

Abstract
Despite the complexity and heterogeneity of mood disorders, basic and clinical research studies have begun to elucidate the pathophysiology of depression and to identify rapid, efficacious antidepressant agents. Stress and depression are associated with neuronal atrophy, characterized by loss of synaptic connections in key cortical and limbic brain regions implicated in depression. This is thought to occur in part via decreased expression and function of growth factors, such as brain-derived neurotrophic factor (BDNF), in the prefrontal cortex (PFC) and hippocampus. These structural alterations are difficult to reverse with typical antidepressants. However, recent studies demonstrate that ketamine, an N-methyl-D-aspartate (NMDA) receptor antagonist that produces rapid antidepressant actions in treatment-resistant depressed patients, rapidly increases spine synapses in the PFC and reverses the deficits caused by chronic stress. This is thought to occur by disinhibition of glutamate transmission, resulting in a rapid but transient burst of glutamate, followed by an increase in BDNF release and activation of downstream signaling pathways that stimulate synapse formation. Recent work demonstrates that the rapid-acting antidepressant effects of scopolamine, a muscarinic receptor antagonist, are also associated with increased glutamate transmission and synapse formation. These findings have resulted in testing and identification of additional targets and agents that influence glutamate transmission and have rapid antidepressant actions in rodent models and in clinical trials. Together these studies have created tremendous excitement and hope for a new generation of rapid, efficacious antidepressants.
Keywords: BDNF, glutamate, ketamine, mTORC1, prefrontal cortex, rapamycin, scopolamine, stress, synaptogenesis
Abstract
A pesar de la complejidad y heterogeneidad de los trastornos del ánimo, los estudios de investigación básicos y clínicos han comenzado a aclarar la fisio patología de la depresión y a identificar agentes antidepresivos rápidos y eficaces. El estrés y la depresión están asociados con atrofia neural, caracterizada por pérdida de conexiones sinápticas en regiones corticales y Iímbicas claves que están implicadas en la depresión. Se cree que esto ocurre en parte a través de la reducción de la expresión y función de los factores de crecimiento, como el factor neurotrófico derivado del cerebro (BDNF) en la corteza prefrontal (CPF) y en el hipocampo. Estas alteraciones estructurales son difíciles de revertir con antidepresivos típicos. Sin embargo; estudios recientes demuestran que la ketamina, un antagonista del receptor N-metil-D-aspártico (NMDA) que produce rápidas acciones anti-depresivas en los pacientes con depresión resistente al tratamiento, aumenta rápidamente las espinas sinápticas en la CPF y revierte los déficits causados por el estrés crónico. Se cree que esto ocurre por desinhíbición de la transmisión glutamatérgica, lo que se traduce en un incremento rápido, pero transitorio de glutamato, seguido de un aumento de la liberación de BDNF y activación de las vías de señales hacia abajo que estimulan la formación de sinapsis. Un trabajo reciente demuestra que los efectos antidepresivos de rápida acción de la escopolamina, un antagonista del receptor muscarínico, también están asociados con un aumento de la transmisión glutamatérgíca y la formación de sinapsís. Estos hallazgos se han traducido en pruebas e identificación de objetivos adicionales y agentes que afectan la transmisión glutamatérgica, y tienen rápida acción antidepresiva en modelos de roedores y en ensayos clínicos. En conjunto estos estudios han generado grandes ilusiones y esperanzas para una nueva generación de antídepresivos rápidos y eficaces.
Abstract
Malgré la complexité et l'hétérogénéité des troubles de l'humeur, des études de recherche clinique et fondamentale ont commencé à élucider la physiopathologie de la dépression et à identifier des antidépresseurs rapides et efficaces. Le stress et la dépression sont associés à une atrophie neuronale, caractérisée par la perte des connexions synaptiques dans les régions cérébrales limbiques et corticales impliquées dans la dépression, ce qui se manifeste en partie par une diminution de l'expression et de la fonction des facteurs de croissance, comme le facteur neurotrophique dérivé du cerveau (BDNF), dans le cortex préfrontal (CPF) et l'hippocampe. Ces altérations structurales sont difficiles à supprimer avec les antidépresseurs classiques. Cependant, d'après des études récentes, la kétamine, un antagoniste du récepteur du N-méthyl-D-aspartate (NMDA) qui induit une action antidépressive rapide chez des patients déprimés résistants au traitement, augmente rapidement la formation des synapses avec les épines dendritiques dans le CPF et s'oppose aux déficits causés par le stress chronique. Ce mécanisme intervient par désinhibition de la transmission du glutamate, aboutissant à sa stimulation rapide mais brève, suivie d'une augmentation de la libération du BDNF et d'une activation des voies de signalisation en aval, qui stimulent la formation des synapses. Un travail récent démontre que les effets antidépresseurs d'action rapide de la scopolamine, un antagoniste du récepteur muscarinique, s'associent également à une augmentation de la transmission du glutamate et de la formation des synapses. Ces résultats ont conduit à vérifier et identifier dans un modèle murin et dans des études cliniques, les produits et les cibles supplémentaires influant sur la transmission du glutamate et ayant une action antidépressive rapide. Ces études ont toutes suscité un espoir et une excitation considérables pour une nouvelle génération d'antidépresseurs rapides et efficaces.
Introduction
Depression is a widespread, devastating illness, affecting approximately 17% of the population at some point in life, resulting in enormous personal suffering and economic loss.1 Moreover, currently available treatments have significant limitations (ie, low response rates, treatment resistance, high incidence of relapse, and a time-lag of weeks to months for a response), highlighting a major unmet need for more efficacious and faster-acting antidepressants, particularly with the high suicide rates in depression.2 Further confusing matters, depression is a heterogeneous syndrome, making it difficult to identify the underlying pathophysiology, and thereby complicating the diagnosis and treatment of depression and related mood disorders.
Despite these problems, significant progress is being made in identifying the brain regions and circuits that control emotion, mood, and anxiety, and the neurochemical and cellular alterations underlying depression and stress-related disorders. Emerging from these basic research and clinical studies is evidence that stress and depression cause neuronal atrophy and loss in brain regions that control mood and emotion, resulting in disconnection and loss of function.3-5 This includes a reduction in the number of spine synapses, the key points of connection between neurons. There is also evidence of hypertrophy and increased function of other brain regions that could contribute to dysregulation of mood and anxiety. Progress has also been made in characterizing the pathophysiological processes that contribute to the structural alterations in the brain and to depressive behaviors. In addition to the well-established role of elevated hypothalamic-pituitary-adrenal (HPA) axis activity, a hallmark of stress responses, disruptions of neurotrophic/growth factors have been the focus of work in recent years and could be involved in the atrophy and loss of neurons in response to stress and depression.
Although the development of novel therapeutic agents has proven extremely difficult, recent studies have found that previously known psychotropic drugs are capable of producing startling antidepressant responses. Most notably, N-methyl-D-aspartate (NMDA) receptor antagonists (eg, ketamine) produce rapid and long-lasting antidepressant actions in treatment-resistant patients.6,7 Similarly, the muscarinic receptor antagonist scopolamine also produces rapid antidepressant actions.8,9 These rapid effects, by mechanisms completely different from typical monoamine reuptake inhibitors, represent the most significant findings in the field of depression in over 60 years. Moreover, we have found that the actions of ketamine and scopolamine are dependent on the induction of new spine synapses, which rapidly reverse the synaptic atrophy caused by stress and depression, resulting in reconnection of critical cortical-limbic circuits. These findings represent a fundamental shift in our understanding of the mechanisms underlying the rapid actions of NMDA and muscarinic receptor blockade, and identify novel targets for the treatment of depression.
Together, these studies support a hypothesis that alterations of synaptogenesis and neural plasticity that result in functional disconnections underlie the pathophysiology and treatment of depression. Negative factors that decrease synaptogenesis in a circuit-specific manner (eg, stress, excess glucocorticoids, reduced trophic support, and increased inflammatory cytokines) increase vulnerability and/or cause depression, while treatments or conditions that enhance synaptogenesis and increase connectivity (eg, neurotrophic factors, NMDA antagonists) have antidepressant actions and reduce vulnerability. The current review will focus on the signaling pathways and targets that underlie neuronal atrophy, and conversely those that contribute to the reversal of synaptic deficits caused by stress and depression, notably the role of neurotrophic factors. However, there are also other dysregulated systems of interest that will not be covered in this review, including proinflammatory cytokines, sex steroids (notably estrogen), and metabolic/feeding factors (ie, insulin/diabetes, leptin, and grehlin) to name a few that play an important role in the pathophysiology of depression and that could provide new therapeutic targets.
Disrupted connectivity of cortical and limbic depression circuitry
Brain imaging and postmortem studies have identified key structures involved in the regulation of mood and depression, including the prefrontal cortex (PFC), hippocampus, cingulate cortex, amygdala, and basal ganglia.5,10 Blood flow and functional imaging studies have identified regions with reduced (PFC and hippocampus) or increased (subcallosal cingulate cortex and amygdala) activity. Together these studies have contributed to the emergence of a functional depression circuit. Altered activity and functional connections between the regions within this circuit correlate with unbalanced behavior that characterizes depression (see refs 4,5,10 for a complete description and discussion of these models). One model is based on extensive evidence of decreased function and activity of the PFC and connections to and from other limbic and subcortical structures implicated in depression.5 Decreased function of the PFC in depressed patients underlies certain abnormal responses (eg, decreased reaction time and cognitive function), as well as a corresponding gain of function of the amygdala (loss of control of emotion and mood, and increased fear anxiety and IIPA axis reactivity), which is negatively controlled by PFC. Disruption of connections between the PFC and amygdala, as well as the ventral striatum, could also contribute to decreased motivation and reward in depression, which could underlie disrupted eating, sleeping, and libido.
While this circuit model may be oversimplified, there is sufficient evidence from human, as well as animal studies (see below) to support the hypothesis that there is loss of function, in part from decreased connections between PFC and other brain regions. Disconnection between these regions could occur at various levels, including reductions in white matter tracts,5 but also more subtle changes in synapse number, which cannot be observed using current brain imaging approaches. Novel treatment strategies, including NMDA receptor antagonists, could act via reversal of these synaptic deficits.
Stress and depression cause neuronal atrophy and loss: circuit-specific effects
Among the findings of altered brain structure and function in depression, the most consistent is reduced volume of the PFC and hippocampus (Figure 1).11,12 Reduced volume is inversely correlated with length of illness, time of treatment, and severity of depression. Postmortem studies also demonstrate reduced neuronal cell body size, atrophy of processes, and a reduction of glia in PFC.13-15 Although ultrastructural studies, such as spine synapse analysis, have been difficult in postmortem tissue, a recent report demonstrates a decrease in the number of synapses in the PFC in a small cohort of depressed subjects.16 Findings in rodent models have extended the human studies, and confirm that exposure to chronic stress, like depression, causes atrophy and loss of neurons and glia in the PFC and hippocampus, which could contribute to the decreased volume reported in patients.3,13,17,18 The atrophy of neurons is characterized by a decrease in the number and length of dendrite branches, as well as a decrease in the density of spine synapses.18-20
Figure 1. Chronic stress causes neuronal atrophy: a decreased number of spine synapses. Basic research studies demonstrate that repeated stress causes atrophy of neurons in the prefrontal cortex and hippocampus of rodents. Shown on the left is a diagram of a segment of a dendrite that is decorated with spines, and the reduction in spine number after exposure to repeated stress. On the right are examples of two photon laser microscopy images of neurobiotin-labeled dendrites from layer V pyramidal neurons in the prefrontal cortex of rats housed under control conditions or after exposure to immobilization stress (7 days, 45 minutes per day).
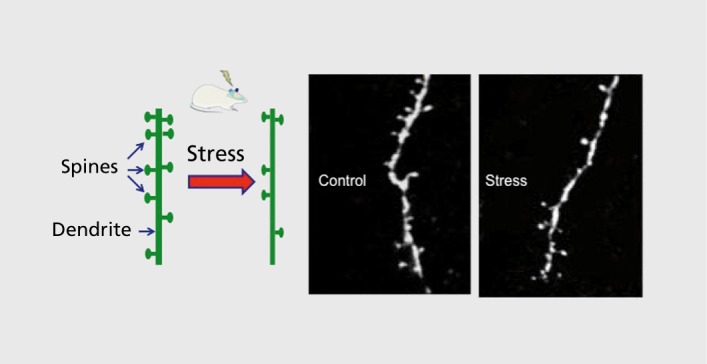
In contrast to the atrophy observed in PFC and hippocampus, stress causes hypertrophy of neurons in the amygdala, which could contribute to increased emotional valence and anxiety in depression.21 Hypertrophy and increased function of amygdala could result from atrophy and decreased function of PFC, as this region provides inhibitory control of amygdala and could thereby contribute to depressive symptoms. Similarly, atrophy and decreased function of PFC neurons could contribute to abnormal function of other aspects of the depression circuit (eg, increased activity of basal ganglia and subcallosal cingulate cortex).
Stress decreases neuronal and glial proliferation
In addition to decreased neuronal arborization and synapse number, stress also exerts a significant impact on the birth of new neurons and glia. Acute and chronic stress decreases neurogenesis in the dentate gyrus of adult hippocampus.22 There have also been several studies of cell proliferation in the hippocampus of postmortem depressed subjects, although there is no evidence to date of decreased neurogenesis, indicating that loss of neurons does not contribute to depression.23 Rather, these postmortem studies have reported that depressed patients treated with antidepressants have increased levels of newborn cells in the hippocampus. This is consistent with preclinical studies demonstrating that chronic administration of most classes of antidepressants increase neurogenesis in the adult hippocampus.
Decreased glial number has been one of the most consistent findings of postmortem studies.24,25 This includes decreased numbers of astrocytes and oligodendrocytes in PFC. Preclinical studies also demonstrate that stress decreases glial fibrillary acidic protein (GFAP)-labeled astrocytes as well as the proliferation of oligodendrocytes.24
Conversely, chronic antidepressant administration increases the proliferation of oligodendrocytes in the PFC.24 Glial cells play an important role in providing metabolic support for neurons, and loss of glia could contribute to the atrophy and loss of neurons caused by stress and depression. This is an interesting, yet understudied, area of research that has important implications for elucidating the pathophysiology of stress and depression.
Stress decreases brain-derived neurotrophic factor expression
One of the mechanisms that has been studied for the atrophy and loss of neurons caused by stress and depression is disruption of neurotrophic/growth factor support, most notably brain-derived neurotrophic factor (BDNF) (Figure 2). Neurotrophic factors were first identified and characterized for their role during development, including guidance, maturation, and survival of neurons, but it is now well-established that these factors continue to play an important role in the adult brain, including activity-dependent synaptic plasticity as well as survival.26 BDNF, a member of the nerve growth factor family, is one of the most highly expressed neurotrophic factors in the brain, and can be regulated at the level of gene expression as well as activity-stimulated release at synapses. Stress significantly impacts the expression of BDNF, decreasing levels of messenger RNA (mRNA) and protein in the hippocampus and PFC.3,4 A possible role for BDNF in depression is supported by studies demonstrating that levels of this factor are decreased in postmortem cerebral cortex of depressed subjects.3 Surprisingly, blood levels of BDNF are also decreased in depressed subjects.27 Studies of BDNF heterozygous deletion mutant mice have not revealed an outright depressive phenotype as might be expected, although BDNF deletion increases vulnerability to depressive behavior in rodents, measured in models of despair, anxiety, and anhedonia.28 The finding that the BDNF deletion mutants do not display depressive behavior could be due to region-specific effects of BDNF (ie, BDNF is prodepressant in the mesolimbic dopamine system, but antidepressant in the PFC and hippocampus).29 This possibility is supported by recent studies demonstrating that hippocampus-specific knockdown of BDNF produces depressive behavior.30
Figure 2. Stress and depression decrease, while rapid-acting antidepressants (eg, ketamine) increase, synaptic connections. Under normal, nonstress conditions, synapses of glutamate terminals are maintained and regulated by circuit activity and function, including activity-dependent release of brain derived neurotrophic factor (BDNF) and downstream signaling pathways. Stress and depression are associated with neuronal atrophy and decreased synaptic connections in the prefrontal cortex and hippocampus. This is thought to occur via decreased expression and release of BDNF as well as increased levels of adrenal glucocorticoids. This decrease has been compared with long-term depression (LTD). Rapid-acting antidepressants, notably ketamine, cause a burst of glutamate that results in an increase in synaptogenesis that has been compared with long-term potentiation (LTP). The increase in glutamate is thought to occur via blockade of N-methyl-Daspartate (NMDA) receptors located on inhibitory γ-aminobutyric acid (GABA)--ergic neurons, resulting in disinhibition of glutamate transmission. The burst of glutamate increases BDNF release and causes activation of mammalian target of rapamycin (mTOR) signaling, which then increases the synthesis of synaptic proteins required for new spine synapse formation. These new connections allow for proper circuit activity and normal control of mood and emotion. However, the new synapses are unstable and are lost after about 10 days, which coincides with depression relapse in patients. Akt, protein kinase b; AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionicacid; ERK, extracellular signal-regulated kinases; GABA, γ-aminobutyric acid; GSK, glycogen synthase kinase; PP1, phosphoprotein phosphatase 1; TrkB, tropomyosin receptor kinase B.

Studies of neuronal morphology demonstrate the importance of BDNF in maintaining normal dendrite and spine integrity. BDNF heterozygous deletion mutants have decreased dendrite length and branching in the hippocampus, similar to the effects of chronic stress; moreover, the effects of stress are occluded in these mice.31,32 Similar dendrite and synaptic deficits in the hippocampus, as well as in the PFC, have been observed in mutant mice with a knock-in of a loss of function BDNF polymorphism, Val66Met.32-34 This polymorphism, found in approximately 25% of the human population, decreases the processing, transport, and activity-dependent release of BDNF.35 Carriers of the Met allele have a decrease in hippocampal volume and are at increased risk of developing depression when exposed to early life stress or trauma.12,36 These studies demonstrate that stress decreases BDNF expression and that loss of BDNF, or mutation that reduces release, negatively influences dendrite morphology and behavior.
New targets to reverse neuronal atrophy: remodeling synaptic connections
The evidence that major depressive disorder (MDD) is associated with decreased volume of cortical and limbic brain regions, atrophy of neurons, and decreased number of synaptic connections, indicates that depression- and stress-related illnesses are mild neurodegenerative disorders. There is also evidence that the magnitude of the volume reduction is inversely correlated with the length of antidepressant treatment, and some direct evidence that treatment can reverse the deficit, indicating that the atrophy is reversible.3,12,37 This is supported by preclinical studies demonstrating that administration of a typical antidepressant can block or reverse the loss of synapses caused by chronic stress exposure,38,39 as well as enhance synaptic plasticity.40,41 However, the ability of typical antidepressants to reverse neuronal atrophy is limited and requires chronic treatment.
The requirement for chronic administration of typical antidepressants that influence monoamines is not surprising, as the neurotransmitter systems are considered to be modulators of synaptic activity. In contrast, drugs that target glutamate, the major excitatory neurotransmitter in the brain that controls rapid as well as longterm synaptic plasticity, could produce more profound and rapid alterations. The rapid actions of glutamate are mediated via regulation of glutamate ionotropic receptors, including the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and NMDA subtypes, which are required for cellular and behavioral models of short- and long-term learning and memory, including regulation of the number and function of spine synapses.42,43
Recent studies have provided compelling evidence that the glutamate neurotransmitter system is an important and relevant target for novel and rapid-acting antidepressants. Discussed below are the clinical and preclinical studies demonstrating that NMDA receptor antagonists are rapid-acting antidepressants, as well as evidence for other glutamate-related targets, including metabotropic glutamate 2/3 (mGlu2/3) receptor antagonists and AMPA receptor modulators.
NMDA receptor antagonists produce rapid, efficacious antidepressant actions
Given that glutamate is the major excitatory neurotransmitter system in the brain, it is not surprising that it has been implicated in a number of psychiatric illnesses, including depression and schizophrenia. This has led to clinical and preclinical studies of agents like ketamine, an NMDA antagonist, which when administered at a high dose is a dissociative anesthetic, but at low doses has mild psychotomimetic effects. In what is now considered a seminal study Berman and colleagues discovered that a low dose of ketamine (0.5 mg/kg IV, a dose that produces mild dissociative and psychotomimetic effects) produces a rapid antidepressant response within 4 hours of treatment, and that this response was sustained for at least 3 days.6 This finding was replicated in a second study, which reported an even more rapid antidepressant effect (within 2 hours) and was sustained for up to 7 days after a single dose of ketamine.7 The rapid and sustained antidepressant actions of ketamine have now been replicated in several independent studies from different groups.44 These studies have included patients both on and off other antidepressant medications. Moreover, most of these studies were conducted in patients who had failed to respond to two or more typical antidepressants, and were therefore considered to be treatment-resistant. In addition, ketamine is reported to rapidly reverse suicide ideation and bipolar depression.45,46 The discovery that ketamine produces a rapid response, that is relatively long-lasting, in treatment-resistant depressed patients represents one of the most significant advances in the field of affective illnesses in over 60 years, since the discovery of the monoaminergic antidepressants.
Despite this promise, ketamine has significant limitations for widespread use for the treatment of depression. Low doses of ketamine produce rapid, mild, psychotomimetic effects and euphoria in normal subjects and depressed patients, leading to the use of ketamine as a pharmacological tool to investigate the role of NMDA hypofunction in schizophrenia.6,7,44 Ketamine is also an abused street drug, referred to as “Special K”, and there are reports that chronic ketamine abusers display disruptions of white matter integrity and cortical atrophy.47,48 In addition, basic research studies have demonstrated that repeated, daily ketamine dosing causes neurotoxic effects.49,50 However, characterization of the mechanisms underlying the actions of ketamine are leading to the development of safer ketamine-like agents with potentially fewer side effects.
Ketamine increases synapse formation in the PFC
The impact of the fast antidepressant actions of ketamine are further highlighted by the discovery that a single dose of ketamine causes a rapid induction of the number and function of spine synapses in medial PFC of naïve animals (Figure 2).51 Ketamine also increases the function of these new synapses shown by an increase in the amplitude and frequency of 5-hydroxytyramine (5-HT)- and hypocretin-induced excitatory postsynaptic currents in layer V pyramidal neurons in the medial PFC. Ketamine increases levels of synaptic proteins after 2 hours, indicating an increase in synapse number, a time that corresponds to the initial therapeutic response in humans.51 Ketamine also produces antidepressant behavioral responses in the forced swim test (decreased immobility or despair) and novelty suppressed feeding (decreased latency to feed in an open field). Moreover, a single dose of ketamine reverses the deficit in the number of synapses caused by exposure to 3 weeks of chronic unpredictable stress (CUS).52 This corresponds with the rapid reversal of the CUS-precipitated behavioral deficit, anhedonia, which is measured by a decrease in preference for sweetened solution. Together, these findings demonstrate a novel rapid action of ketamine, the induction of synaptic connections that reverses the effects of CUS and corresponds to an antidepressant behavioral response that requires chronic administration of a typical antidepressant. Assuming that a similar synaptogenic response occurs in humans, a question of intense interest, ketamine would then reverse the atrophy and synaptic deficits that are a critical pathophysiological component of depression.
Indirect support for this hypothesis is provided by a recent clinical study, using magnetoencephalographic recordings, before and after ketamine infusions, in depressed patients and during somatosensory cortex activation (ie, tactile stimulation of index fingers).53 The results show that cortical excitability is increased in responders during the period of antidepressant response (≈4 hours after ketamine), but not in those patients who did not respond to ketamine. There was no effect of ketamine on baseline γ-band activity, suggesting that the acute NMDA receptor-blocking effects of ketamine do not account for the antidepressant responses. Rather, the results are consistent with the possibility that increased glutamate activity and enhanced synaptic potentiation underlie the increase in cortical excitability, as well as the antidepressant response to ketamine.53 It is possible that NMDA receptor blockade of glutamate transmission contributes to the therapeutic actions of ketamine, although this would be rapid and transient (ie, when the drug is present in the brain and during the time frame for the psychotomimetic effects, 30 to 60 minutes after dosing) and therefore would not correspond to the delayed time of the therapeutic response (80 to 120 minutes).
Role of mammalian target of rapamycin complex 1 signaling in the actions of ketamine
The induction of synapse formation has been studied and documented in cellular models of long-term memory and has been shown to require the synthesis of new synaptic proteins, including glutamate receptors (eg, GluR1) and structural proteins (eg, postsynaptic density protein 95 [PSD95]). These studies describe the signaling pathways that control synaptic plasticity in long-term potentiation (FTP) and long-term depression (LTD), notably the cycling of glutamate receptor subtypes and synaptic structural proteins to and from the membrane (Figure 2).42,43,54
Ketamine also increases levels of these synaptic proteins and reverses the deficits caused by CUS, consistent with the increased number and function of synapses.51,52 The mechanisms underlying the induction of protein synthesis-dependent synapse formation have also been examined in learning and memory studies, and have demonstrated a role for the mammalian target of rapamycin complex 1 (mTORC1), a protein kinase complex that regulates p70S6 kinase (S6K), 4E binding protein 1 (4EBP1), and de novo protein synthesis at the synapse. Protein synthesis by mTORC1 is tightly regulated by neuronal activity as well as metabolic and endocrine factors, and ketamine rapidly increases levels of the phosphorylated and activated form of mTORC1 signaling proteins.51 A role for mTORC1 in the actions of ketamine is supported by studies demonstrating that the behavioral actions of ketamine are blocked by pretreatment with rapamycin, a selective mTORC1 inhibitor.51,55
Role of BDNF release in the actions of ketamine
Another important link in the induction of mTORC1 signaling and synapse formation is BDNF (Figures 2 and 3). The antidepressant response to ketamine is blocked in BDNF deletion mutant mice56 and in mice with a knock-in of the human BDNF Val66Met polymorphism. As the Met allele blocks activity dependent release of BDNF, this finding indicates that BDNF release is required for the actions of ketamine. This possibility is supported by studies demonstrating that infusion of a BDNF antibody into the medial PFC, which neutralizes the BDNF that is released into the extracellular space, also blocks the behavioral effects of ketamine (Duman, unpublished data). The significance of these preclinical findings has also been examined in depressed patients, since the BDNF Met polymorphism is found in approximately 25% of the population. An examination of patients treated with ketamine reveals that those carrying the Met allele have a significantly decreased response to ketamine,57 indicating that the BDNF Val66Met allele is a marker of treatment response and further demonstrating a requirement for BDNF release.
Figure 3. Glutamatergic targets for rapid-acting antidepressants. Basic research studies demonstrate that ketamine causes a rapid and transient burst of glutamate in the prefrontal cortex, in part via disinhibition of γ-aminobutyric acid (GABA)-ergic neurons that exert negative control over glutamatergic firing. Recent basic and clinical studies have demonstrated a number of related glutamatergic, as well as muscarinic, cholinergic targets with the potential to produce rapid-acting antidepressant effects. In addition to ketamine, the nonselective Nmethyl-D-aspartate (NMDA) antagonist AZD6765 and the selective NR2b antagonists CP-101,606 and Ro 25-6981 have shown efficacy in clinical trials and/or rodent models. A highly novel tetrapeptide, GLYX-13, which is a partial agonist/antagonist at the glycine binding site on the NMDA receptor also produces rapid antidepressant responses in rodents and in clinical trials. The metabotropic glutamate receptor 2/3 (mGluR2/3) antagonists LY341495 and MGS0039 have also been shown to increase glutamate and produce rapid, mammalian target of rapamycin (mTOR)-dependent antidepressant effects in rodent models. The nonselective muscarinic receptor antagonist scopolamine, as well as telenzapine, which has modest M1 selectivity, also increase glutamate and produce rapid mTOR-dependent antidepressant effects. It is important to point out that these agents may also act at postsynaptic sites to enhance synapse formation and produce antidepressant responses. Also acting at postsynaptic sites are α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor potentiating agents, although studies are still underway to determine the efficacy of these agents as rapid-acting drugs in rodent models. Inhibition of GSK3 contributes to the actions of ketamine, and the nonselective GSK3 antagonist lithium and selective agent SB216763 enhance the behavioral and synaptic responses to ketamine. Akt, protein kinase b; ERK, extracellular signal-regulated kinases; GABA, γ-aminobutyric acid; GSK, glycogen synthase kinase; PP1, phosphoprotein phosphatase 1 ; TrkB, tropomyosin receptor kinase B.

The requirement for BDNF release and activation of downstream signaling pathways indicates that a BDNF agonist would also be an effective antidepressant approach. Indeed, direct infusion of BDNF into the hippocampus, or even peripheral administration of BDNF, produces antidepressant behavioral responses.27,58 However, the development of small molecular BDNF agonists has been extremely difficult and has met with little success. There have been reports of agents that act via BDNF-tropomyosin receptor kinase B (TrkB) signaling, although the ability of these agents to directly stimulate TrkB receptors is still in question. In addition, BDNF is known to cause depressive behaviors when infused or expressed in the mesolimbic dopamine system,4,59 raising some questions about systemic administration of a direct acting agonist. However, we have found that peripheral administration of recombinant BDNF increases signaling in the brain and produces antidepressant actions in rodent models, indicating that an antidepressant response is the predominant effect of systemic administration.60
Novel NMDA receptor antagonists for the treatment of depression: new concepts for development of glutamatergic agents
The exciting studies of ketamine and the potential for development of an entirely new class of antidepressants with a novel mechanism and rapid, efficacious onset of action have motivated the field to identify additional NMDA receptor agents. Listed below are a few of the most promising agents under development. In addition, studies of ketamine demonstrate a different conceptual framework for pharmacological actions in the treatment of depression: namely a drug with rapid, but transient acute actions on glutamate, which is critical to avoid excitotoxic damage, followed within a few hours by a therapeutic antidepressant response. Importantly, ketamine also produces a relatively long-lasting synaptogenic and antidepressant behavioral response. This differs from current drug development approaches to produce high-affinity agents that engage and occupy the target-binding site for extended time periods. This possibility is supported by anecdotal evidence using low doses of ketamine and bolus vs slow infusions.61
Although the prevailing theory holds that the therapeutic response occurs via blockade of NMDA receptors, it is also known that ketamine acts at other neurotransmitter receptors and ion channels. This includes blockade of dopamine D2 receptors62 and cholinergic nicotinic receptors.63 These findings raise the possibility that the actions of ketamine occur through disruption of multiple neurotransmitter systems. It is also possible that disruption of these other receptors could contribute to the side effects of ketamine. These possibilities will require further investigation, including studies of more selective NMDA receptor antagonists as described below.
N-methyl-D-aspartate receptor subtype b (NR2b) selective antagonists
Ketamine is a nonselective NMDA receptor antagonist; and it is possible that a more selective agent would retain the antidepressant actions, but not have the side effects of ketamine. NMDA receptors are comprised of two major subunits, NR1 and NR2, with 8 different splice variants of NR1 and 4 different genes for NR2 (a-d). Functional NMDA receptors are tetramers and are comprised of two NR1 subunits and two NR2 subunits. Glutamate binds to the NR2 subunit and a coagonist binding site for glycine is also located on the NR2 subunit.
NR2b has been studied as a potential target for blocking glutamate excitoxicity, as this subtype is found at extrasynaptic locations that contribute to excitotoxic damage.64 Previous studies have demonstrated that the NR2b selective antagonist Ro 25-6981 produces antidepressant actions in rodent models of depression, including the forced swim and learned helplessness paradigms. Moreover, we have reported that Ro 25-6981 stimulates mTORC1 signaling and increases synaptic protein synthesis in the PFC, and that the antidepressant behavioral actions of Ro 25-6981 are blocked by rapamycin (Figure 3). A single dose of Ro 25-6981 produces a rapid antidepressant response in the CUS paradigm, causing a rapid reversal of anhedonia as well as a longer latency to feed in novelty suppressed feeding; these effects of Ro 25-6981 are also blocked by rapamycin. Another study also reports that the antidepressant actions of Ro 25-6981 are not associated with hyperactivity, which is thought to correlate with the psychotomimetic effects of other NMDA receptor antagonists.65 Studies are currently being conducted to determine the clinical actions of Ro 25-6981 and related compounds in depressed patients.
In addition to these preclinical studies, there is a clinical report of a rapid response to an NR2b selective antagonist, CP-101,606.66 This double -blind placebo-controlled study reports that CP-101,606 produces a significant antidepressant response in treatment-resistant depressed patients. Moreover, the side effects of CP101,606, particularly the dissociative effects, were relatively minor. Although additional studies are needed to confirm the antidepressant actions of CP-101,606 and other selective agents, these studies indicate that the NR2b receptor subtype may be a viable target for producing ketamine like rapid actions with reduced side effects.
Other NMDA antagonists
There are several other NMDA antagonists that have been examined as potential antidepressants, and a brief overview of these agents is provided. In addition, the channel-blocking properties of these compounds relative to ketamine is discussed, particularly with regard to the psychotomimetic side effects of these agents. These channel blocking agents enter the channel when the neuron is activated and the channel is opened, allowing Ca2+ ions to flow in, carrying the antagonist further into the channel at the same time. Ketamine is trapped inside the channel at a relatively high rate of approximately 82%, and there is evidence that the psychotomimetic action of NMDA receptor blockers is positively correlated with the degree of trapping.
Memantine
Memantine is a nonselective NMDA receptor antagonist that is reported to have antidepressant actions in rodent models64,67; there are no data on the effects of memantine on mTORC1 signaling or synapse formation. Memantine is approved for use in humans (ie, Alzheimer's disease) for which it has modest effects.68 Although there have also been clinical studies of memantine in depressed patients, the results have not been promising. In a double-blind, placebo-controlled study, memantine was found to have no significant antidepressant effect in MDD patients.7 The reasons for the lack of response are unclear, but could be related to the dose of memantine, or the route and time course of administration. For example, memantine was administered orally at an escalating dose over several weeks; it would be interesting to determine if intravenous dosing, similar to that for ketamine, would be more efficacious and rapid, ft is also notable that memantine is a low-affinity open-channel antagonist that is trapped at a lower rate (70%) compared with ketamine.64
AZD6765
Another nonselective NMDA antagonist that has been tested as an antidepressant is AZD6765. This compound is reported to have antidepressant actions in rodent models.69 Moreover, AZD6765 has a reasonable safety profile in humans and does not induce psychotomimetic side effects. This compound was developed as a neuroprotective agent for the treatment of stroke, but lack of efficacy halted further development. However, a recent clinical study demonstrates that AZD6765 produces a rapid antidepressant response in depressed patients.69 fn this study, which was conducted with patients considered treatment-resistant (ie, based on their lack of response to typical antidepressants), approximately one third showed a rapid and significant antidepressant response within 80 minutes of a single treatment. A recent study has reported that repeated dosing of AZD6765 (3 times a week for 3 weeks) produces an antidepressant response after 1 to 2 weeks of treatment.70 The only side effects reported were mild, transient dizziness and headaches. This relatively mild side-effect profile, particularly with regard to psychotomimetic and dissociative effects, could be related to the low proportion of AZD6765 that is trapped in the pore (54%) relative to ketamine (82 %).69 Together, these studies indicate that AZD6765 has a relatively rapid onset of action, with fewer side effects than ketamine.
GLYX-13
GLYX-13 is a tetrapeptide that acts as a partial agonist at the glycine site of the NMDA receptor complex, making this agent unique among the drugs acting at NMDA receptors that are being investigated for antidepressant activity. Originally designed to enhance learning and memory, subsequent studies have demonstrated that GLYX-13 produces rapid antidepressant actions in rodent models and in depressed patients. A single dose of GLYX-13 produces an antidepressant response in the forced swim, learned helplessness, and novelty induced suppression paradigms; these effects are blocked by an AMPA receptor antagonist, similar to the blockade of ketamine.71 GLYX-13 enhances LTP in hippocampal slices, demonstrating synaptic plasticity inducing effects of this agent. Recent studies demonstrate that GLYX-13 also produces a rapid antidepressant response in the CUS/anhedonia model of depression (Moskal J, personal communication). The potential of GLYX-13 is also supported by clinical studies. Representatives from Naurex, the company developing GLYX-13, have reported at a recent American College of Neuropsychopharmacology meeting (2012) that a single intravenous dose of GLYX13 produces a significant antidepressant response within 24 hours of treatment and that the effects last on average for 7 days.
The cellular mechanisms underlying the actions of GLYX-13 are being examined. Although GLYX-13 is a partial agonist of the glycine site, it is also possible that it acts as a partial antagonist depending on the binding of endogenous glycine (ie, at higher levels of endogenous glycine, GLYX-13 could antagonize binding) (Figure 3) . This would be consistent with the possibility that GLYX-13 increases glutamate transmission via blockade of tonic firing γ-aminobutyric acid (GABA) neurons (see below) and that the effects of GLYX-13 require AMPA receptor activation. Studies are currently underway to determine if GLYX-13 increases mTORC1 signaling, similar to the actions of ketamine.
Ketamine stimulates a “glutamate burst”: metabotropic (mGluR) and AMPA receptors as rapid antidepressant targets
The induction of synapse formation by ketamine, an NMDA antagonist, is unexpected as synaptic plasticity in cellular models of learning requires NMDA receptor activation, not inhibition. Studies of glutamate transmission and regulation of GABA neurons have helped clarify this apparent paradox. Microdialysis studies demonstrate that ketamine administration causes a rapid (~ 30 minutes), but transient (~ 90 minutes) elevation of extracellular glutamate in the medial PFC.72 In addition, subsequent studies demonstrate that ketamine blocks the tonic firing of GABAergic interneurons, leading to the hypothesis that the glutamate burst results from disinhibition of glutamate terminals.73 These studies indicate that agents that increase glutamate release or act directly on postsynaptic AMPA receptors may also have rapid-acting antidepressant effects. Several targets that could influence glutamate transmission are discussed.
mGluR2/3 antagonists
The metabotropic glutamate receptors (mGluRs) represent a diverse class that has been targeted for the treatment of depression as well as other psychiatric illnesses. There are eight different mGluR receptor subtypes that are divided into three major groups, with Group II receiving attention for the treatment of depression. Group fl includes mGluR2 and mGluR3 receptors, which are located at presynaptic terminals and serve as autoreceptors, providing inhibitory control of glutamate transmission (Figure 3).74 Both receptors are also located at postsynaptic sites, with mGluR3 found on glia. Importantly, studies have shown that antagonists of mGluR2/3 receptors increase the release of extracellular glutamate72,75 supporting the possibility that these agents could produce ketamine-like rapid antidepressant responses.
Indeed, previous studies have demonstrated that mGluR2/3 antagonists produce antidepressant actions in the standard behavioral paradigms such as the forced swim test.76-78 Moreover, recent studies have extended these findings and demonstrate that mGluR2/3 antagonists, including LY341495 and MGS0039, increase mTORC1 signaling in the medial PFC and show that the behavioral actions of these agents are blocked by administration of the selective mTORC1 inhibitor rapamycin.55,79 LY341495 also increases levels of the synaptic proteins GluRf , PSD95, and synapsin I in the medial PFC, indicating that mGluR2/3 antagonists increase synaptic connections.79 Moreover, we have recently reported that a single dose of LY34f 495 rapidly reverses the anhedonia caused by CUS exposure, providing the first evidence that mGluR2/3 antagonists, like ketamine, have rapid antidepressant actions in a chronic stress model that requires long-term (3 weeks) administration of a typical antidepressant.80
AMPA receptor potentiating agents
The induction of glutamate release indicates that activation of postsynaptic glutamate receptors contributes to the antidepressant actions of ketamine. This is supported by studies demonstrating that pretreatment with an AMPA receptor antagonist blocks the behavioral actions of ketamine, as well as the induction of mTORC1 signaling and synaptic proteins.51,81 These studies suggest that agents that stimulate postsynaptic AMPA receptors could have antidepressant efficacy. While direct-acting AMPA agonists would be expected to have serious excitotoxic side effects due to overactivation of neurons, there has been progress in developing agents, that potentiate AMPA receptor activation (Figure 3). Positive AMPA receptor-modulating agents have been developed largely for use as cognitive enhancing drugs and are reported to have positive actions on cellular and behavioral models of learning and memory, and to increase the expression of BDNF.82,83 In general, AMPA receptor-potentiating drugs influence the kinetics of AMPA receptor channel kinetics by decreasing receptor desensitization or inactivation.84,85 While there are no reports on the in vivo actions of AMPA potentiating drugs on mTORC1 signaling and synaptogenesis, there is an in vitro study demonstrating the effects of one such agent. This study found that the AMPA potentiating agent CX614 increases the release of BDNF, activates mTORC1 signaling, and increases synapse formation in primary neuronal cultures.86
There are several different types of AMPA agents, including those referred to as either high- or low -impact potentiating drugs depending on the ability of these agents to increase the flow of current in AMPA receptors. For example, CX614 and LY451646 are high-impact agents, and CX1739 and Org26575 are low-impact agents. In addition, there is evidence that AMPA receptor potentiating agents have antidepressant actions in behavioral models as well as cellular targets of treatment response.87,88 Other than the in vitro studies of CX614 there is no direct evidence that AMPA receptor potentiating drugs produce ketamine-like effects, such as activation of mTORC1 signaling and increased synaptogenesis. Further studies are required to determine if AMPA receptor potentiating agents influence these pathways and targets in vivo, and produce a rapid antidepressant response in models like CUS.
While these novel targets, including AMPA receptor potentiating drugs and mGluR2/3 antagonists may prove to be rapid and effective antidepressant agents, it remains to be determined if they will also produce unwanted side effects. In addition, it is possible that the actions of these agents are closely tied to synaptic levels of glutamate, since the efficacy of both depends on either blocking presynaptic glutamate actions at mGluR2/3 receptors, or potentiating postsynaptic actions of glutamate at AMPA receptors.
Role of glycogen synthase kinase-3 (GSK-3) in the actions of ketamine: GSK-3 antagonists enhance the response to ketamine
Another mechanism implicated in the actions of ketamine is regulation of GSK-3β. GSK-3β is a serine/threonine kinase that is inhibited by lithium and is thought to play a significant role in the therapeutic actions of lithium in bipolar patients. Levels of GSK-3β are also increased in bipolar depressed patients.89 The function of GSK3 is inhibited by phosphorylation of specific amino acid residues. Interestingly, a recent study has demonstrated that ketamine increases the phosphorylation of these inhibitory sites and that mice with a knockin of GSK-β that is resistant to phosphorylation do not show an antidepressant response to ketamine.90 These studies demonstrate that inhibition of GSK-3β significantly contributes to the actions of ketamine. This may occur in part by blocking the LTD-promoting actions of GSK-3β, which would enhance ketamine induction of the LTP-like synaptogenic effects (Figure 3).
The interaction between ketamine and GSK-β inhibitors is further demonstrated in a recent report. This study found that coadministration of ketamine and a selective GSK-3β inhibitor, SB216763, at low doses that have no effect when used alone, produce a significant antidepressant behavioral effect, as well as induction of mTORC1 signaling and synaptogenesis in the medial PFC.91 Similar effects were found with lithium, raising the possibility that ketamine plus lithium combination therapy could have reduced side effects compared with the higher dose of ketamine that is currently used. Another possibility is that addition of lithium may extend or sustain the therapeutic actions of ketamine beyond the period of approximately 1 week currently observed after a single dose of ketamine. Studies are currently under way to test this possibility.
Scopolamine and muscarinic targets as rapid-acting antidepressants
The acetylcholine or cholinergic hypothesis of depression and antidepressant response has been a topic of discussion for several decades, but only recently has there been strong evidence that cholinergic agents are capable of producing rapid antidepressant actions. Two recent placebo-controlled crossover studies have demonstrated that the cholinergic antagonist scopolamine produces a rapid antidepressant response in depressed patients.8,92 These studies observed antidepressant actions at the first clinical assessment conducted 3 days after a single intravenous low dose (4 μg per kg) of scopolamine, with anecdotal reports of an improvement in mood 24 hours after treatment. Additional doses produced a further improvement in rating scales (32% reduction in depression rating scale after first dose, 53% after second dose), indicating an additive effect. Another study found that the antidepressant actions of scopolamine are greater in women than in men.9 These findings indicate that cholinergic antagonists like scopolamine produce relatively rapid antidepressant actions.
Scopolamine increases mTORC1 signaling and synaptogenesis
Based on these findings we examined the possibility that scopolamine also influences the mTORC1 signaling system and synapse formation. We found that a single low dose of scopolamine (25 μg per kg) significantly increases mTORC1 signaling and increases the number and function of new spine synapses in rat medial PFC.89 A single dose of scopolamine also produces an antidepressant behavioral response in the forced swim test, that is blocked by inhibition of mTORC1 signaling or by blockade of AMPA receptors. In addition, a role for enhanced glutamate transmission is supported by preliminary microdialysis studies, demonstrating that scopolamine increases extracellular glutamate levels in the medial PFC.89 Together these studies indicate that increased glutamate transmission, mTORC1 signaling, and increased synaptogenesis are common targets and functional responses to different classes of rapid-acting antidepressant agents.
Given these findings, it is interesting to discuss the mechanisms involved in the actions of other treatments that have efficacy as rapid-acting antidepressants. There is anecdotal evidence that electroconvulsive therapy (ECT) produces a rapid antidepressant response, although the typical course of treatment is 3 sessions per week for 2 weeks. ECT causes depolarization throughout the central nervous system and thereby causes a burst of glutamate transmission. However, preliminary studies have failed to demonstrate an effect of a single electro-convulsive seizure on mTORC1 signaling in rodent PFC.51 Another treatment possibility is sleep deprivation, which is reported to influence mTORC1 signaling in the mouse hippocampus, but mTORC1 was decreased, not increased.93 Further studies will be necessary to identify the molecular mechanisms underlying the antidepressant actions of these treatments, which could lead to additional targets for the treatment of depression.
Cellular mechanisms and muscarinic receptor subtypes underlying the actions of scopolamine: novel drug targets
The pre- and postsynaptic targets underlying the actions of scopolamine remain to be determined. Scopolamine is a nonselective muscarinic (M) receptor antagonist that blocks all 5 receptor subtypes with high affinity. These receptors are located at pre- and postsynaptic sites for cholinergic, as well as glutamatergic synapses. Postsynaptic M1 receptors are reported to regulate LTD via enhanced internalization of AMPA and/or NMDA receptors94; blockade of these receptors by scopolamine would inhibit this process and could thereby enhance synaptic plasticity and possibly increase synaptogenesis. The ability of scopolamine to increase extracellular glutamate raises the possibility that muscarinic receptor subtypes are expressed on GABAergic interneurons and that, like NMDA receptors, are capable of regulating GABA firing. This possibility is supported by studies demonstrating that Ml receptor agonist incubation increases GABA overflow in slices of striatum,95 suggesting that scopolamine could act by blocking muscarinic activation of GABA firing and disinhibit glutamate release (Figure 3). We have found that telenzapine, an antagonist with limited selectivity for Ml receptors, also increases mTORC1 signaling and produces antidepressant responses in the forced swim test.89 Preliminary studies with a more selective Ml antagonist, VU0255035, support these findings, although we cannot rule out the possibility that other muscarinic receptor subtypes are involved in the actions of scopolamine.
Conclusions and future directions
The discovery of the rapid antidepressant actions of ketamine and scopolamine, and the role of glutamate transmission, represent major breakthroughs for the development of novel, rapid acting, and efficacious therapeutic agents. This comes at a time when the pharmaceutical industry is pulling back efforts to develop new medications for major psychiatric illnesses because of the challenges associated with predicting which new drug targets will prove successful, the repeated failures in clinical trials, and the high placebo response rates. The promise of these new antidepressant targets will hopefully reinvigorate central nervous system drug discovery and development.
In addition to the glutamatergic and muscarinic receptor targets discussed above, there are additional possibilities to investigate with regard to synaptogenesis. Of particular interest are the estrogen receptors (ER) α and β. Early studies have demonstrated that estrogens can regulate dendrite length and synapse formation, induce intracellular signaling pathways that regulate synaptogenesis, and produce antidepressant actions in behavioral models of depression.96,97 Disruption of normal cycling of ovarian hormones has been implicated in the higher rates of depression in women relative to men, and indicates that abnormal estrogen levels and/or signaling could be involved in the pathophysiology of depression. Novel and selective estrogen ligands could prove useful for reversing the atrophy of neurons caused by stress, as well as the behavioral symptoms of depression.
Another area that has received attention is the role of elevated inflammatory responses in depression. This hypothesis is supported by studies demonstrating that serum levels of proinflammatory cytokines, most notably IL-1β, IL6, and TNFα, are increased in depressed patients.98,99 In addition, cytokines can produce sickness behavior, including fatigue and decreased appetite that could account for some symptoms of depression.100 Elevated inflammatory cytokines also contribute to other illnesses, including cardiovascular disease, diabetes, and obesity, that have high rates of comorbidity with depression.101 Together these studies demonstrate a role of inflammatory cytokines in the pathophysiology of depression, and identify novel therapeutic targets. This includes the use of antagonists and agents that block the production of cytokines. For example, there are now reports demonstrating that peripheral administration of an antibody (Inflixamab), or TNF α receptor-fusion protein (Etanercept) that neutralizes TNFα, produces antidepressant responses.102,103 Preclinical studies also show that blocking or neutralizing IL-1β produces antidepressant actions in cellular and behavioral models.101,104 There is also evidence that blockade of purinergic 2X7 (P2X7) receptor, which activates the inflammasome to stimulate the processing and release of IL-iβ, produces antidepressant effects.101 ft is not clear at this point if induction of inflammatory cytokines contributes to neuronal atrophy, but there is evidence that activated microglia participate in dendrite pruning in support of this possibility.105
The targets and concepts discussed represent the major areas related to the pathophysiology of depression that are currently being investigated for drug development. Taken together, these breakthroughs represent significant potential for a new era of novel therapeutic target development for the treatment of depression.
Acknowledgments
The research was supported by grants from the NIMH MH089983, MH96891, and the Dowshen Program for Neuroscience.
REFERENCES
- 1.Kessler R., Chiu WT., Dernier O., Merikangas KR., Walters EE. Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62:617–627. doi: 10.1001/archpsyc.62.6.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Trivedi M., Rush AJ., Wisniewski SR., et aI STAR*D Study Team. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry. 2006;163:28–40. doi: 10.1176/appi.ajp.163.1.28. [DOI] [PubMed] [Google Scholar]
- 3.Duman R., Monteggia LM. A neurotrophic model for stress-related mood disorders. Biol Psychiatry. 2006;59:1116–1127. doi: 10.1016/j.biopsych.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 4.Krishnan V., Nestler EJ. The molecular neurobiology of depression. Nature. 2008;455:894–902. doi: 10.1038/nature07455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Savitz J., Drevets WC . Bipolar and major depressive disorder: neuroimaging the developmental-degenerative divide. Neurosci Biobehav Rev. 2009;33:699–771. doi: 10.1016/j.neubiorev.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berman R., Cappiello A., Anand A., et al Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47:351–354. doi: 10.1016/s0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- 7.Zarate CA Jr., Singh JB., Quiroz JA., et al A double-blind, placebo-controlled study of memantine in the treatment of major depression. Am J Psychiatry. 2006;163:153–155. doi: 10.1176/appi.ajp.163.1.153. [DOI] [PubMed] [Google Scholar]
- 8.Furey M., Drevets WC. Antidepressant efficacy of the antimuscarinic drug scopolamine: a randomized placebo-controlled clinical trial. Arch Gen Psychiatry. 2006;63:1121–1129. doi: 10.1001/archpsyc.63.10.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Furey M., Khanna A., Hoffman EM., Drevets WC. Scopolamine produces larger antidepressant and antianxiety effects in women than in men. Neuropsychopharmacology. 2010;35:2479–2488. doi: 10.1038/npp.2010.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mayberg H. Targeted electrode-based modulation of neural circuits for depression. J Clin Invest. 2009;119:717–725. doi: 10.1172/JCI38454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Drevets W., Price JL., Furey ML. Brain structural and functional abnormalities in mood disorders: implications for neurocircuitry models of depression. Brain Struct Fund. 2008;213:93–118. doi: 10.1007/s00429-008-0189-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.MacQueen G., Frodl T. The hippocampus in major depression: evidence for the convergence of the bench and bedside in psychiatric research? Mol Psychiatry. 2011;16:252–264. doi: 10.1038/mp.2010.80. [DOI] [PubMed] [Google Scholar]
- 13.Banasr M., Valentine GW., Li XY., Gourley S., Taylor J., Duman RS. Chronic stress decreases cell proliferation in adult cerebral cortex of rat: Reversal by antidepressant treatment. Biol Psychiatry. 2007;62:496–504. doi: 10.1016/j.biopsych.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 14.Rajkowska G., Miguel-Hidalgo JJ., Wei J., et al Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression. Biol Psychiatry. 1999;45:1085–1098. doi: 10.1016/s0006-3223(99)00041-4. [DOI] [PubMed] [Google Scholar]
- 15.Rajkowska G., Miguel-Hidalgo JJ. Gliogenesis and glial pathology in depression. CMS Neurol Disord Drug Targets. 2007;6:219–233. doi: 10.2174/187152707780619326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kang H., Voleti B., Hajszan T., et al Decreased expression of synapse-related genes and loss of synapses in major depressive disorder. Nat Med. 2012;18:1413–1417. doi: 10.1038/nm.2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McEwen B. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev. 2007;87:873–904. doi: 10.1152/physrev.00041.2006. [DOI] [PubMed] [Google Scholar]
- 18.Shansky R., Morrison JH. Stress-induced dendritic remodeling in the medial prefrontal cortex: effects of circuit, hormones and rest. Brain Res. 2009;1293:108–113. doi: 10.1016/j.brainres.2009.03.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu RJ., Aghajanian GK. Stress blunts serotonin- and hypocretin-evoked EPSCs in prefrontal cortex: role of corticosterone-mediated apical dendritic atrophy. Proc Natl Acad Sci USA. 2008;105:359–364. doi: 10.1073/pnas.0706679105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McEwen B. The ever-changing brain: cellular and molecular mechanisms for the effects of stressful experiences. Dev Neurobiol. 2012;72:878–890. doi: 10.1002/dneu.20968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roozendaal B., McEwen BS., Chattarji S. Stress, memory and the amygdala. Nat Rev Neurosci. 2009;10:423–433. doi: 10.1038/nrn2651. [DOI] [PubMed] [Google Scholar]
- 22.Sahay A., Hen R. Hippocampal neurogenesis and depression. Novartis Found Symp. 2008;289:152–160. doi: 10.1002/9780470751251.ch12. [DOI] [PubMed] [Google Scholar]
- 23.Boldrini M., Underwood MD., Hen R., et al Antidepressants increase neural progenitor cells in the human hippocampus. Neuropsychopharmacology. 2009;34:2376–2389. doi: 10.1038/npp.2009.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Banasr M., Dwyer JM., Duman RS. Cell atrophy and loss in depression: reversal by antidepressant treatment. Curr Opin Cell Biol. 2012;23:730–737. doi: 10.1016/j.ceb.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rajkowska G., O'Dwyer G., Teleki Z., Stockmeier CA., Miguel-Hidalgo JJ. GABAergic neurons immunoreactive for calcium binding proteins are reduced in the prefrontal cortex in major depression. Neuropsychopharmacology. 2007;32:471–482. doi: 10.1038/sj.npp.1301234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duman R., Voleti B. Signaling pathways underlying the pathophysiology and treatment of depression: novel mechanisms for rapid-acting agents. Trends Neurosci. 2012;35:47–56. doi: 10.1016/j.tins.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmidt H., Shelton RC., Duman RS. Functional markers of depression: diagnosis, treatment and pathophysiology. Neuropsychopharmacology. 2011;36:2375–2394. doi: 10.1038/npp.2011.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu H., Wang DD., Wang Y., Liu T., Lee FS., Chen ZY. Variant brain-derived neurotrophic factor Val66Met polymorphism alters vulnerability to stress and response to antidepressants. J Neurosci. 2012;32:4092–4101. doi: 10.1523/JNEUROSCI.5048-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krishnan V., Han MH., Mazei-Robison M., et al AKT signaling within the ventral tegmental area regulates cellular and behavioral responses to stressful stimuli. Biol Psychiatry. 2008;64:691–700. doi: 10.1016/j.biopsych.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taliaz D., Stall N., Dar DE., Zangen A. Knockdown of brain-derived neurotrophic factor in specific brain sites precipitates behaviors associated with depression and reduces neurogenesis. Mol Psychiatry. 2010;15:80–92. doi: 10.1038/mp.2009.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen Z-Y., Jing D., Bath KG., et al Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science. 2006;314:140–143. doi: 10.1126/science.1129663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Magarihos A., Li CJ., Gal Toth J., et al Effect of brain-derived neurotrophic factor haploinsufficiency on stress-induced remodeling of hippocampal neurons. Hippocampus. 2011;21:253–264. doi: 10.1002/hipo.20744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen H., Pandey GN., Dwivedi Y. Hippocampal cell proliferation regulation by repeated stress and antidepressants. Neuroreport. 2006;17:863–867. doi: 10.1097/01.wnr.0000221827.03222.70. [DOI] [PubMed] [Google Scholar]
- 34.Liu R., Lee FS., Li XY., Bambico F., Duman RS., Aghajanian GK. Brain-derived neurotrophic factor val66met allele impairs basal and ketamine-stimulated synaptogenesis in prefrontal cortex. Biol Psychiatry. 2012;71:996–1005. doi: 10.1016/j.biopsych.2011.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chiaruttini C., Vicario A., Li Z., et al Dendritic trafficking of BDNF mRNA is mediated by translin and blocked by the G196A (Val66Met) mutation. Proc Natl Acad Sci U S A. 2009;106:16481–16486. doi: 10.1073/pnas.0902833106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gatt J., Nemeroff CB., Dobson-Stone C., et al Interactions between BDNF Val66Met polymorphism and early life stress predict brain and arousal pathways to syndromal depression and anxiety. Mol Psychiatry. 2009;14:681–695. doi: 10.1038/mp.2008.143. [DOI] [PubMed] [Google Scholar]
- 37.Sheline Y., Gado MH., Kraemer HC. Untreated depression and hippocampal volume loss. Am J Psychiatry. 2003;160:1516–1518. doi: 10.1176/appi.ajp.160.8.1516. [DOI] [PubMed] [Google Scholar]
- 38.Bessa J., Ferreira D., Melo I., et al Hippocampal neurogenesis induced by antidepressant drugs: an epiphenomenon in their mood-improving actions. Mol Psychiatry. 2009;14:764–773. doi: 10.1038/mp.2008.119. [DOI] [PubMed] [Google Scholar]
- 39.Magarinos A., McEwen BS. Stress-induced atrophy of apical dendrites of hippocampal CA3c neurons: involvement of glucocorticoid secretion and excitatory amino acid receptors. Neurosci. 1995;69:89–98. doi: 10.1016/0306-4522(95)00259-l. [DOI] [PubMed] [Google Scholar]
- 40.Karpova N., Pickenhagen A., Lindholm J., et al Fear erasure in mice requires synergy between antidepressant drugs and extinction training. Science. 2011;334:1731–1734. doi: 10.1126/science.1214592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maya Vetencourt J., Sale A., Viegi A., et al The antidepressant fluoxetine restores plasticity in the adult visual cortex. Science. 2008;320:385–388. doi: 10.1126/science.1150516. [DOI] [PubMed] [Google Scholar]
- 42.Holtmaat A., Svoboda K. Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci. 2009;10:647–658. doi: 10.1038/nrn2699. [DOI] [PubMed] [Google Scholar]
- 43.Kessels H., Malinow R. Synaptic AMPA receptor plasticity and behavior. Neuron. 2009;61:340–350. doi: 10.1016/j.neuron.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aan Het Rot M., Zarate CA Jr., Charney DS., Mathew SJ. Ketamine for depression: where do we go from here? Biol Psychiatry. 2012;72:537–547. doi: 10.1016/j.biopsych.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Diazgranados N., Ibrahim L., Brutsche NE., et al A randomized add-on trial of an N-methyl-D-aspartate antagonist in treatment-resistant bipolar depression. Arch Gen Psychiatry. 2010;67:793–802. doi: 10.1001/archgenpsychiatry.2010.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Larkin G., Beautrais AL. A preliminary naturalistic study of low-dose ketamine for depression and suicide ideation in the emergency department. Int J Neuropsychopharmacol. 2011;14:1127–1131. doi: 10.1017/S1461145711000629. [DOI] [PubMed] [Google Scholar]
- 47.Roberts E., Curran HV., Friston KJ., Morgan CJ. Abnormalities in white matter microstructure associated with chronic ketamine use. Neuropsychopharmacology. 2014;39:329–338. doi: 10.1038/npp.2013.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang C., Zheng D., Xu J., Lam W., Yew DT. Brain damages in ketamine addicts as revealed by magnetic resonance imaging. Front Neuroanat. 2013;7:23. doi: 10.3389/fnana.2013.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Behrens M., Ali SS., Dao DN., et al Ketamine-induced loss of phenotype of fast-spiking interneurons is mediated by NADPH-oxidase. Science. 2007;318:1645–1647. doi: 10.1126/science.1148045. [DOI] [PubMed] [Google Scholar]
- 50.Behrens M., Sejnowski TJ. Does schizophrenia arise from oxidative dysregulation of parvalbumin-interneurons in the developing cortex? Neuropharmacology. 2009;57:193–200. doi: 10.1016/j.neuropharm.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li N., Lee BY., Liu RJ., et al mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329:959–964. doi: 10.1126/science.1190287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li N., Liu R-J., Dwyer J., et al Glutamate N-methyl-D-aspartate receptor antagonists rapidly reverse behavioral and synaptic deficits caused by chronic stress exposure. Biol Psychiatry. 2011;69:754–761. doi: 10.1016/j.biopsych.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cornwell B., Salvadore G., Furey M., et al Synaptic potentiation is critical for rapid antidepressant response to ketamine in treatment-resistant major depression. Biol Psychiatry. 2012;72:55–61. doi: 10.1016/j.biopsych.2012.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yoshihara Y., De Roo M., Muller D. Dendritic spine formation and stabilization. Curr Opin Neurobiol. 2009;19:146–153. doi: 10.1016/j.conb.2009.05.013. [DOI] [PubMed] [Google Scholar]
- 55.Koike H., Lijima M., Chaki S. Involvement of the mammalian target of rapamycin signaling in the antidepressant-like effect of group II metabotropic glutamate receptor antagonists. Neuropharmacology. 2011;61:1419–1423. doi: 10.1016/j.neuropharm.2011.08.034. [DOI] [PubMed] [Google Scholar]
- 56.Autry A., Adachi M., Nosyreva E., et al NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature. 2011;475:91–95. doi: 10.1038/nature10130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Laje G., Lally N., Mathews D., et al Brain-derived neurotrophic factor Val66Met polymorphism and antidepressant efficacy of ketamine in depressed patients. Biol Psychiatry. 2012;72:27–28. doi: 10.1016/j.biopsych.2012.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shirayama Y., Ishida H., Iwata M., Hazama G-i., Kawahara R., Duman R. Stress increases dynorphin immunoreactivity in limbic brain regions and dynorphin blockade produces antidepressant effects. J Neurochem. 2004;90:1258–1268. doi: 10.1111/j.1471-4159.2004.02589.x. [DOI] [PubMed] [Google Scholar]
- 59.Russo S., Nestler EJ. The brain reward circuitry in mood disorders. Nat Rec Neurosci. 2013;14:609–625. doi: 10.1038/nrn3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schmidt H., Duman RS. Peripheral BDNF produces antidepressant-like effects in cellular and behavioral models. Neuropsychopharmacology. 2010;35:2378–2391. doi: 10.1038/npp.2010.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Blier P. Exploiting N-methyl-d-aspartate channel blockade for a rapid antidepressant response in major depressive disorder. Biol Psychiatry. 2013;74:238–239. doi: 10.1016/j.biopsych.2013.05.029. [DOI] [PubMed] [Google Scholar]
- 62.Kapur S., Seeman P. NMDA receptor antagonists ketamine and PCP have direct effects on the dopamine D(2) and serotonin 5-HT(2)receptors-implications for models of schizophrenia. Mol Psychiatry. 2002;7:837–844. doi: 10.1038/sj.mp.4001093. [DOI] [PubMed] [Google Scholar]
- 63.Coates K., Flood P. Ketamine and its preservative, benzethonium chloride, both inhibit human recombinant alpha7 and alpha4beta2 neuronal nicotinic acetylcholine receptors in Xenopus oocytes. Br J Pharmacol. 2001;134:871–879. doi: 10.1038/sj.bjp.0704315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sanacora G., Zarate CA., Krystal JH., Manji HK. Targeting the glutamatergic system to develop novel, improved therapeutics for mood disorders. Nat Rev Drug Discov. 2008;7:426–437. doi: 10.1038/nrd2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lima-Ojeda J., Vogt MA., Pfeiffer N., et al Pharmacological blockade of GluN2B-containing NMDA receptors induces antidepressant-like effects lacking psychotomimetic action and neurotoxicity in the perinatal and adult rodent brain. Prog Neuropsychopharmacol Biol Psychiatry. 2013;45:28–33. doi: 10.1016/j.pnpbp.2013.04.017. [DOI] [PubMed] [Google Scholar]
- 66.Preskorn S., Baker B., Omo K., Kolluri S., Menniti FS., Landen JW. A placebo-controlled trial of the NR2B subunit specific NMDA antagonist CP101,606 plus paroxetine for treatment resistant depression (TRD). Paper presented at the American Psychological Association meeting; 2007. [Google Scholar]
- 67.Zarate CA Jr., Du J., Quiroz J., et al Regulation of cellular plasticity cascades in the pathophysiology and treatment of mood disorders: role of the glutmatergic system. Ann N Y Acad Sci. 2003;1003:273–291. doi: 10.1196/annals.1300.017. [DOI] [PubMed] [Google Scholar]
- 68.Reisberg B., Doody R., Stoffler A., Schmitt F., Ferris S., Mobius HJ. Memantine Study Group. Memantine in moderate-to-severe Alzheimer's disease. N Engl J Med. 2003;348:1333–1341. doi: 10.1056/NEJMoa013128. [DOI] [PubMed] [Google Scholar]
- 69.Zarate CJ., Mathews D., Ibrahim L., et al A randomized trial of a lowtrapping nonselective N-methyl-d-aspartate channel blocker in major depression. Biol Psychiatry. 2013;74:257–264. doi: 10.1016/j.biopsych.2012.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sanacora G., Smith MA., Pathak S., et al Lanicemine: a low-trapping NMDA channel blocker produces sustained antidepressant efficacy with minimal psychotomimetic adverse effects. Mol Psychiatry. 2013. Epub ahead of print. doi: 10.1038/mp.2013.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Burgdorf J., Zhang XL., Nicholson KL., et al GLYX-13, a NMDA receptor glycine-site functional partial agonist, induces antidepressant-like effects without ketamine-like side effects. Neuropsychopharmacology. 2012;38:729–742. doi: 10.1038/npp.2012.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Moghaddam B., Adams B., Verma A., Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci. 1997;17:2912–2917. doi: 10.1523/JNEUROSCI.17-08-02921.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Homayoun H., Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci. 2007;27:11496–11500. doi: 10.1523/JNEUROSCI.2213-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shigemoto R., Kinoshita A., Wada E., et al Differential presynaptic localization of metabotropic glutamate receptor subtypes in the rat hippocampus. J Neurosci. 1997;17:7503–7522. doi: 10.1523/JNEUROSCI.17-19-07503.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hascup E., Hascup KN., Stephens M., et al Rapid microelectrode measurements and the origin and regulation of extracellular glutamate in rat prefrontal cortex. J Neurochem. 2010;115:1608–1620. doi: 10.1111/j.1471-4159.2010.07066.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bespalov A., van Gaalen MM., Sukhotina IA., et al Behavioral characterization of the mGlu group ll/lll receptor antagonist, LY-341495, in animal models of anxiety and depression. Eur J Pharmacol. 2008;592:96–102. doi: 10.1016/j.ejphar.2008.06.089. [DOI] [PubMed] [Google Scholar]
- 77.Chaki S., Yoshikawa R., Hirota S., et al MGS0039: a potent and selective group II metabotropic glutamate receptor antagonist with antidepressant-like activity. Neuropharmacology. 2004;46:457–467. doi: 10.1016/j.neuropharm.2003.10.009. [DOI] [PubMed] [Google Scholar]
- 78.Marek G. Metabotropic glutamate2/3 (mGlu2/3) receptors, schizophrenia and cognition. Eur J Pharmacol. 2010;639:81–90. doi: 10.1016/j.ejphar.2010.02.058. [DOI] [PubMed] [Google Scholar]
- 79.Dwyer JM., Lepack AE., Duman RS. mTOR activation is required for the antidepressant effects of mGluR2/3 blockade. Int J Neuropsychopharmacol. 2012;15:429–434. doi: 10.1017/S1461145711001702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dwyer J., Lepack AE., Duman RS. mGluR2/3 blockade produces rapid and long-lasting reversal of anhedonia caused by chronic stress exposure. J Mol Psych. In press. doi: 10.1186/2049-9256-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Maeng S., Zarate CA Jr., Du J., et al Cellular mechanisms underlying the antidepressant effects of ketamine: role of alpha-amino-3-hydroxy-5methylisoxazole-4-propionic acid receptors. Biol Psychiatry. 2008;63:349–352. doi: 10.1016/j.biopsych.2007.05.028. [DOI] [PubMed] [Google Scholar]
- 82.Arai A., Kessler M., Rogers G., Lynch G. Effects of the potent ampakine CX614 on hippocampal and recombinant AMPA receptors: interactions with cyclothiazide and GYKI 52466. Mol Pharmacol. 2000;58:802–813. doi: 10.1124/mol.58.4.802. [DOI] [PubMed] [Google Scholar]
- 83.Lauterborn J., Lynch G., Vanderklish P., Arai A., Gall CM. Positive modulation of AMPA receptors increases neurotrophin expression by hippocampal and cortical neurons. J Neurosci. 2000;20:8–21. doi: 10.1523/JNEUROSCI.20-01-00008.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Arai A., Kessler M. Pharmacology of ampakine modulators: from AMPA receptors to synapses and behavior. Curr Drug Targets. 2007;8:583–602. doi: 10.2174/138945007780618490. [DOI] [PubMed] [Google Scholar]
- 85.Lynch A., Walsh C., Delaney A., Nolan Y., Campbell VA., Lynch MA. Lipopolysaccharide-induced increase in signalling in hippocampus is abrogated by IL-10— a role for IL-1b? J Neurochem. 2004;88:635–646. doi: 10.1046/j.1471-4159.2003.02157.x. [DOI] [PubMed] [Google Scholar]
- 86.Jourdi H., Hsu YT., Zhou M., Qin Q., Bi X., Baudry M. Positive AMPA receptor modulation rapidly stimulates BDNF release and increases dendritic mRNA translation. J Neurosci. 2009;29:8688–8697. doi: 10.1523/JNEUROSCI.6078-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bai F., Li X., Clay M., Lindsrom T., Skolnick P. Intra- and interstrain differences in models of “behavior despair”. Pharmacol Biochem Behav. 2001;70:187–192. doi: 10.1016/s0091-3057(01)00599-8. [DOI] [PubMed] [Google Scholar]
- 88.Bai F., Bergeron M., Nelson DL. Chronic AMPA receptor potentiator (LY451646) treatment increases cell proliferation in adult rat hippocampus. Neuropharmacology. 2003;44:1013–1021. doi: 10.1016/s0028-3908(03)00104-7. [DOI] [PubMed] [Google Scholar]
- 89.Voleti B., Navarria A., Liu RJ., et al Scopolamine rapidly increases mTORC1 signaling, synaptogenesis, and antidepressant behavioral responses. Biol Psychiatry. 2013;74:742–749. doi: 10.1016/j.biopsych.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Beurel E., Song L., Jope RS. Inhibition of glycogen synthase kinase-3 is necessary for the rapid antidepressant effect of ketamine in mice. Mol Psychiatry. 2011;16:1068–1070. doi: 10.1038/mp.2011.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liu RJ., Fuchikami M., Dwyer JM., Lepack AE., Duman RS., Aghajanian GK. GSK-3 inhibition potentiates the synaptogenic and antidepressant-like effects of subthreshold doses of ketamine. Neuropsychopharmacology. 2013;38:2268–2277. doi: 10.1038/npp.2013.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Drevets W., Furey ML. Replication of scopolamine's antidepressant efficacy in major depressive disorder: a randomized, placebo-controlled clinical trial. Biol Psychiatry. 2010;67:432–438. doi: 10.1016/j.biopsych.2009.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Vecsey C., Peixoto L., Choi JH., et al Genomic analysis of sleep deprivation reveals translational regulation in the hippocampus. Physiol Genomics. 2012;44:981–991. doi: 10.1152/physiolgenomics.00084.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Caruana D., Warburton EC., Bashir Zl. Induction of activity-dependent LTD requires muscarinic receptor activation in medial prefrontal cortex. J Neurosci. 2011;31:18464–18478. doi: 10.1523/JNEUROSCI.4719-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Harsing L., Zigmond MJ. Postsynaptic integration of cholinergic and dopaminergic signals on medium-sized GABAergic projection neurons in the neostriatum. Brain Res Bull. 1998;45:607–613. doi: 10.1016/s0361-9230(97)00460-7. [DOI] [PubMed] [Google Scholar]
- 96.Hughes Z., Liu F., Marquis K., et al Estrogen receptor neurobiology and its potential for translation into broad spectrum therapeutics for CNS disorders. Curr Mol Pharmacol. 2009;2:215–236. doi: 10.2174/1874467210902030215. [DOI] [PubMed] [Google Scholar]
- 97.Woolley C., McEwen BS. Estradiol regulates hippocampal dendritic spine density via an N-methyl-D-aspartate receptor-dependent mechanism. J Neurosci. 1994;14:7680–7687. doi: 10.1523/JNEUROSCI.14-12-07680.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Dowlati Y., Herrmann N., Swardfager W., et al A meta-analysis of cytokines in major depression. Biol Psychiatry . 2010;67:446–457. doi: 10.1016/j.biopsych.2009.09.033. [DOI] [PubMed] [Google Scholar]
- 99.Howren M., Lamkin DM., Suls J. Associations of depression with C-reactive protein, IL-1, and IL-6: a meta-analysis. Psychosom Med. 2009;71:171–186. doi: 10.1097/PSY.0b013e3181907c1b. [DOI] [PubMed] [Google Scholar]
- 100.Schiepers O., Wichers MC., Maes M. Cytokines and major depression. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29:201–217. doi: 10.1016/j.pnpbp.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 101.lwata M., Ota KT., Duman RS. The inflammasome: pathways linking psychological stress, depression, and systemic illnesses. Brain Behav irnmun. 2013;31:105–114. doi: 10.1016/j.bbi.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Krishnan V., Han M-H., Graham DL., et al Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell. 2007;131:391–404. doi: 10.1016/j.cell.2007.09.018. [DOI] [PubMed] [Google Scholar]
- 103.Raison C., Rutherford RE., Woolwine BJ., et al A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: the role of baseline inflammatory biomarkers .JAMA Psychiatry. 2013;70:31–41. doi: 10.1001/2013.jamapsychiatry.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Koo J., Duman RS. IL-1B is an essential mediator of the anti-neurogenic and anhedonic effects of stress. Proc Natl Acad Sci USA. 2008;105:751–756. doi: 10.1073/pnas.0708092105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Paolicelli R., Bolasco G., Pagani F., et al Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333:1456–1458. doi: 10.1126/science.1202529. [DOI] [PubMed] [Google Scholar]