1.98 (m, 2H), 1.62–1.90 (m, 3H). 13C NMR (125.8 MHz, CDCl3) δ 209.4, 69.6, 50.3, 40.7, 32.7, 20.51.
(R)-3-Hydroxycyclohexane-1-one (7e-OH)
Following the procedure described above for oxidation using the enantioselective borylated intermediate, the title compound was obtained as a yellow oil with spectra in accordance with that described above for the racemate. [α]D20 = −1.4 (c = 0.1, MeOH). Both (R)- and (S)-MTPA esters were prepared according to general procedure B, and the enantiomeric ratio was determined by 19F NMR (338.8 MHz, CDCl3): (R)-MTPA derivative of (R) enantiomer δ = −71.89, (S) enantiomer δ = −71.33. The enantiomeric ratio was measured to be 26:74, the major enantiomer having an absolute configuration of R.
General Procedure for the Suzuki-Miyaura Cross-Coupling with Aryl Electrophiles
To a Biotage microwave vial was added precatalyst (0.0125 mmol, 5 mol %), K2CO3 (0.1037 g, 0.75 mmol, 3 equiv), aryl electrophile (0.25 mmol, 1 equiv), and potassium β-trifluoroboratoamide (0.25 mmol, 1 equiv). This mixture was sealed in the microwave vial and purged with Ar (three times). To the vial was added CPME (1.0 mL) and H2O (0.15 mL), and then the reaction was heated to 100 °C for 24 h. The reaction mixture was allowed to cool to rt and diluted with H2O (1 mL). The reaction mixture was extracted with EtOAc (3 × 3 mL) and dried (MgSO4). The solvent was removed in vacuo, and the product was purified by flash column chromatography on silica gel.
Determination of Absolute Configuration and Enantiomeric Ratio of Enantioenriched Cross-Coupled Products: N-(4-Methoxyphenyl)-3-(pyridin-3-yl)butanamide
Using the general procedure described for the Suzuki-Miyaura cross-coupling of potassium N-(4-methoxyphenyl)-3-(trifluoroborato)butanamide and 3-chloropyridine, the title compound was obtained in 56% yield (38 mg) as a pale yellow oil after silica gel column chromatography. 1H NMR (300 MHz, CDCl3) δ 8.42–8.58 (m, 2H), 7.76 (br s, 1H), 7.59 (d, J = 7.8 Hz, 1H), 7.33 (d, J = 8.9 Hz, 2H), 7.22–7.27 (m, 1H), 6.82 (d, J = 9.0 Hz, 2H), 3.78 (s, 3H), 3.36–3.51 (m, 1H), 2.60 (d, J = 7.4 Hz, 2H), 1.40 (d, J = 7.0 Hz, 3H). 13C NMR (75.4 MHz, CDCl3) δ 169.1, 156.3, 148.3, 147.7, 141.1, 134.7, 130.7, 123.5, 121.8, 114.0, 55.3, 45.7, 34.5, 21.0. Using SFC analysis (Column OJ-H, 10% i-PrOH, 2 mL, 10 MPa); (S)-isomer retention time = 16.82 min and (R)-isomer retention time = 14.60 min.
(S)-N-(4-Methoxyphenyl)-3-(pyridin-3-yl)butanamide (12a)
Using the general procedure described for the Suzuki-Miyaura cross-coupling of potassium (R)-N-(4-methoxyphenyl)-3-(trifluoroborato)butanamide (2a) and 3-chloropyridine, the title compound was obtained as a pale yellow oil in 56% yield (38 mg), with spectral data in accordance with that described for the racemate. [α]D20 = +33.8 (c = 0.54, MeOH). The title compound was found to have an enantiomeric ratio of 9:91 with an absolute configuration of S for the major enantiomer using SFC analysis (Column OJ-H, 10% i-PrOH, 2 mL, 10 MPa); (S)-isomer retention time= 16.59 min and (R)-isomer retention time= 14.99 min.
N-(4-Methoxyphenyl)-3-(thiophen-3-yl)butanamide
Using the general procedure described for the Suzuki-Miyaura cross-coupling of potassium N-(4-methoxyphenyl)-3-(trifluoroborato)butanamide and 3-chlorothiophene, the title compound was obtained in 60% (41 mg) yield as a pale yellow solid after silica gel column chromatography. mp = 102–105 °C. 1H NMR (300 MHz, CDCl3) δ 7.23–7.36 (m, 3H), 7.03–7.05 (m, 3H), 6.83 (d, J=8.9 Hz, 2H), 3.79 (s, 3H), 3.51 (q, J = 7.2 Hz, 1H), 2.44–2.65 (m, 2H), 1.38 (d, J = 6.9 Hz, 3H). 13C NMR (75.4 MHz, CDCl3) δ 169.6, 156.3, 146.5, 130.6, 126.4, 125.9, 121.8, 119.6, 113.9, 55.3, 46.3, 32.3, 21.2. IR (neat) 3279, 2961, 2933, 2835, 1649, 1603, 1531, 1510, 1245, 1032, 828, 776, 640 cm−1. HRMS (ESI) calcd. for C15H18NO2S [M+H]+ 276.1058, found 276.1051. Using SFC analysis (Column OJ-H, 10% i-PrOH, 2 mL, 10 MPa); (S)-isomer retention time= 10.2 min and (R)-isomer retention time= 11.8 min.
(S)-N-(4-Methoxyphenyl)-3-(thiophen-3-yl)butanamide (12b)
Using the general procedure described for the Suzuki-Miyaura cross-coupling of potassium (R)-N-(4-methoxyphenyl)-3-(trifluoroborato)butanamide (2a) and 3-chlorothiophene, the title compound was obtained as a pale yellow oil in 60% (41 mg) yield, with spectral data in accordance with that described for the racemate. mp = 102–104 °C. [α]D20 = +9.5 (c = 0.23, MeOH). The title compound was found to have an enantiomeric ratio of >99:1 with an absolute configuration of S for the major enantiomer using SFC analysis (Column OJ-H, 10% i-PrOH, 2 mL, 10 MPa); (S)-isomer retention time = 10.2 min.
N-(4-Methoxyphenyl)-3-(thiophen-2-yl)butanamide
Using the general procedure described for the Suzuki-Miyaura cross-coupling of potassium N-(4-methoxyphenyl)-3-(trifluoroborato)butanamide and 2-chlorothiophene, the title compound was obtained in 53% yield (37 mg) as a pale yellow solid after silica gel column chromatography. mp = 84–86 °C. 1H NMR (360 MHz, CDCl3) δ 7.28 (d, J = 9.0 Hz, 2H), 7.17 (d, J = 6.1 Hz, 1H), 7.07 (br s, 1H), 6.92–6.94 (m, 1H), 6.87–6.92 (m, 1H), 6.82 (d, J = 9.0 Hz, 2H), 3.82 (s, 3H), 3.67–3.75 (m, 1H), 2.55–2.68 (m, 2H), 1.44 (d, J = 6.9 Hz, 3H). 13C NMR (90.5 MHz, CDCl3) δ 169.3, 156.5, 149.7, 130.7, 126.8, 123.3, 123.1, 122.0, 114.0, 55.4, 47.4, 32.6, 22.6. IR (neat) 3281, 2961, 2933, 2831, 1648, 1605, 1528, 1508, 1247, 1034, 825, 695, 686, 633 cm−1. HRMS (ESI) calcd. For C15H17NO2NaS [M+Na]+ 298.0878, found 298.0876. Using SFC analysis (Column OJ-H, 10% i-PrOH, 2 mL, 10 MPa); (S)-isomer retention time = 9.5 min and (R)-isomer retention time = 11.5 min.
(S)-N-(4-Methoxyphenyl)-3-(thiophen-2-yl)butanamide (12c)
Using the general procedure described for the Suzuki-Miyaura cross-coupling of potassium (R)-N-(4-methoxyphenyl)-3-(trifluoroborato)butanamide (2a) and 2-chlorothiophene, the title compound was obtained as a pale yellow solid in 56% yield (37 mg), with spectral data in accordance with that described for the racemate. mp = 82–85 °C. [α]D20 = +15.0 (c = 0.1, MeOH). The title compound was found to have an enantiomeric ratio of >99:1 with an absolute configuration of S for the major enantiomer using SFC analysis (Column OJ-H, 10% i-PrOH, 2 mL, 10 MPa); (S)-isomer retention time = 9.5 min.
N-(4-Methoxyphenyl)-3-(quinolin-6-yl)butanamide
Using the general procedure described for the Suzuki-Miyaura cross-coupling of potassium N-(4-methoxyphenyl)-3-(trifluoroborato)butanamide and 6-chloroquinoline, the title compound was obtained in 58% yield (47 mg) as a pale yellow oil after silica gel column chromatography. mp = 132–135 °C. 1H NMR (500 MHz, CDCl3) δ 8.87 (br s, 1H), 8.03–8.14 (m, 2H), 7.62–7.70 (m, 2H), 7.37–7.45 (m, 1H), 7.23 (d, J = 9.0 Hz, 2H), 7.09 (s, 1H), 6.78 (d, J = 9.0 Hz, 2H), 3.76 (s, 3H), 3.62 (q, J = 7.0 Hz, 1H), 2.66–2.69 (m, 2H), 1.47 (d, J = 7.0 Hz, 3H). 13C NMR (125.8 MHz, CDCl3) δ 169.6, 156.3, 149.7, 147.0, 144.1, 135.9, 130.8, 129.3, 128.8, 128.2, 125.0, 121.9, 121.1, 114.0, 113.9, 55.3, 45.9, 36.7, 21.4. IR (neat) 3290, 3054, 2965, 2924, 1654, 1601, 1510, 1243, 1173, 1035, 833, 685, 635 cm−1. HRMS (ESI) calcd. for C20H21N2O2 [M+H]+ 321.1603, found 321.1606. Using SFC analysis (Column OJ-H, 10% i-PrOH, 2 mL, 10 MPa); (S)-isomer retention time= 7.75 min and (R)-isomer retention time= 8.51 min.
(S)-N-(4-Methoxyphenyl)-3-(quinolin-6-yl)butanamide (12d)
Using the general procedure described for the Suzuki-Miyaura cross-coupling of potassium (R)-N-(4-methoxyphenyl)-3-(trifluoroborato)butanamide (2a) and 6-chloroquinoline, the title compound was obtained as a white solid in 58% yield (47 mg), with spectral data in accordance with that described for the racemate. mp = 133–135 °C. [α]D20 = +47.3 (c = 0.15, MeOH). The title compound was found to have an enantiomeric ratio of 92:7 with an absolute configuration of S for the major enantiomer using SFC analysis (Column OJ-H, 10% i-PrOH, 2 mL, 10 MPa); (S)-isomer retention time= 7.73 min and (R)-isomer retention time= 8.51 min.
tert-Butyl 5-(4-((4-Methoxyphenyl)amino)-4-oxobutan-2-yl)-1H-indole-1-carboxylate
Using the general procedure described for the Suzuki-Miyaura cross-coupling of potassium N-(4-methoxyphenyl)-3- (trifluoroborato)butanamide and tert-butyl 5-chloro-1H-indole-1-carboxylate, the title compound was obtained in 43% yield (44 mg) as a pale yellow oil after silica gel column chromatography. 1H NMR (500 MHz, CDCl3) δ 8.10 (br s, 1H), 7.59 (d, J = 3.7 Hz, 1H), 7.45 (s, 1H), 7.16–7.26 (m, 3H), 6.83–6.98 (m, 1H), 6.79 (d, J = 8.9 Hz, 2H), 6.52 (d, J = 3.7 Hz, 1H), 3.76 (s, 3H), 3.46–3.49 (m, 1H), 2.61–2.66 (m, 2H), 1.68 (s, 9H), 1.42 (d, J = 6.9 Hz, 3H). 13C NMR (90.5 MHz, CDCl3) δ 169.9, 156.3, 149.7, 140.1, 134.0, 130.9, 130.7, 126.2, 123.1, 121.9, 118.9, 115.3, 114.8, 114.0, 107.2, 83.6, 55.4, 47.0, 37.0, 28.2, 22.2. IR (neat) 3277, 2973, 2933, 1734, 1656, 1511, 1450, 1368, 1345, 1244, 1157, 1130, 1083, 1023, 835, 789, 764, 725, 686, 636 cm−1. HRMS (ESI) calcd. for C24H29N2O4 [M+H]+ 409.2127, found 409.2119. Using SFC analysis (Column AS-H, 10% i-PrOH, 2 mL, 10 MPa); (S)-isomer retention time = 14.4 min and (R)-isomer retention time = 15.3 min.
tert-Butyl (S)-5-(4-((4-Methoxyphenyl)amino)-4-oxobutan-2-yl)-1H-indole-1-carboxylate (12e)
Using the general procedure described for the Suzuki-Miyaura cross-coupling of potassium (R)-N-(4-methoxyphenyl)-3- (trifluoroborato)butanamide (2a) and tert-butyl 5-chloro-1H-indole-1-carboxylate, the title compound was obtained as a yellow oil in 43% yield (44 mg), with spectral data in accordance with that described for the racemate. [α]D20 = +38.8 (c = 0.43, MeOH). The title compound was found to have an enantiomeric ratio of >99:1 with an absolute configuration of S for the major enantiomer using SFC analysis (Column AS-H, 10% i-PrOH, 2 mL, 10 MPa); (S)-isomer retention time= 15.3 min.
N-(4-Methoxyphenyl)-3-phenylbutanamide[2]
Using the general procedure described for the Suzuki-Miyaura cross-coupling of potassium N-(4-methoxyphenyl)-3-(trifluoroborato)butanamide and chlorobenzene, the title compound was obtained in 85% yield (57 mg) as a white solid after silica gel column chromatography. mp = 125–128 °C. 1H NMR (360 MHz, CDCl3) δ 7.21–7.25 (m, 7H), 6.98 (br s, 1H), 6.80 (d, J = 9.0 Hz, 2H), 3.76 (s, 3H), 3.34–3.40 (m, 1H), 2.55–2.59 (m, 2H), 1.37 (d, J = 7.0 Hz, 3H). 13C NMR (90.5 MHz, CDCl3) δ 169.8, 156.6, 145.7, 130.7, 128.7, 126.8, 126.6, 122.0, 113.9, 55.4, 46.6, 37.1, 21.6. Using SFC analysis (Column OJ-H, 10% i-PrOH, 2 mL, 10 MPa); (S)-isomer retention time = 11.19 min and (R)-isomer retention time = 12.13 min.
(S)-N-(4-Methoxyphenyl)-3-phenylbutanamide (8)
Using the general procedure described for the Suzuki-Miyaura cross-coupling of potassium (R)-N-(4-methoxyphenyl)-3-(trifluoroborato)butanamide (2a) and chlorobenzene the title compound was obtained as a white solid in 85% yield (57 mg) with spectral data in accordance with that described for the racemate. mp = 123–126 °C. [α]D20 = +57.5 (c = 0.2, MeOH). The title compound was found to have an enantiomeric ratio of 8:92 with an absolute configuration of S for the major enantiomer using SFC analysis (Column OJ-H, 10% i-PrOH, 2 mL, 10 MPa); (S)-isomer retention time = 10.61 min and (R)-isomer retention time = 11.35 min.
3-(4-Fluorophenyl)-N-(4-methoxyphenyl)butanamide[2]
Using the general procedure described for the Suzuki-Miyaura cross-coupling of potassium N-(4-methoxyphenyl)-3-(trifluoroborato)butanamide and 4-chlorofluorobenzene, the title compound was obtained in 89% yield (64 mg) as a pale yellow solid after silica gel column chromatography. mp = 107–109 °C. 1H NMR (300 MHz, CDCl3) δ 7.27 (d, J = 8.9 Hz, 2H), 7.18–7.25 (m, 2H), 7.10 (s, 1H), 7.00 (t, J = 8.7 Hz, 2H), 6.82 (d, J = 9.0 Hz, 2H), 3.78 (s, 3H), 3.33–3.44 (m, 1H), 2.54 (d, J = 7.4 Hz, 2H), 1.35 (d, J = 7.0 Hz, 3H). 13C NMR (75.4 MHz, CDCl3) δ 169.5, 156.4, 141.3, 130.5, 128.1 (d, J = 7.8 Hz), 128.0, 121.9, 115.4, 115.1, 113.9, 55.3, 46.5, 36.2, 21.6. 19F NMR (282.4 MHz, CDCl3) δ −116.58. Using SFC analysis (Column AS-H, 10% i-PrOH, 2 mL, 10 MPa); (S)-isomer retention time= 19.29 min and (R)-isomer retention time= 20.82 min.
(S)-3-(4-Fluorophenyl)-N-(4-methoxyphenyl)butanamide (13a)
Using the general procedure described for the Suzuki-Miyaura cross-coupling of potassium (R)-N-(4-methoxyphenyl)-3-(trifluoroborato)butanamide (2a) and 4-chlorofluorobenzene, the title compound was obtained as a white solid in 89% yield (64 mg) with spectral data in accordance with that described for the racemate. mp = 107–110 °C. [α]D20 = +26.5 (c = 0.2, MeOH). The title compound was found to have an enantiomeric ratio of 9:91 with an absolute configuration of S for the major enantiomer using SFC analysis (Column AS-H, 10% i-PrOH, 2 mL, 10 MPa); (S)-isomer retention time = 19.47 min and (R)-isomer retention time = 21.15 min.
3-(4-Fluorophenyl)-N-phenylbutanamide
Using the general procedure described for the Suzuki-Miyaura cross-coupling of potassium 3-(trifluoroborato)-N-phenylbutanamide and 4-chlorofluorobenzene, the title compound was obtained in 88% yield (57 mg) as a yellow solid after silica gel column chromatography. mp = 80–82 °C. 1H NMR (500 MHz, CDCl3) δ 7.29–7.44 (m, 2H), 7.13–7.28 (m, 2H), 7.04–7.08 (m, 1H), 6.97–7.03 (m, 2H), 6.81–6.87 (m, 2H), 3.36–3.40 (m, 1H), 2.57 (dd, J = 7.4, 3.1 Hz, 2H), 1.34 (d, J = 6.8 Hz, 3H). 13C NMR (125.8 MHz, CDCl3) δ 169.8, 161.3 (d, J = 243.2 Hz), 141.2, 137.5, 128.8, 128.1 (d, J = 7.8 Hz), 124.3, 120.0, 115.3 (d, J = 21.3 Hz), 46.6, 36.1, 21.6.19F NMR (470.8 MHz, CDCl3) δ −116.51. IR (neat) 3241, 3196, 3135, 3075, 2965, 1649, 1598, 1555, 150 1442, 1311, 1224, 833, 757, 692, 658 cm− 1. HRMS (ESI) calcd. for C16H17NOF [M+H]+ 258.1294, found 258.1291. Using SFC analysis (Column OJ-H, 10% i-PrOH, 2 mL, 10 MPa); (S)-isomer retention time= 11.19 min and (R)-isomer retention time= 11.85 min.
(S)-3-(4-Fluorophenyl)-N-phenylbutanamide (13b)
Using the general procedure described for the Suzuki-Miyaura cross-coupling of potassium (R)-3-(trifluoroborato)-N-phenylbutanamide (2c) and 4-chlorofluorobenzene, the title compound was obtained as a white solid in 88% yield (57 mg), with spectral data in accordance with that described for the racemate. mp = 80–82 °C. [α]D20 = +17.5 (c = 0.2, MeOH). The title compound was found to have an enantiomeric ratio of 7:93 with an absolute configuration of S for the major enantiomer using SFC analysis (Column OJ-H, 10% i-PrOH, 2 mL, 10 MPa); (S)-isomer retention time = 11.18 min and (R)-isomer retention time = 11.83 min.
N-Ethyl-3-(4-fluorophenyl)-N-(o-tolyl)butanamide
Using the general procedure described for the Suzuki-Miyaura cross-coupling of potassium N-ethyl-3-(trifluoroborato)-N-(o-tolyl)butanamide and 4-chlorofluorobenzene, the title compound was obtained in 81% yield (61 mg) as a colorless oil. 1H NMR (360 MHz, asterisk denotes minor rotamer peaks, CDCl3) δ 6.90–7.24 (m, 8H), 6.46–6.48* (m, 1H), 4.05–4.09 (m, 1H), 3.37–3.41 (m, 1H), 3.09–3.15 (m, 1H), 2.20 (s, 3H), 2.07–2.17 (m, 2H), 1.93* (s, 3H), 1.16–1.19 (m, 3H), 1.08–1.12 (t, J = 7.1 Hz, 3H), 1.00–1.04* (t, J = 7.1 Hz, 3H). 13C NMR (90.5 MHz, asterisk denotes rotamer peaks, CDCl3) δ 171.0, 170.9*, 140.7, 135.9, 131.4, 131.2*, 129.4, 129.3*, 128.5, 128.4*, 128.3, 128.2*, 126.9, 126.8*, 115.1, 115.0*, 114.8, 114.7*, 43.0, 42.9*, 42.8, 42.7*, 35.8, 35.5*, 21.6, 21.5*, 17.5, 17.2*, 12.8. 19F NMR (338.8 MHz, CDCl3) δ −117.30. IR (neat) 2965, 2929, 2864, 1712, 1651, 1510, 1361, 1220, 769, 727, 686, 639 cm−1. HRMS (ESI) calcd. for C19H23NOF [M+H]+ 300.1764, found 300.1759. Using SFC analysis (Column OD-H, 10% i-PrOH, 2 mL, 10 MPa); (S)-isomer retention time = 14.4 min and (R)-isomer retention time = 15.3 min.
(S)-N-Ethyl-3-(4-fluorophenyl)-N-(o-tolyl)butanamide (13c)
Using the general procedure described for the Suzuki-Miyaura cross-coupling of potassium (R)-N-ethyl-3-(trifluoroborato)-N-(o-tolyl)butanamide (2j) and 4-chlorofluorobenzene, the title compound was obtained as a colorless oil in 81% yield (61 mg) with spectral data in accordance with that described for the racemate. [α]D20 = −3.2 (c = 0.7, MeOH). The title compound was found to have an enantiomeric ratio of 15:85 with an absolute configuration of S for the major enantiomer using SFC analysis (Column OD-H, 10% i-PrOH, 2 mL, 10 MPa); (S)-isomer retention time = 11.75 min and (R)-isomer retention time = 14.4 min.
N-Cyclohexyl-3-(4-fluorophenyl)butanamide[2]
Using the general procedure described for the Suzuki-Miyaura cross-coupling of potassium N-cyclohexyl-3-(trifluoroborato)butanamide and 4-chlorofluorobenzene, the title compound was obtained in 83% yield (55 mg) as a white solid after silica gel column chromatography. mp = 132–134 °C. 1H NMR (300 MHz, CDCl3) δ 7.12–7.26 (m, 2H), 6.90–7.04 (m, 2H), 5.32 (d, J = 9.1 Hz, 1H), 3.60–3.81 (m, 1H), 3.26–3.30 (m, 1H), 2.31–2.47 (m, 2H), 1.79–1.93 (m, 1H), 1.50–1.76 (m, 4H), 1.27 (d, J-7.0 Hz, 3H), 0.83–1.25 (m, 5H). 13C NMR (75 MHz, CDCl3) δ 170.2, 162.9 (d, J = 243.5 Hz), 141.4, 128.10 (d, J = 7.8 Hz), 115.0 (d, J=21.1 Hz), 47.8, 46.0, 36.3, 32.9, 32.8, 25.3, 24.7, 24.6, 21.5. 19F NMR (282.4 MHz, CDCl3) δ −116.94. Using SFC analysis (Column AS-H, 10% i-PrOH, 2 mL, 10 MPa); (S)-isomer retention time = 8.41 min and (R)-isomer retention time = 9.81 min.
(S)-N-Cyclohexyl-3-(4-fluorophenyl)butanamide (13d)
Using the general procedure described for the Suzuki-Miyaura cross-coupling of potassium (R)-N-cyclohexyl-3-(trifluoroborato)butanamide (2d) and 4-chlorofluorobenzene, the title compound was obtained as a white solid in 83% yield (55 mg) with spectral data in accordance with that described for the racemate. mp = 131–134 °C. [α]D20 = −41.3 (c = 0.15, MeOH). The title compound was found to have an enantiomeric ratio of 5:95 with an absolute configuration of S for the major enantiomer using SFC analysis (Column AS-H, 10% i-PrOH, 2 mL, 10 MPa); (S)-isomer retention time = 8.19 min and (R)-isomer retention time = 9.64 min.
N-Cyclohexyl-3-(4-fluorophenyl)pentanamide
Using the general procedure described for the Suzuki-Miyaura cross-coupling of potassium N-cyclohexyl-3-(trifluoroborato)pentanamide and 4-chlorofluorobenzene, the title compound was obtained in 85% yield (59 mg) as a white solid after silica gel column chromatography. mp = 102–104 °C. 1H NMR (360 MHz, CDCl3) δ 7.14–7.18 (m, 2H), 6.90–7.06 (m, 2H), 4.96 (s, 1H), 3.65–3.69 (m, 1H), 2.95–3.03 (m, 1H), 2.48 (dd, J = 13.7, 6.0 Hz, 1H), 2.25 (dd, J = 13.7, 9.1 Hz, 1H), 1.51–1.84 (m, 8H), 1.21–1.36 (m, 2H), 0.90–1.18 (m, 2H), 0.79 (t, J = 7.4 Hz, 3H). 13C NMR (90.5 MHz, CDCl3) δ 170.4, 161.3 (d, J = 239 Hz), 139.7, 128.9 (d, J = 8.1 Hz), 115.18 (d, J = 20.9 Hz), 47.8, 44.9, 44.0, 33.0, 29.1, 25.4, 24.7, 11.9. 19F NMR (338.8 MHz, CDCl3) δ −117.44. IR (neat) 3302, 2930, 2851, 1637, 1542, 1508, 1445, 1223, 1160, 834, 684, 649, 638 cm−1. HRMS (ESI) calcd. For C17H25NOF [M+H]+ 278.1920, found 278.1916. Using SFC analysis (Column AS-H, 10% i-PrOH, 2 mL, 10 MPa); (S)-isomer retention time = 13.9 min and (R)-isomer retention time = 14.4 min.
(S)-N-Cyclohexyl-3-(4-fluorophenyl)pentanamide (13e)
Using the general procedure described for the Suzuki-Miyaura cross-coupling of potassium (R)-N-cyclohexyl-3-(trifluoroborato)pentanamide (2f) and 4-chloroflourobenzene, the title compound was obtained as a white solid in 85% yield (59 mg), with spectral data in accordance with that described for the racemate. mp = 102–104 °C. [α]D20 = +109.5 (c = 0.2, MeOH). The title compound was found to have an enantiomeric ratio of 10:90 with an absolute configuration of (S) for the major enantiomer using SFC analysis (Column AS-H, 10% i-PrOH, 2 mL, 10 MPa); (S)-isomer retention time = 19.11 min and (R)-isomer retention time = 14.4 min.
Supplementary Material
Acknowledgments
This research was supported by the NIGMS (R01 GM-081376) and an NSF GOALI grant. Melvin Lim and Dr. Sarah Trice (University of Pennsylvania) are acknowledged for their assistance in the initial optimization with Taniaphos T001. BASF and AllyChem Co. Ltd. are acknowledged for their generous donations of bisboronic acid. BASF is also acknowledged for their generous donation of tetrakis(dimethylamino)diboron. Frontier Scientific is acknowledged for their donation of palladium salts. Dr. Rakesh Kohli (University of Pennsylvania) is acknowledged for obtaining HRMS data.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/adsc.201######.
References
- 1.Chea H, Sim HS, Yun J. Adv Synth Catal. 2009;351:855. [Google Scholar]
- 2.Sandrock DL, Jean-Gerard L, Chen CY, Dreher SD, Molander GA. J Am Chem Soc. 2010;132:17108. doi: 10.1021/ja108949w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Kabalka GW, Das BC, Das S. Tetrahedron Lett. 2002;43:2323. [Google Scholar]; b) Shiomi T, Adachi T, Toribatake K, Zhou L, Nishiyama H. Chem Commun. 2009:5987. doi: 10.1039/b915759j. [DOI] [PubMed] [Google Scholar]
- 4.a) Lillo V, Geier MJ, Westcott SA, Fernandez E. Org Biomol Chem. 2009;7:4674. doi: 10.1039/b909341a. [DOI] [PubMed] [Google Scholar]; b) Bonet A, Gulyas H, Koshevoy IO, Estevan F, Sanau M, Ubeda MA, Fernandez E. Chem Eur J. 2010;16:6382. doi: 10.1002/chem.200903095. [DOI] [PubMed] [Google Scholar]
- 5.a) Lawson Y, Lesly G, Marder T, Norman N, Rice C. Chem Commun. 1997:2051. [Google Scholar]; b) Bell J, Cox A, Cameron N, Evans J, Marder T, Duin M, Elsevier C, Baucherel X, Tulloch A, Tooze R. Chem Commun. 2004:1854. doi: 10.1039/b406052k. [DOI] [PubMed] [Google Scholar]
- 6.Hirano K, Yorimitsu H, Oshima K. Org Lett. 2007;9:5031. doi: 10.1021/ol702254g. [DOI] [PubMed] [Google Scholar]
- 7.a) Bonet A, Gulyas H, Fernandez E. Angew Chem Int Ed. 2010;49:5130. doi: 10.1002/anie.201001198. [DOI] [PubMed] [Google Scholar]; b) Pubill-Ulldemolins C, Bonet A, Gulyas H, Bo C, Fernandez E. Org Biomol Chem. 2012;10:9677. doi: 10.1039/c2ob26899j. [DOI] [PubMed] [Google Scholar]; c) Pubill-Ulldemolins C, Bonet A, Bo C, Gulyas H, Fernandez E. Chem Eur J. 2012;18:1121. doi: 10.1002/chem.201102209. [DOI] [PubMed] [Google Scholar]
- 8.Wu H, Radomkit S, O’Brien JM, Hoveyda AH. J Am Chem Soc. 2012;134:8277. doi: 10.1021/ja302929d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fischer C, Koenig B. Beilstein J Org Chem. 2011;7:59. doi: 10.3762/bjoc.7.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Molander GA, Trice SLJ, Dreher SD. J Am Chem Soc. 2010;132:17701. doi: 10.1021/ja1089759. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Molander GA, Trice SLJ, Kennedy SM, Dreher SD, Tudge MT. J Am Chem Soc. 2012;134:11667. doi: 10.1021/ja303181m. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Molander GA, McKee SA. Org Lett. 2011;13:4684. doi: 10.1021/ol201900d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Molander GA, Trice SLJ, Kennedy SM. Org Lett. 2013;14:4814. doi: 10.1021/ol302124j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ishiyama T, Murata M, Ahiko T, Miyaura N. Org Synth. 2004;10:115. [Google Scholar]
- 13.Bagutski V, Ros A, Aggarwal V. Tetrahedron. 2009;65:9956. [Google Scholar]
- 14.Bull JA. Angew Chem Int Ed. 2012;51:8930. doi: 10.1002/anie.201203876. [DOI] [PubMed] [Google Scholar]
- 15.For reviews on potassium organotrifluoroborates, see: Molander GA, Figueroa R. Aldrichimica Acta. 2005;38:49.Molander GA, Ellis N. Acc Chem Res. 2007;40:275. doi: 10.1021/ar050199q.Stefani HA, Cella R, Vieira AS. Tetrahedron. 2007;63:3623.Darses S, Genet JP. Chem Rev. 2008;108:288. doi: 10.1021/cr0509758.Molander GA, Jean-Gerard L. Organotrifluoroborates: Organoboron Reagents for the Twenty-First Century. In: Hall DG, editor. Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials. Vol. 2. Wiley-VCH; Weinheim: 2011. pp. 507–550.Molander GA, Jean-Gerard L. Cross-Coupling Reactions of Organotrifluoroborate Salts. In: Denmark SE, editor. Organic Reactions. Vol. 79. John Wiley & Sons. Inc; New York: 2013. pp. 1–316.
- 16.a) Joule JA, Mills K. Heterocyclic Chemistry. 5. Wiley; West Sussex: 2010. pp. 48–50. [Google Scholar]; b) Hall DG, editor. Boronic Acids. Wiley-VCH; Weinheim: 2005. [Google Scholar]
- 17.Molander GA, Wisniewski SR. J Am Chem Soc. 2012;134:16856. doi: 10.1021/ja307861n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.See Supporting Information for HTE information.
- 19.a) Li W, Zhang Z, Xiao D, Zhang X. Tet Lett. 1999;40:6701. [Google Scholar]; b) Li W, Zhang Z, Xiao D, Zhang X. J Org Chem. 2000;65:3489. doi: 10.1021/jo000066c. [DOI] [PubMed] [Google Scholar]; c) RajanBabu TV, Yan Y-Y, Shin S. J Am Chem Soc. 2001;123:10207. doi: 10.1021/ja011500a. [DOI] [PubMed] [Google Scholar]
- 20.a) Ireland T, Grossheimann G, Wieser-Jeunesse C, Knochel P. Angew Chem Int Ed. 1999;38:3212. [PubMed] [Google Scholar]; b) Almassy A, Skvorcova A, Horvath B, Bilcik F, Bariak V, Rakovsky E, Sebesta R. Eur J Org Chem. 2013:111. [Google Scholar]
- 21.Thadani AN, Ntaganda R, Beshai MM. Chiral Acyclic Diaminocarbene Ligands, Precursors, Therefore and Their Use In Organic Synthesis Reactions. WO2010/003226 A1. 2010 Jan 14;
- 22.See Supporting Information for optimization with Taniaphos T001
- 23.See Supporting Information for enantiomeric ratios.
- 24.For reviews of the Suzuki-Miyaura cross-coupling reaction, see: Miyaura N, Suzuki A. Chem Rev. 1995;95:2457.Bellina F, Carpita A, Rossi R. Synthesis. 2004:2419.Kotha S, Lahiri K, Kashinath D. Tetrahedron. 2002;58:9633.Sandrock DL. Alkylboron Reagents. In: Molander GA, editor. Science of Synthesis, Cross Coupling and Heck-Type Reactions 1. Vol. 1 Georg Thieme Verlag; Stuttgart, Germany: 2013.
- 25.For examples of the transition metal-catalyzed hydrogenation, see Shang J, Han Z, Li Y, Wang Z, Ding K. Chem Commun. 2012;48:5172. doi: 10.1039/c2cc30812f.Ikeno T, Kimura T, Ohtsuka Y, Yamada T. Synlett. 1999:96.Yamada T, Ohtsuka Y, Ikeno T. Chem Lett. 1998;11:1129.Alimardanov A, Nikitenko A, Potoski JR, Springer DM, Kreft AF, Tadayon S, Ho DM, Connolly TJ, Feigelson G, Chan AW, Ding Z, Ghosh M, Shi X, Ren J, Hansen E, Farr R, MacEwan M. Org Proc Res Dev. 2009;13:1161.
- 26.Gianolli E, Giannini E, Bigini L, Piccolo O, Holmberg P, Lundholm T. Process For the Preparation of N,N-Diisopropyl-3-(2-Hydroxy-5-Methylphenyl)-3-Phenylpropylamine and its Salts Starting From a Novel Intermediate. WO2012/098044 A1. 2012 Jul 26;
- 27.a) Cordova A. Catalytic Asymmetric Conjugate Reactions. Wiley-VCH; Weinheim, Germany: 2010. [Google Scholar]; b) Shao C, Yu HJ, Wu NY, Tian P, Wang R, Feng CG, Lin GQ. Org Lett. 2011;13:788. doi: 10.1021/ol103054a. [DOI] [PubMed] [Google Scholar]; c) Gendrineau T, Chuzel O, Eijsberg H, Genet JP, Darses S. Angew Chem Int Ed. 2008;47:7669. doi: 10.1002/anie.200803230. [DOI] [PubMed] [Google Scholar]
- 28.a) Ohmura T, Awano T, Suginome M. J Am Chem Soc. 2010;132:13191. doi: 10.1021/ja106632j. [DOI] [PubMed] [Google Scholar]; b) Awano T, Ohmura T, Suginome M. J Am Chem Soc. 2011;133:20738. doi: 10.1021/ja210025q. [DOI] [PubMed] [Google Scholar]
- 29.Kinzel T, Zhang Y, Buchwald SL. J Am Chem Soc. 2010;132:14073. doi: 10.1021/ja1073799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.For other references about the synthesis of aminobiphenylpalladium precatalysts, see: Albert J, Granell J, Zafrilla J, Font-Bardia M, Solans X. J Organomet Chem. 2005;690:422.Albert J, Granell J, Luque A, Minguez J, Moragas R, Font-Bardia M, Solans X. J Organomet Chem. 1996;522:87.
- 31.a) Labadie JW, Stille JK. J Am Chem Soc. 1983;105:6129. [Google Scholar]; b) Kells KW, Chong JM. J Am Chem Soc. 2004;126:15666. doi: 10.1021/ja044354s. [DOI] [PubMed] [Google Scholar]
- 32.Hatanaka Y, Hiyama T. J Am Chem Soc. 1990;112:7793. [Google Scholar]
- 33.a) Ridgway BH, Woerpel KA. J Org Chem. 1998;63:458. doi: 10.1021/jo970803d. [DOI] [PubMed] [Google Scholar]; b) Matos K, Soderquist JA. J Org Chem. 1998;63:461. doi: 10.1021/jo971681s. [DOI] [PubMed] [Google Scholar]
- 34.a) Amatore C, Jutand A, Le Duc G. Chem Eur J. 2011;17:2492. doi: 10.1002/chem.201001911. [DOI] [PubMed] [Google Scholar]; b) Carrow BP, Hartwig JF. J Am Chem Soc. 2011;133:2116. doi: 10.1021/ja1108326. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Amatore C, Le Duc G, Jutand A. Chem Eur J. 2013;19:10082. doi: 10.1002/chem.201300177. [DOI] [PubMed] [Google Scholar]; d) Lennox AJJ, Lloyd-Jones GC. Angew Chem Int Ed. 2013;52:7362. doi: 10.1002/anie.201301737. [DOI] [PubMed] [Google Scholar]
- 35.a) Bergbreiter DE, Rainville DP. J Organomet Chem. 1976;121:19. [Google Scholar]; b) Kabalka GW, Gooch EE., III J Org Chem. 1980;45:3578. [Google Scholar]; c) Larouche-Gauthier R, Elford TG, Aggarwal VK. J Am Chem Soc. 2011;133:16794. doi: 10.1021/ja2077813. [DOI] [PubMed] [Google Scholar]
- 36.Tang S, Sun Y, He L, She X, He J. J Org Chem. 2010;6:1961. doi: 10.1021/jo1000065. [DOI] [PubMed] [Google Scholar]
- 37.Dong-Sheng L. Tetrahedron: Asymmetry. 2009;17:2014. [Google Scholar]
- 38.Li W. Org Lett. 2011;15:3876. doi: 10.1021/ol201406w. [DOI] [PubMed] [Google Scholar]
- 39.Annunziata R, Cinquini M, Cozzi F, Montanari F, Restelli A. Tetrahedron. 1984;19:3815. [Google Scholar]
- 40.Wan X, Sun Y, Luo Y, Li D, Zhang Z. J Org Chem. 2005;70:1070. doi: 10.1021/jo048466d. [DOI] [PubMed] [Google Scholar]
- 41.Rehdorf J, Lengar A, Bornscheuer UT, Mihovilovic MD. Bioorg Med Chem Lett. 2009;19:3739. doi: 10.1016/j.bmcl.2009.05.014. [DOI] [PubMed] [Google Scholar]
- 42.Rahaim RJ, Maleczka RE. Org Lett. 2011;13:584. doi: 10.1021/ol102757v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Singh P, Bhardwaj A. J Med Chem. 2010;53:3707. doi: 10.1021/jm1001327. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.